

Abstract
Background
Brassica napus is of substantial economic value for vegetable oil, biofuel, and animal fodder production. The breeding of yellow-seeded B. napus to improve seed quality with higher oil content, improved oil and meal quality with fewer antinutrients merits attention. Screening the genes related to this phenotype is valuable for future rapeseed breeding.
Results
A total of 85,407 genes, including 4317 novel genes, were identified in the developing seeds of yellow- and black-seeded B. napus, and yellow rapeseed was shown to be an introgression line between black-seeded B. napus and yellow-seeded Sinapis alba. A total of 15,251 differentially expressed genes (DEGs) were identified among all the libraries, and 563 and 397 common DEGs were identified throughout black and yellow seed development, including 80 upregulated and 151 downregulated genes related to seed development and fatty acid accumulation. In addition, 11 up-DEGs and 31 down-DEGs were identified in all developmental stages of yellow rapeseed compared with black seed. Enrichment analysis revealed that many DEGs were involved in biosynthetic processes, pigment metabolism, and oxidation-reduction processes, such as flavonoid and phenylpropanoid biosynthesis, phenylalanine metabolism, flavone and flavonol biosynthesis, and fatty acid biosynthesis and metabolism. We found that more than 77 DEGs were related to flavonoid and lignin biosynthesis, including 4CL, C4H, and PAL, which participated in phenylalanine metabolism, and BAN, CHI/TT5, DFR, F3H, FLS, LDOX, PAP, CHS/TT4, TT5, bHLH/TT8, WD40, MYB, TCP, and CYP, which were involved in flavonoid biosynthesis. Most of these DEGs were downregulated in yellow rapeseed and were consistent with the decreased flavonoid and lignin contents. Both up- and down-DEGs related to fatty acid biosynthesis and metabolism were also analyzed, which could help to explain the improved oil content of yellow rapeseed.
Conclusion
This research provided comprehensive transcriptome data for yellow-seeded B. napus with a unique genetic background, and all the DEGs in comparison with the black-seeded counterpart could help to explain seed quality differences, such as lower pigmentation and lignin contents, and higher oil content.
Electronic supplementary material
The online version of this article (10.1186/s12870-019-1821-z) contains supplementary material, which is available to authorized users.
Keywords: Brassica napus, Seed coat color, Flavonoid biosynthesis, Fatty acid, Gene expression
Background
Brassica napus, as the third leading oilseed crop grown worldwide, greatly contributes to providing edible oils, biofuels, and animal fodder [1]. Rapeseed with increased oil content, better oil and meal quality, and improved yield has been the main breeding drive during the past decades [2]. Yellow-seeded B. napus was improved with better seed oil and meal quality due to fewer pigments and polyphenols; thus, breeding yellow rapeseed has been preferred for decades, and studies on the molecular mechanism of this phenotype have been reported as well [3]. Fewer antinutrients, including phenolic compounds, tannins, proanthocyanins (PAs), and lignin, in yellow rapeseed were correlated with the flavonoid biosynthetic, phenylpropanoid, and phenylalanine/tyrosine metabolic pathways with common substrates, such as coumaroyl CoA, caffeoyl CoA, and feruloyl CoA [4–6]. Hitherto, yellow-seeded B. napus has been selected from interspecific hybridization of Brassica species and intergeneric hybridization [7, 8]. As reported in Arabidopsis thaliana, mutations in transparent testa (TT) genes are responsible for seed coat color variations, including early (EBGs) and late (LBGs) biosynthesis genes [9]. The EBGs include TT4/CHS, TT5/CHI, TT6/F3H, and TT7/F3’H, and LBGs include TT3/DFR, TT18/LDOX/ANS, BAN/ANR, TT12, TT19/GSTF12/GST26, and AHA10. In addition, the MYB-bHLH-WD40 (MBW) complex has been reported to have important regulatory functions in anthocyanin accumulation. Four MBW have been reported, including TT2-TT8-TTG1, MYB5-TT8-TTG1, TT2-EGL3-TTG1, and TT2-GL3-TTG1 [10]. Appelhagen et al. (2011) found that PAP1/MYB75 and PAP2/MYB90 participated in the formation of MBW [11]. Homologous TTs related to seed coat variation in Brassicaceae have been delineated, including 95 copies of 21 TTs in B. napus [3]. Previously, F3’H, TT2, PAL, BAN, TTG1, TT10, and TT1 were cloned and shown to have functions in flavonoid biosynthesis [12–19]. Yu (2013) reviewed the molecular mechanism of manupulating seed coat color in Brassica species, including the homologous TTs cloned in Brassicas [20]. Besides, molecular markers have also been developed for yellow-seeded B. rapa [21–23], B. juncea [24, 25] and B. napus. Liu et al. (2012) reported a lignin biosynthesis gene, BnCCR1, associated with yellow seed character, indicating a strong correlation between seed color and lignin content [26]. Stein et al. (2013) found that BnAHA10 had effects on seed color and lignin content [27]. Wang et al. (2017) identified 22 single nucleotide polymorphisms (SNPs) on 7 chromosomes associated with seed coat color using a genome wide association study (GWAS) [28]. Functional analysis of BnTT10 and BnTT1 revealed that they are involved in PA metabolism, lignin synthesis, seed coat pigmentation, and fatty acid (FA) biosynthesis [18, 19]. However, due to the genome complexity of B. napus and the sensitivity of yellow seed color to environmental influences (e.g., light, temperature, fertilizers, and harvesting time), the molecular mechanisms of this phenotype remained unclear until now [29, 30].
The short domestication history of B. napus after interspecific hybridization between B. rapa and B. oleracea and the artificial selection during rapeseed breeding have greatly narrowed the genetic background [31]. No yellow-seed germplasm has been found in natural B. napus, and most of the yellow-seeded B. napus were created by crossing between Brassica species, such as hybrids between B. napus and B. juncea, B. napus and B. carinata, B. juncea and B. oleracea [32, 33]. Wild species in Brassicaceae, such as Sinapis alba, S. arvensis, Camelina sativa, Crambe abyssinica, and Descurainia sophia, exhibit many desirable characteristics, such as yellow seed coat, high erucic acid, pod shattering resistance, high unsaturated FA contents, and resistance to various diseases and abiotic stresses [34–36]. Introducing gene resources from these wild species would help to enrich the genetic background of B. napus, accompanying desirable traits. Zhang et al. (2009) obtained yellow-seeded rapeseed from intergeneric hybrids between B. napus and D. sophia [37]. Previously, Wang et al. (2005) created somatic hybrids between B. napus and S. alba [38] and selected yellow-seeded B. napus from hybrid progenies with improved oil content and decreased antinutrients [8, 39, 40]. In the present study, we used RNA-Seq to identify the expression differences at different seed developmental stages of yellow- and black-seeded B. napus, revealing the expression patterns of genes involved in various biological pathways related to seed coat pigmentation, FA biosynthesis and metabolism.
Results
Transcriptome sequencing and read mapping of yellow- and black-seeded B. napus
RNA-Seq was performed at three weeks after flowering (3 WAF), 4 WAF, 5 WAF, 6 WAF, and mature stage of yellow- and black-seeded B. napus to investigate the transcriptome difference that might be related to the quality variation between two rapeseeds. After quality control, ~ 30.3 to ~ 37.6 million reads from the libraries were uniquely mapped to the B. napus genome (Table 1). A total of 85,407 genes, including 4317 novel genes, were expressed in the developing seeds of two rapeseeds (Fig. 1, Additional file 1: Table S1). We found 52,452 and 51,777 common genes were expressed in the developing seeds of yellow and black rapeseed, respectively. In addition, 67,629 (3 WAF), 64,070 (4 WAF), 64,775 (5 WAF), 62,143 (6 WAF), and 54,257 (mature stage) overlapping genes were expressed at each stage in two rapeseed lines. Besides, 47,912 genes were coexpressed in all the developmental stages of yellow and black rapeseed lines.
Table 1.
Summary of read mapping for RNA-seq
| Sample name | B3 | B4 | B5 | B6 | BM | Y3 | Y4 | Y5 | Y6 | YM |
|---|---|---|---|---|---|---|---|---|---|---|
| Raw reads | 47,536,866 | 42,753,820 | 44,341,556 | 51,507,518 | 51,272,946 | 48,704,282 | 45,694,038 | 44,797,562 | 45,996,584 | 52,804,244 |
| Clean reads | 45,555,018 | 41,801,216 | 40,620,518 | 43,833,034 | 48,665,348 | 46,813,976 | 43,798,322 | 42,843,336 | 43,736,106 | 50,268,482 |
| Total mapped | 36,499,205 (80.12%) | 34,214,293 (81.85%) | 32,844,328 (80.86%) | 35,032,820 (79.92%) | 39,325,179 (80.81%) | 38,116,991 (81.42%) | 35,893,586 (81.95%) | 35,376,347 (82.57%) | 36,029,106 (82.38%) | 40,839,634 (81.24%) |
| Multiple mapped | 2,175,877 (4.78%) | 2,332,136 (5.58%) | 2,561,972 (6.31%) | 3,271,514 (7.46%) | 3,151,365 (6.48%) | 2,327,452 (4.97%) | 2,287,911 (5.22%) | 2,274,889 (5.31%) | 2,451,932 (5.61%) | 3,198,280 (6.36%) |
| Uniquely mapped | 34,323,328 (75.34%) | 31,882,157 (76.27%) | 30,282,356 (74.55%) | 31,761,306 (72.46%) | 36,173,814 (74.33%) | 35,789,539 (76.45%) | 33,605,675 (76.73%) | 33,101,458 (77.26%) | 33,577,174 (76.77%) | 37,641,354 (74.88%) |
| Reads map to ‘+’ | 17,137,064 (37.62%) | 15,873,247 (37.97%) | 15,138,033 (37.27%) | 15,900,632 (36.28%) | 18,123,297 (37.24%) | 17,872,927 (38.18%) | 16,769,787 (38.29%) | 16,508,902 (38.53%) | 16,764,771 (38.33%) | 18,866,098 (37.53%) |
| Reads map to ‘-’ | 17,186,264 (37.73%) | 16,008,910 (38.3%) | 15,144,323 (37.28%) | 15,860,674 (36.18%) | 18,050,517 (37.09%) | 17,916,612 (38.27%) | 16,835,888 (38.44%) | 16,592,556 (38.73%) | 16,812,403 (38.44%) | 18,775,256 (37.35%) |
| Non-splice reads | 21,266,684 (46.68%) | 21,368,313 (51.12%) | 21,381,177 (52.64%) | 22,323,651 (50.93%) | 24,534,251 (50.41%) | 22,491,873 (48.05%) | 22,291,894 (50.9%) | 24,114,006 (56.28%) | 24,559,779 (56.15%) | 26,061,989 (51.85%) |
| Splice reads | 13,056,644 (28.66%) | 10,513,844 (25.15%) | 8,901,179 (21.91%) | 9,437,655 (21.53%) | 11,639,563 (23.92%) | 13,297,666 (28.41%) | 11,313,781 (25.83%) | 8,987,452 (20.98%) | 9,017,395 (20.62%) | 11,579,365 (23.04%) |
B3, B4, B5, B6 and BM indicate 3 ~ 6 WAF and mature black seed. Y3, Y4, Y5, Y6 and YM indicate 3 ~ 6 WAF and mature yellow seed
Fig. 1.

