

Abstract
Described here is a chemo-selective enrichment strategy, termed Semi-Tryptic Peptide Enrichment Strategy for Proteolysis Procedures (STEPP), to isolate the semi-tryptic peptides generated in mass spectrometry-based proteome-wide applications of limited proteolysis methods. The strategy involves reacting the ε-amino groups of lysine side chains and any N-termini created in the limited proteolysis reaction with isobaric mass tags. A subsequent digestion of the sample with trypsin and the chemo-selective reaction of the newly exposed N-termini of the tryptic peptides with N-hydroxysuccinimide (NHS) activated agarose resin removes the tryptic peptides from solution leaving only the semi-tryptic peptides with one non-tryptic cleavage site generated in the limited proteolysis reaction for subsequent LC-MS/MS analysis. As part of this work the STEPP technique is interfaced with two different proteolysis methods including the Pulse Proteolysis (PP) and Limited Proteolysis (LiP) methods. The STEPP-PP workflow is evaluated in two proof-of-principle experiments involving the proteins in a yeast cell lysate and two well-studied drugs, cyclosporin A and geldanamycin. The STEPP-LiP workflow is evaluated in a proof-of-principle experiment involving the proteins in two cell culture models of human breast cancer, MCF-7 and MCF-10A cell lines. The STEPP protocol increased the number of semi-tryptic peptides detected in the LiP and PP experiments by 5- to 10-fold. The STEPP protocol not only increases the proteomic coverage, but also increases the amount of structural information that can be gleaned from limited proteolysis experiments. Moreover, the protocol also enables the quantitative determination of ligand binding affinities.
Graphical Abstract
INTRODUCTION
Limited proteolysis methods have long been utilized to obtain information about the structure and function of proteins.1 Recently, several limited proteolysis methods have been developed that enable such protein structure and function studies to be performed on the proteomic scale. The methods have included the Limited Proteolysis (LiP),2 Drug Affinity Responsive Target Stability (DARTS),3 and Pulse Proteolysis (PP)4–5 techniques. The LiP approach involves the cleavage of solvent exposed regions of protein structure by a protease using buffer conditions under which proteins are generally folded into their native three-dimensional structure. The LiP approach has proven useful for identifying protein conformational changes related to environmental changes, disease states, post-translational modifications as well as small molecule binding.2, 6–8 The DARTS approach is very similar to LiP in experimental design but specialized in drug-binding detection.3, 9 The pulse proteolysis (PP) approach, which involves measuring the denaturant dependence of a limited proteolysis reaction using a protease with broad specificity, has proven useful for the quantitative analysis of protein folding stabilities and protein-ligand binding affinities.4–5, 10
While limited proteolysis methods for studying the conformational properties of purified proteins and protein-ligand complexes are well established, their recent extension to the proteomic-scale using the DARTS, LiP, and PP approaches has been challenging. Current proteome-wide applications of these techniques have typically relied on either gel-3–5 or solution-based2, 6–10 proteomics strategies. Unfortunately, the relatively low resolving power of gel electrophoresis methods can complicate applications of the LiP, DARTS and PP techniques to complex protein mixtures. Higher resolution LC-MS/MS strategies using bottom-up proteomics methods have been utilized to read out the results of proteome-wide limited proteolysis experiments.2, 6–10 However, the large number of tryptic peptides generated in the bottom-up proteomics readout can make identification of the cleavage sites in the limited proteolysis experiment especially challenging, due to the relatively low abundance of the semi-tryptic peptides that result from such cleavages. The semi-tryptic peptides generated in a limited proteolysis experiment are those that have a non-tryptic cleavage site at one end as a result of the limited proteolysis reaction and a tryptic cleavage site at the other end as a result of the trypsin digestion step used to prepare the sample for bottom-up proteomics analysis.
Recently, attempts have been made to improve the bottom-up proteomics readout in proteome-wide limited proteolysis experiments by separating the intact proteins from the smaller peptides generated in the limited proteolysis reaction. For example, the use of high molecular weight (e.g., 100,000 Da) cut-off filters has been reported to isolate the intact proteins prior to their trypsin digestion for bottom-up proteomics analysis.11 A protein precipitation reaction involving trichloroacetic acid (TCA) has also been used to remove the undigested, intact proteins from the limited proteolysis reaction so that the remaining peptide fragments could be digested with trypsin and subject to a quantitative bottom-up proteomics analysis.12 A drawback to the proteomics readouts employed in the filtration and precipitation approaches is that they do not provide any structural information about the site(s) of conformational changes, as they are both protein-centered readouts.
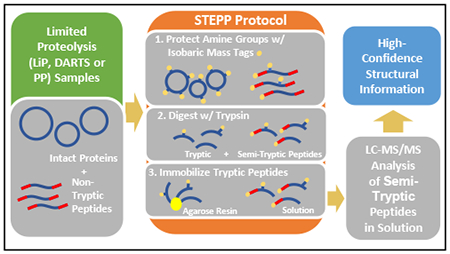
Described here is a general strategy termed, semi-tryptic peptide enrichment for proteolysis procedures (STEPP), to facilitate detection of the limited proteolysis cleavage sites generated in proteome-wide limited proteolysis experiments. The semi-tryptic peptide-centered proteomics readout in the STEPP protocol significantly enhances the information content of limited proteolysis experiments by facilitating the acquisition of more detailed structural information about the site(s) of conformational changes observed in the limited proteolysis experiment. The results of the proof-of-principle studies performed here demonstrate the ability of the STEPP protocol to generate domain- and amino acid-specific structural information about the conformational properties of proteins using the PP and LiP techniques, respectively. In combination with PP the STEPP protocol can also be used to evaluate the dissociation constants, Kd values, of protein-ligand complexes.
EXPERIMENTAL SECTION
Materials
The following materials were from Sigma-Aldrich (St. Louis, MO): guanidine hydrochloride (GdmCl), S-methylmethanesulfonate (MMTS), dimethyl sulfoxide (DMSO), urea, centrifugal filter units (Amicon Ultra-0.5, 10k), tris(hydroxymethyl)aminomethane hydrochloride (Tris•HCl), thermolysin from geobacillus stearothermophilus, proteinase K from tritirachium album, phenylmethylsulfonyl fluoride (PMSF), trifluoroacetic acid (TFA), triethylammonium bicarbonate buffer (TEAB, 1 M, pH 8.5), methacrylic acid N-hydroxysuccinimide(NHS) ester, cytochrome c from equine heart. The following materials were from Thermo-Scientific (Waltham, MA): acetonitrile (ACN, LC-MS grade), 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF), Bestatin, E-64, Leupeptin, Pepstatin A, EDTA solution (Corning, 0.5M, pH 8.0), TMT10plex isobaric label reagent set, N-hydroxysuccinimide(NHS)-activated agarose dry resin (Pierce), trypsin protease MS grade (Pierce). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was from Santa Cruz Biotechnology (Dallas, TX). Phosphate-buffered saline (PBS, pH 7.4, 1X) was from Gibco (Gaithersburg, MD). Geldanamycin (≥98 wt%) was from Chem-Impex International, Inc. (Wood Dale, IL). Cyclosporin A (CsA) was from LKT Laboratories, Inc. MacroSpin columns (silica C4 and C18) were from Nest Group (Southborough, MA).
