

Abstract
Background
The amide derivatives of nonsteroidal anti-inflammatory drugs have been reported to possess antitumor activity. The present work describes the synthesis of dexibuprofen amide analogues (4a–j) as potential anticancer agents.
Methods
The title amides (4a–j) were obtained by simple nucleophilic substitution reaction of dexibuprofen acid chloride with substituted amines in good yield and chemical structures were confirmed by FTIR, 1H NMR, 13C NMR and mass spectral data.
Results
The brine shrimp lethality assay results showed that all of the synthesized compounds are non-toxic to shrimp larvae. The inhibitory effects on tumor growth were evaluated and it was observed that N-(2,5-dichlorophenyl)-2-(4-isobutylphenyl) propionamide (4e) and N-(2-chlorophenyl)-2-(4-isobutylphenyl) propionamide (4g) exhibited excellent antitumor activity compared to all other derivatives. The compound 4e bearing 2,5-dichloro substituted phenyl ring and 4g possesses 2-chloro substituted phenyl ring exhibited 100% inhibition of the tumor growth. The anticancer activity was evaluated against breast carcinoma cell line (MCF-7) and it was observed that derivative 4e exhibited excellent growth inhibition of cancer cells with IC50 value of 0.01±0.002 µm, which is better than the standard drugs. The docking studies against breast cancer type 1 susceptibility protein BRCA1 (PDBID 3K0H) exhibited good binding affinities, which are in good agreement with the wet lab results. The compounds 4e and 4g showed the binding energy values of −6.39 and −6.34 Kcal/mol, respectively. The molecular dynamic (MD) simulation was also carried out to evaluate the residual flexibility of the best docking complexes of compounds 4e and 4g. The MD simulation analysis assured that the 4e formed a more stable complex with the target protein than the 4g. The synthesized amide derivatives exhibited were devoid of gastrointestinal side effects and no cytotoxic effects against human normal epithelial breast cell line (MCF-12A) were found.
Conclusion
Based upon our wet lab and dry lab findings we propose that dexibuprofen analogue 4e may serve as a lead structure for the design of more potent anticancer drugs.
Keywords: dexibuprofen analogues, preparation, computational studies, cytotoxicity, anticancer activity
Introduction
Malignant cancer is the leading cause of human deaths worldwide, accounting for 8.2 million deaths in 2012 and it is expected that annual cases will continuously increase.1 Cancer arises due to the accumulation of mutations in critical genes that alter normal programs of cell proliferation, differentiation and death.2 The deaths due to cancer gradually increased, despite the enormous amount of research and rapid development seen during the past decade.3 Many options for the treatment of cancer exist including radiotherapy and chemotherapy. A number of different types of cells inside the body are proliferating into tumor cells such as alimentary tract, bone marrow and epidermal cells. Thus patients receiving anticancer drugs suffer from side effects such as hair loss and anemia.4 During the last few decades, researchers made efforts to discover effective clinical approaches for the treatment of cancer and searched for novel anticancer agents, but the management of malignancies in humans still constitutes a major concern for contemporary medicine.5–8 Therefore, it is vital to find novel anticancer drugs with new chemical entity.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used class of therapeutic agents. The mode of action of NSAIDs is the inhibition of the cyclooxygenase enzymes ie, COX-1 and COX-2.9–11 It has been well established that NSAIDs exert their analgesic effects not only through peripheral inhibition of prostaglandin synthesis but also through a variety of other peripheral and central mechanisms. Some NSAIDs inhibit the lipoxygenase pathway, which may itself result in the production of algogenic metabolites. Interference with G-protein-mediated signals transduction by NSAIDs may form the basis of an analgesic mechanism unrelated to inhibition of prostaglandin synthesis. A central mechanism of action for NSAIDs has also been proposed that augments the peripheral mechanism. This effect may be the result of interference with the formation of prostaglandins within the central nervous system (CNS). The central mechanism of action may be mediated by endogenous opioid peptides or blockade of the release of serotonin (5-hydroxytryptamine). The inhibition of excitatory amino acids or N-methyl-D-aspartate receptor activation has also been reported to be the reason for NSAIDs action.12
In recent years, several clinical and experimental studies have shown that NSAIDs also exhibit anticancer properties; though the mechanisms in anti-tumorigenesis are not fully understood, both cyclooxygenase (COX)-dependent and -independent pathways play a role.13 Several groups have postulated that both anti-inflammatory and anticancer activities are due to reduction in products formed by COX or lipoxygenase-catalyzed reactions, which are overexpressed in many tumors.14–18 However, several other groups reported the COX-independent mechanism of antitumor activity of NSAIDs. Gurpinar in 2014 reported that apoptosis induction and suppression of β-catenin-dependent transcription are important aspects of their antineoplastic activity. Studies showed that the latter involves phosphodiesterase (PDE) inhibition and the elevation of intracellular cyclic guano-sine monophosphate (GMP) levels.19 Tinsley and Piazza also reported that cyclic GMP (cGMP) PDEs, which are responsible for negative regulation of cGMP signaling, are an attractive COX-independent target. cGMP signaling is aberrantly suppressed in cancer cells and its activation appears to be sufficient to inhibit tumor cell growth.20 It has also been postulated that NSAIDs mediate breast cancer inhibition as an immune modulator and use of combination of immunotherapy with NSAIDs to prevent cancer has also been reported.21,22
COX-2 has been reported to contribute to tumorigenesis and malignant phenotype of tumor cells through one or more of the following mechanisms: increasing the production of prostaglandins, converting procarcinogens to carcinogens, inhibiting apoptosis, promoting angiogenesis, increasing the invasiveness of cancer cells, modulating inflammation and immune responsiveness.23–25 It has been observed that ester and amide derivatives of the NSAIDs had both anti-oxidative and antiproliferative activities. It was further investigated that amides were more potent inhibitors of cell proliferation than parent NSAID itself and the corresponding esters.26 Keeping in view the antitumorigenesis potential of the NSAIDs and their derivatives, the present study is planned to synthesize amide derivatives of dexibuprofen with the aim of possessing potent anticancer activity. The computational studies have been performed to design potent amide derivatives as it is helpful in drug designing. Computational software suggests the prospective interactions between receptors and drug molecules. This method can be used to identify the binding energy and binding strength when comparing a group of compounds.27–29
Dexibuprofen S-(+) isomer is a pharmacologically active enantiomer of racemic ibuprofen. Racemic ibuprofen and dexibuprofen differ in their physicochemical properties and R-(−) ibuprofen can exhibit pharmacological action only after conversion into dexibuprofen. In the present study dexibuprofen amide derivatives were synthesized as potential anticancer agents. The structures of the synthesized compounds were confirmed by FTIR, 1H NMR and 13C NMR spectral data. Brine shrimp lethality assay is a simple method for preliminary assessment of toxicity activity by using Artemia. This method has been successfully employed to determine preliminary toxicity because of its reliability, low cost as brine shrimps are commercially available as dry cysts, easy to hatch, grow and maintain.30–32 In vitro antitumor activity of the synthesized amides was determined by following the potato disc method. It has been reported that inhibition of crown gall tumor on potato discs has good agreement with compounds to be active in the 3PS (in vivo, mouse leukemia) antitumor assay.33 Anticancer activity was evaluated against breast carcinoma cell line (MCF-7) to check their potential inhibition on cancer cell growth. The ulcerogenic activity and cytotoxic effects against human normal epithelial breast cell line (MCF-12A) have also been evaluated. Computational studies were performed to compare the dry lab results with wet lab findings.
Materials and methods
Instrumentation
Melting points were recorded using a digital Gallenkamp (SANYO; Loughborough, UK) apparatus. FTIR spectra were recorded using PerkinElmer spectrophotometer (PerkinElmer Inc., Waltham, MA, USA), using potassium bromide (KBr) pellet technique. 1H NMR and 13C NMR spectra were determined in CDCl3 solution at 300 MHz on Bruker AM-300 spectrometer (Bruker AXS Inc., Madison, WI, USA). The breast carcinoma cell line (MCF-7) was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Mass spectra were performed on an Agilent 6460 Series Triple Quadrupole instrument (Agilent Technologies, Santa Clara, CA, USA). The ionization was achieved by electrospray ionization in the positive ion mode (ESI+). The capillary voltage was set to 4.0 kV. The source temperature was 120°C, and the desolvation temperature was 350°C. Nitrogen was used as a desolvation gas (flow 600 L/hour). Elemental analysis (C, H) was carried out on a Flash 2000 series elemental analyzer with TCD detector system (PerkinElmer Inc., Waltham, MA, USA) and results are with ±0.3%.
General procedure
Dexibuprofen (1) (0.3 g, 0.001 mol) was refluxed and stirred with freshly distilled thionyl chloride (2 mL) for 8 hours. Excess of thionyl chloride was removed under reduced pressure to afford the dexibuprofen acid chloride (2). To a solution of an amine (3a–j) (0.001 mol) and pyridine (0.2 mL) in acetone (2.5 mL) maintained at 10°C, dexibuprofen acid chloride (2) (0.001 mol) was added with stirring over aperiod of 1 hour. The reaction mixture was stirred for 8 hours at room temperature. After the completion of the reaction, the mixture was poured into ice cold water. The residue thus obtained was filtered, dissolved in chloroform, washed with 5% hydrochloric acid (3×50 mL), 5% sodium bicarbonate (3×50 mL) and finally with brine solution (2×50 mL). The organic layer was rotary-evaporated to yield dexibuprofen amides (4a–j). The title amides were purified by flash column chromatography by using petroleum ether:ethyl acetate (3:1) as eluents. This general procedure was adopted for the preparation and purification of dexibuprofen amides (4a–j). Chiral HPLC analysis was performed for the identification of enantiomers using a normal-phase HPLC method coupled with UV detector and a chiral column. The analysis was performed using PerkinElmer Series 200 HPLC (PerkinElmer Inc.) equipped with UV detector and using Navchrome software (Shelton, CT, USA). Chiral column was a Lux 5u Cellulose-1 250×460 mm. The n-hexane and isopropyl alcohol (60:40) was used as mobile phase.