Genes commonly and specifically expressed in developing seeds of yellow- and black-seeded B. napus. a-e Venn diagram of genes expressed between each developmental stage of yellow and black seed. f Venn diagram of genes in black seed. g Venn diagram of genes in yellow seed. B3, B4, B5, B6 and BM indicate 3 ~ 6 WAF and mature black seed. Y3, Y4, Y5, Y6 and YM indicate 3 ~ 6 WAF and mature yellow seed
Identification of differentially expressed genes
Global changes in differentially expressed genes were identified using DESeq, and a total of 15,251 DEGs (including 523 novel genes) were identified among all the libraries. We found 563 common DEGs in B4 (4 WAF of black seed), B5 (5 WAF of black seed), B6 (6 WAF of black seed), and BM (mature stage of black seed) compared with B3 (3 WAF of black seed), including 141 upregulated and 331 downregulated genes at each developmental stage of the black seed. A total of 397 common DEGs were identified in Y4 (4 WAF of yellow seed), Y5 (5 WAF of yellow seed), Y6 (6 WAF of yellow seed), and YM (mature stage of yellow seed) compared with Y3 (3 WAF of yellow seed), including 92 upregulated and 220 downregulated genes throughout yellow seed development (Fig. 2a, b; Additional file 2: Table S2). Generally, 80 upregulated and 151 downregulated genes were identified during the seed development of two B. napus lines, which might be related to seed development and FA accumulation (Additional file 3: Table S3). Those DEGs related to cruciferin, oleosin, caleosin, late embryogenesis abundant (LEA) hydroxyproline, lipid transfer protein, myrosinase-binding protein, embryo-specific protein, and alpha-tonoplast intrinsic protein were upregulated, and those related to galactosyltransferase, glycosyl hydrolase, lipid transfer protein, basic leucine zipper protein, cytochrome P450 (CYP), laccase, chalcone synthase, delta vacuolar processing enzyme, jasmonic acid carboxyl methyltransferase, and senescence were downregulated with seed development. In addition, 42 DEGs were identified between yellow and black rapeseeds at each stage, including 11 upregulated and 31 downregulated genes in yellow-seeded B. napus (Fig. 2c, Additional file 4: Table S4). These common DEGs were mainly related to the RNA-binding family protein, nascent polypeptide-associated complex (NAC), calcium binding EF-hand family protein, and metallothionein protein that were upregulated and dormancy/auxin associated protein, S-adenosylmethionine synthetase, pectin lyase-like superfamily protein, insulinase family protein that were downregulated in all stages of yellow seed compared with black seed. The number of DEGs increased with seed development in both rapeseed lines, whereas the downregulated DEGs at the mature stage dramatically increased to ~ 8000 (Fig. 2d, e). The up-regulated and downregulated DEGs between Y4 and B4 were higher than those in the other stages of the two rapeseed lines (Fig. 2f). Hierarchical cluster analysis of all the DEGs was performed using the log10(RPKM+ 1) value (Fig. 2g). H-cluster and SOM-cluster of the DEGs from five developmental stages of two rapeseed lines were also performed using the log2(Fold change), and the DEGs in all the clusters showed similar patterns between two rapeseed lines, except for subcluster_1_1, which includes DEGs with significant differences between Y4 and B4 (Fig. 2h, Additional file 5: Figure S1).
Fig. 2.

Expressional analysis of DEGs in developing seeds of yellow- and black-seeded B. napus. (a) DEGs with black seed development. (b) DEGs with yellow seed development. (c) DEGs between yellow and black rapeseed. (d-f) Number of up- and down-DEGs. (g-h) Clustering of DEGs
Enrichment analysis of DEGs
To acknowledge the putative functions and pathways of the DEGs, GO and KEGG enrichment analysis of DEGs was proceeded with GOseq and KOBAS 2.0. Among all the identified DEGs, 11,772 were annotated with GO terms and assigned to three categories: cellular component, biological process, and molecular function. Most of the up- and downregulated DEGs between the same stage of yellow and black seeds were assigned to catalytic activity, binding, metabolic process, cellular process, followed by cell, cell part, membrane, organelle part, macromolecular complex, regulation of biological process, and biological regulation (Fig. 3, Additional file 6: Table S5). In detail, among all the downregulated genes (1435 out of 1945 with GO annotations) between yellow and black rapeseed, 425, 39, 211, 89, and 65 DEGs were assigned to biosynthetic process (GO: 0009058), pigment metabolic process (GO: 0042440), oxidation-reduction process (GO: 0055114), cellular response to stimulus (GO: 0051716), and signal transduction (GO: 0007165), respectively. In contrast, 181 upregulated genes between yellow and black seed were assigned to biosynthetic process (GO: 0009058), 95 up-DEGs related to oxidation-reduction process (GO: 0055114), and 181 up-DEGs related to biosynthetic process (GO: 0009058).
Fig. 3.

GO enrichment of up- and down-DEGs between yellow- and black-seeded B. napus
KEGG pathway analysis showed that flavonoid biosynthesis was most significantly enriched with downregulated DEGs in comparisons of Y3 vs B3, Y4 vs B4, Y5 vs B5, and Y6 vs B6. Phenylpropanoid biosynthesis was also enriched with downregulated genes between the same stages of the two rapeseed lines, except for the mature stage. Phenylalanine metabolism, flavone and flavonol biosynthesis was enriched with downregulated DEGs in comparisons of Y3 vs B3 and Y4 vs B4 (Additional file 7: Figure S2). In contrast, the upregulated DEGs between yellow and black rapeseed were most significantly enriched in photosynthesis, followed by carbon metabolism, FA biosynthesis and metabolism (Additional file 8: Figure S3, Additional file 9: Table S6). The pathway enrichment analysis helps to determine the functions of DEGs and complemented secondary metabolisms that were not specified by GO terms. Using the log2(Fold change) values between yellow and black rapeseed (Y3 vs. B3, Y4 vs. B4, Y5 vs. B5, Y6 vs. B6, and YM vs. BM), we assigned all the DEGs to all the metabolism pathways using the Arabidopsis TAIR9 version as mapping reference. We also found many DEGs were enriched in metabolism (flavonoids, terpenes, phenylpropanoids and phenolics) and lipids (Additional file 10: Figure S4).
