

SUMMARY
Regulated secretion is critical for diverse biological processes ranging from immune and endocrine signaling to synaptic transmission. Botulinum and tetanus neurotoxins, which specifically proteolyze vesicle fusion proteins involved in regulated secretion, have been widely used as experimental tools to block these processes. Genetic expression of these toxins in the nervous system has been a powerful approach for disrupting neurotransmitter release within defined circuitry, but their current utility in the brain and elsewhere remains limited by lack of spatial and temporal control. Here we engineered botulinum neurotoxin B so that it can be activated with blue light. We demonstrate the utility of this approach for inducibly disrupting excitatory neurotransmission, providing a first-in-class optogenetic tool for persistent, light-triggered synaptic inhibition. In addition to blocking neurotransmitter release, this approach will have broad utility for conditionally disrupting regulated secretion of diverse bioactive molecules, including neuropeptides, neuromodulators, hormones, and immune molecules.
In Brief
Few tools in neuroscience allow long-term synapse inhibition with light. We engineered botulinum neurotoxin B so that it can be switched on with blue light. Light activation results in robust proteolysis of VAMP2, a SNARE protein universally required for neurotransmitter release.
INTRODUCTION
Tools for rapidly and locally silencing distinct populations of neurons have been indispensable for assigning circuit function to in vivo behaviors. Microbial ion pumps (e.g., halorhodopsin or archaerhodopsin), which hyperpolarize neurons during illumination, have been ideal for neural silencing on millisecond to second timescales (Chow et al., 2010; Zhang et al., 2007). However, many experiments require longer (minutes to hours) silencing, which can be difficult to achieve with the current optogenetic toolkit. Most opsin-based silencing strategies require continuous illumination, making photodamage and tissue heating a concern for long-term silencing. Moreover, persistent activation of widely used proton or chloride-conducting microbial opsin pumps leads to perturbations in intracellular pH and buildup of intracellular chloride to levels where activation of GABAA receptors can cause depolarization rather than hyperpolarization (Alfonsa et al., 2015; Raimondo et al., 2012; Wiegert et al., 2017). Complementary chemogenetic approaches for longer-term silencing have been developed, including ivermectin-gated chloride channels, allatostatin-activated receptors, designer receptors exclusively activated by designer drugs (DREADDs), and engineered inhibitory neurotransmitter receptors, but these approaches lack the fine spatial and temporal control of optogenetics (Alexander et al., 2009; Armbruster et al., 2007; Lechner et al., 2002; Lerchner et al., 2007; Magnus et al., 2011; Slimko et al., 2002; Wulff et al., 2007). Thus, there is an unmet demand for tools that are biologically orthogonal and combine the sustained silencing qualities of chemogenetic approaches with the spatial control of optogenetics. Indeed, mechanistically diverse silencing approaches that can complement and extend existing capabilities would have wide utility.
A classic experimental approach for long-term disruption of synaptic transmission is through genetic expression or direct application of Clostridium botulinum or tetanus neurotoxins (Brooks et al., 1957; Molgo et al., 1990; Sweeney et al., 1995; Yamamoto et al., 2003). The catalytic light chains of these toxins are zinc-dependent endoproteases that cleave conserved soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) family proteins that are critical for vesicle docking and fusion with the plasma membrane (Blasi et al., 1993; Link et al., 1992; Schiavo et al., 1992). Although some degree of temporal control can be achieved using inducible expression of these toxins via regulated promoter or recombinase systems (Kim et al., 2009; Nakashiba et al., 2008; Yamamoto et al., 2003; Yu et al., 2004), rapid and local control is currently not possible.
In this work, we engineered a photoactivatable form of botulinum neurotoxin serotype B (BoNT/B) light chain protease. BoNT/B cleaves vesicle-associated membrane proteins (VAMPs) required for diverse forms of regulated secretion, including VAMP2, involved in neurotransmitter release from inhibitory and excitatory neurons (Schiavo et al., 1992). We demonstrate that photoactivatable BoNT/B is activated by light to cleave VAMP2 and suppress neurotransmission within minutes in hippocampal and cortical circuits and can suppress coordinated movement behavior in Caenorhabditis elegans.
