

Abstract
Nitric oxide is now universally recognized as an extracellular signaling molecule. Nitric oxide, produced in one cell, diffuses across the extracellular space and acts with targets in an adjoining cell. In this study, we present proof that hydrogen peroxide – like nitric oxide – acts as a true first (intercellular) messenger for a multitude of pro-inflammatory ligands. RAW 264.7 macrophages were activated with three different ligands, lipopolysaccharide, interferon-gamma or advanced glycation end products in the presence of increasing concentrations of (hydrogen peroxide scavenging) catalase. As inflammatory readouts, nitric oxide and tumor necrosis factor were determined. We hypothesize that hydrogen peroxide travels between cells propagating the signal, then a certain percentage of the readout should be inhibited by catalase in a concentration-dependent manner. The experiment showed concentration-dependent inhibition of nitric oxide and tumor necrosis factor-α production in response to all three ligands/ligand combinations (interferon-gamma, lipopolysaccharide, and chicken egg albumin-derived advanced glycation end product) in the presence of increasing concentration of catalase. For example, catalase inhibited 100% of nitric oxide and 40% of tumor necrosis factor-α production at its highest concentration. Our results suggest that hydrogen peroxide travels through cell membranes into the extracellular space and enters and activates adjacent cells. Like nitric oxide, we suggest that it is a ubiquitous first messenger, able to transmit cell-to-cell pro-inflammatory signals such as nitric oxide and tumor necrosis factor-α. In a therapeutic setting, our data suggest that compounds acting as hydrogen peroxide scavengers might not even need to enter the cell to act as anti-inflammatory drugs.
Keywords: inflammation, glycation, redox signaling, cytokines, anti-inflammatory drugs, hydrogen peroxide, membrane permeable, lipopolysaccharide, Interferon-gamma, signaling
Chinese Library Classification No. R453; R364
Introduction
Inflammation, predominantly caused by activated macrophages and microglia (Ransohoff, 2016), has been suggested to be a major contributing factor for many chronic diseases including neurodegenerative diseases like Alzheimer’s disease (Heneka, 2006). Microglia release cytotoxic substances such as nitric oxide (NO), proteases, prostaglandins, and cytokines as well as reactive oxygen species, such as superoxide, hydrogen peroxide and the hydroxyl radical (HO•). The first reactive oxygen species in the chain, superoxide, is generated from nicotinamide adenine dinucleotide phosphate (NADPH) by NADPH oxidase, a phenomenon known as “respiratory burst” (de Rooij et al., 2009). In their milestone publication in 1975, Root et al. have shown that the respiratory burst is a mechanism by which macrophages attack invading microorganism in the human body (Root et al., 1975). O2 – is then converted to hydrogen peroxide by superoxide dismutase (Noor et al., 2002), and hydrogen peroxide is subsequently converted to water by catalase. Amphipathic hydrogen peroxide easily passes in and out of cells, and although it was long suspected to do so by simple diffusion, aquaporin water channels also have a role in hydrogen peroxide transport across plasma membranes (Vieceli Dalla Sega et al., 2014) (Figure 1).
Figure 1.
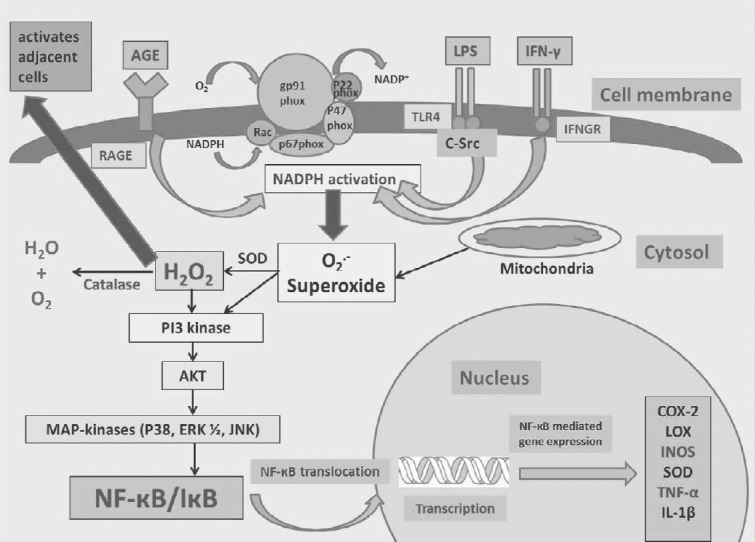
Signal pathways involved in LPS, IFN-γ, and CEA-AGE induced proinflammatory mediator expression via hydrogen peroxide
After binding to their respective receptors, LPS, IFN-γ, and AGEs lead to the activation of NADPH oxidase and subsequent superoxide production and conversion to hydrogen peroxide. ROS activate downstream signaling pathway components including PI3K/Akt and MAPK. Subsequently, MAPK (p38, ERK 1/2, and JNK) activation induces the phosphorylation of NF-κB. Hydrogen peroxide can also travel through the membrane into the extracellular space and activate adjacent cells. LPS: Lipopolysaccharide; IFN-γ: Interferon-gamma; NADPH: nicotinamide adenine dinucleotide phosphate; ROS: reactive oxygen species; PI3K: phosphoinositide-3-kinase; MAPK: mitogen-activated protein kinase; ERK1/2: extracellular-signal-regulated kinase 1/2; NF-κB: nuclear factor kappa B; JNK: c-Jun N-terminal protein kinase; COX-2: cyclooxygenase-2; LOX: lysyl oxidase; iNOS: inducible nitric oxide synthase; SOD: superoxide dismutase; TNF: tumor necrosis factor; IL: interleukin; IκB: inhibitor of NF-κB; CEA-AGE: chicken egg albumin-derived advanced glycation end product; RAGE: receptor for AGEs; IFNGR: interferon-gamma receptor.