DEGs associated with flavonoid and lignin biosynthesis
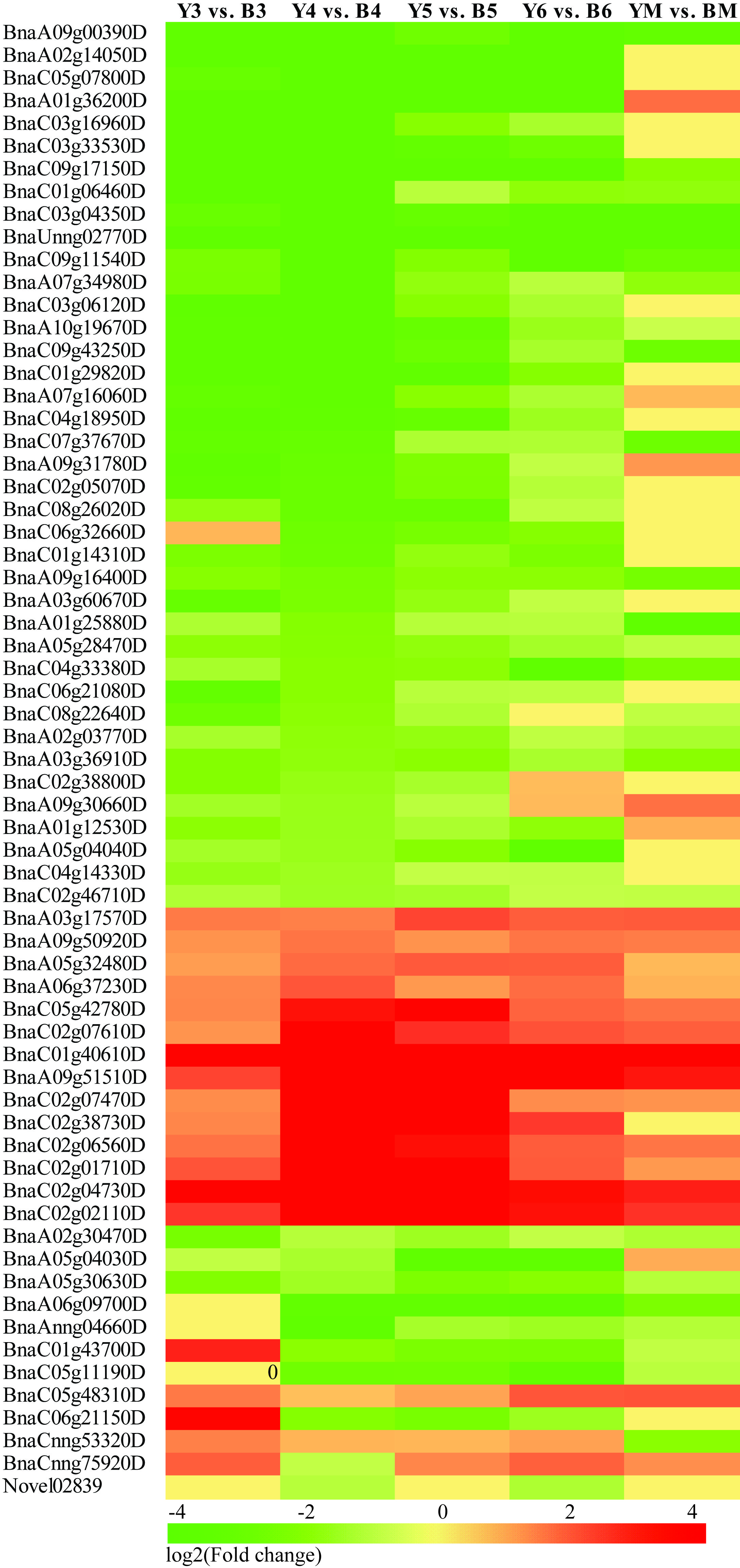
As mentioned above, yellow seed color is associated with flavonoid and lignin biosynthesis. We found 77 DEGs related to phenylpropanoid and flavonoid biosynthesis and the phenylalanine metabolic pathway. As shown in Fig. 4, genes encoding 4-coumarate CoA ligase (4CL), cinnamate 4-hydroxylase (C4H), and phenylalanine ammonia-lyase (PAL) were downregulated in yellow rapeseed compared with black seed, except for a homolog of PAL (BnaC05g42780D). These genes play important roles in lignin biosynthesis, and the expression changes should be related to the reduced lignin in yellow seed. Genes involved in the flavonoid pathway were also downregulated in yellow seed compared to black seed, including BAN, CHI/TT5, DFR, F3H, FLS, LDOX, PAP, CHS/TT4, TT5, bHLH/TT8, WD40, and MYB, which participate in the biosynthesis of chalcones, flavanones, flavonols, anthocyanins, and PAs. However, we found a MATE (BnaC01g40630D) and UGT84A1 (BnaA01g18540D) were upregulated at the early developmental stages of yellow seed compared with black seed. Comparing with other two down-regulated MATEs in yellow-seeded B. napus, BnaA01g24940D (a homologous of AT3G03620) and BnaA07g18120D (a homologous of AtTT12), we found BnaC01g40630D with lower expression throughout rapeseed development than that of BnaA01g24940D and BnaA07g18120D. Expressional differences of TCPs and CYPs were also identified, which might be related to flavonoid biosynthesis, since TCP could interact with R2R3-MYB [41], and CYPs (e.g., CYP71/75/93/81) have been reported to function in the biosynthesis of anthocyanin pigments and condensed tannins [42]. In addition, we compared the expression pattern of all the genes involved in secondary metabolism and found DEGs encoding peroxidase superfamily protein (BnaA02g02010D, BnaC06g32660D, BnaA02g14050D, and BnaC06g21080D), glycosyl hydrolase family protein (BnaUnng02770D), beta glucosidase 19/29/25 (BnaA06g09700D, BnaA05g04040D, BnaC05g11190D, BnaC01g43700D, and BnaC01g40610D), thioglucoside glucohydrolase 1 (BnaCnng53320D) were also assigned to phenylpropanoid biosynthesis and the phenylalanine metabolic pathway (Additional file 11: Figure S5). Generally, most of the genes involved in secondary metabolism were downregulated in yellow seed compared with black seed.
Fig. 4.
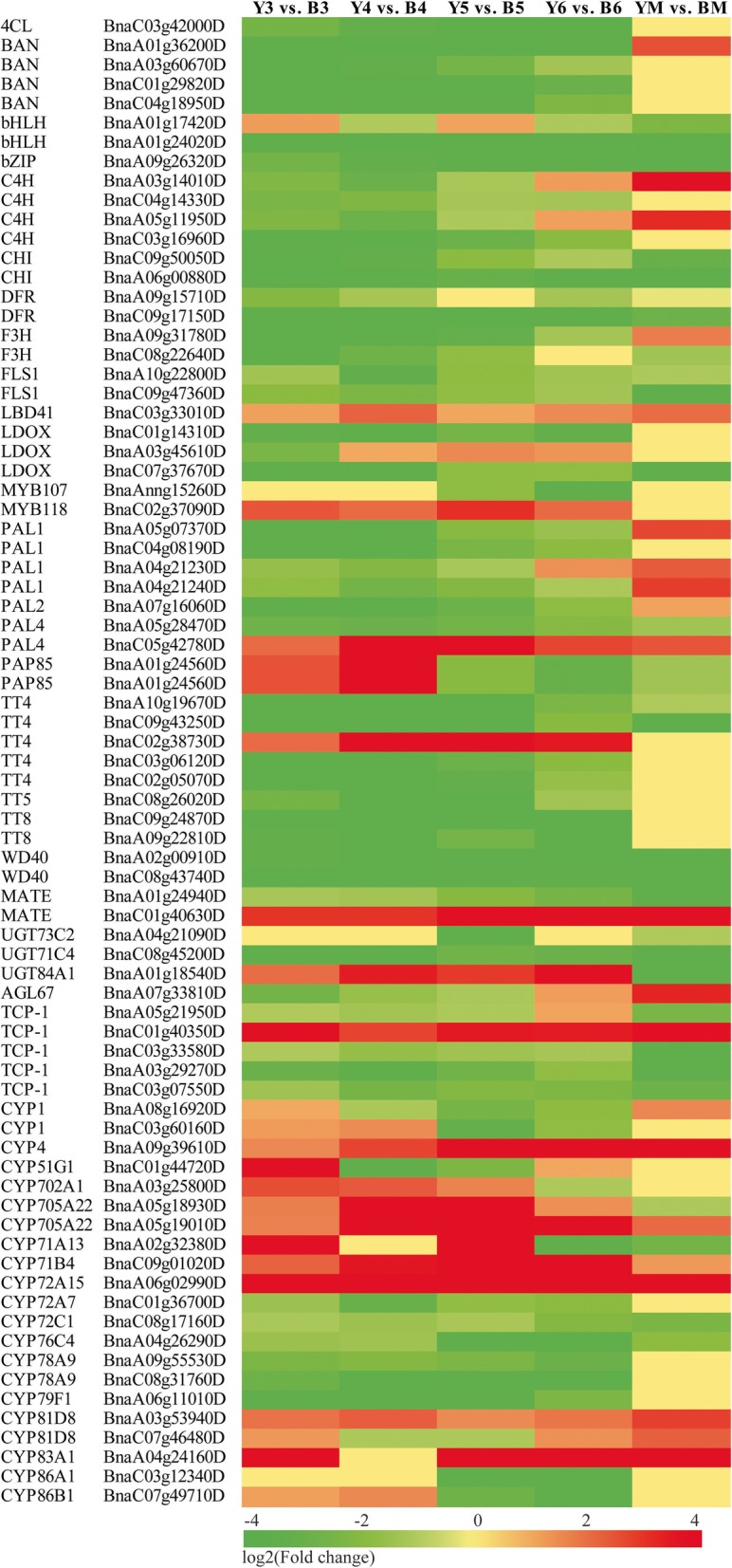
Heatmap of DEGs related to flavonoid and lignin biosynthesis
DEGs associated with fatty acid biosynthesis and metabolism
We found that genes involved in FA biosynthesis, metabolism, and degradation were also changed (Fig. 5). For instance, five up-DEGs in yellow seed were involved in FA biosynthesis, encoding an NAD(P)-binding Rossmann-fold superfamily protein (BnaC01g40290D), a thioesterase superfamily protein (BnaA03g02830D), acetyl Coenzyme A carboxylase carboxyltransferase (BnaA03g17570D), stearoyl-acyl-carrier-protein desaturase (BnaA03g28050D), and chloroplastic acetylcoenzyme A carboxylase 1 (BnaC02g06560D). However, another homolog of the thioesterase superfamily protein (BnaC03g04180D) was downregulated in yellow seed compared with black seed. Five up-DEGs (BnaA03g02830D, BnaA03g17570D, BnaA03g28050D, BnaC01g40290D, and BnaC02g06560D) and six down-DEGs (BnaA02g30470D, BnaA05g30630D, BnaA08g28600D, BnaC02g38800D, BnaC03g04180D, and BnaC03g04350D) were involved in FA metabolism. BnaC03g04350D was also related to FA elongation. In addition, four down-DEGs (BnaC06g21150D, BnaA02g30470D, BnaA05g30630D, and BnaC02g38800D) were related to FA degradation. These DEGs might be related to the different oil content between yellow- and black-seeded B. napus.
Fig. 5.