RESULTS
Engineering and Optimizing a Split BoNT/B
The light chain of BoNT/B (BoNT/B-LC, amino acids 1–441) is an ~50-kD endoprotease that forms a compact catalytic core. To regulate BoNT/B-LC (hereafter called BoNT/B) protease activity with light, we used a split protein complementation approach wherein a protein is split into two fragments that can be functionally reconstituted when fused to inducible protein dimerizer modules (Figure 1A). We split BoNT/B at solvent-exposed loops to minimize disturbance to the protein structure, targeting five initial sites (Figure 1B). To assay protease activity, we generated a 68-kDa GFP-VAMP-glutathione S-transferase (GST) reporter that yields a 33-kDa fragment when cleaved by BoNT/B. Co-expression of full-length BoNT/B resulted in near-complete conversion of the full-length reporter to the smaller cleavage product (Figure 1C, left).
Figure 1. Split BoNT/B Can Be Reconstituted with Photodimerizers.
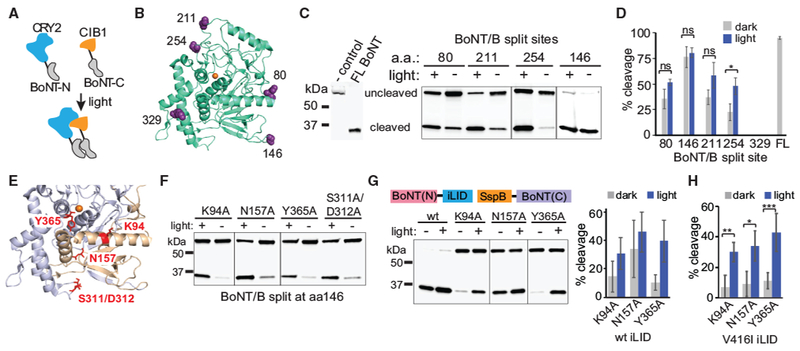
(A) Schematic illustrating reconstitution of split BoNT/B light chain N- and C-terminal fragments mediated by blue light-actuated interaction between CRY2 (blue) and CIBN (orange) photodimerizers.
(B) Location of split sites (purple) within BoNT/B light chain structure (PDB: 2ETF; green). The Zn2+ cofactor is shown in orange.
(C and D) Quantification of light-induced reconstitution of protease activity. HEK293T cells were transfected with a GFP-VAMP-GST cleavage reporter and BoNT/B N- and C-terminal fragments split at the indicated sites. Cells were treated with 461-nm blue light (2-s pulse every 3 min, 15.6 μW/cm2) or kept in dark for 4–5 hr and then analyzed by immunoblot for reporter cleavage. Representative immunoblot results are shown in (C). The line between 211 and 254 samples indicates that the intervening lane was removed; sample 146 was on a separate blot. A summary of reporter cleavage results (average and SD of 3 independent experiments) is shown in (D). ns, not significant; *p < 0.05, 2-tailed Student’s t test.
(E) Amino acids targeted for mutagenesis. Residues predicted to disrupt interaction between the 1–146 (gold) and 147–441 (purple) BoNT/B fragments are indicated in red (the Zn2+ cofactor is indicated in orange).
(F) Immunoblot showing BoNT/B (146-147) interface-disrupting mutations with low dark background and significant light-regulated activity when reconstituted with CRY2/CIB1 photodimerizers. Cells were treated as in (C). The line between N157A/Y365A indicates that the intervening lane was removed; K94A samples were on a separate blot.
(G) Quantification of BoNT/B 146-147 split fragments for reconstitution with iLID-SspBmilli. HEK293T cells were transfected with GFP-VAMP-GST and the indicated BoNT/B constructs and then treated as in (C). Light-treated samples were exposed to a 2-s pulse every 30 s for 4 h. Left: a representative blot; lines indicate that the intervening lanes were removed. Right: average percent cleavage and range of two independent experiments.
(H) Quantification of cleavage using BoNT/B 146-147, SspBmilli, and the long-lived V416I iLID variant. HEK293T cells were treated and analyzed as in (G) except for a 2-s light pulse every 3 min. Data show average and error (SD) from 3-6 independent experiments. *p < 0.05, **p < 0.005, ***p < 0.0005, 2-tailed Student’s t test.