Synthesis of N-(4-chlorophenyl)-2-(4-isobutylphenyl) propionamide (4a)
Yield 75%; m.p. 82°C–84°C; FTIR (KBr, νmax cm−1): 3,297 (N−H), 2,954 (Ar C−H), 1,660 (C=O, amide), 1,541 (C=C aromatic); ESI-MS: m/z 338.5[M+23] (M+Na)+; 1H-NMR, (δ-ppm; CDCl3): 7.39 (2H, d, J=8.7 Hz, H-2″,6″), 7.31 (2H, d, J=8.7 Hz, H-3″,5″), 7.2 5 (2H, d, J=8.4 Hz, H-2′,6′), 7.19 (2H, d, J=8.1 Hz, H-3′,5′), 7.02 (1H, s, N−H), 3.71 (1H, q, J=7.2 Hz, H-2), 2.48 (2H, d, J=7.2 Hz, H-4), 1.86 (1H, m, H-5), 1.59 (3H, d, J=6.6 Hz, H-3), 0.91 (6H, d, J=6.6 Hz, H-6,7); 13C-NMR, (δ-ppm) (CDCl3): 172.8 (C-1), 142.2 (C-1″), 141.2 (C-4″), 137.8 (C-4′), 136.4 (C-1′), 129.9 (C-3″,5″), 128.9 (C-3′,5′), 127.4 (C-2′,6′), 120.8 (C-2″,6″), 47.7 (C-2), 45.0 (C-4), 30.1 (C-5), 22.3 (C-6,7), 18.4 (C-3). Anal. calcd for C19H22ClNO: C, 72.26 H, 6.97; found: C, 72.13 H, 6.82.
Synthesis of N-(benzyl)-2-(4-isobutylphenyl) propionamide (4b)
Yield 76%; m.p. 92°C–94°C; FTIR (KBr, νmax cm−1): 3,285 (N−H), 2,946 (Ar C−H), 1,665 (C=O, amide), 1,534 (C=C aromatic); ESI-MS: m/z 318[M+23] (M+Na)+; 1H-NMR, (δ-ppm, CDCl3): 7.29 (2H, d, J=7.8 Hz, H-2′,6′), 7.24 (2H, d, J=8.1 Hz, H-3′,5′), 7.12–7.16 (5H, m, H-2″-6″), 5.61 (1H, s, N−H), 4.42 (2H, d, J=5.7 Hz, H-8), 3.61 (1H, q, J=7.2 Hz, H-2), 2.48 (2H, d, J=7.2 Hz, H-4), 1.89 (1H, m, H-5), 1.58 (3H, d, J=7.2 Hz, H-3), 0.92 (6H, d, J=6.6 Hz, H-6,7); 13C-NMR, (δ-ppm, CDCl3): 174.4 (C-1), 140.8 (C-1′), 138.4 (C-4′), 138.3 (C-1″), 129.6 (C-2′,6′), 128.6 (C-3′,5′), 127.4 (C-2″,3″,5″,6″), 127.3 (C-4″), 46.8 (C-8), 45.0 (C-4), 43.5 (C-2), 30.2 (C-5), 22.3 (C-6,7), 18.4 (C-3); Anal. calcd for C20H25NO: C, 81.35 H, 8.47; found: C, 81.22 H, 8.38.
Synthesis of N-(cyclohexyl)-2-(4-isobutylphenyl) propionamide (4c)
Yield 72%, amorphous solid, m.p. 94°C–96°C; FTIR (KBr, νmax cm−1): 3,296 (N−H), 2,946 (Ar C−H), 1,656 (C=O, amide), 1,542 (C=C aromatic); ESI-MS: m/z 310 [M+23] (M+Na)+; 1H NMR, (δ-ppm) (CDCl3): 7.26 (2H, d, J=8.1 Hz, H-2′,6′), 7.14 (2H, d, J=8.1 Hz, H-3′,5′), 5.27 (1H, s, N−H), 3.55 (1H, m, H-1″), 3.53 (1H, q, J=7.2 Hz, H-2), 2.48 (2H, d, J=7.2 Hz, H-4), 1.88 (1H, m, H-5), 1.76 (10H, m, H-2″-6″), 1.53 (3H, d, J=7.2 Hz, H-3), 0.91 (6H, d, J=6.6 Hz, H-6, 7); 13C NMR, (δ-ppm) (CDCl3): 180.5 (C-1), 140.6 (C-1′), 138.5 (C-4′), 129.6 (C2′,6′), 127.3 (C-3′,5′), 48.1 (C-1″), 46.7 (C-2), 45.0 (C-4), 30.2 (C-5), 25.7 (C-2″,6″), 24.6 (C-3″,5″), 24.5 (C-4″), 22.3 (C-6, 7), 18.1 (C-3); Anal. calcd for C19H29NO: C, 79.44 H, 10.10; found: C, 79.33 H, 9.97.
Synthesis of N-(2,4-dichlorophenyl)-2-(4-isobutylphenyl) propionamide (4d)
Yield 78%; m.p. 66°C–68°C; FTIR (KBr, νmax cm−1): 3,277 (N−H), 2,922 (Ar C−H), 1,663 (C=O amide), 1,540 (C=C aromatic); ESI-MS: m/z 373[M+23] (M+Na)+; 1H-NMR, (δ-ppm, CDCl3): 8.40 (1H, s, H-3″), 7.63 (1H, s, N−H), 7.59 (1H, d, J=8.4 Hz, 6″), 7.53 (1H, d, J=8.4 Hz, H-5″), 7.30 (2H, d, J=7.9, H-2′,6′), 7.19 (2H, d, J=7.9 Hz, H-3′,5′), 3.82 (1H, q, J=7.2 Hz, H-2), 2.53 (2H, d, J=7.2 Hz, H-4), 1.88 (1H, m, H-5), 1.6 (3H, d, J=7.2 Hz, H-3), 0.92 (6H, d, J=6.6 Hz, H-6, 7); 13C-NMR, (δ-ppm, CDCl3): 172.8 (C-1), 141.5 (C-4″), 137.1 (C-2″), 133.4 (C-3″), 130.9 (C-5″), 129.7 (C-1″), 128.7 (C-1′), 128.3 (C-4′), 127.6 (C-3′,5′), 123.0 (C-6″), 127.6 (C-2′,6′), 48.0 (C-2), 45.0 (C-4), 30.2 (C-5), 22.3 (C-6,7), 17.8 (C-3); Anal. calcd for C19H21ClNO: C, 65.14 H, 6.00; found: C, 65.03 H, 5.91.
Synthesis of N-(2,5-dichlorophenyl)-2-(4-isobutylphenyl) propionamide (4e)
Yield 74%; m.p. 63°C–65°C; ESI-MS: m/z 373[M+23] (M+Na)+; FTIR (KBr, νmax cm−1): 3,297 (N−H), 2,952 (Ar C-H), 1,664 (C=O, amide), 1,512 (C=C aromatic); 1H-NMR, (δ-ppm, CDCl3): 8.52 (1H, s, H-6″), 7.64 (1H, s, N−H), 7.30 (2H, d, J=8.1 Hz, H-2′,6′), 7.22 (2H, d, J=8.1 Hz, H-3′,5′), 7.14 (1H, d, J=8.1 Hz, H-3″), 6.99 (1H, d, J=8.1 Hz, H-4″), 3.80 (1H, q, J=7.2 Hz, H-2), 2.49 (2H, d, J=7.2 Hz, H-4), 1.88 (1H, m, H-5), 1.66(3H, d, J=7.2 Hz, H-3), 0.93 (6H, d, J=6.6 Hz, H-6, 7); 13C-NMR, (δ-ppm) (CDCl3): 172.0 (C-1), 141.5 (C-1″), 137.0 (C-4′), 135.4 (C-1′), 133.4 (C-5″), 130.1 (C-2′,6′), 129.4 (C-3″), 127.6 (C-3′,5′), 127.2 (C-2″), 124.0 (C-4″), 120.5 (C-6″), 48.0 (C-2), 45.0 (C-4), 30.2 (C-5), 22.3 (C-6,7), 17.8 (C-3); Anal. calcd for C19H21ClNO: C, 65.14 H, 6.00; found: C, 65.03 H, 5.91.
Synthesis of N-(phenyl)-2-(4-isobutylphenyl) propionamide (4f)
Yield 84%; m.p. 104°C–106°C; FTIR (KBr, νmax cm−1): 3,294 (N−H), 2,950 (Ar C−H), 1,658(C=O, amide), 1,545 (C=C aromatic); ESI-MS: m/z 281[M+23] (M+Na)+; 1H-NMR, (δ-ppm, CDCl3): 7.53 (1H, s, N−H), 7.26–7.44 (5H, m, H-2″-6″), 7.19 (2H, d, J=8.1, H-2′,6′), 7.10 (2H, d, J=8.1, H-3′,5′), 3.72 (1H, q, J=7.2 Hz, H-2), 2.51 (2H, d, J=7.2 Hz, H-4), 1.88 (1H, m, H-5), 1.62 (3H, d, J=7.2 Hz, H-3), 0.94 (6H, d, J=6.6 Hz, H-6,7); 13C-NMR, (δ-ppm) (CDCl3): 172 (C-1), 141 (C-1″), 138.0 (C-4′), 137.9 (C-1′), 129.9 (2′,6′), 128.9 (3′, 5′), 127.4 (C-3″,5″), 124.1 (C-4″), 119.5 (C-2″,6″), 48.0 (C-2), 45.0 (C-4), 30.2 (C-5), 22.4 (C-6, 7), 18.5 (C-3); Anal. calcd for C19H23NO: C, 81.13 H, 8.18; found: C, 81.03 H, 8.09.
Synthesis of N-(2-chlorophenyl)-2-(4-isobutylphenyl) propionamide (4g)
Yield 82%; m.p. 68°C–70°C; FTIR (KBr, νmax cm−1): 3,275 (N−H), 2,952 (Ar C−H), 1,658 (C=O, amide), 1,523 (C=C aromatic); ESI-MS: m/z 338.5[M+23] (M+Na)+; 1H-NMR, (δ-ppm, CDCl3): 8.42 (1H, d, J=8.4, H-3″), 7.66 (1H, s, N−H), 7.40 (1H, d, J=8.1 Hz, H-6″), 7.28 (1H, t, J=8.1 Hz, H-5″), 7.22 (2H, d, J=8.1 Hz, H-2′,6′), 7.02 (2H, d, J=8.1 Hz, H-3′,5′), 6.97 (1H, t, J=7.8 Hz, H-4″), 3.53 (1H, q, J=7.2 Hz, H-2), 2.52 (2H, d, J=7.2 Hz, H-4), 1.88 (1H, m, H-5), 1.67 (3H, d, J=7.2 Hz, H-3), 0.91 (6H, d, J=6.6 Hz, H-6,7); 13C NMR, (δ-ppm) (CDCl3): 172.8 (C-1), 141.4 (C-1″), 137.4 (C-2″), 137.1 (C-4′), 134.6 (C-1′), 130.0 (C-3′,5′), 128.3 (C-3″), 127.6 (C-2′,6′), 124.3 (C-4″), 122.6 (C-5″), 121.0 (C-6″), 48.0 (C-2), 44.6 (C-4), 24.5 (C-5), 22.3 (C-6, 7), 17.9 (C-3); Anal. calcd for C19H22ClNO: C, 72.26 H, 6.97; found: C, 72.13 H, 6.82.