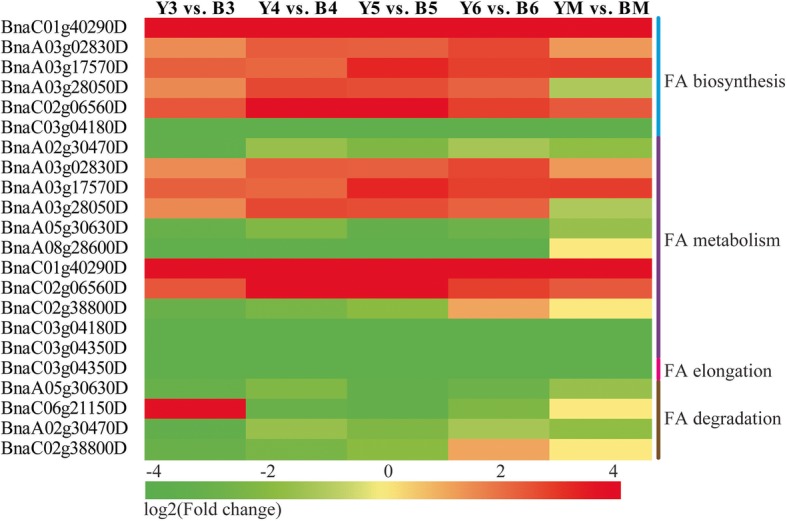
Heatmap of DEGs related to fatty acid biosynthesis and metabolism
Real-time qPCR validates gene expression profiles
To verify the sequencing results, DEGs involved in flavonoid and lignin biosynthesis, FA biosynthesis and metabolism were randomly selected for qPCR detection, including BnaC01g40290D encoding NAD(P)-binding Rossmann-fold superfamily protein/short-chain dehydrogenase/reductase (SDR), BnaA03g17570D encoding acetyl CoA carboxylase carboxyltransferase alpha subunit (CAC3), BnaC03g04350D encoding 3-hydroxyacyl-CoA dehydratase PASTICCINO 2 (PAS2), 4CL (BnaC03g42000D), C4H (BnaC04g14330D, BnaA05g11950D), LBD41 (BnaC03g33010D), PAL (BnaC04g08190D), BAN (BnaA01g36200D), CHI (BnaC09g50050D), LDOX (BnaC01g14310D) and CHS (BnaA10g19670D). The results showed that the qPCR data of the DEGs were in accordance with the sequencing results throughout seed development of yellow- and black-seeded B. napus (Fig. 6).
Fig. 6.

qPCR confirmation of DEGs between yellow- and black-seeded B. napus
Discussion
Due to the important economic value of rapeseed worldwide, research on improving seed quality has been carried out over the past half century, including reducing the erucic acid and glucosinolate contents and increasing the oleic acid content [43, 44]. However, breeding of rapeseed has been greatly hindered by the narrow genetic background either due to the short history or the artificial selection [31]. Previously, Wang et al. created yellow-seeded B. napus from the backcrossing progenies of B. napus-S. alba hybrids, including S. alba specific fragments in the 38 chromosomes [8, 34, 45]. RNA-Seq is a reliable method in analyzing the gene expressional level on the whole transcriptome level, and it also helps to reveal the expressional differences among different samples, which could somehow explain the different characteristics among samples. In the past decade, RNA-Seq has been broadly applied in expressional comparison among plant samples, and in finding alternative splicing, novel transcripts, SNP and InDels [46–48]. To identify the gene expression differences related to yellow seed character and accompanying quality variations, we carried out comparative transcriptome analysis between different developmental stages of yellow- and black-seeded B. napus. As reported by Qu et al. (2013), accumulation of polymeric phenolic compounds were similar in yellow and black rapeseeds, but the flavonoids were more accumulated in black seed since 3 WAF [49]. Thus, we collected the seeds at 3 WAF, 4 WAF, 5 WAF, 6 WAF and mature stage for RNA-Seq analysis. We found a total of 39,632 SNPs in ten libraries, including 1142 SNPs common in five developmental stages of black seed and 1543 SNPs common in five developmental stages of yellow seed, indicating that genomic differences exist between yellow- and black-seeded B. napus. We found a total of 15,251 DEGs among all the libraries, including 80 upregulated and 151 downregulated genes identified throughout yellow and black seed development, which might be related to seed development and FA accumulation (Additional file 2: Table S2). Regarding the DEGs between yellow and black rapeseed, we found that most were annotated to biosynthetic, pigment metabolic, and oxidation-reduction processes, such as flavonoid and phenylpropanoid biosynthesis, phenylalanine metabolism, flavone and flavonol biosynthesis, and FA biosynthesis and metabolism (Additional file 8: Figure S3, Additional file 9: Table S6). This agreed with Hong et al. who found that the downregulated DEGs in yellow seed coats were enriched in phenylpropanoid and flavonoid biosynthesis using yellow- and brown-seeded near-isogenic lines (NILs) of rapeseed as research materials [50]. Similar expression changes in TT genes have been reported in B. juncea seed coat [51].
Flavonoid biosynthesis has been considered to be the main pathway related to plant pigmentation, and TT genes related to it have been clarified in A. thaliana. Recently, homologs of TTs in Brassica have been comprehensively identified by Qu et al. [3], as well as the expression patterns in different yellow- and black-seeded B. napus inbred lines. In the present study, we compared the expression pattern of genes related to flavonoid biosynthesis in developing seeds of B. napus cv. ‘Yangyou 6’ and yellow-seeded B. napus selected from somatic hybrids of B. napus-S. alba (Fig. 4). Four homologs of BnBAN (BnaA01g36200D, BnaA03g60670D, BnaC01g29820D and BnaC04g18950D) were downregulated at two to four consecutive developmental stages of yellow rapeseed compared with black rapeseed. Two homologs of BnDFR (BnaA09g15710D and BnaC09g17150D) were also expressed at lower levels in yellow seed, which encodes an enzyme catalyzing dihydroflavanones to leucoanthocyanidins that was then converted to anthocyanidin [52]. BnLDOX (BnaC01g14310D, BnaA03g45610D and BnaC07g37670D) was downregulated in yellow seed, which encodes leucoanthocyanidin reductase and catalyzes the formation of anthocyanidins. Genes encoding chalcone synthase (BnaA10g19670D, BnaC09g43250D, BnaC03g06120D, BnaC02g05070D and BnaC08g26020D) were also downregulated throughout yellow seed development, which catalyzes the first step of flavonoid biosynthesis. BnTT5 (BnaC08g26020D) was also downregulated in yellow seed compared with black seed at specific stages, encoding chalcone isomerase that is redundant and responsive to lights in Brassicaceae [53]. BnTT8 (BnaC09g24870D and BnaA09g22810D) was also downregulated in yellow seed compared with black seed at specific stages, encoding the transcription factor bHLH as an important regulator throughout flavonoid biosynthesis. Another two bHLHs (BnaA01g17420D and BnaA01g24020D) were also downregulated, which have been proven to have functions in regulating jasmonate responses [54] and glucose homeostasis, and may affect ABA or salinity response in Arabidopsis [55]. In the present study, 21 CYPs were differentially expressed between yellow and black rapeseeds, although they were not shown to have functions in the secondary metabolism of B. napus. Previously, CYP71/75/93/81 have been reported to have functions in the biosynthesis of anthocyanin pigments and condensed tannins [42]. Lam et al. found that CYP75B4 and CYP93G1 promote tricin accumulation in Arabidopsis and O. sativa, which functions in generating 3′-hydroxylated flavonoids and flavone aglycones [56, 57]. Recently, Lei et al. found that a group of CYPs, including members of CYP71/72/77/78/81/85/86/90/93, were differentially expressed in Dendrobium catenatum from different locations and with different outlooks, which might be related to flavonoid biosynthesis [58]. CYP86 has been reported to be involved in suberin monomer biosynthesis in Arabidopsis [59]. Expressional differences of five TCPs were also identified, which might be related to flavonoid biosynthesis since TCP3 could interact with R2R3-MYB [41]. In addition, TCP1 directly activates DWARF4 and promotes brassinosteroid biosynthesis [60].
Asides from flavonoids, lignin has been revealed to have a positive correlation with seed coat color [26]. We found that 4CL, C4H and PAL were downregulated in yellow rapeseed compared with black seed, which play important roles in the less lignin in yellow seed (Fig. 4). PAL is involved in the first step of the phenylalanine metabolic pathway by catalyzing phenylalanine into cinnamic acid. Cinnamic acid was then transformed into coumaric acid by C4H. 4CL plays an important role in several steps of lignin biosynthesis and participates in the formation of coumaroyl-CoA, caffeoyl-CoA and feruloyl-CoA. Previously, Jiang et al. (2013) and Wang et al. (2018) reported that sinapic acid was more accumulated in yellow seed than black seed [39, 61]. Down-regulation of 4CL in yellow rapeseed indicated that caffeic acid and ferulic acid might be less transformed into caffeoyl-CoA and feruloyl-CoA. Thus, sinapis acid as another downstream chemical of caffeic acid and ferulic acid was more accumulated (Fig. 7). All these compounds are necessary intermediates of lignin biosynthesis [5]. Hong et al. found that the expression of genes involved in lignin biosynthesis were slightly but not markedly changed in yellow-seeded NILs [50]. As to the other DEGs related to secondary metabolism, we found genes encoding S-adenosylmethionine synthetase 2 (BnaA09g00390D), peroxidase (BnaA02g14050D, BnaC06g32660D, BnaC06g21080D), branched-chain amino acid transaminase 1 (BnaA01g36200D), methionine synthase 2 (BnaC03g33530D), aspartate aminotransferase 5 (BnaC01g06460D), phosphoglycerate kinase (BnaA07g34980D, BnaC02g46710D), lactate/malate dehydrogenase (BnaA09g16400D), glucose-1-phosphate adenylyltransferase family protein (BnaC04g33380D), beta glucosidase 19/28 (BnaA05g04030D, BnaC01g43700D, BnaC05g11190D, BnaA06g09700D), and pyruvate kinase (BnaA03g36910D) were downregulated in yellow rapeseed (Additional file 11: Figure S5). Peroxidase is one of three major enzymes involved in flavonoid oxidation through the associated reduction of hydrogen peroxide in the peroxidative cycle [62]. Hong et al. assigned peroxidase superfamily protein (e.g., peroxidase 4/7/25/52/53) to phenylpropanoid and flavonoid pathways, which were differentially expressed between yellow and black rapeseed NILs [50]. The DEGs related to flavonoid and lignin biosynthesis confirmed that they contribute to the different testa color.