To conditionally reconstitute BoNT/B, N- and C-terminal BoNT/B fragments were fused to nuclear localization signal (NLS)-deleted versions of Arabidopsis cryptochrome 2 (CRY2) and its binding partner CIBN (residues 1–170 of Arabidopsis CIB1), which dimerize upon blue light exposure (Kennedy et al., 2010). CRY2- and CIBN-fused BoNT fragments were expressed in HEK293T cells along with the cleavage reporter. Reporter cleavage was monitored in cells maintained in the dark or after 4 h of light exposure. Of five split sites tested, one site (split at residue 254) showed significant light-regulated activity (Figures 1C and 1D), and another site (329) showed no activity (Figure 1D). BoNT/B split at residue 146-147 showed no light dependence but near-complete reporter cleavage (76.4% ± 4.8% cleavage in the dark, 80.3% ± 10.4% in light) (Figures 1C and 1D), indicating that these fragments reassemble into an active enzyme independent of the fused dimerizer modules. Fragments split at this site but expressed alone showed no detectable reporter cleavage (Figure S1A).
We chose the 146-147 split fragments for further manipulation, given the potent activity of the BoNT/B split at this location. As demonstrated previously with other split proteins (Johnsson and Varshavsky, 1994), we hypothesized that we could identify mutations in the interface between the two fragments that reduce affinity sufficiently to block fragment self-assembly but allow reconstitution of activity upon induction of photodimerizer interaction with light. Analysis of the crystal structure of the intact BoNT/B-LC (Swaminathan and Eswaramoorthy, 2000) revealed extensive electrostatic and hydrogen bonding interactions at the interface between BoNT/B(1–146) and BoNT/B(147–441) (Figures 1E, S1B, and S1C). Of nine interface-disrupting mutants tested (Table S1), four (K94A, N157A, Y365A, and S311A/D312A) showed greatly reduced dark activity but could be activated to varying degrees with light (Figure 1F).
In parallel with testing the CRY2-CIBN dimerizers for reconstitution of split BoNT/B, size limitations for viral packaging motivated us to test other photodimerizer systems. The improved light-induced dimerizer (iLID)-SspB photodimerizer system uses smaller fusions—a light, oxygen, voltage (LOV) domain-peptide fusion (AsLOV2-SsrA, “iLID”) and a domain of E. coli SspB—that are triggered to interact with blue light (Guntas et al., 2015; Zimmerman et al., 2016). In addition to their smaller size, the iLID-SspB system has been engineered for dynamic light control over a range of expression levels by generating mutations in SspB that reduce affinity to LOV2-SsrA (Guntas et al., 2015; Zimmerman et al., 2016). Upon testing the 146-147 split fragments in the iLID-SspB system, we found the optimal fusion configuration to be BoNT(N)-iLID and SspB-BoNT(C), with other configurations not functional (Figure S1D). Our initial studies used SspBnano, which binds with high affinity to iLID, even in the dark (Guntas et al., 2015), and resulted in high background with minimal light-dependent differences in cleavage (Figure S1D). Substitution of SspBmilli, a lower-affinity version of SspB (binding affinity >1 mM in the dark and 56 μM in the light) (Zimmerman et al., 2016) resulted in robust light-dark differences (Figure 1G). Because the wild-type AsLOV2 domain used to generate the iLID component has a short photoactivation lifetime (half-life, ~27 s), we also included a mutation (V416I, using AsLOV2 domain nomenclature) found previously to slow the dark reversion rate ~10-fold (Strickland et al., 2012), enabling use of less frequent light pulse treatments (Figure 1H). The Y365A BoNT mutant, which showed the highest dynamic range in initial experiments, showed low background under a range of expression levels, with both dark- and light-induced activity increasing at higher expression (Figures S1E and S1F). We also tested the Y365A variant with SspBmicro, a R73Q SspB variant that has intermediate affinity between SspBnano and SspBmilli (affinity for the iLID partner of 47 μM in the dark and 800 nM in the light). Use of SspBmicro also yielded good light-dark differences but showed an optimal dynamic range of activity at lower levels of expression compared with SspBmilli (Figure S1G). To enable dual expression of the two fragments, a combined bicistronic construct separated with an internal ribosome entry site (IRES) element worked well (Figure S1F); however, a version separated by a P2A cleavable peptide showed high background, presumably because of the generation of a residual amount of full-length protein composed of both toxin fragments (Figure S1H).