Hydrogen peroxide has long been known to act as a second messenger in intracellular signal transduction pathways, including nuclear factor kappa B (NF-κB)-dependent gene expression, e.g. for pro-inflammatory mediators, including tumor necrosis factor (TNF-α) and inducible nitric oxide synthase (Han et al., 2001). Oxidation dependent, redox-sensitive signal transduction pathways can be decreased by different types of “anti-inflammatory” antioxidants such as α-lipoic acid and polyphenols (Zhang et al., 2007). Further evidence for this intracellular function of hydrogen peroxide comes from the fact that exposure to exogenous hydrogen peroxide induces NF-κB translocation (Takada et al., 2003). This property of hydrogen peroxide to behave as a second messenger is termed “redox-sensitive” signal transduction (de Rooij et al., 2009). Although the exact targets of hydrogen peroxide are not known, it most likely activates a variety of redox-sensitive kinases, upstream of NF-κB through oxidations of critical cysteines (Hancock, 2009).
All the previous examples show that hydrogen peroxide can act as an intracellular signaling molecule. In this study, we provide evidence that hydrogen peroxide can act as an extracellular signaling molecule, as it can diffuse through cell membranes and is able to transfer pro-inflammatory signals from one cell to another, thus acting as a true first (intercellular) messenger. The mechanisms and sequence of events behind our hypothesis (“hydrogen peroxide is an intercellular messenger”) are as follows:
-
a)
hydrogen peroxide is produced in reaction to a pro-inflammatory ligand, e.g. by NADPH oxidase (“respiratory burst”) (Johnson and Sung, 1987).
-
b)
Hydrogen peroxide diffuses through cell membranes into adjacent target cells, e.g. via aquaporin channels (Bienert et al., 2007).
-
c)
Hydrogen peroxide reacts with target molecules such as redox-sensitive kinases in the target cells.
In this study, we present evidence for this novel paradigm in cell-to-cell communication using three quite different pro-inflammatory activators, and two readouts, NO, and TNF-α.
Materials and Methods
Preparation of chicken egg-albumin-derived advanced glycation end products
Chicken egg albumin (CEA)-derived advanced glycation end products (AGEs) were prepared by incubating 903 mg glucose with 50 mg CEA in 100 mM sodium phosphate-buffered saline (PBS) (pH 7.4). After sterilizing the samples through a 0.45 Acrodisc syringe filter into a 50 mL autoclaved glass jar, they were incubated for 37 days at 60°C. The lids were loosened to allow air exchange. The pH was checked every week and was adjusted to around 7.6 by adding sterile 100 mM NaOH. Since a steady and nearly linear increase in both fluorescence (370 nm excitation/440nm emission) and absorbance at 400 nm was observed during the 6 weeks and the increase thereafter was only marginal, the CEA was assumed to be maximally modified”. After incubation, unreacted sugars were removed before the assay by extensive dialysis against PBS. The CEA-AGE solutions were stored at –80°C (Dukic-Stefanovic et al., 2003).
Maintenance of RAW 264.7 macrophages
RAW 264.7 macrophages (Sigma-Aldrich, Castle Hill, NSW, Australia) were grown in 175 cm2 flasks on Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, Mulgrave, VIC, Australia) containing 5% fetal bovine serum (FBS) that was supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL) and L-glutamine (2 mM). The cell line was maintained in 5% CO2 at 37°C, with media being replaced every 3–4 days. Once cells had grown to confluence in the culture flask, they were removed using a rubber policeman, as opposed to using trypsin, which can remove membrane-bound receptors (Gunawardena et al., 2015).
Activation of RAW 264.7 macrophages
For assays, 90 μL of each concentration of catalase (eight concentrations made by serial dilution from 0–2000 U/mL catalase in DMEM; Sigma-Aldrich) were added an hour prior to addition of 10 µL of activator solution. The activator solution consisted of a combination of 10 μg/mL Lipopolysaccharide (LPS) (Salmonella serotype; Sigma-Aldrich) and 10 U/mL (1 U = 0.1 ng/mL) interferon-gamma (IFN-γ), IFN-γ along (25 U/mL) or 400 µg/mL CEA-AGE in DMEM. After activation, the cells were incubated for 24 hours at 37°C and then NO, TNF-α and cell viability were determined. Non-activated cells (exposed to media alone) were used as negative control and activated cells as a positive control (Gunawardena et al., 2015). Recombinant IFN-γ was purchased from PeproTech Asia (Rehovot, Israel).
Determination of NO scavenging activity of catalase
In a 96-well plate, 50 μL of a freshly dissolved 5 mM sodium nitroprusside solution in 20 mM sodium phosphate solution, pH 7.4, 0.09% NaCl was added to 50 μL of a solution of catalase in various concentrations ranging from 2000 U/mL to 31.25 U/mL for 1 hour at 50°C. Nitrite formed was analyzed with the Griess reagent (freshly made from sulfanilamide and 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride) as described below (Gunawardena et al., 2015).