Fig. 7.
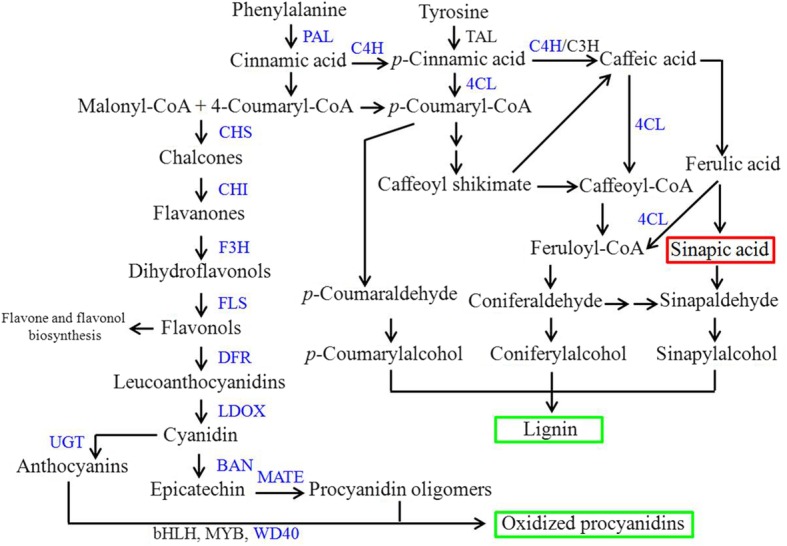
Proposed model for DEGs related to yellow seed quality. Blue font, downregulated genes in yellow seed compared with black seed. Red frame, increased chemical in yellow seed. Green frame, decreased chemical in yellow seed
Since the oil content of yellow seed was higher than that of black seed, we found 12 DEGs related to FA biosynthesis, metabolism, elongation and degradation (Fig. 5). In addition, we found that BnaA09g51510D (encoding pyruvate dehydrogenase E1 alpha) was upregulated throughout yellow seed development compared with black seed (Additional file 11: Figure S5). Its homolog in A. thaliana is important for seed oil biosynthesis. Plastidic pyruvate kinase (PKP) provides most of the pyruvate for plastidic FA synthesis, and the mutation of PKP severely impairs seed storage lipid synthesis [63]. BnaA03g17570D (acetyl Coenzyme A carboxylase carboxyltransferase alpha subunit) was upregulated in yellow seed. Its homolog in A. thaliana has been confirmed to be a subunit of heteromeric acetyl-CoA carboxylase (ACCase), which catalyzes the carboxyltransferase (CT) reaction. ACCase is responsible for the first step of FA synthesis [64]. Additionally, homolog of UDP-glucose pyrophosphorylase (UGP) (BnaA05g32480D), a sucrose-regulated protein, was upregulated in yellow seed, which is required for fumonisin B1-induced cell death [65]. A homolog of beta glucosidase 25 (BnaC01g40610D) was also upregulated in yellow seed, which is involved in the carbohydrate metabolic process [66]. Upregulation of cytochrome B5 reductase 1 (CB5R1) (BnaC02g02110D) in yellow seed might also be related to the FA variation, since CB5R is a microsomal membrane-bound protein that functions as part of the microsomal electron transfer system in FA desaturation [67]. CB5R can interact with ankyrin repeat-containing protein 2A (AKR2A), which interacts with ascorbate peroxidase 3 (APX3). APX3 can target peroxisomes [68]. This might be related to the above-mentioned expressional changes in genes encoding peroxidases. The expression changes of these genes may be helpful in explaining the FA variation in yellow seed and its adaptability to environments after flavonoid reduction.
Conclusions
In the present study, whole transcriptome gene expression was analyzed in developing seeds of yellow-seeded B. napus derived from hybrids of B. napus-S. alba, and black-seeded B. napus. We identified the DEGs with seed development, which might be related to the development and biosynthetic process. In addition, DEGs related to the quality difference between yellow and black rapeseed have been identified, which mainly participate in flavonoid biosynthesis, phenylpropanoid biosynthesis, phenylalanine metabolism, flavone and flavonol biosynthesis, fatty acid biosynthesis and metabolism. These down-regulated genes are helpful to explain the less pigmentation (e.g. CHS, CHI, F3H, FLS, DFR, LDOC, BAN) and lignin (PAL, C4H and 4CL), and higher oil content in yellow rapeseed compared with black seed (Fig. 7). Future functional analysis of these genes would contribute to the molecular dissection of yellow seed character in B. napus.
Methods
Plant material
Yellow-seeded B. napus (line W82) was preserved in our lab and was selected from back-crossing progenies of somatic hybrids of B. napus-S. alba. The black-seeded rapeseed (B. napus cv. ‘Yangyou 6’) was obtained from the Jiangsu Lixiahe Region Agricultural Research Institute, China [45]. Both rapeseed lines were grown in the experimental field of Yangzhou University, Yangzhou, China. In addition to the visible difference in seed coat color, the flavonoid content in yellow seed was lower than that in black seed, and the seed FA composition and content were different between yellow- and black-seeded B. napus. The oil content of W82 was 6% higher than that of Yangyou 6. Higher protein and sucrose contents, less dietary fiber and crude fiber, and fewer glucosinolates accumulated in the seed meal of yellow rapeseed compared with black rapeseed (Additional file 12: Table S7) [8, 39, 61, 69]. The developing seeds at 3 WAF, 4 WAF, 5 WAF, 6 WAF, and mature seeds were collected from three pods each of ten individual plants for comparative analysis. During the seed development, differentially accumulated pigments were visual since 5 WAF of yellow and black rapeseeds. Proanthocyanidins (PAs) were less accumulated throughout yellow seed development than black seed [39, 70].
RNA extraction, library construction, and RNA sequencing
For each developmental stage of each rapeseed line, five RNA samples were separately isolated from polled seeds of ten plants using Trizol Reagent Solution (Invitrogen, USA). Each library was pooled by mixing equal quantities of five RNA samples. The RNA quality was validated using agarose gel electrophoresis, Nanodrop, Qubit, and Agilent 2100 to confirm the purity, concentration, and integrity, respectively. mRNA was purified using beads with Oligo (dT), and cDNA was then synthesized with random hexamers after fragmentation of mRNA. After purification of cDNA with AMPure XP beads, unique adaptors and indexes were ligated. Certain fragments were selected with beads and enriched using PCR amplification. Finally, ten cDNA libraries for five developmental stages (3 WAF, 4 WAF, 5 WAF, 6 WAF and mature seeds) of yellow- and black-seeded B. napus were normalized based on a Qubit assessment, and the insert size was validated by Agilent 2100. Then, the polled libraries were sequenced by the Illumina HiSeq™ 2000 platform.
Bioinformatics analysis
After removing adaptor sequences and low-quality sequences, clean reads were mapped to the B. napus cv. Darmor-bzh genome (version 5) using TopHat2 [71, 72]. Based on the predicted gene models of Cufflinks, classification and statistics of alternative splicing were carried out using AS profile [73, 74]. Novel transcripts were predicted using Cufflinks to assemble the mapped reads on the genome and compare it with the known gene models [75]. The abundance of reads mapped to reference was estimated and normalized using Reads Per Kilo bases per Million reads (RPKM), and all the transcripts with RPKM value> 1 were used for further analysis. Differential gene expression analysis was performed using DESeq [76, 77], and DEGs between libraries were screened with a threshold of | (FoldChange)| > 1 and q value< 0.005. All the DEGs among different libraries were clustered base on the log10(RPKM+ 1) and log2(Fold Change) value. Gene ontology (GO) enrichment of the DEGs was performed using GOseq with a corrected p-value < 0.05 [78], and KEGG pathway enrichment of DEGs was performed using KOBAS 2.0 with a corrected p-value < 0.05 [79]. An overview of pathways related to these DEGs was predicted by MapMan (version 3.6.0) analysis [80].
cDNA synthesis and qRT-PCR validation
Subsamples of RNA-Seq were reverse transcribed into cDNA for real-time qPCR validation using the Revert Aid First Strand cDNA Synthesis Kit and SYBR Green Real-Time PCR Master Mixes (Thermo, USA). qRT-PCR was performed on a fluorescence quantitative system Mx3005P (Agilent, USA). Genes, primers and size of the amplicon are listed in Additional file 13: Table S8. B. napus β-actin (NCBI AF111812) was used as an endogenous control to generate the △Ct for three technological replicates.