Cell Culture and Lysis
Yeast strain YDR155C was obtained from Open Biosystems (Lafayette, CO) and cultured in YEPD medium according to standard protocols, which are further described in the Supporting Information. The MCF-7 and MCF-10A cell lines were purchased from American Type Culture Collection (ATCC) (Manassas, VA) and cultured according to ATCC guidelines. The yeast cells in the STEPP-PP experiments were lysed in PBS in the presence of the following protease inhibitors: 1 mM AEBSF, 20 µM leupeptin, 10 µM pepstatin A, 500 µM bestatin and 15 µM E-64. The human cells in the STEPP-LiP experiments were lysed in PBS. Cell lysis was accomplished using 0.5 mm glass beads (in the case of yeast) or 1mm zirconia/silica beads (in the case of the human cell lines) and mechanical disruption at 4 °C with 25 s of disruption and 1 min intervals on ice for 15 cycles. The lysates were centrifuged at 14,000 x g for 10 min at 4 °C. The supernatants were separated and used for subsequent analyses. The total concentration of protein in each lysate was determined using a Bradford assay and adjusted to 5.0 mg/ml. For ligand-binding experiments using PP, the yeast lysate was divided into two equal aliquots. One aliquot was spiked with a solution of the test ligand (CsA or geldanamycin) prepared in DMSO and the other one was spiked with DMSO as vehicle. The concentration of drug in each cell lysate aliquot was 500 µM and 50 µM in the CsA and geldanamycin binding experiments, respectively. The final concentration of DMSO in each cell lysate aliquot was 5% v/v in all experiments. The solutions were equilibrated for either 1 (CsA) or 24 hours (geldanamycin).
STEPP-PP Analysis
In both CsA and geldanamycin-binding experiments, 10 µL aliquots of the (+) and (−) ligand-containing lysate samples were distributed into a series of 30 µL urea-containing buffers (PBS buffer, pH 7.4) where the final urea concentration ranged from 0 to 6.8 M. The final concentration of ligand was 125 µM in the CsA-binding experiment, 12.5 µM in the geldanamycin binding experiment. The samples in the urea-containing buffers were equilibrated for ~2 h at room temperature. The proteolysis reaction was initiated by adding thermolysin to the protein and protein–ligand samples in each denaturant-containing buffer. The thermolysin to total protein ratio was approximately 1:10 (w/w). The thermolysin proteolysis reactions were allowed to proceed for 1 min at room temperature before each reaction was quenched with the addition of 60 µL urea/EDTA solution (~0.2 M EDTA, 9 M urea, pH 8.0). These thermolysin proteolysis reaction conditions were similar to those previously described.5, 10
The proteolyzed samples were subject to the STEPP protocol developed here. As part of the STEPP protocol, the proteolyzed samples were reacted with 1.5 mM TCEP for 1 h at 30 °C and then with 2.5 mM MMTS for 10 min at room temperature. Subsequently, the thermolysin digested samples were labeled with TMT reagents by adding 41uL of ACN containing 0.5 unit of each TMT reagent and incubating for 1.5 hours at room temperature. In the CsA-binding (+) ligand samples, the urea concentrations, from low to high, were labeled with TMT tags 130N, 130C, 126, 127N, 127C, 128N, 128C, 129N, 129C, 131. The (−) ligand samples were labeled in the reverse manner (i.e., the urea concentrations, from high to low, were labeled with TMT tags 130N, 130C, 126, 127N, 127C, 128N, 128C, 129N, 129C, 131). In the geldanamycin-binding experiment (+) ligand samples, the urea-concentrations, from low to high, were labeled with TMT tags 126, 127N, 127C, 128N, 128C, 129N, 129C, 130N, 130C, 131. The (−) ligand samples were labeled in the reverse manner. The labeling reaction was quenched by the addition of 4 µL of a 5% v/v hydroxylamine for 15 min at room temperature. In both experiments, the (+) and (−) ligand samples of the 1st, 3rd, 5th, 7th and 9th (i.e., the odd) urea concentration (from low to high) were combined into one sample, and the samples from the other 5 concentrations (i.e., the even urea concentrations) were combined into another sample. The resulting samples were dried, re-dissolved in 2% v/v TFA solution, and desalted using C18 columns according to the manufacturer’s protocol. The desalted samples were dried, re-dissolved in 100 µL of 0.1 M TEAB solution (pH 8.5), and digested with trypsin overnight at 37 °C. NHS-activated agarose resin and 50 µL of 0.5M NaCl were added to the trypsin-digested samples such that the NHS-activated agarose resin to total peptide ratio was approximately 150:1 (w/w). The tryptic peptides mixture was allowed to react with the NHS-activated agarose resin for 1.5 hours at room temperature before the samples were acidified with 2% v/v TFA and the soluble peptides desalted using C18 columns. The desalted samples were subject to the LC-MS/MS analysis described below.
STEPP-LiP Analysis
Cell lysates generated from five MCF-7 and five MCF-10A cell cultures were each divided into two 10 µL aliquots and each aliquot was combined with 30 µL of PBS. Five of the 10 resulting samples generated from each cell line were treated with proteinase K at an enzyme/substrate ratio of 1/100 (w/w) for 5 min at room temperature. The proteinase K proteolysis reaction was quenched with 5 mM PMSF and the samples were subjected to the same STEPP protocol used in the STEPP-PP Analysis (with the exception that C4 columns were used to desalt the TMT labelled samples instead of C18 columns), which included a trypsin digestion step, to generate 5 “double digestion” samples for each cell line. The other 5 aliquots generated for each cell line were reduced with 1.5 mM TCEP for 1 h at 55 °C, modified with 2.5 mM MMTS for 10 min at room temperature and then treated with trypsin at 37 °C overnight to generate 5 “single digestion” samples for each cell line. In total, five biological replicates of the double and single digestion samples were generated from the MCF-7 and MCF-10A cell lines. The 10 double digested samples were labeled with TMT tags in the STEPP procedure such that one TMT 10-plex contained 5 biological replicates of the MCF-7 double digested samples and 5 biological replicates of the MCF-10A double digest samples. The 10 singly digested samples were also labelled with TMT tags and combined in the same manner as that described above for the double digested samples.
Quantitative LC-MS/MS Analyses
LC-MS/MS analyses were performed on a Q-Exactive HF high-resolution mass spectrometer (Thermo) with a nano-Acquity UPLC system (Waters) and a nano-electrospray ionization source fitted with a SilicaTip emitter (New Objective). Samples were trapped on a 2D Symmetry C18 trapping column with dimensions 180 μm x 20 mm and particle diameter of 5 μm, pore size 100 Å. The trapping time was 5 minutes at 5 μL/minute (99.9:0.1 v/v water/ACN 0.1% formic acid). The samples were separated on a 75 μm x 250 mm high strength silica (HSS) T3 column with 1.8 μm particle diameter (Waters) heated to 55 °C. Peptides were eluted using a gradient of 3–30% acetonitrile with 0.1% formic acid over 90 minutes at a flow rate of 0.3 μL/min. LC-MS/MS data were collected using a top 20 data-dependent acquisition (DDA) method including MS1 at 120k and MS2 at 30k resolution. The AGC target for MS1 was 3 × 106 ions with a max IT of 50 msec. The AGC target for MS2 was 1 × 105 ions with a max IT of 45 msec. The normalized collision energy (NCE) was set to 30 V and the scan range was 375–1600 m/z. The isolation window was 0.7 m/z and the dynamic exclusion time was set to 20.0 seconds.