Synthesis of N-dodecyl-2-(4-isobutylphenyl) propionamide (4h)
Yield 78%, amorphous solid, m.p. 102°C–104°C; FTIR (KBr, νmax cm−1): 3,287 (N−H), 2,952 (Ar C−H), 1,656 (C=O, amide), 1,540 (C=C aromatic); ESI-MS: m/z 368[M+23] (M+Na)+; 1H NMR, (δ-ppm) (CDCl3): 7.21 (2H, d, J=8.1, H-2′,6′), 7.19 (2H, d, J=8.1, H-3′,5′), 5.31 (1H, s, N−H), 3.18 (1H, q, J=6.2, H-2), 2.71 (2H, t, J=8.1, H-1″), 2.44 (2H, d, J=7.2, H-4), 1.93 (2H, m, H-2″), 1.88 (1H, m, H-5), 1.53 (3H, d, J=7.2, H-3), 1.26 (20H, m, H-3″-11″), 0.91 (6H, d, J=6.7, H-6, 7), 0.87 (3H, t, J=6.9, H-12″); 13C NMR, (δ-ppm) (CDCl3): 174.0 (C-1), 140.7 (C-1′), 138.6 (C-4′), 129.6 (C-2′,6′), 127.3 (C-3′,5′), 46.0 (C-2), 45.0 (C-4), 39.5 (C-5), 31.9 (C-1″), 30.2 (C-2″), 29.6 (C-3″,4″), 29.5 (C-5″), 29.4 (C-6″), 29.3 (C-7″), 29.2 (C-8″), 29.1 (C-9″), 26.7 (C-10″, 11″), 22.3 (C-6,7), 18.4 (C-3), 14.1 (C-12″); Anal. calcd for C23H39NO: C, 80.00 H, 11.30; found: C, 79.92 H, 11.21.
Synthesis of N-butyl-2-(4-isobutylphenyl) propionamide (4i)
Yield 75%; m.p. 70°C–72°C; ESI-MS: m/z 284[M+23] (M+Na)+; FTIR (KBr, νmax cm−1): 3,297 (N–H), 2,954 (Ar C−H), 1,660 (C=O, amide), 1,541 (C=C aromatic); 1H NMR, (δ-ppm) (CDCl3): 7.26 (2H, d, J=8.1, H-2′,6′), 7.19 (2H, d, J=8.1, H-3′,5′), 5.48 (1H, s, N–H), 3.18 (1H, q, J=6.2, H-2), 2.71 (2H, t, J=8.1, H-1″), 2.44 (2H, d, J=7.2, H-4), 1.9 (4H, m, H-2″,3″), 1.88 (1H, m, H-5), 1.53 (3H, d, J=7.2, H-3), 0.93 (6H, d, J=6.7, H-6, 7), 0.87 (3H, t, J=6.9, H-4″); 13C NMR, (δ-ppm) (CDCl3): 174.0 (C-1), 140.7 (C-1′), 138.4 (C-4′), 129.6 (C-2′,6′), 127.3 (C-3′,5′), 46.7 (C-4), 45.0 (C-2), 39.4 (C-1″), 31.4 (C-2″), 30.2 (C-5), 22.4 (C-3), 22.3 (C-6,7), 18.3 (C-3″), 13.7 (C-4″); Anal. calcd for C17H27NO: C, 78.16 H, 10.34; found: C, 78.07 H, 10.25.
Synthesis of N-(4-pyridyl)-2-(4-isobutylphenyl) propionamide (4j)
Yield 72%; m.p. 97°C–99°C; FTIR (KBr, νmax cm−1): 3,275 (N−H), 2,961 (Ar C−H), 1,651 (C=O, amide), 1,549 (C=C aromatic); ESI-MS: m/z 305[M+23] (M+Na)+; 1H-NMR, (δ-ppm; CDCl3): 7.78 (2H, d, J=6.6 Hz, H-3″,5″), 7.54 (2H, d, J=6.6 Hz, H-2″,6″), 7.33 (2H, d, J=7.6 Hz, H-2′,6′), 7.23 (2H, d, J=7.6 Hz, H-3′,5′), 7.12 (1H, s, N–H), 3.74 (1H, q, J=6.4 Hz, H-2), 2.45 (2H, d, J=6.4 Hz, H-4), 1.75 (1H, m, H-5), 1.63 (3H, d, J=6.8 Hz, H-3), 0.91 (6H, d, J=6.8 Hz, H-6,7); 13C-NMR, (δ-ppm) (CDCl3): 168.6 (C-1), 144.3 (C-1″), 140.5 (C-4″), 136.3 (C-4′), 134.2 (C-1′), 130.6 (C-3″, 5″), 129.2 (C-3′, 5′), 127.7 (C-2′, 6′), 121.3 (C-2″, 6″), 51.2 (C-2), 47.8 (C-4), 29.7 (C-5), 20.2 (C-6, 7), 16.8 (C-3); Anal. calcd for C18H22N2O: C, 76.59 H, 7.80; found: C, 76.51 H, 7.73.
Cytotoxic studies
The brine shrimp cytotoxicity test of the synthesized amides was performed against freshly hatched nauplii of Artemia salina.34 Brine-shrimp (Artemia salina) eggs (San Francisco Bay Brand, Inc., New York, USA) were hatched in artificial seawater (40 g sea salts/L) at room temperature (22°C–29°C). The hatched shrimps after two days were transferred to vials (10 shrimps per vial) containing artificial sea water. The synthesized amides (4a–j) were also added to the vials with final concentrations 1,000, 100 and 10 µg/mL taken from their stock solution of 10 mg/mL in dimethyl sulfoxide (DMSO). The surviving nauplii were counted with the aid of a magnifying glass after 24 hours. The mean mortality at three dose levels for amides (4a–j) was determined and repeated three times. Potassium dichromate was used as positive control; data were analyzed; and LD50 values were determined by Probit analysis.
Antitumor activity
Antitumor activity of the synthesized dexibuprofen amides (4a–j) was determined by the crown gall tumor inhibition assay (potato disc assay) with slight modification.35 The fresh red-skinned potatoes were sterilized with 0.1% mercuric chloride for 10 minutes. The potatoes discs (0.5 cm thickness) were formed by using a metallic cork borer (8 mm) and a special cutter under complete aseptic conditions. These potato discs were then placed into petri plates each containing 1.5% agar (25 mL) solution. Five potato discs were placed on each plate and three plates were used for each test sample along with same number of plates for control. Each of the synthesized dexibuprofen amide (10 mg) was dissolved in DMSO (1.0 mL) in separate test tubes as a stock solution. Then the stock solution of the test sample (0.5 mL, 10 mg/mL) was added to a broth culture of Agrobacterium tumefaciens (2.0 mL, at 10 and 48 hours culture containing 5×109 cells/mL) and autoclaved distilled water (2.5 mL) to yield 1,000 µg/mL final concentration. These cultures (50 µL) were poured onto each potato disc. The petri plates were wrapped with parafilm to make the plates air tight and were incubated at 28°C for 21 days. Lugol’s solution was prepared by mixing equal amount of 5% iodine and 10% potassium iodide. After 21 days incubation, the discs were then covered with Lugol’s solution for staining purpose and were allowed for 15 minutes to diffuse. The number of tumors was counted with a dissecting microscope with side illumination of light. The destained portion of discs were actually tumors, and the number of tumors per disc was counted. The following formula was used for the determination of percentage of inhibition;
Anticancer activity
In vitro anticancer activity of the synthesized amides (4a–j) was tested against breast carcinoma cell line (MCF-7) and human normal epithelial breast cell line (MCF-12A) by sulforhodamine B (SRB) assay.36 Cells were plated in 96-mul-tiwell plate (104 cells/well) for 24 hours before performing anticancer activity to allow attachment of cells to the wall of the plate. The anticancer activity of the amides (4a–j) at various concentrations (0, 5, 12.5, 25 and 50 µm/mL) was assessed by the addition of tested compounds to the cell monolayer triplicate wells prepared for each individual dose. The cells were then incubated with synthesized amides for 48 hours at 37°C and in atmosphere of 5% CO2. The cells were fixed, washed and stained with SRB after the incubation time. Acetic acid (1%) was used to wash the excess stain and color intensity was measured spectrophotometrically with an ELISA reader. Anticancer activity was expressed as the mean IC50 of three independent experiments and compared with the IC50 of the standard drugs (erlotinib and doxorubicin).