Additional files
Table S1. Summary of read mapping in RNA-Seq analysis of yellow and black rapeseeds. B3, B4, B5, B6 and BM indicate 3 ~ 6 WAF and mature black seed. Y3, Y4, Y5, Y6 and YM indicate 3 ~ 6 WAF and mature yellow seed. (XLSX 34577 kb)
Table S2. DEGs identified with black or yellow seed development and between same developmental stages of two rapeseeds. (XLSX 11030 kb)
Table S3. DEGs related to seed development. (XLSX 94 kb)
Table S4. Up- and downregulated genes among all the developmental stages of yellow- and black-seeded B. napus. (XLSX 25 kb)
Figure S1. The SOM clusters of DEGs from five developmental stages of two rapeseed lines. (XLSX 173 kb)
Table S5. All the up- and down-DEGs with GO annotation. (XLSX 63 kb)
Figure S2. KEGG enrichment of DEGs (Y3 vs. B3) in flavonoid and phenylpropanoid biosynthesis. (XLSX 24 kb)
Figure S3. KEGG enrichment of up- and down-DEGs between yellow- and black-seeded B. napus. (XLS 31 kb)
Table S6. KEGG pathways of DEGs between yellow- and black-seeded B. napus. (PDF 519 kb)
Figure S4. Overview of pathways related to the DEGs between yellow- and black-seeded B. napus. (PDF 981 kb)
{kind=link}
Figur S5. Heatmap of DEGs involved in secondary metabolism. (JPG 6164 kb)
{kind=link}
Table S7. Quality differences between yellow- and black-seeded B. napus. (JPG 1896 kb)
{kind=link}
Table S8. Primers for qPCR validation of DEGs (JPG 3234 kb)
Acknowledgements
Not applicable.
Funding
This study was supported by the National Key Basic Research Program of China (2015CB150201), the National Natural Science Foundations (31330057, 31771825, 31401414), the National Key Research and Development Program of China (2016YFD0101000, 2016YFD0102000), the Natural Science Foundation of Jiangsu Province (BK20180101, BK20140478), China Postdoctoral Science Foundation (2015 T80591, 2014 M561719), Jiangsu Postdoctoral Science Foundation (1401078B), the Postgraduate Research & Practice Innovation Program of Jiangsu Province (XKYCX17_066), the Priority Academic Program Development of Jiangsu Higher Education Institutions, and Yangzhou University for Excellent Talent Support Program. The founders did not play any roles in the design, analysis, interpretation of this study or relevant data.
Availability of data and materials
All the data pertaining to the present study have been included in the tables and figures of the manuscript, and the authors are pleased to share all the data and plant materials upon reasonable request.
Abbreviations
- 4CL
4-coumarate CoA ligase
- ACCase
Acetyl-CoA carboxylase
- AKR2A
Ankyrin repeat-containing protein 2A
- APX3
Ascorbate peroxidase
- C4H
Cinnamate 4-hydroxylase
- CB5R1
Cytochrome B5 reductase 1
- CT
Carboxyltransferase
- CYP
Cytochrome P450
- DEG
Differentially expressed gene
- EBG
Early biosynthesis gene
- FA
Fatty acid
- GO
Gene ontology
- GWAS
Genome wide association study
- KEGG
Kyoto encyclopedia of genes and genomes
- LBG
Late biosynthesis gene
- LEA
Late embryogenesis abundant hydroxyproline
- MBW
MYB-bHLH-WD40
- NIL
Near-isogenic line
- PA
Proanthocyanin
- PAL
Phenylalanine ammonia-lyase
- PKP
Pyruvate kinase
- qRT-PCR
Quantitative real-time polymerase chain reaction
- RPKM
Reads per kilo bases per million reads
- SNP
Single nucleotide polymorphism
- TT
Transparent testa
- UGP
UDP-Glucose pyrophosphorylase
- WAF
Weeks after flowering
Authors’ contributions
JJ performed the experiments, analyzed the RNA-seq data, and drafted the manuscript. SZ, YY, YW and LZ sampled the materials and performed qRT-PCR analysis. JB and YPW revised the manuscript. All the authors approved the final manuscript.
Ethics approval and consent to participate
This study has not directly involved humans or animals. The yellow-seeded B. napus, which originated from somatic hybridization between B. napus and S. alba, was created and preserved by our group. The black-seeded B. napus was obtained from the Jiangsu Lixiahe Region Agricultural Research Institute, China. No specific permission was required for use of these materials for experimental purposes. The seedlings were grown in the experimental field of Yangzhou University, Yangzhou, China as per standard practices, and samples were harvested at the required time. We comply with the Convention on the Trade in Endangered Species of Wild Fauna and Flora.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jinjin Jiang, Email: jjjiang@yzu.edu.cn.
Shuang Zhu, Email: 1696459031@qq.com.
Yi Yuan, Email: 1010508485@qq.com.
Yue Wang, Email: 954778771@qq.com.
Lei Zeng, Email: 812377664@qq.com.
Jacqueline Batley, Email: jacqueline.batley@uwa.edu.aul.
You-Ping Wang, Phone: +86-514-87997303, Email: wangyp@yzu.edu.cn.
References
- 1.Wittkop B, Snowdon RJ, Friedt W. New NIRS calibrations for fiber fractions reveal broad genetic variation in Brassica napus seed quality. J Agric Food Chem. 2012;60(9):2248–2256. doi: 10.1021/jf204936f. [DOI] [PubMed] [Google Scholar]
- 2.Abbadi A, Leckband G. Rapeseed breeding for oil content, quality, and sustainability. Eur J Lipid Sci Technol. 2011;113(10):1198–1206. [Google Scholar]
- 3.Qu C, Zhao H, Fu F, Wang Z, Zhang K, Zhou Y, Wang X, Wang R, Xu X, Tang Z, et al. Genome-wide survey of flavonoid biosynthesis genes and gene expression analysis between black- and yellow-seeded Brassica napus. Front Plant Sci. 2016;7:1755. doi: 10.3389/fpls.2016.01755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lepiniec L, Debeaujon I, Routaboul JM, Baudry A, Pourcel L, Nesi N, Caboche M. Genetics and biochemistry of seed flavonoids. Annu Rev Plant Biol. 2006;57:405–430. doi: 10.1146/annurev.arplant.57.032905.105252. [DOI] [PubMed] [Google Scholar]
- 5.Liu Q, Luo L, Zheng L. Lignins: biosynthesis and biological functions in plants. Int J Mol Sci. 2018;19(2):335–350. doi: 10.3390/ijms19020335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mittasch J, Bottcher C, Frolov A, Strack D, Milkowski C. Reprogramming the phenylpropanoid metabolism in seeds of oilseed rape by suppressing the orthologs of reduced epidermal fluorescence1. Plant Physiol. 2013;161(4):1656–1669. doi: 10.1104/pp.113.215491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahman MH, Joersbo M, Poulsen MH. Production of yellow-seeded Brassica napus of double low quality. Plant Breed. 2001;120:473–478. [Google Scholar]
- 8.Li A, Wei C, Jiang J, Zhang Y, Snowdon RJ, Wang Y. Phenotypic variation in progenies from somatic hybrids between Brassica napus and Sinapis alba. Euphytica. 2009;170(3):289–296. [Google Scholar]
- 9.Appelhagen I, Thiedig K, Nordholt N, Schmidt N, Huep G, Sagasser M, Weisshaar B. Update on transparent testa mutants from Arabidopsis thaliana: characterisation of new alleles from an isogenic collection. Planta. 2014;240(5):955–970. doi: 10.1007/s00425-014-2088-0. [DOI] [PubMed] [Google Scholar]
- 10.Xu W, Grain D, Bobet S, Le Gourrierec J, Thevenin J, Kelemen Z, Lepiniec L, Dubos C. Complexity and robustness of the flavonoid transcriptional regulatory network revealed by comprehensive analyses of MYB-bHLH-WDR complexes and their targets in Arabidopsis seed. New Phytol. 2014;202(1):132–144. doi: 10.1111/nph.12620. [DOI] [PubMed] [Google Scholar]
- 11.Appelhagen I, Jahns O, Bartelniewoehner L, Sagasser M, Weisshaar B, Stracke R. Leucoanthocyanidin Dioxygenase in Arabidopsis thaliana: characterization of mutant alleles and regulation by MYB-BHLH-TTG1 transcription factor complexes. Gene. 2011;484(1–2):61–68. doi: 10.1016/j.gene.2011.05.031. [DOI] [PubMed] [Google Scholar]
- 12.Xu BB, Li JN, Zhang XK, Wang R, Xie LL, Chai YR. Cloning and molecular characterization of a functional flavonoid 3′-hydroxylase gene from Brassica napus. J Plant Physiol. 2007;164(3):350–363. doi: 10.1016/j.jplph.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Wei YL, Li JN, Lu J, Tang ZL, Pu DC, Chai YR. Molecular cloning of Brassica napus TRANSPARENT TESTA 2 gene family encoding potential MYB regulatory proteins of proanthocyanidin biosynthesis. Mol Biol Rep. 2007;34(2):105–120. doi: 10.1007/s11033-006-9024-8. [DOI] [PubMed] [Google Scholar]
- 14.Ni Y, Jiang H-L, Lei B, Li J-N, Chai Y-R. Molecular cloning, characterization and expression of two rapeseed (Brassica napus L.) cDNAs orthologous to Arabidopsis thaliana phenylalanine ammonia-lyase 1. Euphytica. 2007;159(1–2):1–16. [Google Scholar]
- 15.Chai YR, Lei B, Huang HL, Li JN, Yin JM, Tang ZL, Wang R, Chen L. TRANSPARENT TESTA 12 genes from Brassica napus and parental species: cloning, evolution, and differential involvement in yellow seed trait. Mol Gen Genomics. 2009;281(1):109–123. doi: 10.1007/s00438-008-0399-1. [DOI] [PubMed] [Google Scholar]