Split BoNT Cleaves VAMP2 and Impairs Neurotransmitter Release
We chose three top candidates from our initial HEK293T screening, CRY2-CIBN BoNT/B K94A and N157A and iLID-SspBmilli Y365A (using BoNT/B fragments split at 146-147), for characterization of endogenous VAMP2 cleavage in dissociated hippocampal neurons. Because proteins expressed in neurons or delivered through viral transduction may express at much lower levels than in HEK293T cells, we also tested the medium-affinity iLID-SspBmicro Y365A version. We transfected neurons with dimerizer-fused BoNT fragments along with synap-tophysin (syph)-GFP as a marker of presynaptic terminals and exposed cells to dark or light for 4 h. Neurons were fixed and immunostained for endogenous VAMP2 using a monoclonal antibody that labels full-length VAMP2 but does not recognize BoNT/B VAMP2 cleavage products. Neurons transfected with mCherry (mCh) alone or mCh plus intact full-length BoNT/B-LC served as negative and positive controls, respectively. Cells expressing full-length BoNT/B showed nearly undetectable levels of VAMP2 (Figure 2A). The CRY2-CIBN K94A and N157A variants and iLID-SspBmilli Y365A exhibited VAMP2 levels in the dark equivalent to the negative control (Figures 2A and S2A). Use of SspBmicro resulted in slightly decreased dark levels of VAMP2 staining (70% ± 5% relative to the negative control), indicating some background activity. Light exposure resulted in a substantial loss of VAMP2 immunoreactivity for all variants tested except CRY2/CIB K94A (Figure 2A). The kinetics and extent of VAMP2 cleavage for iLID-SspB Y365A milli and micro SspB variants are shown in Figure 2B.
Figure 2. Functional Characterization of Split BoNT Variants in Neurons.
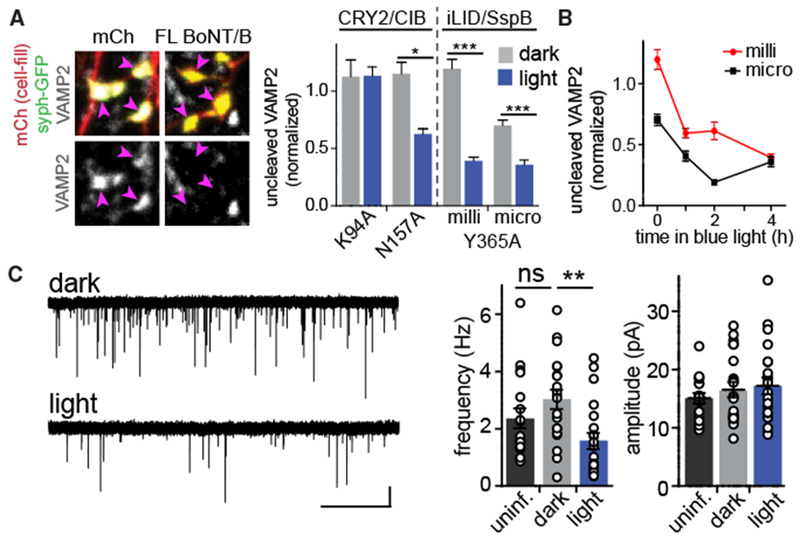
(A) Light triggers reconstitution of split BoNT/B activity. Cultured hippocampal neurons were transfected with syph-GFP (to label presynaptic terminals, pink arrowheads) and either mCherry (mCh) or mCh along with full-length BoNT/B. Note the absence of VAMP2 staining in BoNT/B-expressing terminals. Right: quantification of VAMP2 staining in synaptic terminals from neurons expressing split BoNT/B variants. Cells were kept in the dark (gray bars) or exposed to blue light (1 s every 2 min, 4.5 μW/cm2) for 4 h (blue bars). Values (mean ± SEM from at least 12 cells/condition) were normalized between 0 and 1 relative to positive (full-length BoNT/B) and negative (mCh) controls. *p < 0.05, ***p < 0.0001.
(B) Time course of VAMP2 cleavage in cells transfected with sPA-BoNT with SspBmilli (red circles) or SspBmicro (black squares). VAMP2 levels were normalized as in (A). Mean ± SEM are reported from at least 8 cells from 2 independent experiments.