Determination of nitrite (as a measure of NO production) by the Griess assay
NO was determined by Griess reagent quantification of nitrite. Griess reagent was freshly made up of equal volumes of 1% sulfanilamide (Sigma-Aldrich) and 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride (Sigma-Aldrich) in 5% HCl. From each well, 50 µL of supernatant was transferred to a fresh 96-well plate and mixed with 50 µL of Griess reagent and measured at 540 nm in a POLARstar Omega microplate reader (BMG Labtech, Mornington, Australia). The concentration of nitrite was calculated using a standard curve with sodium nitrate (0–500 µM; Sigma-Aldrich) and linear regression analysis (Gunawardena et al., 2015).
Determination of NO2 – scavenging activity of catalase
50 µL of a freshly prepared sodium nitrate solution (Sigma-Aldrich; 150 μM NO2 –) was added to 50 μL of a solution of catalase in various concentrations ranging from 2000 U/mL to 31.25 U/mL for 1 hour at 37°C. Nitrite in the media was analyzed with the Griess reagent as described below.
Reduction of nitrate to nitrite
A freshly prepared vanadium chloride solution (Sigma-Aldrich; 50 µL; 200 mg per 25 mL in 1 M HCl) was added to 50 µL of cell culture supernatant to achieve the reduction of nitrate to nitrite. Nitrite in the cell culture media was then analyzed with the Griess reagent as described previously (Egan et al., 2004).
Determination of TNF-α by enzyme-linked immunosorbent assay
The diluted supernatants were used for determination of TNF-α using a commercial sandwich enzyme-linked immunosorbent assay (Peprotech, Rehovot, Israel) according to the manufacturer’s protocol with a few modifications. In brief, the capture antibody was used at a concentration of 1.5 μg/mL in PBS (1.9 mM NaH2PO4, 8.1 mM Na2HPO4, 154 mM NaCl) (pH 7.4). Serial dilutions of TNF-α standard from 0 to 10,000 pg/mL in diluent (0.05% Tween-20, 0.1% bovine serum albumin (Sigma-Aldrich) in PBS) were used as internal standard. TNF-α was detected with a biotinylated second antibody and an avidin-peroxidase conjugate with tetramethyl benzidine as the detection reagent. The color development was monitored at 655 nm, taking readings every 5 minutes. After about 30 minutes the reaction was stopped using 0.5 M sulphuric acid and the absorbance was measured at 450 nm using a POLARstar Omega microplate reader (BMG Labtech, Mornington, Australia) and expressed as a percentage of that in control cells after conversion of the concentrations by using a standard curve constructed with defined concentrations of TNF-α. Curve fitting of this standard curve and extrapolation of experimental data were performed using non-linear regression analysis (Gunawardena et al., 2015).
Determination of cell viability by the Alamar Blue assay
Alamar Blue solution (100 µL; 10% Alamar Blue (Resazurin; Sigma-Aldrich) in DMEM media) was added to each well and incubated at 37°C for 1 hour. After incubation, fluorescence intensity was measured with the microplate reader (excitation at 530 nm and emission at 590 nm) and results were expressed as a percentage of the intensity of that in control cells (Gunawardena et al., 2015).
Determination of hydrogen peroxide in cell culture medium via perchloric acid-ferrous oxidation-xylenol orange assay
To measure hydrogen peroxide in the medium, 100 μL DMEM (with 0.1% FBS) containing 200 µM hydrogen peroxide was added to each well and supernatants (10 µL) were collected in intervals indicated. Supernatants were then diluted nine times with DMEM (total volume 90 µL).10 μL Fox-reagent (2.5 mM ferrous ammonium sulfate, 2.5 mM xylenol orange, 1.10 M HClO4) was added to the diluted supernatants and mixed. The plate was incubated for 20 minutes and the absorbance was measured at 540 nm in a POLARstar Omega microplate reader (BMG Labtech, Mornington, Australia). The concentration of hydrogen peroxide was calculated using a standard curve with hydrogen peroxide (0–125 µM), and linear regression analysis (Maczurek, 2011).
Statistical analysis
Calculations were performed using MS-Excel 2010 software (Microsoft, Redmond, Washington, USA). The Concentration of an inhibitor where the response (is reduced by half (IC50) values were obtained by using the sigmoidal concentration-response function in GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). The results were expressed as the mean ± standard error of the mean (SEM) or standard deviation (SD). Statistical analysis was carried out using a one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison tests. A value of P < 0.05 was considered statistically significant.
Results
Pro-inflammatory activation patterns of different ligands (LPS/IFN-γ, IFN-γ alone and CEA-AGE)
In a first experiment, RAW264.7 macrophages were activated in a concentration-dependent manner with LPS (up to 10 μg/mL) + IFN-γ (up to 10 U/mL (1 U = 0.1 ng), IFN-γ (25 U/mL) or CEA-AGE (up to 400 μg/mL) (Gunawardena et al., 2014a, b, 2015). NO and TNF-α production, as well as cell viability, were determined after 24 hours (Figure 2). All pro-inflammatory ligands produced similar maximal amounts of NO (corresponding to 80 μM nitrite). For TNF-α, CEA-AGE and the combination of LPS + IFN-γ produced about 80 ng/mL, while IFN-γ alone only produced about 60 ng/mL at the maximum concentration (Figure 2). LPS alone did produce less than 5 µM NO and 5 ng/mL TNF-α (data not shown), and was therefore not used for further experiments as a sole activator.
Figure 2.

Dose-response curves for (A) LPS/IFN-γ, (B) IFN-γ and (C) CEA-AGE for NO production with cell viability and (D) LPS/IFN-γ, (E) IFN-γ and (F) CEA-AGE for TNF-α production with cell viability.