- 16.Auger B, Baron C, Lucas MO, Vautrin S, Berges H, Chalhoub B, Fautrel A, Renard M, Nesi N. Brassica orthologs from BANYULS belong to a small multigene family, which is involved in procyanidin accumulation in the seed. Planta. 2009;230(6):1167–1183. doi: 10.1007/s00425-009-1017-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu J, Li JN, Lei B, Wang SG, Chai YR. Molecular cloning and characterization of two Brassica napus TTG1 genes reveal genus-specific nucleotide preference, extreme protein-level conservation and fast divergence of organ-specificity. Genes Genomics. 2009;31:129–142. [Google Scholar]
- 18.Zhang K, Lu K, Qu C, Liang Y, Wang R, Chai Y, Li J. Gene silencing of BnTT10 family genes causes retarded pigmentation and lignin reduction in the seed coat of Brassica napus. PLoS One. 2013;8(4):e61247. doi: 10.1371/journal.pone.0061247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lian J, Lu X, Yin N, Ma L, Lu J, Liu X, Li J, Lu J, Lei B, Wang R, et al. Silencing of BnTT1 family genes affects seed flavonoid biosynthesis and alters seed fatty acid composition in Brassica napus. Plant Sci. 2017;254:32–47. doi: 10.1016/j.plantsci.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 20.Yu CY. Molecular mechanism of manipulating seed coat coloration in oilseed Brassica species. J Appl Genet. 2013;54(2):135–145. doi: 10.1007/s13353-012-0132-y. [DOI] [PubMed] [Google Scholar]
- 21.Kebede B, Cheema K, Greenshields DL, Li C, Selvaraj G, Rahman H. Construction of genetic linkage map and mapping of QTL for seed color in Brassica rapa. Genome. 2012;55(12):813–823. doi: 10.1139/g2012-066. [DOI] [PubMed] [Google Scholar]
- 22.Rahman M, McVetty PB, Li G. Development of SRAP, SNP and multiplexed SCAR molecular markers for the major seed coat color gene in Brassica rapa L. Theor Appl Genet. 2007;115(8):1101–1107. doi: 10.1007/s00122-007-0636-8. [DOI] [PubMed] [Google Scholar]
- 23.Zhao H, Basu U, Kebede B, Qu C, Li J, Rahman H. Fine mapping of the major QTL for seed coat color in Brassica rapa var. yellow Sarson by use of NIL populations and transcriptome sequencing for identification of the candidate genes. PLoS One. 2019;14(2):e0209982. doi: 10.1371/journal.pone.0209982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahmood T, Rahman MH, Stringam GR, Raney JP, Good AG. Molecular markers for seed colour in Brassica juncea. Genome. 2005;48(4):755–760. doi: 10.1139/g04-122. [DOI] [PubMed] [Google Scholar]
- 25.Mahmood T, Rahman MH, Stringam GR, Yeh F, Good AG. Identification of quantitative trait loci (QTL) for oil and protein contents and their relationships with other seed quality traits in Brassica juncea. Theor Appl Genet. 2006;113:1211–1220. doi: 10.1007/s00122-006-0376-1. [DOI] [PubMed] [Google Scholar]
- 26.Liu L, Stein A, Wittkop B, Sarvari P, Li J, Yan X, Dreyer F, Frauen M, Friedt W, Snowdon RJ. A knockout mutation in the lignin biosynthesis gene CCR1 explains a major QTL for acid detergent lignin content in Brassica napus seeds. Theor Appl Genet. 2012;124(8):1573–1586. doi: 10.1007/s00122-012-1811-0. [DOI] [PubMed] [Google Scholar]
- 27.Stein A, Wittkop B, Liu L, Obermeier C, Friedt W, Snowdon RJ, Chevre AM. Dissection of a major QTL for seed colour and fibre content in Brassica napus reveals colocalization with candidate genes for phenylpropanoid biosynthesis and flavonoid deposition. Plant Breed. 2013;132(4):382–389. [Google Scholar]
- 28.Wang J, Xian X, Xu X, Qu C, Lu K, Li J, Liu L. Genome-wide association mapping of seed coat color in Brassica napus. J Agric Food Chem. 2017;65(26):5229–5237. doi: 10.1021/acs.jafc.7b01226. [DOI] [PubMed] [Google Scholar]
- 29.Jaakola L. New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci. 2013;18(9):477–483. doi: 10.1016/j.tplants.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 30.Chalhoub B, Denoeud F, Liu S, Parkin IA, Tang H, Wang X, Chiquet J, Belcram H, Tong C, Samans B, et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 2014;345(6199):950–953. doi: 10.1126/science.1253435. [DOI] [PubMed] [Google Scholar]
- 31.Wei D, Cui Y, He Y, Xiong Q, Qian L, Tong C, Lu G, Ding Y, Li J, Jung C, et al. A genome-wide survey with different rapeseed ecotypes uncovers footprints of domestication and breeding. J Exp Bot. 2017;68(17):4791–4801. doi: 10.1093/jxb/erx311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rashid A, Rakov G, Downey RK. Development of yellow seeded Brassica napus through interspecific crosses. Plant Breed. 1994;112:127–134. [Google Scholar]
- 33.Wen J, Zhu L, Qi L, Ke H, Yi B, Shen J, Tu J, Ma C, Fu T. Characterization of interploid hybrids from crosses between Brassica juncea and B. oleracea and the production of yellow-seeded B. napus. Theor Appl Genet. 2012;125:19–32. doi: 10.1007/s00122-012-1813-y. [DOI] [PubMed] [Google Scholar]
- 34.Wang YP, Zhao XX, Sonntag K, Wehling P, Snowdon RJ. Behaviour of Sinapis alba chromosomes in a Brassica napus background revealed by genomic in-situ hybridization. Chromosom Res. 2005;13(8):819–826. doi: 10.1007/s10577-005-1017-2. [DOI] [PubMed] [Google Scholar]
- 35.Jiang JJ, Zhao XX, Tian W, Li TB, Wang YP. Intertribal somatic hybrids between Brassica napus and Camelina sativa with high linolenic acid content. Plant Cell, Tissue and Organ Culture (PCTOC) 2009;99(1):91–95. [Google Scholar]
- 36.Warwick SI, Simard MJ, Legere A, Beckie HJ, Braun L, Zhu B, Mason P, Seguin-Swartz G, Stewart CN, Jr.: Hybridization between transgenic Brassica napus L. and its wild relatives: Brassica rapa L., Raphanus raphanistrum L., Sinapis arvensis L., and Erucastrum gallicum (Willd.) O.E. Schulz. Theor Appl Genet 2003, 107(3):528–539. [DOI] [PubMed]
- 37.Zhang L, Jiang S, Xin R, Zhang F, Wang J, Guan R. Obtaining of yellow-seeded rapeseed germplasm from intergeneric hybridization between Brassica napus and Descurainia sophia. Chinese Journal of Oil Crop Sciences. 2009;31:434–439. [Google Scholar]
- 38.Wang Y, Sonntag K, Rudloff E, Chen J. Intergeneric somatic hybridization between Brassica napus L. and Sinapis alba L. J Integr Plant Biol. 2005;47(1):8. [Google Scholar]
- 39.Jiang J, Shao Y, Li A, Lu C, Zhang Y, Wang Y. Phenolic composition analysis and gene expression in developing seeds of yellow- and black-seeded Brassica napus. J Integr Plant Biol. 2013;55(6):537–551. doi: 10.1111/jipb.12039. [DOI] [PubMed] [Google Scholar]
- 40.Jiang J, Shao Y, Li A, Zhang Y, Wei C, Wang Y. FT-IR and NMR study of seed coat dissected from different colored progenies of Brassica napus-Sinapis alba hybrids. J Sci Food Agric. 2013;93(8):1898–1902. doi: 10.1002/jsfa.5986. [DOI] [PubMed] [Google Scholar]
- 41.Li S, Zachgo S. TCP3 interacts with R2R3-MYB proteins, promotes flavonoid biosynthesis and negatively regulates the auxin response in Arabidopsis thaliana. Plant J. 2013;76(6):901–913. doi: 10.1111/tpj.12348. [DOI] [PubMed] [Google Scholar]
- 42.Ayabe S, Akashi T. Cytochrome P450s in flavonoid metabolism. Phytochem Rev. 2006;5(2–3):271–282. [Google Scholar]
- 43.Wu G, Wu Y, Xiao L, Li X, Lu C. Zero erucic acid trait of rapeseed (Brassica napus L.) results from a deletion of four base pairs in the fatty acid elongase 1 gene. Theor Appl Genet. 2008;116(4):491–499. doi: 10.1007/s00122-007-0685-z. [DOI] [PubMed] [Google Scholar]
- 44.Schierholt A, Becker HC, Ecke W. Mapping a high oleic acid mutation in winter oilseed rape (Brassica napus L.) Theor Appl Genet. 2000;101(5–6):897–901. [Google Scholar]
- 45.Li A, Jiang J, Zhang Y, Snowdon RJ, Liang G, Wang Y. Molecular and cytological characterization of introgression lines in yellow seed derived from somatic hybrids between Brassica napus and Sinapis alba. Mol Breed. 2012;29:209–219. [Google Scholar]
- 46.Zhu J, Wang X, Xu Q, Zhao S, Tai Y, Wei C. Global dissection of alternative splicing uncovers transcriptional diversity in tissues and associates with the flavonoid pathway in tea plant (Camellia sinensis) BMC Plant Biol. 2018;18(1):266. doi: 10.1186/s12870-018-1497-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang G, Sun M, Wang J, Lei M, Li C, Zhao D, Huang J, Li W, Li S, Li J, et al. PacBio full-length cDNA sequencing integrated with RNA-seq reads drastically improves the discovery of splicing transcripts in rice. Plant J. 2019;97(2):296–305. doi: 10.1111/tpj.14120. [DOI] [PubMed] [Google Scholar]