(C) Virally delivered sPA-BoNT reduces mEPSC frequency in a light-dependent manner. Left: representative AMPA mEPSC traces from cultures infected with AAVs encodings PA-BoNTmicro maintained in the dark (top) or exposed to blue light (1-s pulse every 2 min) for at least 1 h prior to recording (bottom). Right:quantification of AMPA mEPSC frequency (**p < 0.001, one-way ANOVA) and amplitude (n.s.) from uninfected cells (black, n = 18 cells) or infected cells maintained in darkness (gray, n = 20 cells) or exposed to 1–4h of blue light (blue, n = 21 cells). Scale bars, 10 pA, 5 s.
Although light-activated toxins cleaved a significant fraction of VAMP2, the remaining uncleaved VAMP2 could still contribute to vesicular fusion (van den Bogaart et al., 2010; Mohrmann et al., 2010). Thus, we functionally tested whether iLID-SspB variants could impair neurotransmitter release in a light-dependent manner. We first measured vesicle fusion and subsequent uptake of the styryl fluorescent dye FM1-43 into presynaptic terminals of cultured hippocampal neurons. We transfected neurons with iLID-SspBmicro Y365A BoNT fragments, maintained them in the dark or pre-exposed them to 2 or 4 h of blue light pulses, and then assessed FM1-43 uptake triggered by a brief (60-s) exposure to a high (50-mM) isosmotic extracellular K+ solution. Neurons expressing both constructs but maintained in darkness displayed activity-triggered FM1-43 uptake that was nearly identical to that of control neurons (Figures S2B and S2C). Two hours following onset of light exposure, FM1-43 uptake was reduced ~4-fold.
Based on these results, we generated adeno-associated viral vectors containing the BoNT(1–146)-iLIDV416I and SspB-BoNT(147–441, Y365A) combination, which we refer to as soluble photoactivatable BoNT (sPA-BoNT). Because protein expression from viral vectors is likely to be lower than in transiently transfected cells, we tested both SspBmilli and SspBmicro variants. To confirm the efficacy of virally delivered BoNT, we measured spontaneous miniature excitatory postsynaptic currents (mEPSCs) from transduced dissociated hippocampal cultures. Based on the kinetics of sPA-BoNT VAMP2 cleavage, we exposed neurons to blue light for at least 1 h (and no more than 4 h) to achieve maximum depletion of VAMP2 prior to mEPSC recordings. Following exposure of cells expressing sPA-BoNTmicro (using SspBmicro) to blue light, we observed an ~2-fold reduction in the frequency (2.76 ± 0.447 s−1 dark versus 1.460 ± 0.350 s−1 light, p = 0.031, Student’s t test) but no change in amplitude (16.55 ± 1.490 pA dark versus 17.76 ± 2.13 pA light) of AMPA mEPSCs (Figure 2C). In dark control cultures expressing sPA-BoNT, mEPSC frequency and amplitude were not significantly different from uninfected cultures, indicating that background VAMP2 cleavage in the dark did not impair spontaneous vesicle fusion (Figure 2C). sPA-BoNTmilli was not as effective at impairing mEPSCs in cultured hippocampal neurons, presumably because of lower expression levels of virally delivered sPA-BoNT (data not shown). Together, these results demonstrate that sPA-BoNTmicro effectively cleaves VAMP2 and impairs neurotransmission, albeit with relatively slow kinetics.
Targeting Split BoNT/B to Synaptic Vesicles Enhances the Kinetics and Potency of PA-BoNT
We next tested whether targeting PA-BoNT to synaptic vesicles would increase its efficacy. We fused the BoNT(N)-iLID fragment to the synaptic vesicle protein syph along with an EGFP reporter (Figure 3A). The SspB-fused BoNT(C) fragment (SspBmicro or SspBmilli) was expressed separately as a soluble protein. Thus, light will trigger protease assembly directly on synaptic vesicles, in close proximity to VAMP2. We refer to this combination as vesicular PA-BoNT (vPA-BoNT). We observed a 3-fold increase in the rate of VAMP2 cleavage using vPA-BoNTmicro compared with the soluble sPA-BoNT (vPA-BoNT, τ = 7 ± 2 min; sPA-BoNT, τ = 21 ±4 min; Figure 3B). Importantly, vPA-BoNTmicro showed an increased maximal fraction of VAMP2 cleavage in light compared with sPA-BoNTmicro (90% ± 5%, vPA-BoNTmicro; 65% ± 4%, sPA-BoNTmicro) (Figure 3B).
Figure 3. Targeting PA-BoNT to Vesicles Improves Its Efficacy.

(A) Schematic of constructs used to target PA-BoNT to synaptic vesicles.