RAW264.7 macrophages were activated with LPS + IFN-γ, IFN-γ or CEA-AGE in a concentration-dependent manner and measured the NO and TNF-α inhibition and cell viability after 24 hours. Results represent the mean ± SEM of two experiments in triplicate. Statistical analysis was carried out using a one-way analysis of variance followed by Dunnett’s multiple comparison tests (*P < 0.05, **P < 0.01, and ***P < 0.001, vs. non-activated cells). LPS: Lipopolysaccharide; IFN-γ: interferon-gamma; CEA-AGE: chicken egg albumin-derived advanced glycation end product; NO: nitric oxide; TNF: tumor necrosis factor.
Catalase inhibits NO and TNF-α production
RAW 264.7 macrophages were activated with LPS (10 μg/mL) + IFN-γ (10 U/mL [1 U = 0.1 ng/mL]), IFN-γ (25 U/mL) or CEA-AGE (400 μg/mL) in the presence of increasing doses of catalase (ranging from 0 to 2000 U/mL) for 24 hours, after which NO and TNF-α production were determined. Addition of catalase to LPS/IFN-γ and IFN-γ activated RAW264.7 macrophages led to inhibition of NO production at much lower catalase concentration (IC50 values were 200 ± 13 U/mL and 278 ± 14 U/mL, respectively) than to CEA-AGE activated RAW 264.7 cells (IC50 > 2000 U/mL) (Figure 3). TNF-α production appears to be less dependent on cell-to-cell hydrogen peroxide signaling, as the maximum inhibition upon the addition of catalase was only around 40% (IC50 > 2000 U/mL for all three ligands or ligand combinations). The percentages of TNF-α inhibition of LPS/IFN-γ, IFN-γ and CEA-AGE activated RAW 264.7 macrophages at a maximum concentration of catalase (2000 U/mL) were 64 ± 9%, 59 ± 1% and 57 ± 1%, respectively (Table 1 and Figure 3).
Figure 3.

NO and TNF-α production with cell viability of RAW 264.7 macrophages activated with (A) LPS + IFN-γ, (B) IFN-γ and (C) CEA-AGE.
RAW 264.7 macrophages were activated with LPS (10 μg/mL) + IFN-γ (10 U/mL (1 U = 0.1 ng/mL), IFN-γ (25 U/mL) or CEA-AGE (400 μg/mL). NO and TNF-α production were determined in the media after 24 hours of incubation with increasing concentrations of catalase. Results represent the mean ± SEM of two experiments in triplicate. Statistical analysis was carried out using a one-way analysis of variance followed by Dunnett’s multiple comparison tests (*P < 0.05, **P < 0.01, and ***P < 0.001, vs. positve control (activated cells in the absence of catalase). NO: Nitric oxide; TNF: tumor necrosis factor; LPS: lipopolysaccharide; IFN-γ: interferon-gamma; CEA-AGE: chicken egg albumin-derived advanced glycation end product.
Table 1.
NO and TNF-α production by RAW 264.7 macrophages activated with LPS/IFN-γ, IFN-γ, and CEA-AGE in the presence of catalase
| Activator | Inhibition of NO production (IC50 in U/mL) | Inhibition of TNF-α production (IC50 in U/mL) | % Cell viability at maximum catalase concentration |
|---|---|---|---|
| LPS/IFN-γ | 200±13 | >2000 (64±9++) | 100±11 |
| IFN-γ | 278±14 | >2000 (59±1++) | 93±6 |
| CEA-AGE | >2000 (73 ±1+) | >2000 (57±1++) | 100±6 |
+ % NO production at maximum concentration of catalase; ++ %TNF-α production at maximum concentration of catalase. Results represent the mean ± SD. NO: Nitric oxide; TNF: tumor necrosis factor; LPS: lipopolysaccharide; IFN-γ: interferon-gamma; CEA-AGE: chicken egg albumin-derived advanced glycation end product; IC50: half-maximal inhibitory concentration.
Direct NO scavenging
To find out if direct NO scavenging is the reason for the decreased amount of nitrite in the presence of catalase, a control experiment in a cell-free setup was created. A freshly dissolved 5 mM sodium nitroprusside solution (50 μL) in 20 mM sodium phosphate solution, pH 7.4, 0.09% NaCl was added to 50 μL of a solution of catalase in various concentrations ranging from 2000 U/mL to 31.25 U/mL for 1 hour at 50°C. NO was generated by the NO donor sodium nitroprusside (Roncaroli et al., 2005), which is then converted to nitrite. After incubation of the NO-producing set-up with catalase, Griess reagent was added and the concentration of nitrite was measured. If catalase scavenges NO generated by the NO donor sodium nitroprusside, then the NO concentration would decrease by increasing concentrations of catalase. To our relief, we could not find evidence that catalase scavenges NO directly (Figure 4A).
Figure 4.
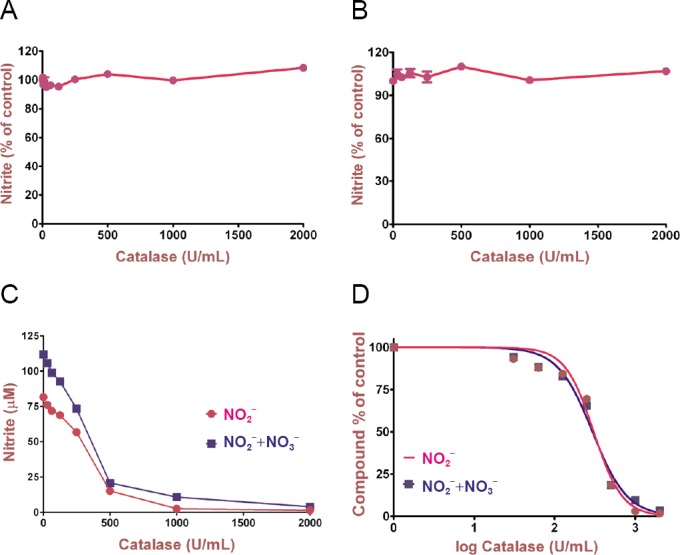
Control experiments investigating possible direct NO and NO2– scavenging activity as well as nitrate oxidation by catalase.