- 48.Thakur O, Randhawa GS. Identification and characterization of SSR, SNP and InDel molecular markers from RNA-Seq data of guar (Cyamopsis tetragonolobaL Taub) roots. BMC genomics. 2018;19(1):951. doi: 10.1186/s12864-018-5205-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qu C, Fu F, Lu K, Zhang K, Wang R, Xu X, Wang M, Lu J, Wan H, Zhanglin T, et al. Differential accumulation of phenolic compounds and expression of related genes in black- and yellow-seeded Brassica napus. J Exp Bot. 2013;64(10):2885–2898. doi: 10.1093/jxb/ert148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong M, Hu K, Tian T, Li X, Chen L, Zhang Y, Yi B, Wen J, Ma C, Shen J, et al. Transcriptomic analysis of seed coats in yellow-seeded Brassica napus reveals novel genes that influence proanthocyanidin biosynthesis. Front Plant Sci. 2017;8:1674. doi: 10.3389/fpls.2017.01674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, Lu Y, Yuan Y, Liu S, Guan C, Chen S, Liu Z. De novo transcriptome of Brassica juncea seed coat and identification of genes for the biosynthesis of flavonoids. PLoS One. 2013;8(8):e71110. doi: 10.1371/journal.pone.0071110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang H, Fan W, Li H, Yang J, Huang J, Zhang P. Functional characterization of Dihydroflavonol-4-reductase in anthocyanin biosynthesis of purple sweet potato underlies the direct evidence of anthocyanins function against abiotic stresses. PLoS One. 2013;8(11):e78484. doi: 10.1371/journal.pone.0078484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou B, Wang Y, Zhan Y, Li Y, Kawabata S. Chalcone synthase family genes have redundant roles in anthocyanin biosynthesis and in response to blue/UV-A light in turnip (Brassica rapa; Brassicaceae) Am J Bot. 2013;100(12):2458–2467. doi: 10.3732/ajb.1300305. [DOI] [PubMed] [Google Scholar]
- 54.Sasaki-Sekimoto Y, Saito H, Masuda S, Shirasu K, Ohta H. Comprehensive analysis of protein interactions between JAZ proteins and bHLH transcription factors that negatively regulate jasmonate signaling. Plant Signal Behav. 2014;9(1):e27639. doi: 10.4161/psb.27639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Min JH, Ju HW, Yoon D, Lee KH, Lee S, Kim CS. Arabidopsis basic helix-loop-helix 34 (bHLH34) is involved in glucose signaling through binding to a GAGA Cis-element. Front Plant Sci. 2017;8:2100. doi: 10.3389/fpls.2017.02100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lam PY, Zhu FY, Chan WL, Liu H, Lo C. Cytochrome P450 93G1 is a flavone synthase II that channels flavanones to the biosynthesis of tricin O-linked conjugates in rice. Plant Physiol. 2014;165(3):1315–1327. doi: 10.1104/pp.114.239723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lam PY, Liu H, Lo C: Completion of Tricin biosynthesis pathway in Rice: cytochrome P450 75B4 is a unique Chrysoeriol 5′-hydroxylase. Plant Physiol 2015, 168(4):1527–1536. [DOI] [PMC free article] [PubMed]
- 58.Lei Z, Zhou C, Ji X, Wei G, Huang Y, Yu W, Luo Y, Qiu Y. Transcriptome analysis reveals genes involved in flavonoid biosynthesis and accumulation in Dendrobium catenatum from different locations. Sci Rep. 2018;8(1):6373. doi: 10.1038/s41598-018-24751-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma B, Luo Y, Jia L, Qi X, Zeng Q, Xiang Z, He N. Genome-wide identification and expression analyses of cytochrome P450 genes in mulberry (Morus notabilis) J Integr Plant Biol. 2014;56(9):887–901. doi: 10.1111/jipb.12141. [DOI] [PubMed] [Google Scholar]
- 60.Li S. Transcriptional control of flavonoid biosynthesis: fine-tuning of the MYB-bHLH-WD40 (MBW) complex. Plant Signal Behav. 2014;9(1):e27522. doi: 10.4161/psb.27522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Y, Meng G, Chen S, Chen Y, Jiang J, Wang YP. Correlation analysis of phenolic contents and antioxidation in yellow- and black-seeded Brassica napus. Molecules. 2018;23:1815–1829. doi: 10.3390/molecules23071815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pourcel L, Routaboul JM, Cheynier V, Lepiniec L, Debeaujon I. Flavonoid oxidation in plants: from biochemical properties to physiological functions. Trends Plant Sci. 2007;12(1):29–36. doi: 10.1016/j.tplants.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 63.Lonien J, Schwender J. Analysis of metabolic flux phenotypes for two Arabidopsis mutants with severe impairment in seed storage lipid synthesis. Plant Physiol. 2009;151(3):1617–1634. doi: 10.1104/pp.109.144121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keereetaweep J, Liu H, Zhai Z, Shanklin J. Biotin attachment domain-containing proteins irreversibly inhibit acetyl CoA carboxylase. Plant Physiol. 2018;177(1):208–215. doi: 10.1104/pp.18.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chivasa S, Tome DF, Slabas AR. UDP-glucose pyrophosphorylase is a novel plant cell death regulator. J Proteome Res. 2013;12(4):1743–1753. doi: 10.1021/pr3010887. [DOI] [PubMed] [Google Scholar]
- 66.Xu Z, Escamilla-Trevino L, Zeng L, Lalgondar M, Bevan D, Winkel B, Mohamed A, Cheng CL, Shih MC, Poulton J, et al. Functional genomic analysis of Arabidopsis thaliana glycoside hydrolase family 1. Plant Mol Biol. 2004;55(3):343–367. doi: 10.1007/s11103-004-0790-1. [DOI] [PubMed] [Google Scholar]
- 67.Fukuchi-Mizutani M, Mizutani M, Tanaka Y, Kusumi T, Ohta D. Microsomal electron transfer in higher plants: cloning and heterologous expression of NADH-cytochrome b5 reductase from Arabidopsis. Plant Physiol. 1999;119(1):353–362. doi: 10.1104/pp.119.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen G, Kuppu S, Venkataramani S, Wang J, Yan J, Qiu X, Zhang H. ANKYRIN REPEAT-CONTAINING PROTEIN 2A is an essential molecular chaperone for peroxisomal membrane-bound ASCORBATE PEROXIDASE3 in Arabidopsis. Plant Cell. 2010;22(3):811–831. doi: 10.1105/tpc.109.065979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang J, Wang Y, Xie T, Rong H, Li A, Fang Y, Wang Y. Metabolic characteristics in meal of black rapeseed and yellow-seeded progeny of Brassica napus-Sinapis alba hybrids. Molecules. 2015;20(12):21204–21213. doi: 10.3390/molecules201219761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Y, Rong H, Xie T, Jiang J, Wu J, Wang Y. Comparison of DNA methylation in the developing seeds of yellow- and black-seeded Brassica napus through MSAP analysis. Euphytica. 2016;209:157–169. [Google Scholar]
- 71.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jiang L, Schlesinger F, Davis CA, Zhang Y, Li R, Salit M, Gingeras TR, Oliver B. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011;21(9):1543–1551. doi: 10.1101/gr.121095.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ghosh S, Chan CK. Analysis of RNA-Seq data using TopHat and cufflinks. Methods Mol Biol. 2016;1374:339–361. doi: 10.1007/978-1-4939-3167-5_18. [DOI] [PubMed] [Google Scholar]
- 75.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 77.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mao XCT, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21(19):3787–3793. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
- 80.Usadel B, Poree F, Nagel A, Lohse M, Czedik-Eysenberg A, Stitt M. A guide to using MapMan to visualize and compare Omics data in plants: a case study in the crop species, maize. Plant Cell Environ. 2009;32(9):1211–1229. doi: 10.1111/j.1365-3040.2009.01978.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of read mapping in RNA-Seq analysis of yellow and black rapeseeds. B3, B4, B5, B6 and BM indicate 3 ~ 6 WAF and mature black seed. Y3, Y4, Y5, Y6 and YM indicate 3 ~ 6 WAF and mature yellow seed. (XLSX 34577 kb)
Table S2. DEGs identified with black or yellow seed development and between same developmental stages of two rapeseeds. (XLSX 11030 kb)
Table S3. DEGs related to seed development. (XLSX 94 kb)
Table S4. Up- and downregulated genes among all the developmental stages of yellow- and black-seeded B. napus. (XLSX 25 kb)
Figure S1. The SOM clusters of DEGs from five developmental stages of two rapeseed lines. (XLSX 173 kb)
Table S5. All the up- and down-DEGs with GO annotation. (XLSX 63 kb)
Figure S2. KEGG enrichment of DEGs (Y3 vs. B3) in flavonoid and phenylpropanoid biosynthesis. (XLSX 24 kb)
Figure S3. KEGG enrichment of up- and down-DEGs between yellow- and black-seeded B. napus. (XLS 31 kb)
Table S6. KEGG pathways of DEGs between yellow- and black-seeded B. napus. (PDF 519 kb)
Figure S4. Overview of pathways related to the DEGs between yellow- and black-seeded B. napus. (PDF 981 kb)
Figur S5. Heatmap of DEGs involved in secondary metabolism. (JPG 6164 kb)
Table S7. Quality differences between yellow- and black-seeded B. napus. (JPG 1896 kb)
Table S8. Primers for qPCR validation of DEGs (JPG 3234 kb)
Data Availability Statement
All the data pertaining to the present study have been included in the tables and figures of the manuscript, and the authors are pleased to share all the data and plant materials upon reasonable request.