STEPP-PP Quantitative Proteomic Data Analysis
The raw LC-MS/MS files were searched against the yeast UniProt Knowledgebase (2017–06-07 release) using Proteome Discoverer 2.2 (Thermo). The searches were performed with fixed MMTS modification on cysteine, fixed TMT-10plex labeling of lysine side chains, fixed TMT10plex labeling of peptide N-termini, variable deamidation of asparagine and glutamine, variable oxidation of methionine, and variable acetylation of the protein N-terminus. The precursor ion mass tolerance was set at 10 ppm. The fragment ion mass tolerance was set at 0.02 Da. Trypsin(semi) was set as the enzyme, and up to three missed cleavages were allowed. For peptide quantification, reporter ion abundance was set as “intensity” and the normalization mode and scaling mode were each set as “none.” The peptide FDR confidence cutoff was set as “medium” (i.e., FDR <5%).
The data from different technical replicates of the same sample was exported and combined into one file. PSMs corresponding to tryptic peptides mapping to protein N-termini were filtered out. The TMT reporter ion intensities in all the PSMs recorded for a semi-tryptic peptide sequence were summed. For each unique semi-tryptic peptide, the summed (−) ligand TMT reporter ion intensity was divided by the summed (+) ligand TMT reporter ion intensity to generate a fold-change value for each urea concentration at which TMT reporter ion intensities were recorded. The fold-change values of all the semi-tryptic peptide sequences in each experiment were used to calculate the mean log2(fold-change) and the standard deviation (σ) of its distribution in each experiment (Figure S-1). Hit peptides were identified with two criteria: i) the peptide must have a significantly altered log2(fold-change) (>3σ deviations from the mean log2(fold-change)), for at least two consecutive urea concentrations; ii) these significantly altered log2(fold-change) values must have a fold-change in the same direction for at least two consecutive urea concentrations (e.g., a peptide with fold-changes of >3σ, <−3σ and >3σ at three consecutive urea concentrations was not a hit).
STEPP-LiP Quantitative Proteomic Data Analysis
The raw LC-MS/MS files generated in the STEPP-LiP experiment were searched against the human UniProt Knowledgebase (2017–06-07 release) with the same searching parameters as in STEPP-PP data analysis except that the raw file from the single digestion experiment was searched with enzyme set to trypsin(full). For the double digestion sample, similar to STEPP-PP data analysis, PSMs corresponding to tryptic peptides mapping to protein N-termini were filtered out. The TMT reporter ion intensities in all the PSMs recorded for a semi-tryptic peptide sequence were summed. The sum of each TMT reporter ion recorded for a given semi-tryptic peptide was divided by the average of all 10 of the summed TMT reporter ion intensities for that semi-tryptic peptide to generate a single set of so-called relative TMT reporter ion intensities for each semi-tryptic peptide. For the single digestion sample, TMT reporter ion intensities for each identified protein were generated from Proteome Discoverer and these TMT reporter ion intensities for each protein were also divided by the average of all 10 of the summed TMT reporter ion intensities for that protein to generate a single set of so-called relative TMT reporter ion intensities for each protein.
The data normalization and hit selection procedures used in the STEPP-LiP experiments were directly analogous to those in a previously described LiP experiment using SILAC quantitation.6 Briefly, for each semi-tryptic peptide, the relative TMT reporter ion intensities from the double digestion experiment were divided by the parent protein’s relative TMT reporter ion intensities from the single digestion experiment in order to generate normalized TMT reporter ion intensities, which accounted for protein abundance differences in the two cell lines. These normalized values were used in subsequent analyses and selection of semi-tryptic peptide hits. For each semi-tryptic peptide, a median normalized TMT reporter ion intensity value was determined from the five MCF-7 biological replicates. This value was divided by the median normalized TMT reporter ion intensity value determined from the five MCF-10A biological replicates group to generate a MCF-7/MCF-10A fold-change. The fold-change value was used to determine the proteolysis susceptibility change behavior for each semi-tryptic peptide (e.g., a MCF-7/MCF-10A fold change < 1 meant the semi-tryptic peptide site was less susceptible to proteolysis in MCF-7). Semi-tryptic peptide hits were only selected from those that were identified in at least two biological replicates of both cell lines. For peptide hit selection, a Student’s two-tailed t test was used to identify hit peptides with significantly different normalized TMT reporter ion intensities between the MCF-7 and MCF-10A groups. Hit peptides were selected as those with a p-value less than 0.05.
RESULTS and DISCUSSION
STEPP Workflow
Shown in Figure 1 is the STEPP protocol developed here to enrich the semi-tryptic peptides generated in limited proteolysis experiments performed on the proteomic scale. In the STEPP protocol, any intact proteins and the non-tryptic peptide fragments generated in the limited proteolysis reaction are directly labeled with a set of isobaric mass tags (e.g., TMT 10-plex), which react with the ε-amino groups in lysine side-chains and with the proteins’ and non-tryptic peptides’ N-termini. The isobaric mass tag labeling reactions are quenched, and the samples labeled with different TMT tags are combined. The combined sample (see Figures 2 and 3 for TMT labelling scheme used in STEPP-PP and STEPP-LiP experiments, respectively) is digested with trypsin. This exposes new N-terminal amino groups at the trypsin cleavage sites, which only occur at arginine residues, as lysine cleavage sites are blocked due to the presence of the isobaric mass tag. The trypsin digestion shortens the non-tryptic peptide fragments and generates semi-tryptic peptides, that are suitable for bottom-up shotgun proteomics analysis. However, the trypsin digestion also creates a large number of tryptic peptides. These tryptic peptides are removed from solution through reaction with a N-hydroxysuccinimide (NHS) activated agarose resin. This chemo-selection reaction, while immobilizing tryptic peptides, leaves semi-tryptic peptides in solution and ready for subsequent LC-MS/MS analysis. In theory, each cleavage site resulting from the limited proteolysis reaction in the first digestion step of the STEPP protocol generates two non-tryptic “daughter” peptides. Upon trypsin digestion, these two non-tryptic “daughter” peptides each produce one semi-tryptic peptide that terminates at the non-tryptic digestion site. The semi-tryptic peptide from one of the non-tryptic daughter peptides has a tryptic N-terminus and non-tryptic C-terminus. The semi-tryptic peptide from the other non-tryptic daughter peptide has a non-tryptic N-terminus and tryptic C-terminus. We note that the chemo-selection strategy outlined here only enriches for one of these semi-tryptic peptides (i.e., the semi-tryptic peptide that has a semi-tryptic N-terminus and tryptic C-terminus), as the semi-tryptic peptide with the newly generated tryptic N-terminus is immobilized on the N-hydroxysuccinimide (NHS) activated agarose resin.
Figure 1.