(A) NO production was measured as the concentration of nitrite present in the media after 1 h of incubation of the catalase with 5 mM sodium nitroprusside (SNP) at 50°C. Data points were expressed as % of control and represent the mean of four replicates. (B) The concentration of nitrite present in the media after 1 hour of incubation of the catalase with 150 μM NO2– at 37°C. Data points were expressed as % of control and represent the mean of three replicates. (C, D) To exclude that the downregulation of NO production is caused by oxidation of nitrite to nitrate, a reduction step of nitrate to nitrite with vanadium chloride was performed. C and D contain the same data expressed as a linear plot depicting original data for nitrite concentration and a logarithmic plot normalized to 100%. Results represent the mean ± SEM of five experiments. Statistical analysis was carried out using unpaired t-test. NO: Nitric oxide.
Direct (nitrite) NO2 – scavenging
To examine if direct NO2 – scavenging was the reason for the decreased amount of nitrite levels in the presence of catalase in our experiments, 50 µL of freshly prepared sodium nitrate (equivalent to 150 μM NO2 –) was added to 50 μL of a solution of catalase in different concentrations starting from 2000 U/mL to 31.25 U/mL for 1 hour at 37°C. After incubation of the NO-producing set-up with catalase, Griess reagent was added and the concentration of nitrite was measured. Based on these data, catalase did also not scavenge NO2 – (Figure 4B).
Conversion of nitrite to nitrate
While at baseline (no catalase), the concentration of nitrate + nitrate was 112 µM, the concentration of nitrite alone was only 78 µM, ([nitrite] = 34 µM) was observed, a concentration-dependent downregulation of NO production by increasing concentrations of catalase was still observed (Figure 4C and D).
These control experiments suggest indicating that neither direct NO or NO2 – scavenging or oxidation of nitrite are the major modes of action of catalase, further supporting our hypothesis that its activity on hydrogen peroxide traveling between cells is the main reason for a decrease in the overall pro-inflammatory response.
Potential uptake of catalase into the macrophages
One of the other observations in our study was that catalase concentrations required for eliminating hydrogen peroxide in these assays seemed very high. However, one could argue, that - as an alternative mechanism for this observation- a substantial amount of catalase might have been internalized in the macrophages and that this mechanism would rather be the cause of the downregulation of the pro-inflammatory mediators rather than scavenging extracellular hydrogen peroxide, the mechanism proposed by us.
Therefore, we have conducted a row of experiments to investigate if added catalase (e.g. via internalization) increases the overall capacity of cells to detoxify hydrogen peroxide.
In the first experiment, RAW 264.7 macrophages were exposed to high catalase concentration (1000 U/mL) for 24 hours (we have incubated the 96 well plates for 48 hours with 20% FBS to simulate cell culture conditions and block the walls of the wells to minimize protein absorption before adding catalase). After this time period, the well was washed with PBS to remove the added catalase. After that, 200 µM hydrogen peroxide was added to the cells and samples were taken 2.5, 5, 10 and 20 minutes intervals to determine the hydrogen peroxide concentration (Additional Figure 1 (583.5KB, tif) ). When the cells were incubated with 200 µM hydrogen peroxide, cells without catalase degraded hydrogen peroxide with a first order rate constant of 4.77 ± 0.06 × 10–3/s, whereas the cells with added 1000 U/mL catalase (for 24 hours) degraded hydrogen peroxide with a first-order rate constant of 6.17 ± 0.11 × 10–3/s (increase of 1.40 × 10–3/s = 29.3%) (Additional Table 1).
Additional Table 1.
Half-life and degradation rate of hydrogen peroxide by catalase in the different experimental set-ups
| Experimental set-up | H2O2 half-life (t1/2)(minute) | H2O2 half-life (t1/2)(second) | First order rate constant (k) (10-3/s) |
|---|---|---|---|
| Cells without added catalase | 2.42 ± 0.18 | 145 ± 10.8 | 4.77 ± 0.06 |
| Cells with added catalase | 1.87 ± 0.28 | 112 ± 16.8 | 6.17 ± 0.11 |
| Catalase absorbed at walls of the cell culture plate | 6.50 ±0 .32 | 390 ± 19.5 | 1.78 ± 0.04 |
Data are expressed as the mean ± SD.
However, the increased catalase activity could also be due to catalase being absorbed to the plastic surface of the cell culture walls. Therefore, in a second experiment, we investigated how much of the increased catalase activity is caused by catalase absorption to the walls. Indeed, catalase adsorbed to the wall of the cell culture plates detoxified hydrogen peroxide at a rate of 1.78 ± 0.05 × 10–3/s (Additional Table 1). This value was slightly higher than the difference between cells incubated with or without catalase (1.40 × 10–3/s), indicating that the increased activity of cells incubated with extracellular catalase was due to catalase bound to the walls rather than taken up by the cells.