Ulcerogenic studies
The ulcerogenic activity of compounds 4e and 4g were determined by using the fasted rat model. Dexibuprofen was used as standard for comparison purpose. The animals were categorized into 4 groups, each having 5 animals. Group 1 was control and was given 0.5% carboxymethyl cellulose suspension. The standard dexibuprofen at a dose of 20 mg/kg p.o. was given to group II. The 4e and 4g (20 mg/kg p.o.) were given to groups III and IV, respectively. Further, access to food and water was restricted 24 hours before starting the experiment. After 4 hours administration of dose, the stomach was removed by sacrificing the animal. Stomach was kept on saline-soaked filter paper for inspection. The stomach was opened along greater curvature and cleaned with distilled water. The mucous was removed and the area of ulcers and stomachs were calculated.37
The following formula was used for calculation of ulcer index (UI):
Molecular docking
Selection and preparation of BRCA1 from PDB
The human crystal structure of BRCA1 with PDBID 3K0H was accessed from Protein Data Bank (PDB) (www.rcsb.org). The target structure was minimized and all the water molecules were removed. For energy minimization, we employed conjugate gradient algorithm and amber force field with adjusted 100 steepest descent steps having 0.02 Å step size by using UCSF Chimera 1.10.1.38 Furthermore, the structure validity was confirmed by MolProbity server by observing the residual stereo-chemical properties including bond length and bond angles. Moreover, Ramachandran plot values for BRCA1 were also determined by MolProbity server.39,40 The Ramachandran graph was generated by graphical visualizing tool (Discovery Studio 4.1 Client; BIOVIA, San Diego, CA, USA).41 The protein domains (A and B) residues with graphical depictions were retrieved by PDBsum.42
Sketching of synthesized ligands structures by ACD/ChemSketch
ACD/ChemSketch tool (Advanced Chemistry Development, Inc., Toronto, Ontario, Canada) was employed to create images of synthesized compounds (4a–j). Furthermore, the basic chemoinformatic properties of all synthesized compounds were also predicted by ACD/Chemsketch tool. The online tool like Molsoft (http://www.molsoft.com/) was applied to predict their drug-likeness score values, hydrogen bond acceptors (HBAs) hydrogen bond donors (HBDs) and log P-values and also validate the Lipinski’s rule of five (RO5). Moreover, the mutagenic, tumorigenic and irritant risk assessments were determined by DataWarrior tool (openmolecules.org).43
Molecular docking of synthesized compounds using AutoDock
Molecular docking of all the synthesized ligands (4a–j) against BRCA1 was carried out using diverse AutoDock 4.2 tool (Molecular Graphics Laboratory, Department of Molecular Biology, The Scripps Research Institute, La Jolla, CA, USA) according to the specified instructions.44 In brief, for receptor protein the polar hydrogen atoms and Kollman charges were apportioned and for ligands Gasteiger partial charges were designated and nonpolar hydrogen atoms were merged. The torsion angles for all the ligands were set free to rotate through docking experiments. The docking experiments were carried out by considering receptor as a rigid while ligands as flexible molecules. A grid map of 80 Å × 80 Å × 80 Å was set on the whole protein structure to generate the grid map. The number of runs for each docking experiment was set to 100. The Lamarckian genetic algorithm and empirical free energy function were applied by taking docking parameters default. The docked complexes were further evaluated on lowest binding energy (Kcal/mol) values and hydrogen bond analysis by using Discovery Studio (4.1) and UCSF Chimera 1.10.1.
Molecular dynamic (MD) simulation assay
The best docked energy (Kcal/mol) complexes (4e and 4g) were selected for MD simulations to observe the residual backbone flexibility. All the MD simulations were performed by Groningen Machine for Chemical Simulations (GROMACS, University of Groningen, KTH Royal Institute of Technology, Uppsala Universitet, Uppsala, Sweden) 4.5.4 package with GROMOS 53A6 force field and water model SPC216.45,46 The receptor and ligand topology files were generated by using GROMOS 53A6 force field and online PRODRG server, respectively.47 Furthermore, all the receptor-ligand complexes were solvated and placed in the middle of cubic box with an adjusted 0.8 Å distance. The overall system charge was neutralized by adding ions. The energy minimization (nsteps =50,000) was performed by using steepest descent method (1,000 ps) and energy calculation was done by Particle Mesh Ewald method.48 The final MD run was adjusted to 10,000 ps with nsteps 5,000,000 and analyzed using Xmgrace software (http://plasma-gate.weizmann.ac.il/Grace/).
Results and discussion
Chemistry
The title dexibuprofen amide analogues (4a–j) were synthesized by using a previously described method with slight modifications (Scheme 1).49 Dexibuprofen (1) was converted into corresponding acid chloride (2) by reacting with thionyl chloride in the presence of anhydrous benzene. The dexibuprofen acid chloride (2) in the second step was condensed with substituted amines (3a–j) in the presence of dry acetone to produce desired dexibuprofen amides (4a–j). All of the synthesized amides were purified by flash chromatography and structures were confirmed by their spectroscopic data. The characteristic absorption peak for −NH in FTIR appeared at 3,275–3,297 cm−1 and stretching absorption peak for amide carbonyl (C=O) at 1,651–1,665 cm−1 which confirmed the formation of the amide linkage. The presence of singlet for −NH proton appeared at δ 7.01–7.16 ppm in NMR spectra also confirmed the formation of the desired amides. The formation of the title amides was also ascertained by the presence of signal for amide carbonyl at 150–180 ppm in 13C NMR spectral supplementary data. Chiral HPLC analysis has been performed to determine enantiomers using a normal-phase HPLC method coupled with UV detector and a chiral column. The HPLC analysis further confirmed that no racemization occurs with more than 90%, optical purity in the synthesized compounds.
Bioassay
Cytotoxicity of the synthesized dexibuprofen amide derivatives (4a–j) was determined against brine shrimp larvae because shrimp larvae have been extensively utilized to assess preliminary cytotoxicity. Table 1 presents the results of the cytotoxic effects on shrimp larvae and antitumor activity against tumor formation by Agrobacterium tumefaciens on potato discs. The results showed that all of the synthesized compounds are non-toxic to shrimp larvae. The compounds LD50 range from 270.53±12.3 to 543.46±13.7 µg/mL which is far away from the selected concentration to determine the anticancer activity. The various concentrations of the compounds used to evaluate their anticancer activity are 0, 5, 12.5, 25 and 50 µg/mL. The preliminary cytotoxicity investigations revealed that the compounds were not toxic to brine shrimp larvae and could be further employed to evaluate their anticancer activity. The dexibuprofen amide derivatives (4a–j) have been designed and synthesized to evaluate their inhibitory effects on tumor formation by Agrobacterium tumefaciens on potato discs. The antibacterial activity of synthesized compounds has been performed and it was found that none of them possess antibacterial activity against Agrobacterium tumefaciens. The crown gall tumor inhibition assay was adopted to find the antitumor potential of the synthesized compounds. Crown gall is a neoplasmic disease of plants induced by Agrobacterium tumefaciens due to its tumor-inducing plasmid. The homocyclic and heterocyclic moieties are attached to dexibuprofen through amide linkage to explore their role in antitumor activity. The synthesis of halogen-substituted amides was also carried out to evaluate their significance in the growth inhibition of tumors. Most of the synthesized amides exhibited significant antitumor activity while compounds 4h and 4i were inactive. The dexibuprofen amides substituted with aromatic moieties are more active compared to the aliphatic substituted amides. The presences of halogen substituents additionally on aromatic ring improve the antitumor activity. The chloro substituted dexibuprofen amides 4g and 4e displayed excellent antitumor activity with 100% inhibition of tumor growth. It has been exposed from our bioassay results that the major determining factor of inhibitory activity is the position and not the number of the halogens. The compound 4e bearing 2,5-dichloro substituted phenyl ring and 4g possesses 2-chloro substituted phenyl ring showed the most potent activity against inhibition of tumors compared to all other amides. On the other hand compounds 4a and 4d possess 4-chloro and 2,4-dichloro phenyl substitution, respectively. They exhibited 47% and 69% tumor inhibition in case of compounds 4a and 4d respectively. The presence of chloro substituent at ortho position of phenyl ring plays a vital role in antitumor activity of these derivatives. Additionally if the para position of the phenyl ring was unsubstituted, then derivative showed excellent antitumor activity. Interestingly, compound 4f bearing unsubstituted phenyl ring showed better tumor inhibition than compounds 4a and 4d which possess chloro substation and para position. Based upon our results we propose that dexibuprofen analogues 4g and 4e may serve as a lead structure for the design of more potent anticancer drugs.
Table 1.
Brine shrimp lethality assay and antitumor activity of synthesized dexibuprofen amides (4a–j)
| Compounds | Brine shrimp lethality LD50 (µg/mL) | Antitumor activity | |
|---|---|---|---|
| Average number of tumors ± SE | Inhibition of tumors (%) | ||
| 4a | 356.31±21.4 | 2.15±0.22 | 34.66 |
| 4b | 263.25±16.8 | 1.76±0.20 | 46.51 |
| 4c | 543.46±13.7 | 2.13±0.17 | 35.26 |
| 4d | 390.57±11.3 | 1.02±0.15 | 69 |
| 4e | 270.53±12.3 | 0.0±0.0 | 100 |
| 4f | 412.45±11.3 | 2.72±0.23 | 17.33 |
| 4g | 359.55±9.4 | 0.0±0.0 | 100 |
| 4h | 541.64±13.5 | ND | ND |
| 4i | 434.44±11.6 | ND | ND |
| 4j | 289.67±12.54 | 1.34±0.14 | 59.28 |
| Potassium dichromate | 0.89±0.01 ND | ND | ND |
| Negative control | 1.34±0.14 | 59.28 | |
Notes: Data is based on mean value of three replicates of each for 1,000, 100 and 10 µg/mL concentrations against brine shrimps (in vitro); compound dilutions were prepared with DMSO. Data represents mean value of 15 replicates.
Abbreviations: SE, standard error; DMSO, dimethyl sulfoxide; ND, not determined.
Anticancer activity
The synthesized dexibuprofen amide derivatives (4a–j) were tested for anticancer activity against breast carcinoma cell line MCF-7 using SRB assay and using erlotinib and doxorubicin as standard drugs. The anticancer activity results showed that the presence of chloro substitution at phenyl ring imparts a major role in activity. The derivative 4e exhibited excellent growth inhibition of cancer cells with IC50 value 0.01±0.002 µm while IC50 values of erlotinib and doxorubicin are 0.02±0.003 µm and 0.04±0.006 µm, respectively (Table 2). The presence of 2,5-dichloro substituted phenyl ring in compound 4e plays a vital role in growth inhibition of cancer cells. The compound 4g bearing 2-chloro substituted phenyl ring exhibited comparable activity with standard drug erlotinib which has more potent activity (IC50 0.03±0.004 µm) than doxorubicin. The derivatives 4d and 4j showed significant anticancer activity with IC50 values of 1.02±0.15 µm and 1.7±0.81 µm, respectively. The compounds 4b, 4c, 4h and 4i showed no anticancer activity against breast carcinoma cell line MCF-7. The type and position of the substituent at side chain phenyl ring are the determining factors in anticancer activity of the synthesized compounds. The compound 4e displayed more potent anticancer activity against breast carcinoma cell line MCF-7 among the synthesized compounds. None of the synthesized amide derivatives exhibited cytotoxic effects against human normal epithelial breast cell line (MCF-12A).