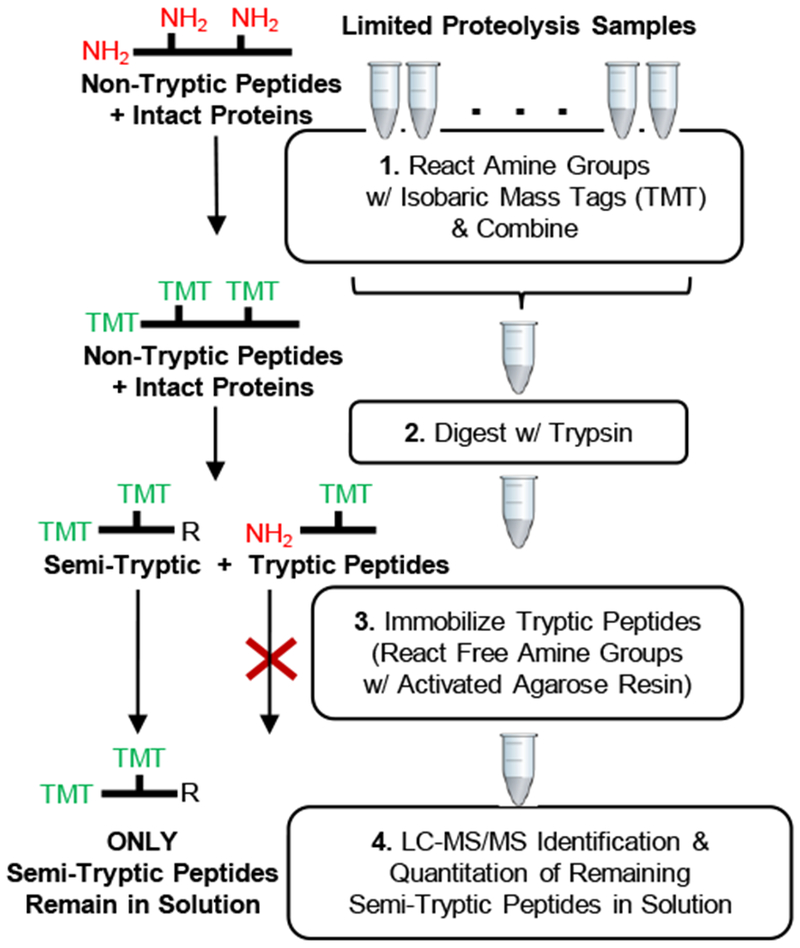
Schematic representation of the STEPP protocol workflow developed in this work.
Figure 2.
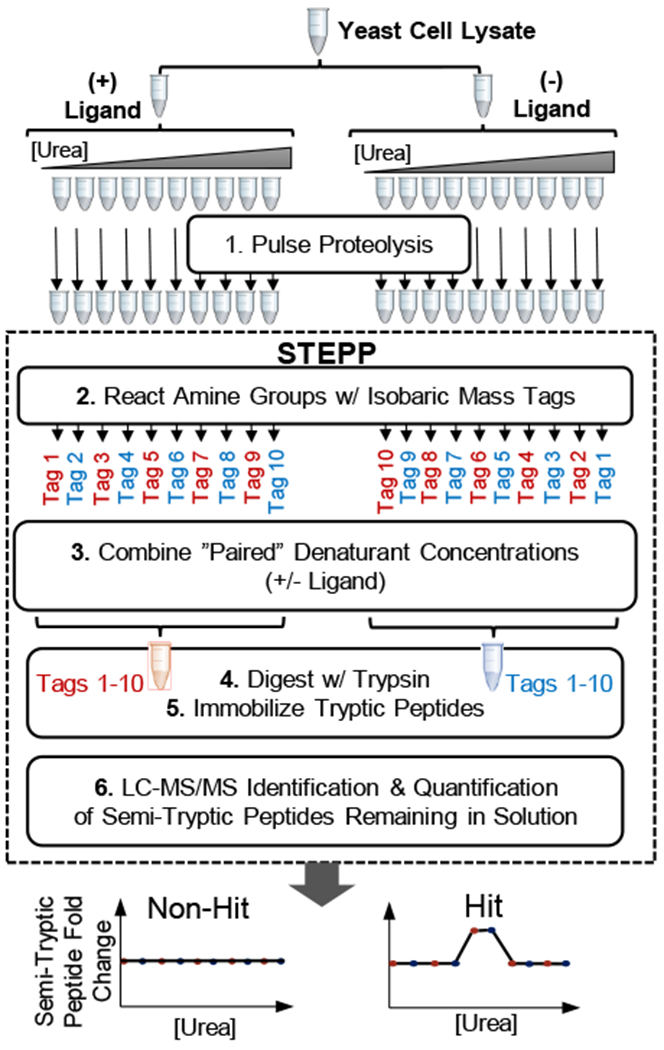
Schematic representation of the STEPP-PP workflow used in this work.
Figure 3.
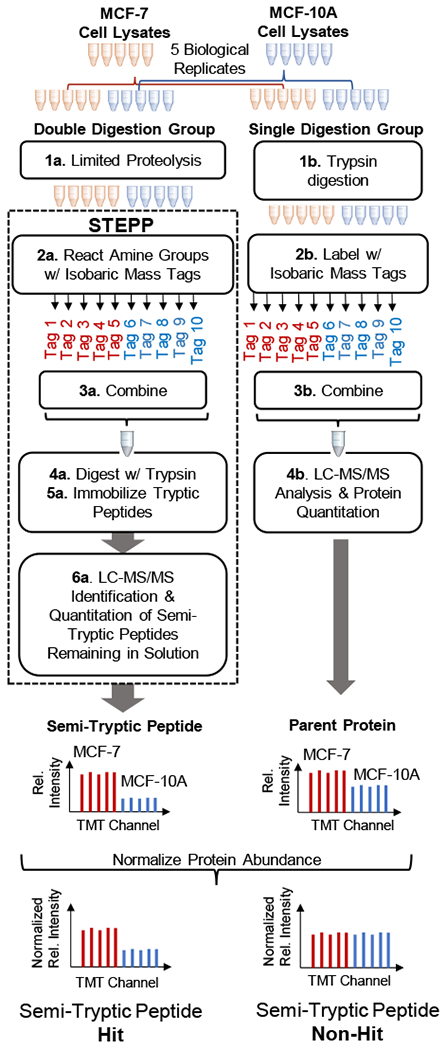
Schematic representation of the STEPP-LiP workflow used in this work.
As part of this work, the ability of the NHS activated agarose resin to immobilize tryptic peptides was tested using tryptic peptides generated from the trypsin digestion of a model protein, cytochrome c (see Supplemental Text). The results of this model study revealed that the resin effectively immobilized all of the tryptic peptides from the sample solution (see Supplemental Figure S-2).
STEPP-PP Analysis of CsA and Geldanamycin Binding to Proteins in a Yeast Cell Lysate
The STEPP protocol was interfaced with PP as illustrated in Figure 2, and the STEPP-PP workflow was used in a proof-of-principle study to identify the protein targets of two well-studied small molecule drugs, CsA and geldanamycin,13–14 using the proteins in a yeast cell lysate. These two model drugs were chosen for these proof-of-principle studies because the binding interactions with their protein targets have been well-studied by conventional methods and by other energetic-based studies, such as chemical denaturation and protein precipitation (CPP), SPROX and SILAC-PP methodology.10, 15–19
In both the CsA and geldanamycin binding experiments close to 900 yeast proteins were identified by more than 4000 unique semi-tryptic peptides (see Table 1, S-2 and S-3). The total number of semi-tryptic peptides was about 10 times more than that observed in a typical proteome-wide LiP experiment with the same number of LC-MS/MS runs.6 The proteomic coverages observed in these STEPP-PP experiments were also significantly greater (~40% larger) than the proteomic coverage observed in similar drug-mode-of action study using SPROX to identify protein targets in the yeast proteome.19
Table 1.