Discussion
It is already established that hydrogen peroxide can act as an intracellular signaling molecule in in redox-active pathways (Gough and Cotter, 2011; Marinho et al., 2014; Sies, 2017). Hydrogen peroxide can carry a redox signal from the site of its generation (such as the membrane-bound NADPH oxidase) to a target protein such as a redox-sensitive kinase or transcription factor. For example, hydrogen peroxide activates pathways leading to activation of a variety of transcription factors, e.g. in bacteria (OxyR and PerR) (Imlay, 2015), in lower eukaryotes (Yap1, Maf1, Hsf1 and Msn2/4) and in mammalian cells (activator protein 1, nuclear resonse factor 2, cyclic-AMP responsive element binding (CREB), heat shock factor protein 1, hypoxia inducible factor 1, tumor protein 53, nuclear factor kappa B, NOTCH, specific protein 1, and sterol regulatory element-binding protein 1) (Marinho et al., 2014). However, the extracellular signaling roles of hydrogen peroxide, particularly in the mediation of cell-cell interactions, are not well defined. As hydrogen peroxide can diffuse through cell membranes, we have hypothesized that it can relay pro-inflammatory signals from one cell to the next, thus acting as a true first (intercellular) messenger. Proving our hypothesis might make a significant contribution to the development of extracellular hydrogen peroxide scavengers as anti-inflammatory drugs. If we can provide evidence that this mechanism is a general signaling mechanism for propagating and distributing an inflammatory response in a tissue, then hydrophilic, large molecular weight compounds which cannot easily enter a cell, can still act as anti-inflammatory drugs.
Many pro-inflammatory ligands lead to superoxide production, followed by conversion to hydrogen peroxide through a mechanism known as the “respiratory burst” (Root et al., 1975). We have selected three ligands, LPS, IFN-γ, and AGEs. Bacterial endotoxins such as LPS bind to Toll-like receptor 4, activate the Proto-oncogene tyrosine-protein kinase Src (Src) kinase/Phosphoinositide 3-kinase (PI3K) pathway, induce activation of p47phox, leading to superoxide production. IFN-γ leads to production of superoxide by binding to the IFN-γ receptor, activation of the JAK/STAT pathway and upregulation of the transcription of various NADPH oxidase subunits, AGEs can also induce superoxide production via the receptor for AGEs and the PI3/MAPK pathway, including ERK1/2.
According to our hypothesis, hydrogen peroxide acting as a first messenger would travel through the extracellular space and diffuse into adjacent cells. Extracellular added catalase, however, converts hydrogen peroxide to water and oxygen in the extracellular environment and therefore hydrogen peroxide as the pro-inflammatory signal would not enter the next cell, resulting in a decrease of the readouts, NO and TNF-α in a concentration-dependent manner.
As the down-regulation of NO was particularly strong, it could be hypothesized that catalase a) scavenges NO directly, b) scavenges nitrite directly, c) facilitates to oxidation of nitrite to nitrate, and that one of these mechanisms could be an alternative cause for its inhibitory effects on nitrite levels.
One other point of criticism might be that - in the presence of catalase - any nitrite formed is being oxidized to nitrate by additional oxygen produced by catalase, and that this phenomenon might be the reason for the apparent downregulation of NO production as nitrite in medium is usually the readout for NO. To exclude this possibility, we have performed a third control experiment where we have included reduction step of nitrate to nitrite and could, therefore, measure total nitrite. If catalase would simply shift the ratio of nitrate/nitrite favor of nitrate, then the total nitrate+ nitrite concentration would not be decreased by increasing concentrations of catalase.
In summary, our two control experiments suggest that extracellular addition of catalase does not lead to a substantial increase of intracellular catalase activity and therefore this mechanism is not the major cause for the downregulation of TNF-α and NO production we have observed.
In this study, we present convincing evidence for this novel paradigm in cell-to-cell communication using three structurally different pro-inflammatory activators, and two readouts, NO and TNF-α. RAW 264.7 macrophages were activated with the three following ligands: LPS (binding to Toll like receptor 4 (Beutler, 2002), interferon-γ (IFN-γ, binding to the interferon-γ receptor) (Griggs et al., 1992) or AGEs (binding to receptor for AGEs) (Srikanth et al., 2011) in the presence of increasing concentrations of (hydrogen peroxide detoxifying) catalase. As inflammatory readouts, NO and TNF-α were used. Consistent with our hypothesis, concentration-dependent inhibition of NO and TNF-α production in response to all three ligands/ligand combinations (IFN-γ, LPS, and CEA-AGE) in the presence of increasing concentration of catalase was observed.
Our results were – at first sight – not consistent with the data published by Nakao et al. (2008). They have reported that hydrogen peroxide induces TNF-α, but not NO production in RAW 264.7 macrophages. Although these data look like a discrepancy, at first sight, the RAW 264.7 macrophages in the study published by Nakao et al. were only exposed to hydrogen peroxide, but not activated by pro-inflammatory extracellular ligands like in our study.
One interesting observation was that the inhibition of AGE-dependent signaling was much less influenced by extracellular hydrogen peroxide scavenging that that of the other ligands. Although the mechanism of AGE-induced activation of NADPH oxidase and superoxide production via the receptor for AGEs has been described in human endothelial cells (Wautier et al., 2001) and polymorphonuclear leukocytes (Bernheim et al., 2001) as for the other ligands, it has also been shown that AGEs can also suppress superoxide production (Bernheim et al., 2001). Furthermore, hydrogen peroxide formation from superoxide might be further compromised by downregulation of Mn-dependent superoxide dismutase by AGEs via the AGE-receptor-1 and p66shc-dependent FKHRL1 phosphorylation (Cai et al., 2007). In summary, it appears that AGE signaling is much less dependent on hydrogen peroxide extracellular signaling that that of the other pro-inflammatory stimuli.