Table 2.
Anticancer activity of synthesized dexibuprofen amides (4a–j) against breast carcinoma cell line (MCF-7) and human normal epithelial breast cell line (MCF-12A)
| Compounds | IC50 (µM/mL) MCF-7 | IC50 in µM/mL MCF-12A |
|---|---|---|
| 4a | 6.57±1.54 | <76 |
| 4b | ND | ND |
| 4c | ND | ND |
| 4d | 1.02±0.15 | <76 |
| 4e | 0.01±0.002 | <80 |
| 4f | 23.34±4.44 | ND |
| 4g | 0.03±0.004 | <76 |
| 4h | ND | ND |
| 4i | ND | ND |
| 4j | 1.7±0.81 | <80 |
| Erlotinib | 0.02±0.003 | ND |
| Doxorubicin | 0.04±0.006 | ND |
Abbreviation: ND, not determined.
The ulcerogenic activity of the 4e and 4g was determined to find out the gastrointestinal side associated with these compounds. It was found that synthesized compounds were devoid of such side effects, having smaller gastrointestinal irritations compared to dexibuprofen. Table 3 presents the ulcerogenic activity results and it was observed that UI of dexibuprofen-treated animals is 2.89 while compounds 4e and 4g exhibited 2.32 and 2.44, respectively.
Table 3.
Ulcerogenic activity of most potent amides 4e and 4g
| Serial no | Compounds | Ulcer index (mean ± SEM) |
|---|---|---|
| Control group | CMC | 0.37±0.37 |
| 1 | Dexibuprofen | 2.89±0.63 |
| 2 | 4e | 2.32±0.35 |
| 3 | 4g | 2.44±0.44 |
Note: Values are expressed as mean ± SEM (N=6).
Abbreviations: CMC, carboxymethyl cellulose; SEM, standard error of the mean.
Structural evaluation of target protein
BRCA1 is a protein coding gene also known as breast cancer type 1 susceptibility protein, is responsible for repairing DNA.50,51 BRCA1 and BRCA2 are unrelated proteins normally expressed in breast cells and other tissue, where they help to repair damaged DNA.52 BRACs proteins are also involved in the repair of chromosomal damage with an important role in the error-free repair of DNA double-strand breaks. However, another study reported that mutations in BRCA1 or BRCA2 increase the risk for breast cancer.53 We selected this protein (BRCA1) having PDBID 3K0H as a target structure in docking experiment because their significance data is available in the literature.54 This study revealed the overall protein architecture including domains and binding pocket by X-ray diffraction analysis. Furthermore, a number of researchers have been using BRCA1 protein as a target molecule to identify the novel anticancer candidates.55,56 Based upon these finding, we may consider BRCA1 as a target protein for cancer study using computational approaches.
Breast cancer type 1 susceptibility protein (BRCA1) is normally expressed in breast cells and functionally involved in DNA repair mechanism. BRCA1 is a single-chain protein which contains 215 residues with couple of domains (A, B) having 106 and 69 residual length, respectively (Figure 1). The single domain with hierarchical pattern is shown in Figure 2. The observed Ramachandran plot indicated that 87.7% of all residues were present in favored regions while 97.2% were adjusted in allowed regions. The predicted Ramachandran graph values showed good accuracy of phi (ϕ) and psi (ψ) angles among the coordinates of targeted protein.
Figure 1.
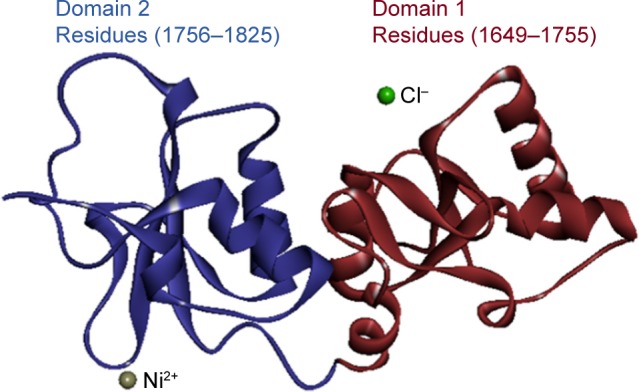
Overview of crystal structure of BRCA1.
Notes: Red color shows domain 1 with residual length while blue color shows domain 2 with residual length. Two circular depictions in green and grey colors represent the chlorine and nickel metals, respectively.
Figure 2.
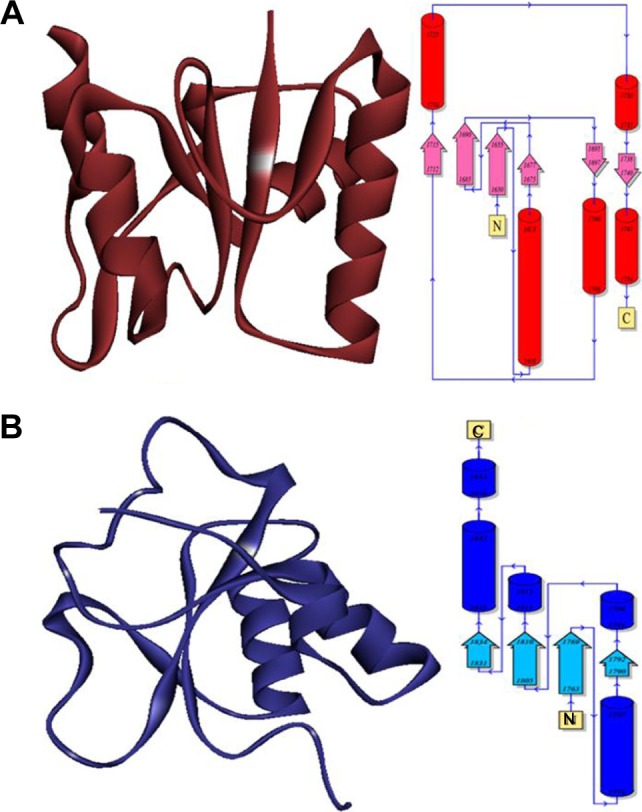
The hierarchical patterns of domains 1 and 2 of target protein BRCA1.
Notes: Red color shows domain 1 while blue color shows domain 2. You can add A with red color and B with blue color.
Chemoinformatic properties and Lipinski’s rule
The biochemical properties of synthesized dexibuprofen amides (4a–j) were predicted using online computational tools. Table 4 shows the predicted properties like molecular weight (g/mol), molar refractivity (MR, cm3), density (g/cm3), polarizability (cm3) and polar surface area (PSA) (Å2) values of synthesized amides. Literature data mining justified that PSA is a very supportive parameter for drug absorption prediction in drug discovery.57 The MR and lipophilic properties of drug molecules are significant in receptor binding, bioavailability and cellular uptake. Ghose and Kadam reported the standard values for MR (40–130 cm3) and molecular weight (160–480 g/mol), respectively.58,59 The compounds (4d–e) and 4h showed higher MR values (98.21, 98.21 and 118.43 cm3) compared to other amides. Moreover, the predicted molecular masses of these compounds were 349.10, 349.10 and 373.33 g/mol which are within the standard values range.
Table 4.
Biological properties of synthesized dexibuprofen amides (4a–j)
| Properties | 4a | 4b | 4c | 4d | 4e | 4f | 4g | 4h | 4i | 4j |
|---|---|---|---|---|---|---|---|---|---|---|
| No. of HBA | 1 | 1 | 1 | 1 | 1 | 3 | 1 | 1 | 1 | 2 |
| No. of HBD | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Mol. log P-value | 5.54 | 4.79 | 5.13 | 6.13 | 6.13 | 1.93 | 5.42 | 8.44 | 4.59 | 3.77 |
| PSA (A2) | 23.33 | 24.65 | 24.38 | 22.63 | 22.63 | 43.86 | 22.63 | 24.67 | 24.76 | 32.76 |
| SC | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 |
| Mol. vol (A3) | 312.83 | 312.42 | 321.46 | 329.29 | 329.29 | 262.08 | 312.02 | 438.47 | 295.22 | 290.93 |
| MR (cm3) | 93.31 | 91.95 | 88.59 | 98.21 | 98.21 | 88.42 | 93.31 | 118.4 | 81.36 | 86.51 |
| Drug score | 1.70 | 1.16 | 1.87 | 1.49 | 1.66 | 0.53 | 1.59 | 0.88 | 1.03 | 1.88 |
Abbreviations: MR, molar refractivity; SC, stereo centers; HBA, hydrogen bond acceptor; HBD, hydrogen bond donor.
The comparative analyses revealed that the predicted values of all the compounds were comparable with standard values. Moreover, irrespective of their higher molecular weight (g/mol) and MR (cm3) values, they showed a very good drug likeness score. The drug score values may reflect the significance of compounds serving as lead structure and possess good therapeutic potential. It has been observed that the higher the drug score values the greater the potential of that compound to act as a lead structure.60 The predicted drug score values of our synthesized compounds ranged from 0.53 to 1.88, which showed that all the compounds are therapeutically active and possess drug-like behavior. The RO5 analysis showed the hydrogen-bonding affinity which is considered as a significant parameter for evaluating the drug permeability. Bakht et al (2010) reported that higher values of HBA (>10) and HBD (>5) in ligands results in poor permeation in the body.61 Our chemoinformatics analyses showed that all the synthesized compounds possess <10 HBA and <5 HBD which may assure their good penetration within the body. Moreover, their log P-values are also in the allowed range when compared with the standard values <5. However, there are plenty of examples available for RO5 violation among the existing drugs.62
Mutagenic risk assessment
DataWarrior tool was employed to predict the mutagenic risk assessment of synthesized compounds (Table 5). Different risk assessments such as mutagenic, tumorigenic, reproductive and irritant effects were observed for amides (4a–j). All compounds showed negative mutagenic behavior in risk assessment. Tumorigenic and reproductive risk effects were high in 4f and low in all other compounds. Furthermore, all compounds showed nonirritant behavior except 4d–e and 4g which depict low irritant behavior. Based on these predicted hypothetical results we may conclude that these compounds have some lead like potential with minimum toxicity risk effects. We further evaluate these compounds on the basis of molecular docking and simulation study to identify the best ligand.
Table 5.