Summary of proteomic coverages and numbers of hits observed in the STEPP-PP CsA and geldanamycin ligand binding experiments using the proteins in a yeast cell lysate.
| Drug Binding Experiment | Protein Assay Criteria | Assayed Peptides (Proteins) | Hit Peptides (Proteins) | Peptides from Known Protein Target(s) | Protein False Positive Rate |
|---|---|---|---|---|---|
| CsA | One peptide | 4265 (812) | 24 (13) | 11 (CPR1), 2 (CPR3) | 1.4% |
| At least two peptides | 3914 (461) | 13 (2) | 0% | ||
| Geldanamycin | One peptide | 4955 (901) | 39 (33) | 4 (HSP82) | 3.6% |
| At least two peptides | 4533 (492) | 10 (4) | 0.6% |
In the CsA binding study, 24 semi-tryptic peptides from 13 proteins were identified as hits, including 13 semi-tryptic peptides from 2 cyclophilin proteins (CPR1 and CPR3) (Figure 4A and B), both of which are known to bind or be inhibited by CsA.20–21 STEPP-PP data from a typical non-hit protein is shown in Figure 4C. In the geldanamycin binding experiment, 39 semi-tryptic peptides from 33 proteins were identified as hits, including 4 semi-tryptic peptides from the known geldanamycin binding protein, HSP82 (yeast HSP90 isoform) (Figure 5A).22 We note that a total of 28 semi-tryptic peptides from HSP82 were assayed. All 23 of the semi-tryptic peptides mapping to the middle and C-terminal domains were not identified as hits, and 4 out of the 5 semi-tryptic peptides mapping to the N-terminal domain of HSP82 were identified as hits. In both the geldanamycin and CsA binding experiments the known binding targets were successfully identified as hits, giving a 0% protein false negative rate.
Figure 4.
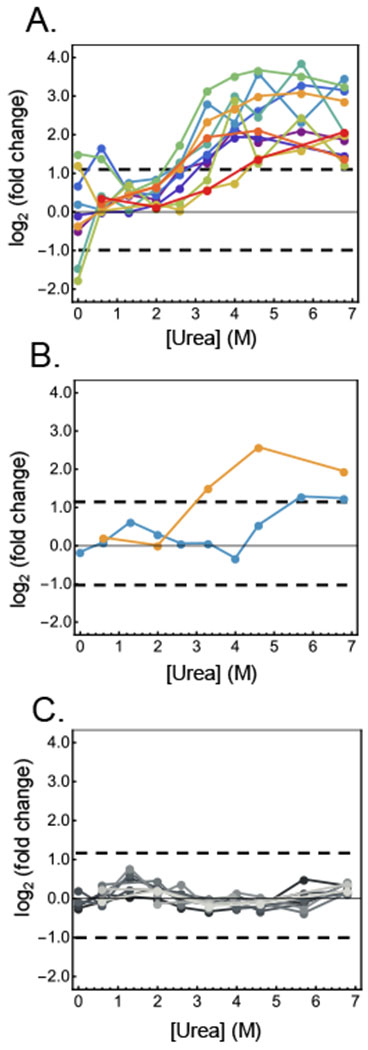
Representative STEPP-PP data obtained in the CsA binding study. (A) Data from the 11 semi-tryptic peptides assayed from the known CsA binding protein, CPR1 (peptidyl-prolyl cis-trans isomerase). (B) Data from the 2 semi-tryptic peptides assayed from the known CsA binding protein, CPR3 (peptidyl-prolyl cis-trans isomerase C, mitochondrial). (C) Data from the 10 semi-tryptic peptides assayed from a non-hit protein, RPL38 (60S ribosomal protein L38). The dashed lines indicate the log2(fold-change) values that are +/− 3σ deviations from the average log2(fold-change) values of all the semi-tryptic peptides assayed in the CsA binding study.
Figure 5.
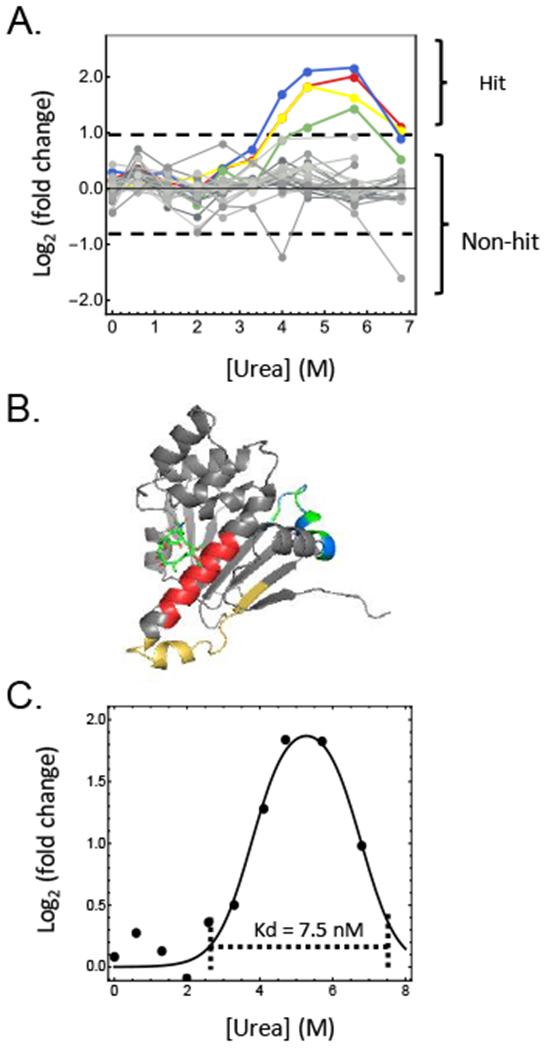
STEPP-PP data obtained in the geldanamycin binding experiment. (A) Data obtained on the 28 semi-tryptic peptides assayed HSP82 peptides in the geldanamycin binding experiment. The 4 hit peptides are shown in blue, red, yellow and green, other non-hit peptides are shown in grey. The dashed lines indicate the log2(fold-change) values that are +/− 3σ deviations from the average log2(fold-change) values of all the semi-tryptic peptides assayed in the geldanamycin binding study. (B) Schematic representation of the HSP82 geldanamycin binding domain with the geldanamycin bound (PDB ID: 1A4H).22 The four hit peptides from HSP82 (highlighted with the same colors as in A, respectively) all mapped to this known geldanamycin binding domain. (C) Data used to calculate the Kd value associated with the HSP82-geldanamycin complex. The data points represent the median data from the four hit peptides in (A). The solid line is the best fit of the data to equation S-1 (see Supplemental Text).