(B) Left: examples of VAMP2 staining in presynaptic terminals (marked by syph-GFP-BoNT-iLID, pink arrows) from neurons transfected with vPA-BoNT and maintained in the dark (left) or exposed to 15 min blue light (1-s pulse every 2 min) (right). Right: quantification of the VAMP2 signal in terminals from transfected cells relative to neighboring terminals in dissociated hippocampal cultures maintained in the dark (0 min) or exposed to blue light for varying times (p < 0.0001, one-way ANOVA). The kinetics of VAMP2 cleavage by sPA-BoNT (dashed gray line) from Figure 2B is replotted for direct comparison.
(C) Representative traces of mEPSCs from infected cultures kept in the dark (top) or exposed to blue light (bottom; 1-s pulse every 2 min, 4.5 μW/cm2). Scale bars, 20 pA, 5 s.
(D) Left: quantification of AMPA mEPSC frequency (left) and amplitude (right) from uninfected cultures (black) or infected cultures kept in the dark (gray) or exposed to blue light for a minimum of 30 min (blue) (frequency, p < 0.05; amplitude, n.s., one-way ANOVA). Right: cumulative distribution of mEPSC inter-event interval (IEI) for cells kept in the dark (gray) or exposed to blue light (blue, *p < 0.0001, Kolmogorov-Smirnov test).
(E) Time course of VAMP2 recovery following cleavage with vPA-BoNT. After 1 h light exposure, hippocampal neurons were maintained in darkness for varying times to assess the recovery of VAMP2 by immunocytochemistry as in (B). Values represent mean ± SEM from at least 11 cells from 2 independent experiments (***p < 0.0001, *p < 0.05, one-way ANOVA).
(F) Time course of functional recovery. Cultured neurons infected with vPA-BoNT were treated with 1 h of blue light, and mEPSC frequency and amplitude were measured immediately following light exposure or after 8 h or 24 h of dark recovery. Data are normalized to neurons expressing vPA-BoNT but maintained in darkness for the duration. Values represent mean ± SEM from at least 9 cells per condition from two independent experiments (*p < 0.05, Student’s t test).
Given its robust light-dependent VAMP2 cleavage, we functionally characterized vPA-BoNTmicro. We first measured spontaneous quantal neurotransmitter release by recording AMPA receptor-mediated mEPSCs in dissociated hippocampal cultures that had been infected with adeno-associated viruses (AAVs) encoding vPA-BoNT (Figures 3C and 3D). The amplitude and frequency of mEPSCs recorded from neurons expressing the toxin constructs but maintained in darkness were not significantly different than control mEPSCs recorded from uninfected neurons (Figure 3D). Subsequent light exposure resulted in a 2-fold decrease in mEPSC frequency compared with neurons maintained in the dark (Figures 3C and 3D). We next measured the time required for recovery of synaptic transmission following vPA-BoNT activation. We measured VAMP2 protein levels and recorded mEPSCs at time intervals from 1–24 h following light exposure. Within 8 h of dark recovery after light exposure, VAMP2 protein levels had recovered to dark control levels (Figure 3E). mEPSC frequency was still reduced at 8 h (but not significantly different from dark controls) and fully recovered after 24 h (Figure 3F). The slight discrepancy between the kinetics of protein recovery and functional recovery may be due to reduced synaptic transmission preventing new VAMP2 from entering the recycling vesicle pool.