Toxicity risk assessment evaluations of dexibuprofen amides (4a–j)
| Compounds | Mutagenic | Tumorigenic | Reproductive | Irritant |
|---|---|---|---|---|
| 4a | None | None | None | None |
| 4b | None | None | None | None |
| 4c | None | None | None | None |
| 4d | None | None | None | Low |
| 4e | None | None | None | Low |
| 4f | None | High | High | None |
| 4g | None | None | None | Low |
| 4h | None | None | None | None |
| 4i | None | None | None | None |
| 4j | None | None | None | None |
Docking energy and binding pocket analysis
To observe the best conformational position of synthesized compounds (4a–j) against BRCA1, docking study was employed using AutoDock 4.2 tool (The Scripps Research Institute, La Jolla, CA, USA). All the docked complexes were analyzed on the basis of lowest energy values (Kcal/mol) and binding interactions. The wet lab results are in good agreement with the dry lab results. The synthesized amides 4e, 4g and 4d showed good binding energy values of −6.39, −6.34 and −6.24 Kcal/mol, respectively, compared with other compounds (Table 6). These binding energy values justified the best conformational position of these molecules within the binding pocket of target protein. It has been observed that ligand efficacy is also an important parameter to evaluate the ligand molecules in docking analysis.63,64 The predicted ligand efficacy values of dexibuprofen amides 4e and 4g also assured their potential to act as lead structures.
Table 6.
Docking energy evaluation of synthesized amides (4a–j) against BRCA1
| Compounds | Binding energy (Kcal/mol) | Ligand efficacy | Intermolecular energy (Kcal/mol) | Torsional energy (Kcal/mol) | Unbound energy (Kcal/mol) |
|---|---|---|---|---|---|
| 4a | −5.67 | −0.26 | −7.46 | 1.79 | −4.43 |
| 4b | −4.65 | −0.21 | −7.04 | 2.39 | −3.19 |
| 4c | −4.41 | −0.21 | −6.20 | 1.79 | −5.07 |
| 4d | −6.24 | −0.30 | −8.03 | 1.19 | −3.92 |
| 4e | −6.39 | −0.28 | −7.58 | 1.19 | −3.38 |
| 4f | −4.61 | −0.22 | −6.40 | 1.79 | −3.51 |
| 4g | −6.34 | −0.29 | −7.54 | 1.19 | −0.97 |
| 4h | −5.21 | −0.19 | −8.49 | 3.28 | −3.24 |
| 4i | −4.44 | −0.23 | −6.53 | 2.09 | −4.08 |
| 4j | −4.61 | −0.22 | −6.40 | 1.79 | −2.95 |
The comparative analysis of binding energy values revealed that compound 4e binds to the target protein with stronger binding interactions compared to all other derivatives. The amide carbonyl oxygen in the same compound forms hydrogen bond with amino acid LEU1701 having bond length of 1.91 Å (Figure 3). In compound 4g the amide carbonyl oxygen interacts through hydrogen bonding with the amino acid residue LEU1701 having bond length of 2.18 Å (Figure 4). The functional groups in our synthesized amides interact with the amino acid residue of the target protein located in the active binding site. Literature also supported our docking results and verified that amides 4e and 4g hits with active binding residues of target protein. The comparative binding energy and SAR analysis showed that compound 4e may serve as potential inhibitor against carcinoma by targeting BRCA1.
Figure 3.
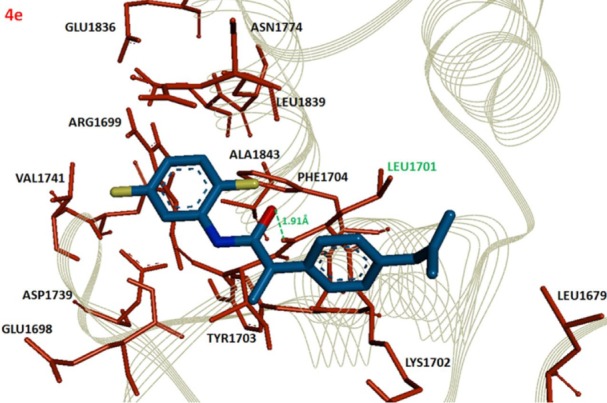
Docking complex of 4e against BRCA1.
Notes: The ligand structure is shown in Berghaus blue while the functional groups such as oxygen, nitrogen and chlorine are displayed in red, blue and yellow colors, respectively. The active site residues are depicted in dark red and labeled with black. The amino acid which participates in hydrogen bonding is highlighted in green along with bonding distance in angstrom (Å). The receptor protein is displayed in line format in light grey.
Figure 4.
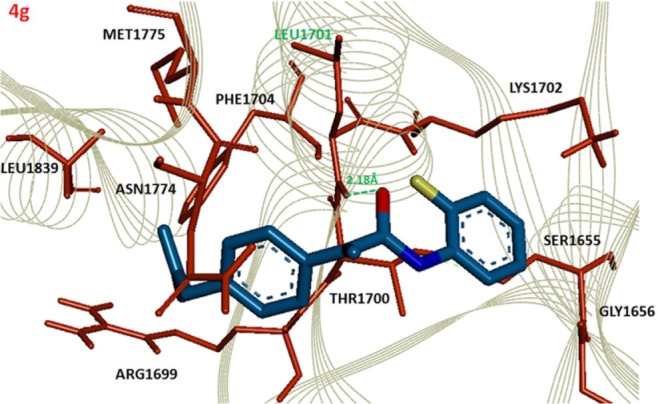
Docking complex of 4g against BRCA1.
Notes: The ligand structure is displayed in Berghaus blue while the functional groups such as oxygen, nitrogen and chlorine are displayed in red, blue and yellow colors, respectively. The active site residues are depicted in dark red and labeled with black. The amino acid which participates in hydrogen bonding is highlighted in green along with bonding distance in angstrom (Å). The receptor protein is displayed in line format in light grey.
Moreover, our standard drugs (erlotinib and doxorubicin) docking results against BRCA1 (Figure 5) also showed the same binding conformational pattern with 4e and 4g with in the active region of target protein. A previously reported data also strengthens our docking results that doxorubicin and both 4e and 4g structures interact with amino acid residues within the binding pocket.65 All the docking complexes were superimposed to analyze the binding conformations of all ligands against the target protein (Figure 6). It has been verified from docking results that our synthesized compounds binds within the active binding site of the target protein.66 In our docking results LEU1701 was the common residue which binds against 4e and 4g compounds and surprisingly the same pattern of interactions was observed for standard drugs (erlotinib and doxorubicin) docking results.
Figure 5.

Docking complexes of standard drugs (erlotinib and doxorubicin) against BRAC1.
Figure 6.
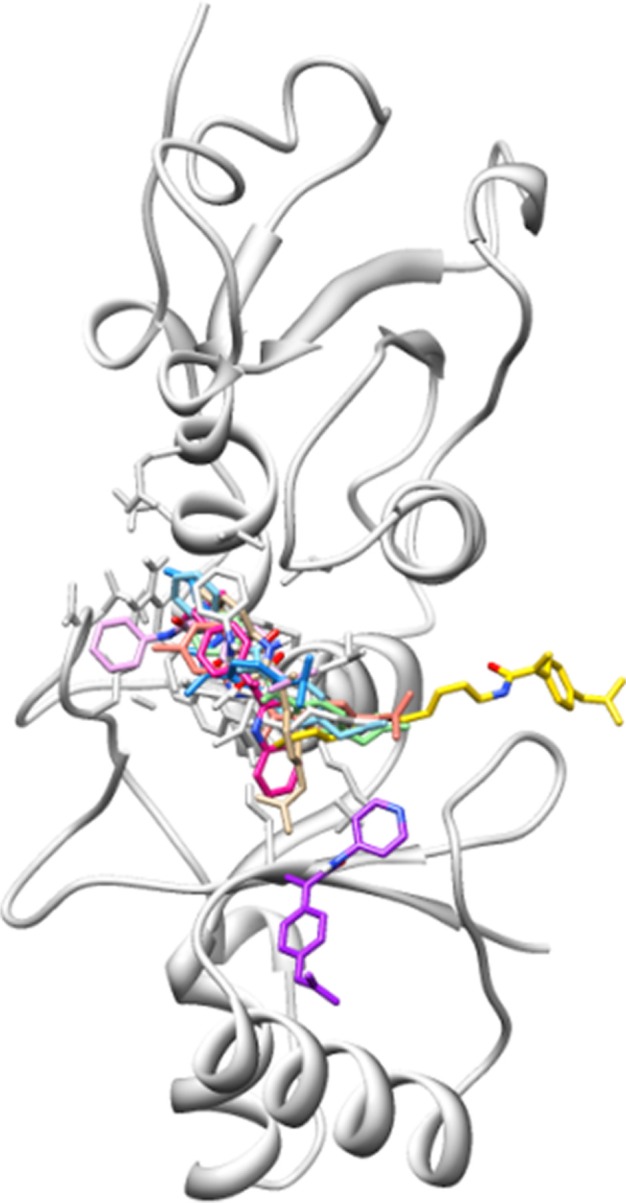
Docking complexes of the synthesized dexibuprofen derivatives 4a–j.
Molecular dynamic simulation analysis
MD simulation analysis was performed to evaluate the residual flexibility of receptor molecule at 10 ns by using GROMACS 4.5.4 tool. Two best docking complexes 4e and 4g were selected to understand reliability of target protein. The root mean square deviation (RMSD), root mean square fluctuations (RMSFs), radius of gyration (Rg) and solvent-accessible surface area (SASA) (A2) graphs were generated to understand the protein structural behavior in docking complexes. The RMSD results of both 4e and 4g complexes showed the protein backbone residual deviation and fluctuations. Initially, both graph lines showed an increasing trend with RMSD values ranging from 0.1 to 0.2 nm at time 0–2,000 ps. From 2,000 ps to 7,000 ps, 4e complex showed stable graph line while within same time frame 4g displayed a fluctuated graph line. Furthermore, from 7,000 ps to 10,000 ps again more fluctuations were observed in 4g as compared to 4e. The RMSD value for 4e remains the same as compared to 4g which confirmed that 4e docking complex simulation graph is more stable throughout the simulation time period as compared to 4g complex (Figure 7).
Figure 7.