Another interesting and unexpected finding of our study was that scavenging of extracellular hydrogen peroxide down-regulated nearly 100% of NO, but only 40% of TNF-α production. This suggests that intercellular signals transmitted by hydrogen peroxide are more relevant for NO than for TNF-α, as TNF-α production appears to be less dependent on cell to cell hydrogen peroxide signaling, as it was less affected than NO by the addition of catalase for all three ligands or ligand combinations. In summary, our results demonstrate that hydrogen peroxide travels between cells and induces a pro-inflammatory signal in an adjacent cell and that it is indeed a true first messenger for pro-inflammatory signaling.
Limitations of the study: However, we also need to point out the limitations of the study. We have used only RAW 264.7 macrophages to demonstrate that hydrogen peroxide – like NO – acts as a true first (intercellular) messenger for a multitude of pro-inflammatory ligands. The results may be different (and the ratio for the downregulation of the NO vs. TNF-α) in other cell lines or primary cells. Moreover, in this experiment, we have used only three different ligands for the activation of RAW 264.7 macrophages, and it would be interesting to observe to what extent hydrogen peroxide is involved in signaling pathways stimulated by other pro-inflammatory receptors such as other Toll-like receptors. Furthermore, it would be possible to use large antioxidant (hydrogen peroxide scavenging) molecules instead of catalase to prove to support our hypothesis.
In addition to cell-based experiments, one of the important questions for a future clinical application is whether scavenging extracellular hydrogen peroxide by introducing catalase into a living organism would be a suitable treatment for inflammation. Indeed, there is some clinical evidence for this approach. For example, it was reported that red blood cells induce hypoxic inflammation by producing reactive oxygen species that diffuse to endothelial cells of adjoining blood vessels in a rat model. The authors demonstrated that all hypoxia-induced responses were completely inhibited when catalase was added to the infusion to neutralize red blood cells-derived reactive oxygen species (Kiefmann et al., 2008).
In conclusion, our results suggest that hydrogen peroxide is a pro-inflammatory first messenger, as it travels through the extracellular space and enters and activates adjacent cells. Like NO, we suggest that it is a ubiquitous first messenger, able to transmit cell-to-cell pro-inflammatory signals such as NO and TNF-α. In a therapeutic setting, our data suggest that compounds acting as hydrogen peroxide scavengers might not even need to enter the cell to act as anti-inflammatory drugs.
Additional files:
Additional Figure 1 (583.5KB, tif) : Time course of H2O2 degradation in cells without added catalase, cells with added catalase, and catalase adsorbed at the 96 well plate’s walls.
Time course of H2O2 degradation in cells without added catalase, cells with added catalase, and catalase adsorbed at the 96 well plate's walls.
Data are expressed as the mean ± SD.
Additional Table 1: Half-life and degradation rate of the hydrogen peroxide by catalase in the different experimental set-ups.
Additional file 1: Open peer review reports 1 (103.3KB, pdf) and 2 (98.6KB, pdf) .
Footnotes
Conflicts of interest: None declared.
Financial support: Australian Postgraduate Award (APA) Ph.D. fellowship by Western Sydney University to DG.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Paulina Carriba, Cardiff University, UK; Attila Köfalvi, University of Coimbra, Portugal.
Funding: Australian Postgraduate Award (APA) Ph.D. fellowship by Western Sydney University to DG.
P-Reviewers: Carriba P, Köfalvi A; C-Editors: Zhao M, Li CH; T-Editor: Liu XL
References
- 1.Bernheim J, Rashid G, Gavrieli R, Korzets Z, Wolach B. In vitro effect of advanced glycation end-products on human polymorphonuclear superoxide production. Eur J Clin Invest. 2001;31:1064–1069. doi: 10.1046/j.1365-2362.2001.00911.x. [DOI] [PubMed] [Google Scholar]
- 2.Beutler B. TLR4 as the mammalian endotoxin sensor. Curr Top Microbiol Immunol. 2002;270:109–120. doi: 10.1007/978-3-642-59430-4_7. [DOI] [PubMed] [Google Scholar]
- 3.Bienert GP, Moller AL, Kristiansen KA, Schulz A, Moller IM, Schjoerring JK, Jahn TP. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007;282:1183–1192. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 4.Cai W, He J, Zhu L, Striker GE, Vlassara H. AGER1 counteracts cellular oxidant stress induced by AGEs via negative regulation of p66shc-dependent FKHRL1 phosphorylation. Am J Physiol Cell Physiol. 2007 doi: 10.1152/ajpcell.00350.2007. [DOI] [PubMed] [Google Scholar]
- 5.de Rooij SR, Nijpels G, Nilsson PM, Nolan JJ, Gabriel R, Bobbioni-Harsch E, Mingrone G, Dekker JM. Low-grade chronic inflammation in the relationship between insulin sensitivity and cardiovascular disease (RISC) population: associations with insulin resistance and cardiometabolic risk profile. Diabetes Care. 2009;32:1295–1301. doi: 10.2337/dc08-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dukic-Stefanovic S, Gasic-Milenkovic J, Deuther-Conrad W, Munch G. Signal transduction pathways in mouse microglia N-11 cells activated by advanced glycation endproducts (AGEs) J Neurochem. 2003;87:44–55. doi: 10.1046/j.1471-4159.2003.01988.x. [DOI] [PubMed] [Google Scholar]