Unfortunately, it is difficult to determine if the peptide hits from proteins other than the cyclophilins in the CsA binding experiment and HSP82 in the geldanamycin binding experiment are from novel CsA or HSP82 binding proteins or are false positives. However, if all such proteins are considered to be false positives, an upper limit for the false positive rates of 0.3–0.7% and 1.4 – 3.6% for peptide and protein hit discovery (respectively) can be established for the STEPP-PP protocol. The false positive rates of protein hit discovery observed here for STEPP-PP are about the same as that previously reported for SILAC-PP with gel fractionation (2.1 – 3.6%);10 and the false positive rates of peptide hit discovery observed here for STEPP-PP are slightly lower than that previously reported for SPROX (~1–2%).23
The false positive rate of protein hit discovery in STEPP-PP can be further reduced if hit proteins are also required to have at least 2 semi-tryptic peptide hits. While this reduced the proteome coverage in our experiments by about 45% (see Table 1), it significantly improved the false positive rate. In the CsA binding experiment, only the two cyclophilins CPR1 and CPR3, passed this more stringent hit selection criteria, giving a 0% false positive rate of protein hit discovery. In the geldanamycin binding experiment, only HSP82 and 3 other proteins passed the multiple-peptide hit selection criteria, reducing the false positive rate of protein hit discovery by 6-fold from 3.6% to 0.6%. Thus, requiring just two-peptide hits for protein hit selection can greatly reduce the false positive rate of protein hit discovery using the STEPP-PP approach. However, proteomic coverage is sacrificed. This is because, even with the STEPP-PP protocol to enrich for semi-tryptic peptides, only a fraction (~55%) of the proteins identified in the bottom-up proteomics analysis are identified with two or more semi-tryptic peptides.
STEPP-PP Generates Domain Specific Ligand-Binding Information
The semi-tryptic peptides generated in the STEPP-PP experiment report on the biophysical properties of the protein folding domains to which they map. In the case of single domain proteins all the semi-tryptic peptides generated in the STEPP-PP experiment should display similar behavior. This was, for example, observed with the semi-tryptic peptides from the cyclophilin proteins in the CsA binding experiment (Figure 4A and B). In the case of large multi-domain proteins, the semi-tryptic peptides from different domains can display different behaviors if they have different interactions with the ligand. This was, for example, observed with the semi-tryptic peptides form HSP82 in the geldanamycin experiment (Figure 5A).
HSP82 has three domains, including the N-terminal ATP-binding domain (residues 1–210 that also encompass the geldanamycin binding site14), the middle domain (residues 267–547), the C-terminal dimerization domain (residues 548–705), and a flexible linker (residues 211–266).22 In the geldanamycin binding experiment, the 4 semi-tryptic peptide hits were all from the N-terminal ATP-binding domain, which contains the known geldanamycin binding site. We note that the 1 semi-tryptic peptide from the N-terminal domain which was not identified as a hit, appears to be a false-negative. Indeed, the fold-change values recorded for this peptide (1.84 and 1.90 at 4 M and 5.7 M urea concentrations, respectively) were very close to the cut-off value (1.93) used for hit selection. As expected, all 23 semi-tryptic peptides from the other domains were not identified as hits (Figure 5A). In the case of large multi-domain proteins like HSP82, the STEPP-PP protocol can provide domain-specific information about ligand-induced conformational changes. Such domain specific information has not been reported using previous PP protocols..,4–5, 10–12 Presumably, the protein-centered readouts employed in these previous protocols complicates the acquisition of such domain-specific binding information.
Dissociation Constant Determination in STEPP-PP
An inherent advantage of PP over other energetics-based approaches such as the thermal proteome profiling (TPP) approach,24 is that it can be used to evaluate the dissociation constants (Kd) of protein-ligand complexes. This is because the chemical denaturation behavior of proteins determined in the presence and in the absence of ligand can be used to evaluate a binding free energy (i.e., a ΔΔGf value). In particular, the denaturant dependence of the fold-change values generated in the STEPP-PP experiment can be fit to equation S-1 (see Supplemental Text) to generate a ΔΔGf value, which can ultimately be used to calculate a Kd value using equation S-2 (see Supplemental Text). Indeed, the STEPP-PP data can be used to obtain a Kd of the geldanamycin-HSP82 binding of ~7.5 nM (Figure 5C), which is in good agreement with (i.e., within 20% of) the previously reported value of 9 nM.25 However, the determination of a Kd value from STEPP-PP data does require data points at denaturant concentrations that are above the transition mid-point of the most stable form (i.e., that the fold-change values return to the baseline value of ~0) (see Figure 4C). For example, the lack of data at higher denaturant concentrations, where the fold-change values are expected to return to 0, precluded the determination of a meaningful Kd value for the CsA - cyclophilin binding interactions (see Figures 4A and B).
STEPP-LiP Study of Breast Cancer-Related Protein Conformational Changes
The STEPP protocol was also interfaced with the LiP method according to the scheme outlined in Figure 3. The STEPP-LiP protocol was used in a proof-of-principle study of breast cancer-related protein conformational changes using a human breast cancer cell line (MCF-7) and a human normal mammary epithelial cell line (MCF-10A). To evaluate the utility of the STEPP-LiP protocol, the STEPP-LiP results reported here were directly compared to the results generated in a previous analysis of the MCF-7 and MCF-10A cell lines using the normal LiP procedure (i.e., without STEPP).6
In the STEPP-LiP experiment about 3000 semi-tryptic peptides from 700 proteins (see Table 2 and Tables S-4 – S-6) were assayed. This is 5 times more semi-tryptic peptides and 1.25 times more proteins than that assayed in reference 6 using a state-of-the-art SILAC-LiP protocol with the same number of biological and technical replicates to study the same MCF-7 and MCF-10A cell lines studied here by STEPP-LiP. The protein hits identified in the STEPP-LiP experiment were also in good agreement with those in the normal LiP experiment. A total of 241 proteins were assayed in both the LiP and STEPP-LiP experiments (see Table S-7). These 241 proteins included 115 hit proteins in LiP and 181 hit proteins in STEPP-LiP, with a large fraction (80%) of the LiP hits overlapping with the STEPP-LiP hits.
Table 2.
Summary of proteomic coverages and numbers of hits observed in the STEPP-LiP MCF-7 vs MCF-10A comparative analysis.
| Double Digestiona | Single Digestionb | |
|---|---|---|
| Total peptide (protein) identified | 3650 (1050) | 16656 (1604) |
| Assayed peptide (protein) | 2977 (688)c | |
| Peptide (protein) hits | 1240 (440)d | |
The identified peptides in the double digestion group are all semi-tryptic.
The identified peptides in the single digestion group are all tryptic.
The number of peptides (proteins) identified in least two biological replicates of both the MCF-7 and MCF-10A cell line analyses and in both the double and single digestion groups.
The number of semi-tryptic peptides (proteins) with significantly different normalized TMT reporter ion intensities between the MCF-7 and MCF-10A groups (p < 0.05).
The increased peptide coverage in the STEPP-LiP experiment not only increases the confidence in protein hits (i.e., protein hits identified with multiple semi-tryptic peptide hits are more likely to be true positives) but it also increases the amount of information that can be gleaned about the conformational properties of hit proteins in the LiP experiment (e.g., see data on HSP90AA1 in Figure 6A and B). The STEPP-LiP data on HSP90AA1 (Figure 6A) indicate a clear picture of the conformational differences in this protein in the MCF-7 and MCF-10A cell lines. The STEPP-LiP data reveal that the N-terminal ATP-binding domain of the protein is more protected against proteolysis in the MCF-7 cell line than in the MCF-10A cell line. This is consistent with HSP90 adopting its closed and active form (Figure 6C) in the MCF-7 cells. This result is also in agreement with the results of other studies that have shown HSP90 is in its closed and active form in cancer cells and in its open and inactive form in normal cells.26
Figure 6.