To more precisely define the onset kinetics of the functional effects of vPA-BoNT, we measured spontaneous quantal neuro-transmission by imaging Ca2+ influx through postsynaptic NMDA receptors at individual synapses using the red Ca2+ indicator jRGECO1a (Dana et al., 2016; Sinnen et al., 2016). This approach allows us to measure spontaneous neurotransmission longitudinally, at the same synapses over longer time periods than what is possible with whole-cell recordings. Dissociated hippocampal cultures infected with AAVs encoding vPA-BoNT were sparsely transfected with jRGECO1a and imaged in extracellular solution containing tetrodotoxin (to block action potential-triggered vesicle fusion) and lacking Mg2+ (to allow Ca2+ entry through NMDA receptors). Under these conditions, robust spontaneous Ca2+ transients can be observed at individual dendritic spines that report quantal glutamate release (Figures 4A and 4B; Videos S1 and S2). Under baseline conditions, we observed no difference between the average number of postsynaptic Ca2+ transients at single synapses in cultures infected with AAVs encoding vPA-BoNT (but not exposed to blue light) compared with uninfected cultures (uninfected: 6.3 ± 0.4 events/synapse/min, n = 105 synapses from 9 neurons; infected dark: 6.3 ± 0.3 events/synapse/min, n = 310 synapses from 26 neurons). Blue light treatment of cultures expressing vPA-BoNT potently suppressed the frequency but not the amplitude of spontaneous Ca2+ transients compared with controls (Figure 4C). The kinetics of Ca2+ transient frequency suppression was similar to VAMP2 cleavage, as measured by immunocytochemistry (Figure 3B). Similar results were observed when we swapped the fragment of split BoNT toxin that was tethered to vesicles and/or used SspBmilli (Figure S3). Finally, we tested whether we could locally activate vPA-BoNT with subcellular spatial resolution. We imaged baseline synaptic Ca2+ transients at the same synapses before and after local illumination of a sub-region of the dendritic arbor (Figure 4D). Synapses in the illuminated region displayed a robust decrease in frequency but not amplitude of postsynaptic Ca2+ transients compared with synapses outside of the illuminated region but on the same neurons or illuminated control cells from cultures that were not infected with vPA-BoNT AAVs (Figures 4E and 4F).
Figure 4. vPA-BoNT Can Locally Inhibit Neurotransmission within Minutes of Activation.
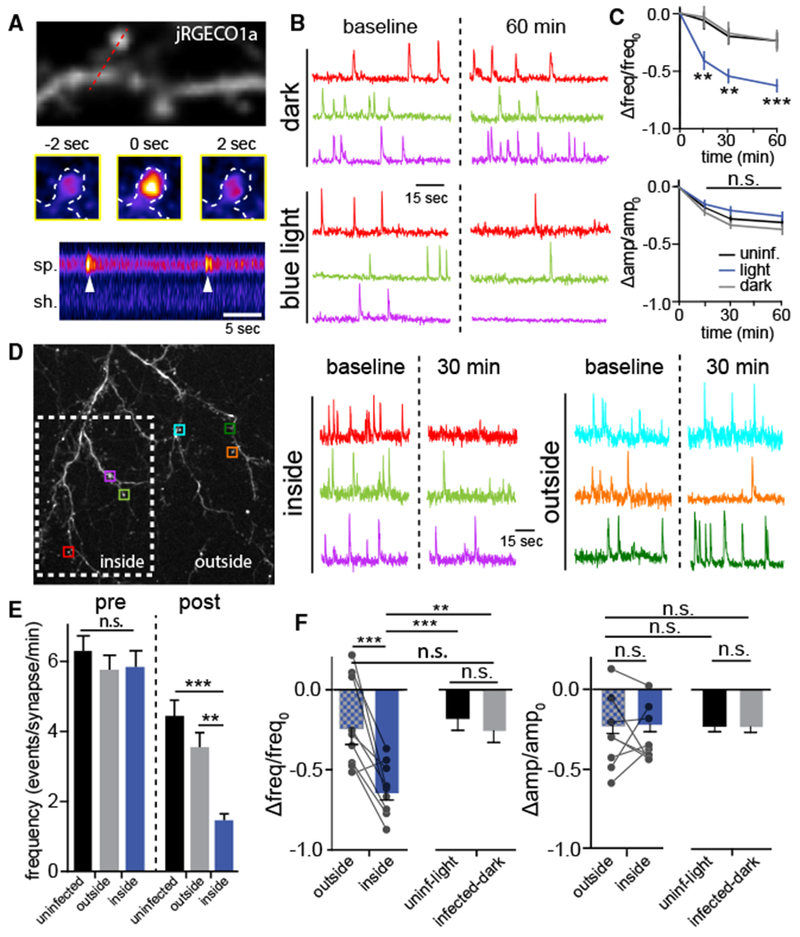
(A) Postsynaptic Ca2+ transients arising from quantal neurotransmitter release can be detected with jRGECO1a. Top: dendritic segment from a cultured hippocampal neuron expressing jRGECO1a. Center: jRGECO1a within a single dendritic spine before, during, and 2 s after a spontaneous Ca2+ transient. Bottom: kymograph generated from the red line drawn through the spine (sp) and shaft (sh) at the top. Two discrete events (arrowheads) can be observed in this example.