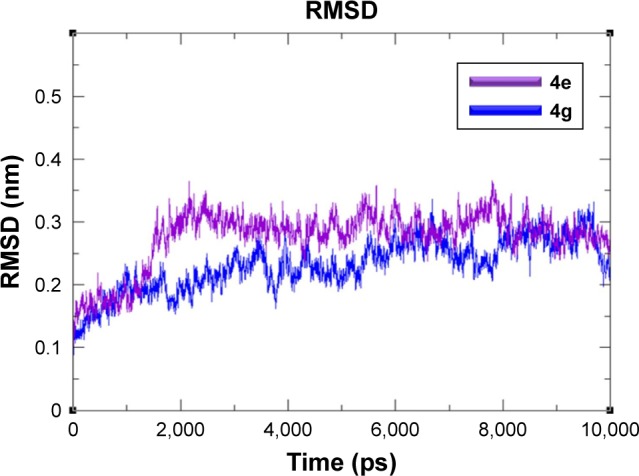
RMSD graphs of 4e and 4g docked complexes are shown in purple and blue respectively from 0–10,000 ps time scale.
Abbreviation: RMSD, root mean square deviation.
Figure 8 exposed the RMSF results which reflect that both C and N-terminal lobes of target protein fluctuated throughout the simulation period. Both RMSD and RMSF graphs showed the significance of 4e docking complex over the 4g complex. The MD simulation behaviors of 4e docked complex throughout MD trajectories thus proved our docking results.
Figure 8.
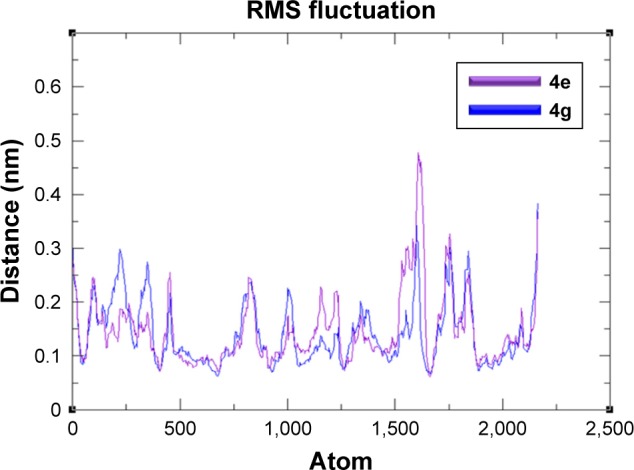
RMSF graphs of 4e and 4g docked complexes are shown in purple and blue respectively from 0–10,000 ps time scale.
Abbreviation: RMSF, root mean square fluctuation.
The Rg was observed to predict the compactness of target protein. The generated results exposed that Rg value is stable between 1.85 and 1.9 nm values throughout the simulation time frame 0–10,000 ps (Figure 9). The SASA was also observed and depicted in Figure 8. Results showed that the 4e and 4g docking complexes co-reside in between 110 and 115 nm2 in the simulation graphs (Figure 10).
Figure 9.
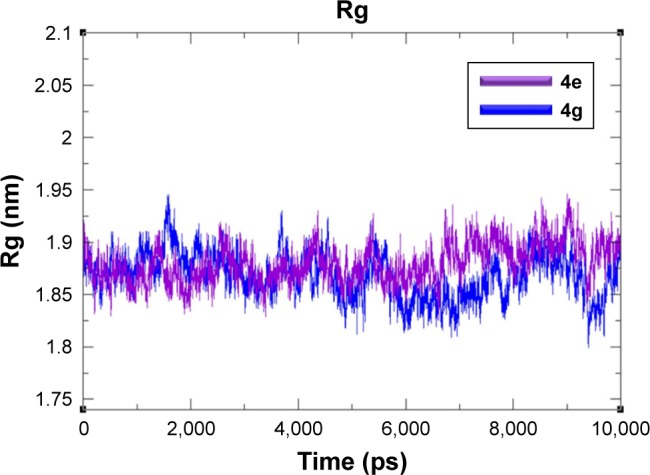
Rg graphs of 4e and 4g docked complexes are shown in purple and blue respectively from 0–10,000 ps time scale.
Abbreviation: Rg, radius of gyration.
Figure 10.
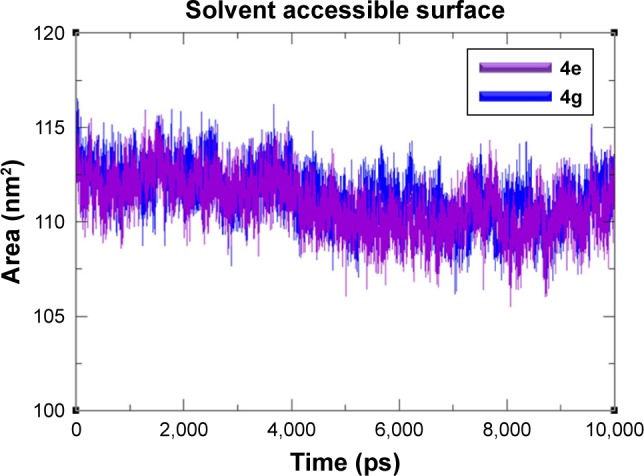
SASA graphs of 4e and 4g docked complexes are shown in purple and blue respectively from 0–10,000 ps time scale.
Abbreviation: SASA, solvent-accessible surface area.
Conclusion
The dexibuprofen amide analogues (4a–j) having alkyl/aryl substitution have been synthesized good to excellent yield. The brine shrimp lethality assay results indicated that all of the synthesized compounds are nontoxic to shrimp larvae. The antitumor activity results indicated that compound 4e bearing 2,5-dichloro substituted phenyl ring and 4g possesses 2-chloro substituted phenyl ring exhibited 100% inhibition of the tumor growth. The anticancer activity results against breast carcinoma cell line (MCF-7) showed that presence of chloro substitution at phenyl ring imparts major role in activity. The derivative 4e exhibited excellent growth inhibition of cancer cells with IC50 value of 0.01±0.002, which is better than the standard drugs. The computational studies revealed that dexibuprofen analogues 4e and 4g possess maximum binding affinity with the target protein PDBID BRCA1 having binding energies −6.39 and −6.34 Kcal/mol, respectively. The MD simulation analysis assured that the 4e formed the most stable complex with the target protein compared to all other derivatives. The synthesized amide derivatives exhibited no cytotoxic effects against human normal epithelial breast cell line (MCF-12A). Based upon our wet lab and dry lab findings we propose that dexibuprofen analogue 4e may serve as a lead structure for the design of more potent anticancer drugs.
Acknowledgments
This research was supported by a grant from from Universiti Kebangsaan Malaysia (DIP-2015-015).
Footnotes
Disclosure
The authors declare that they have no competing interests.
References
- 1.Hung HY, Ohkoshi E, Goto M, Bastow KF, Nakagawa-Goto K, Lee KH. Antitumor agents. 293. nontoxic dimethyl-4,4′-dimethoxy-5,6,5′,6′-dimethy-lenedioxybiphenyl-2,2′-dicarboxylate (DDB) analogues chemosensitize multidrug-resistant cancer cells to clinical anticancer drugs. J Med Chem. 2012;55:5413–5424. doi: 10.1021/jm300378k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012;81:303–311. doi: 10.1111/j.1399-0004.2011.01809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosetti C, Rosato V, Gallus S, Cuzick J, La Vecchia C. Aspirin and cancer risk: a quantitative review to 2011. Ann Oncol. 2012;23:1403–1415. doi: 10.1093/annonc/mds113. [DOI] [PubMed] [Google Scholar]
- 4.Augustin HG. Antiangiogenic tumour therapy: will it work? Trends Pharmacol Sci. 1998;19:216–222. doi: 10.1016/S0165-6147(98)01211-5. [DOI] [PubMed] [Google Scholar]
- 5.Caleta I, Kralj M, Marjanovic M, et al. Novel cyano- and amidinobenzothiazole derivatives: synthesis, antitumor evaluation, and X-ray and quantitative structure-activity relationship (QSAR) analysis. J Med Chem. 2009;52:1744–1756. doi: 10.1021/jm801566q. [DOI] [PubMed] [Google Scholar]
- 6.Ciardiello F, Tortora G. A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res. 2001;7:2958–2970. [PubMed] [Google Scholar]
- 7.Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2004;56:1649–1659. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 8.Aird RE, Cummings J, Ritchie AA, et al. In vitro and in vivo activity and cross resistance profiles of novel ruthenium (II) organometallic arene complexes in human ovarian cancer. Br J Cancer. 2002;86:1652–1657. doi: 10.1038/sj.bjc.6600290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Preetha A, Ajaikumar BK, Chitra S, et al. Cancer is a preventable disease that requires major lifestyle changes. Pharm Res. 2008;25:2097–2116. doi: 10.1007/s11095-008-9661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mei-Hua X, Gui-Ying Z. Effect of indomethacin on cell cycle proteins in colon cancer cell lines. World J Gastroenterol. 2005;21:1693–1696. doi: 10.3748/wjg.v11.i11.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolonel LN, Altshuler D, Henderson BE. The multiethnic cohort study: exploring genes, lifestyle and cancer risk. Nat Rev Cancer. 2004;4:519–527. doi: 10.1038/nrc1389. [DOI] [PubMed] [Google Scholar]
- 12.Cashman JN. The mechanisms of action of NSAIDs in analgesia. Drugs. 1996;52(Supplement 5):13–23. doi: 10.2165/00003495-199600525-00004. [DOI] [PubMed] [Google Scholar]
- 13.Liggett JL, Zhang X, Eling TE, Baek SJ. Anti-tumor activity of non-steroidal anti-inflammatory drugs: cyclooxygenase-independent targets. Cancer Lett. 2014;346:217–224. doi: 10.1016/j.canlet.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konturek PC, Konturek SJ, Bielanski W, et al. Influence of COX-2 inhibition by rofecoxib on serum and tumor progastrin and gastrin levels and expression of PPARgamma and apoptosis-related proteins in gastric cancer patients. Dig Dis Sci. 2003;48:2005–2017. doi: 10.1023/a:1026387908165. [DOI] [PubMed] [Google Scholar]
- 15.Kralj M, Kapitanovic S, Kovacevic D, Lukac J, Spaventi S, Pavelic K. Effect of the nonsteroidal anti-inflammatory drug indomethacin on proliferation and apoptosis of colon carcinoma cells. J Cancer Res Clin Oncol. 2001;127:173–179. doi: 10.1007/s004320000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung WK, Bai AH, Chan VY, et al. Effect of peroxisome proliferator activated receptor gamma ligands on growth and gene expression profiles of gastric cancer cells. Gut. 2004;53:331–338. doi: 10.1136/gut.2003.021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaridze D, Borisova E, Maximovitch D, Chkhikvadze V. Aspirin protects against gastric cancer: results of a case-control study from Moscow, Russia. Int J Cancer. 1999;82:473–476. doi: 10.1002/(sici)1097-0215(19990812)82:4<473::aid-ijc1>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 18.Akre K, Ekstrom AM, Signorello LB, Hansson LE, Nyren O. Aspirin and risk for gastric cancer: a population-based case-control study in Sweden. Br J Cancer. 2001;84:965–968. doi: 10.1054/bjoc.2001.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurpinar E, Grizzle WE, Piazza GA. NSAIDs inhibit tumorigenesis, but how? Clin Cancer Res. 2014;20:1104–1113. doi: 10.1158/1078-0432.CCR-13-1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tinsley HN, Piazza GA. Novel Therapeutics: Sulindac, Derivatives, and Phosphodiesterases. Curr Colorectal Cancer Rep. 2018;4:325–330. doi: 10.1007/s11888-012-0142-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy J. Anti-cancer therapy: non-steroidal anti-inflammatory drugs (NSAIDS) in combination with immunotherapy. MOJ Immunol. 2017;5:00156. doi: 10.15406/moji.2017.05.00156. [DOI] [Google Scholar]
- 22.Yin T, Wang G, Ye T, Wang Y. Sulindac, a non-steroidal anti-inflammatory drug, mediates breast cancer inhibition as an immune modulator. Sci Rep. 2016;6:19534. doi: 10.1038/srep19534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vainio H, Morgan G. Non-steroidal anti-inflammatory drugs and chemoprevention of cancer. Ann Chir Gynaecol. 2000;89:173–176. [PubMed] [Google Scholar]