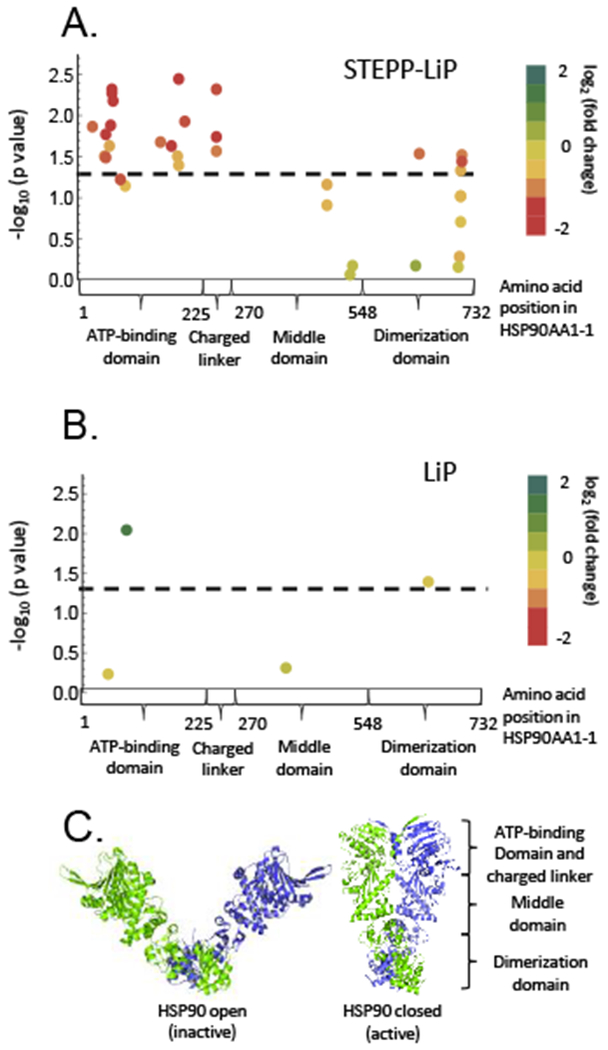
Comparison of the STEPP-LiP and LiP data generated on HSP90AA1 (heat shock protein HSP 90-alpha) in this work and in reference 6 (respectively). (A) The amino acid positions and p-values of the 37 semi-tryptic peptides from HSP90AA1 isoform 1 that were assayed in the STEPP-LiP study of MCF-7 vs MCF-10A protein conformation are plotted. (B) The amino acid positions and p-values of the 4 semi-tryptic peptides from HSP90AA1 isoform 1 that were assayed in the LiP study of MCF-7 vs MCF-10A protein conformation in reference 6 are plotted. In both (A) and (B), the dashed lines correspond to a p-value = 0.05. The color of each data point indicates the MCF-7/MCF-10A TMT reporter ion intensity fold-change of the given semi-tryptic peptide, with more proteolytic susceptibility implicated by a greener color. (C) Schematic representations of the three-dimensional structures associated with the open and closed conformations of the HSP90 homodimer (generated from PDB ID: 2IOQ and 2CG9).29–30
The HSP90AA1 protein was identified as a hit in our previously published normal LiP analysis of the MCF-7 and MCF-10A cell lines. However, the normal LiP data indicated that HSP90AA1 was more protected against proteolysis in the MCF-10A cell line than in the MCF-7 cell line (Figure 6B). One explanation for the apparent discrepancy in the normal LiP and STEPP-LiP results on HSP90AA1 is that the semi-tryptic peptide detected in the normal LiP experiment was actually a “tryptic” semi-tryptic peptide generated from non-specific cleavage during the trypsin digestion step. Such “tryptic” semi-tryptic peptides have been reported to constitute 4–40% of the peptides generated in a trypsin digestion, depending on the trypsin manufacturer.27 While the trypsin used in the normal LiP is expected to only generate a small fraction of semi-tryptic peptides (<5% of total peptides28), the presence of such peptides cannot be ignored in a normal LiP experiment where the number of semi-tryptic peptides is relatively low (i.e., around 13% of the total peptides assayed in the LiP experiment6).
In the normal LiP experiment the presence of “tryptic” semi-tryptic peptides can lead to errant interpretations of the data. Semi-tryptic peptides can be correctly identified as hits in the normal LiP experiment whether they came from the proteinase K or trypsin digestion. However, it becomes ambiguous as to whether or not the hit arises from increased or decreased protection (e.g., a >1-fold change means more susceptible to proteolysis if the semi-tryptic peptide is from proteinase K cleavage but less susceptible if it is from the trypsin digestion step). One advantage of the STEPP-LiP protocol described here is that it is not subject to such ambiguity, as only the N-terminally-TMT-labeled peptides from the proteinase K digestion step are left in solution after the chemo-selection. Therefore, the above ambiguity is effectively eliminated in the STEPP-LiP experiment.
Supplementary Material
ACKNOWLEDGEMENT
The authors thank the Duke Proteomics Facility for collecting the LC-MS/MS data. This work was supported by a grant from the National Institutes of General Medical Sciences at the National Institutes of Health (2RO1GM084174–08) to M.C.F.
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publication website.
Supplementary text providing more details about the yeast culture, on how the chemo-selection efficiency of the STEPP protocol was evaluated, and on how protein-ligand dissociation constants (i.e., Kd values) were determined from the STEPP-PP data as well as Figure S-1, Figure S-2, and Table S-1 (PDF).
Excel spreadsheet summarizing the assayed semi-tryptic peptides, fold changes at each denaturant concentration, and the hit selection results from the STEPP-PP CsA binding experiments (Table S-2).
Excel spreadsheet summarizing the assayed semi-tryptic peptides, fold changes at each denaturant concentration, and the hit selection results from the STEPP-PP geldanamycin binding experiments (Table S-3)
Excel spreadsheet summarizing proteins identified in the STEPP-LiP MCF-7 versus MCF-10A cell line comparison with single digestion (Table S-4).
Excel spreadsheet summarizing semi-tryptic peptides identified in the STEPP-LiP MCF-7 versus MCF-10A cell line comparison with double digestion (Table S-5).
Excel spreadsheet summarizing the proteins identified in the assayed semi-tryptic peptides, normalized fold changes, p-values, and the hit selection results from the STEPP-LiP experiment on the MCF-7 and MCF-10A cell lines (Table S-6).
Excel spreadsheet summarizing the overlapping assayed proteins and protein hits from the STEPP-LiP experiments in this work and the normal LiP experiments in reference 6. (Table S-7).