- 7.Egan CG, Lockhart JC, Ferrell WR. Pathophysiology of vascular dysfunction in a rat model of chronic joint inflammation. J Physiol. 2004;557:635–643. doi: 10.1113/jphysiol.2004.062984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gough DR, Cotter TG. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis. 2011;2:e213–213. doi: 10.1038/cddis.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griggs ND, Jarpe MA, Pace JL, Russell SW, Johnson HM. The N-terminus and C-terminus of IFN-gamma are binding domains for cloned soluble IFN-gamma receptor. J Immunol. 1992;149:517–520. [PubMed] [Google Scholar]
- 10.Gunawardena D, Shanmugam K, Low M, Bennett L, Govindaraghavan S, Head R, Ooi L, Münch G. Determination of anti-inflammatory activities of standardised preparations of plant- and mushroom-based foods. Eur J Nutr. 2014a;53:335–343. doi: 10.1007/s00394-013-0531-9. [DOI] [PubMed] [Google Scholar]
- 11.Gunawardena D, Bennett L, Shanmugam K, King K, Williams R, Zabaras D, Head R, Ooi L, Gyengesi E, Münch G. Anti-inflammatory effects of five commercially available mushroom species determined in lipopolysaccharide and interferon-gamma activated murine macrophages. Food Chem. 2014b;148:92–96. doi: 10.1016/j.foodchem.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 12.Gunawardena D, Karunaweera N, Lee S, van Der Kooy F, Harman DG, Raju R, Bennett L, Gyengesi E, Sucher NJ, Münch G. Anti-inflammatory activity of cinnamon (C. zeylanicum and C.cassia) extracts - identification of E-cinnamaldehyde and o-methoxy cinnamaldehyde as the most potent bioactive compounds. Food Funct. 2015;6:910–919. doi: 10.1039/c4fo00680a. [DOI] [PubMed] [Google Scholar]
- 13.Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J. 2001;353:411. doi: 10.1042/0264-6021:3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hancock JT. The role of redox mechanisms in cell signalling. Mol Biotechnol. 2009;43:162–166. doi: 10.1007/s12033-009-9189-1. [DOI] [PubMed] [Google Scholar]
- 15.Heneka MT. Inflammation in Alzheimer’s disease. Clin Neurosci Res. 2006;6:247–260. [Google Scholar]
- 16.Imlay JA. Transcription factors that defend bacteria against reactive oxygen species. Annu Rev Microbiol. 2015;69:93–108. doi: 10.1146/annurev-micro-091014-104322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson WJ, Sung CP. Rat macrophage treatment with lipopolysaccharide leads to a reduction in respiratory burst product secretion and a decrease in NADPH oxidase affinity. Cell Immunol. 1987;108:109–119. doi: 10.1016/0008-8749(87)90197-3. [DOI] [PubMed] [Google Scholar]
- 18.Kiefmann R, Rifkind JM, Nagababu E, Bhattacharya J. Red blood cells induce hypoxic lung inflammation. Blood. 2008;111:5205–5214. doi: 10.1182/blood-2007-09-113902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maczurek NE. Identification of neuroprotective genes from hydrogen peroxide resistant neuron-like cell lines – an innovative approach to discover novel drug targets for Alzheimer's disease. School of Medicine, Western Sydney University. 2011 [Google Scholar]
- 20.Marinho HS, Real C, Cyrne L, Soares H, Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014;2:535–562. doi: 10.1016/j.redox.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakao N, Kurokawa T, Nonami T, Tumurkhuu G, Koide N, Yokochi T. Hydrogen peroxide induces the production of tumor necrosis factor-alpha in RAW 264. 7 macrophage cells via activation of p38 and stress-activated protein kinase. Innate Immun. 2008;14:190–196. doi: 10.1177/1753425908093932. [DOI] [PubMed] [Google Scholar]
- 22.Noor R, Mittal S, Iqbal J. Superoxide dismutase--applications and relevance to human diseases. Med Sci Monit. 2002;8:RA210–215. [PubMed] [Google Scholar]
- 23.Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–783. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- 24.Roncaroli F, van Eldik R, Olabe JA. Release of NO from reduced nitroprusside ion. Iron-dinitrosyl formation and NO-disproportionation reactions. Inorg Chem. 2005;44:2781–2790. doi: 10.1021/ic050070c. [DOI] [PubMed] [Google Scholar]
- 25.Root RK, Metcalf J, Oshino N, Chance B. H2O2 release from human granulocytes during phagocytosis. I.Documentation, quantitation, and some regulating factors. J Clin Invest. 1975;55:945–955. doi: 10.1172/JCI108024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017;11:613–619. doi: 10.1016/j.redox.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srikanth V, Maczurek A, Phan T, Steele M, Westcott B, Juskiw D, Münch G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging. 2011;32:763–777. doi: 10.1016/j.neurobiolaging.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 28.Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J Biol Chem. 2003;278:24233–24241. doi: 10.1074/jbc.M212389200. [DOI] [PubMed] [Google Scholar]
- 29.Vieceli Dalla Sega F, Zambonin L, Fiorentini D, Rizzo B, Caliceti C, Landi L, Hrelia S, Prata C. Specific aquaporins facilitate Nox-produced hydrogen peroxide transport through plasma membrane in leukaemia cells. Biochim Biophys Acta. 2014;1843:806–814. doi: 10.1016/j.bbamcr.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 30.Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab. 2001;280:E685–694. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]
- 31.Zhang WJ, Wei H, Hagen T, Frei B. Alpha-lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc Natl Acad Sci U S A. 2007;104:4077–4082. doi: 10.1073/pnas.0700305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time course of H2O2 degradation in cells without added catalase, cells with added catalase, and catalase adsorbed at the 96 well plate's walls.
Data are expressed as the mean ± SD.