- 24.Dempke W, Rie C, Grothey A, Schmoll HJ. Cyclooxygenase-2: a novel target for cancer chemotherapy. J Cancer Res Clin Oncol. 2001;127:411–417. doi: 10.1007/s004320000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jolly K, Cheng KK, Langman MJS. NSAIDs and gastrointestinal cancer prevention. Drugs. 2002;62:945–956. doi: 10.2165/00003495-200262060-00006. [DOI] [PubMed] [Google Scholar]
- 26.Marjanovic M, Branka Z, Lana P, Marijana Z, Marijeta K. Fenoprofen and ketoprofen amides as potential antitumor agents. Chem Biol Drug Des. 2007;69:222–226. doi: 10.1111/j.1747-0285.2007.00494.x. [DOI] [PubMed] [Google Scholar]
- 27.Hellberg MR, Namil A, Delgado P. Novel esters and amides of nonsteroidal antiinflammatory carboxylic acids as antioxidants and antiproliferative agents. J Med Chem. 1999;42:267–276. doi: 10.1021/jm980430o. [DOI] [PubMed] [Google Scholar]
- 28.Lengauer T, Rarey M. Computational methods for biomolecular docking. Curr Opin Struct Biol. 1996;6:402–406. doi: 10.1016/s0959-440x(96)80061-3. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Dommert F, Holm C. Optimizing working parameters of the smooth particle mesh Ewald algorithm in terms of accuracy and efficiency. J Chem Phys. 2010;133:034117. doi: 10.1063/1.3446812. [DOI] [PubMed] [Google Scholar]
- 30.Nunes BS, Carvalho FD, Guilhermino LM, Van SG. Use of the genus Artemia in ecotoxicity testing. Environ Pollut. 2006;144:453–462. doi: 10.1016/j.envpol.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 31.Lieberman M. A brine shrimp bioassay for measuring toxicity and remediation of chemicals. J Chem Educ. 1999;76:1689–1691. doi: 10.1021/ed076p1689. [DOI] [Google Scholar]
- 32.Sleet RB, Brendel K. Improved methods for harvesting and counting synchronous populations of artemia nauplii for use in developmental toxicology. Ecotoxicol Environ Saf. 1983;7:435–446. doi: 10.1016/0147-6513(83)90082-9. [DOI] [PubMed] [Google Scholar]
- 33.Galsky AG, Kozimor R, Piotrowski D, Powell RG. The crown-gall potato disk bioassay as a primaryscreen for compounds with antitumor activity. J Natl Cancer Inst. 1981;67:689–692. [PubMed] [Google Scholar]
- 34.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–944. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 35.Meyer BN, Ferrigni NR, Putnam JE, Jacobsen LB, Nichols DE, McLaughlin JL. Brine shrimp: a convenient general bioassay for active plant constituents. Planta Med. 1982;45:31–34. doi: 10.1055/s-2007-971236. [DOI] [PubMed] [Google Scholar]
- 36.Rehman A, Choudhary MI. Bioassay Techniques for Drug Development. Amsterdam: Harwood Academic Publishers; 2001. pp. 9–14. [Google Scholar]
- 37.Ashraf Z, Alamgeer MK, Hassan M, et al. Flurbiprofen-antioxidant mutual prodrugs as safer nonsteroidal anti-inflammatory drugs: synthesis, pharmacological investigation, and computational molecular modeling. Drug Des Devel Ther. 2016;10:2401–2419. doi: 10.2147/DDDT.S109318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2006;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 39.Monk A, Scudiero DA, Skehan P, Shoemaker RH, Paull KD. DTP, DCTD tumor repository a catalog of in vitro cell lines, transplantable animal and human tumors and micro assays. J Natl Cancer Inst. 1991;83:757. [Google Scholar]
- 40.Lovell SC, Davis IW, Arendall WB, III, et al. Structure validation by Cα geometry: φ, ψ and Cβ deviation. Proteins. 2002;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 41.Chen VB, Arendall WB, 3rd, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Studio D. Discovery “Version 21”. San Diego (CA): Accelrys; 2008. [Google Scholar]
- 43.Laskowski RA, Hutchinson EG, Michie AD, Wallace AC, Jones ML, Thornton JM. PDBsum: a web-based database of summaries and analyses of all PDB structures. Trends Biochem Sci. 1997;22:488–490. doi: 10.1016/s0968-0004(97)01140-7. [DOI] [PubMed] [Google Scholar]
- 44.Thomas S, Joel F, Modest VK, Christian R. Data warrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015;55(2):460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- 45.Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDock-Tools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pronk S, Páll S, Schulz R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. 2004;25:1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 48.Schüttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Struct Biol. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 49.Ashraf Z, Imran M, Amin S. Synthesis, characterization and in vitro hydrolysis studies of ester and amide prodrugs of dexibuprofen. Med Chem Res. 2012;21:3361–3368. doi: 10.1007/s00044-011-9866-z. [DOI] [Google Scholar]
- 50.Easton DF. How many more breast cancer predisposition genes are there? Breast Cancer Res. 1999;1(1):14–17. doi: 10.1186/bcr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campeau PM, Foulkes WD, Tischkowitz MD. Hereditary breast cancer: new genetic developments, new therapeutic avenues. Hum Genet. 2008;124(1):31–42. doi: 10.1007/s00439-008-0529-1. [DOI] [PubMed] [Google Scholar]
- 52.Irminger-Finger I, Ratajska M, Pilyugin M. New concepts on BARD1: regulator of BRCA pathways and beyond. Int J Biochem Cell Biol. 2016;72:1–17. doi: 10.1016/j.biocel.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 53.Friedenson B. The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers. BMC Cancer. 2007;7:152. doi: 10.1186/1471-2407-7-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Campbell SJ, Edwards RA, Glover JN. Comparison of the structures and peptide binding specificities of the BRCT domains of MDC1 and BRCA1. Structure. 2010;18(2):167–176. doi: 10.1016/j.str.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 55.Pal S. An insilico drug designing approach to target the BRCA1 protein involved in breast cancer. Helix. 2016:6–1. 761–765. [Google Scholar]
- 56.Thangaraj P. Molecular docking of bioactive compounds against BRCA and COX proteins. In: Jucker EM, editor. Pharmacological Assays of Plant-Based Natural Products Progress in Drug Research. Vol. 71. 2016. pp. 181–183. [PubMed] [Google Scholar]
- 57.Peter E, Bernhard R, Paul S. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43:3714–3717. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 58.Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, Mallamo JP. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem Neurosci. 2012;3:50–3:50. doi: 10.1021/cn300122k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kadam RU, Roy N. Recent trends in drug-likeness prediction: a comprehensive review of in silico methods. Indian J Pharm Sci. 2007;69:609–615. doi: 10.4103/0250-474X.38464. [DOI] [Google Scholar]
- 60.Bakht MA, Yar MS, Abdel-Hamid SG, Al-Qasoumi SI, Samad A. Molecular properties prediction, synthesis and antimicrobial activity of some newer oxadiazole derivatives. Eur J Med Chem. 2010;45:5862–5869. doi: 10.1016/j.ejmech.2010.07.069. [DOI] [PubMed] [Google Scholar]
- 61.Tian S, Wang J, Li Y, Li D, Xu L, Hou T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv Drug Deliv Rev. 2015;86:2–10. doi: 10.1016/j.addr.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 62.Hassan M, Ashraf Z, Abbas Q, Raza H, Seo SY. Exploration of novel human tyrosinase inhibitors by molecular modeling, docking, and simulation studies. Interdiscip Sci. 2016;21 doi: 10.1007/s12539-016-0171-x. [DOI] [PubMed] [Google Scholar]
- 63.Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, Olson AJ. Virtual screening with AutoDock: theory and practice. Expert Opin Drug Discov. 2010;5:597–607. doi: 10.1517/17460441.2010.484460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 65.Ravichandran R, Sundararajan R. In silico-based virtual drug screening and molecular docking analysis of phytochemical derived compounds and FDA approved drugs against BRCA1 receptor. J Cancer Prev Curr Res. 2017;8:00268. doi: 10.15406/jcpcr.2017.08.00268. [DOI] [Google Scholar]
- 66.Periasamy J, Kurdekar VJ, Jasti S, et al. Targeting phosphopeptide recognition by the human BRCA1 tandem BRCT domain to interrupt BRCA1-dependent signaling. Cell Chem Biol. 2018;25(6):677–690. doi: 10.1016/j.chembiol.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]