REFERENCES
- 1.Suckau D; Kohl J; Karwath G; Schneider K; Casaretto M; Bitter-Suermann D; Przybylski M, Molecular epitope identification by limited proteolysis of an immobilized antigen-antibody complex and mass spectrometric peptide mapping. Proc Natl Acad Sci U S A 1990, 87 (24), 9848–9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng Y; De Franceschi G; Kahraman A; Soste M; Melnik A; Boersema PJ; de Laureto PP; Nikolaev Y; Oliveira AP; Picotti P, Global analysis of protein structural changes in complex proteomes. Nat Biotechnol 2014, 32 (10), 1036–1044. [DOI] [PubMed] [Google Scholar]
- 3.Lomenick B; Hao R; Jonai N; Chin RM; Aghajan M; Warburton S; Wang J; Wu RP; Gomez F; Loo JA; Wohlschlegel JA; Vondriska TM; Pelletier J; Herschman HR; Clardy J; Clarke CF; Huang J, Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci U S A 2009, 106 (51), 21984–21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu PF; Kihara D; Park C, Energetics-based discovery of protein-ligand interactions on a proteomic scale. J Mol Biol 2011, 408 (1), 147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang Y; Schlebach JP; VerHeul RA; Park C, Simplified proteomics approach to discover protein-ligand interactions. Protein Sci 2012, 21 (9), 1280–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu F; Fitzgerald MC, Large-Scale Analysis of Breast Cancer-Related Conformational Changes in Proteins Using Limited Proteolysis. J Proteome Res 2016, 15 (12), 4666–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meng H; Fitzgerald MC, Proteome-Wide Characterization of Phosphorylation-Induced Conformational Changes in Breast Cancer. J Proteome Res 2018, 17 (3), 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geiger R; Rieckmann JC; Wolf T; Basso C; Feng Y; Fuhrer T; Kogadeeva M; Picotti P; Meissner F; Mann M; Zamboni N; Sallusto F; Lanzavecchia A, L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167 (3), 829–842 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim D; Hwang HY; Kim JY; Lee JY; Yoo JS; Marko-Varga G; Kwon HJ, FK506, an Immunosuppressive Drug, Induces Autophagy by Binding to the V-ATPase Catalytic Subunit A in Neuronal Cells. J Proteome Res 2017, 16 (1), 55–64. [DOI] [PubMed] [Google Scholar]
- 10.Adhikari J; Fitzgerald MC, SILAC-pulse proteolysis: A mass spectrometry-based method for discovery and cross-validation in proteome-wide studies of ligand binding. J Am Soc Mass Spectrom 2014, 25 (12), 2073–2083. [DOI] [PubMed] [Google Scholar]
- 11.Zeng L; Shin WH; Zhu X; Park SH; Park C; Tao WA; Kihara D, Discovery of Nicotinamide Adenine Dinucleotide Binding Proteins in the Escherichia coli Proteome Using a Combined Energetic- and Structural-Bioinformatics-Based Approach. J Proteome Res 2017, 16 (2), 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trindade RV; Pinto AF; Santos DS; Bizarro CV, Pulse Proteolysis and Precipitation for Target Identification. J Proteome Res 2016, 15 (7), 2236–2245. [DOI] [PubMed] [Google Scholar]
- 13.Handschumacher RE; Harding MW; Rice J; Drugge RJ; Speicher DW, Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 1984, 226 (4674), 544–547. [DOI] [PubMed] [Google Scholar]
- 14.Stebbins CE; Russo AA; Schneider C; Rosen N; Hartl FU; Pavletich NP, Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 1997, 89 (2), 239–250. [DOI] [PubMed] [Google Scholar]
- 15.Meng H; Ma R; Fitzgerald MC, Chemical Denaturation and Protein Precipitation Approach for Discovery and Quantitation of Protein-Drug Interactions. Anal Chem 2018, 90 (15), 9249–9255. [DOI] [PubMed] [Google Scholar]
- 16.West GM; Tucker CL; Xu T; Park SK; Han X; Yates JR 3rd; Fitzgerald MC, Quantitative proteomics approach for identifying protein-drug interactions in complex mixtures using protein stability measurements. Proc Natl Acad Sci U S A 2010, 107 (20), 9078–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeArmond PD; West GM; Huang HT; Fitzgerald MC, Stable isotope labeling strategy for protein-ligand binding analysis in multi-component protein mixtures. J Am Soc Mass Spectrom 2011, 22 (3), 418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tran DT; Adhikari J; Fitzgerald MC, StableIsotope Labeling with Amino Acids in Cell Culture (SILAC)-based strategy for proteome-wide thermodynamic analysis of protein-ligand binding interactions. Mol Cell Proteomics 2014, 13 (7), 1800–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Y; Wallace MA; Fitzgerald MC, Thermodynamic Analysis of the Geldanamycin-Hsp90 Interaction in a Whole Cell Lysate Using a Mass Spectrometry-Based Proteomics Approach. J Am Soc Mass Spectrom 2016, 27 (10), 1670–1676. [DOI] [PubMed] [Google Scholar]
- 20.Haendler B; Keller R; Hiestand PC; Kocher HP; Wegmann G; Movva NR, Yeast cyclophilin: isolation and characterization of the protein, cDNA and gene. Gene 1989, 83 (1), 39–46. [DOI] [PubMed] [Google Scholar]
- 21.Friedman J; Weissman I, Two cytoplasmic candidates for immunophilin action are revealed by affinity for a new cyclophilin: one in the presence and one in the absence of CsA. Cell 1991, 66 (4), 799–806. [DOI] [PubMed] [Google Scholar]
- 22.Prodromou C; Roe SM; O’Brien R; Ladbury JE; Piper PW; Pearl LH, Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 1997, 90 (1), 65–75. [DOI] [PubMed] [Google Scholar]
- 23.Strickland EC; Geer MA; Hong J; Fitzgerald MC, False-positive rate determination of protein target discovery using a covalent modification- and mass spectrometry-based proteomics platform. J Am Soc Mass Spectrom 2014, 25 (1), 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Savitski MM; Reinhard FB; Franken H; Werner T; Savitski MF; Eberhard D; Martinez Molina D; Jafari R; Dovega RB; Klaeger S; Kuster B; Nordlund P; Bantscheff M; Drewes G, Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346 (6205), 1255784. [DOI] [PubMed] [Google Scholar]
- 25.Gooljarsingh LT; Fernandes C; Yan K; Zhang H; Grooms M; Johanson K; Sinnamon RH; Kirkpatrick RB; Kerrigan J; Lewis T; Arnone M; King AJ; Lai Z; Copeland RA; Tummino PJ, A biochemical rationale for the anticancer effects of Hsp90 inhibitors: slow, tight binding inhibition by geldanamycin and its analogues. Proc Natl Acad Sci U S A 2006, 103 (20), 7625–7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamal A; Thao L; Sensintaffar J; Zhang L; Boehm MF; Fritz LC; Burrows FJ, A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425 (6956), 407–410. [DOI] [PubMed] [Google Scholar]
- 27.Burkhart JM; Schumbrutzki C; Wortelkamp S; Sickmann A; Zahedi RP, Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics. J Proteomics 2012, 75 (4), 1454–1462. [DOI] [PubMed] [Google Scholar]
- 28.Thermo Fisher Scientific. Pierce™ Trypsin Protease, MS Grade. https://www.thermofisher.com/order/catalog/product/90057 (accessed Sept 3).
- 29.Shiau AK; Harris SF; Southworth DR; Agard DA, Structural Analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 2006, 127 (2), 329–340. [DOI] [PubMed] [Google Scholar]
- 30.Ali MM; Roe SM; Vaughan CK; Meyer P; Panaretou B; Piper PW; Prodromou C; Pearl LH, Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440 (7087), 1013–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.