(B) Representative traces showing spontaneous Ca2+ transients at the same synapses before (left, baseline) and 60 min following (right) continuous darkness (top traces) or blue light exposure (bottom traces).
(C) The frequency (top) and amplitude (bottom) of spontaneous Ca2+ transients were monitored at the same synapses over time. Data for each synapse were subtracted from its baseline (pre-light exposure) value, and the difference was divided by its baseline value. Cultures infected with vPA-BoNT (blue) show significantly reduced frequency, but not amplitude, of spontaneous Ca2+ transients compared with uninfected control neurons treated with light (black) or vPA-BoNT expressing cultures not exposed to blue light (gray). Values represent mean and SEM from 204 spines from 17 neurons (uninfected), 107 spines from 9 neurons (infected, dark), and 168 spines from 14 neurons (infected, light). **p < 0.01, ***p < 0.001, two-way ANOVA.
(D) Local activation of vPA-BoNT. Cultures infected with vPA-BoNT were locally photoactivated (white box, dashed line). Representative traces show Ca2+ signals from the synapses outlined by colored squares either inside (left) or outside (right) of the illuminated region before and 30 min following local illumination.
(E) Quantification of the absolute frequency of spontaneous synaptic Ca2+ transients in uninfected light-treated cultures (black) and infected cultures, with synapses quantified from the same cells either “outside” (gray) or “inside” (blue) of the illuminated region. Bars to the left of the dashed line display the baseline (pre-illumination) event frequency at individual synapses; bars to the right of the dashed line display the event frequency 30 min following local illumination. Values represent mean ± SEM from 105 spines from 9 neurons (uninfected), 108 spines from 9 neurons (outside), and 106 spines from 9 neurons (inside). **p < 0.001, ***p < 0.0001, one-way ANOVA.
(F) Normalized data from (E) comparing the frequency (left) and amplitude (right) of Ca2+ transients at the same synapses before and 30 min following local illumination. The line pairings represent synapses from the same neuron either “inside” (blue) or “outside” (blue-gray checkered) of the photoactivated region. Results are compared with separate control cultures that were not expressing PA-BoNT but treated with light (black) or cultures expressing vPA-BoNT but not illuminated (gray). **p < 0.001, ***p < 0.0001, one-way ANOVA.
Because spontaneous and evoked neurotransmitter release may arise from different pools of vesicles, we tested the efficacy of vPA-BoNT in regulating vesicle fusion in response to depolarization (Ramirez and Kavalali, 2011). We measured the rate and extent of depolarization-triggered FM4-64 dye unloading in dissociated hippocampal neurons transfected with vPA-BoNT (Figure S4). Prior to blue light exposure, we stimulated FM4-64 loading into presynaptic terminals with a brief (30-s) exposure to extracellular solution containing 50 mM K+. Following dye loading, we either maintained cells in darkness or exposed them to blue light pulses and then triggered vesicle fusion (and FM4-64 unloading) with a second exposure to 50 mM K+ while imaging the FM dye channel at 2 Hz (Figure S4A). The rate of dye unloading from terminals expressing vPA-BoNT was ~2-fold slower (vPA-BoNT: τ = 18 ± 1 s; control, τ = 10 ± 2 s) and incomplete compared with terminals in the same imaging fields that did not express the toxin (dye signal remaining 60 s following stimulation: vPA-BoNT, 45% ± 9%; control, 26% ± 3%; p < 0.05, Student’s t test) (Figure S4B). We observed no difference in the rate or extent of FM dye unloading from vPA-BoNT-expressing terminals when the cells were not exposed to blue light prior to unloading, indicating minimal dark baseline activity of the toxin (Figure S4C). It should be noted that this approach may underestimate vPA-BoNT efficacy because a fraction of dye-loaded vesicles are likely docked prior to light exposure, with a fraction of VAMP2 inaccessible to the reconstituted toxin (Otto et al., 1997).
vPA-BoNT/B Is Effective for Regulating Excitatory Neurotransmission in Intact Circuits
Following validation in dissociated hippocampal neurons, we tested whether vPA-BoNT could be used for controlling neurotransmission in intact circuits. Hippocampal CA1 pyramidal neurons project to the subiculum, providing an ideal circuit to test the effectiveness of vPA-BoNT for disrupting presynaptic neurotransmitter release. We co-injected two AAVs, each encoding one of the vPA-BoNT fragments, into the hippocampal CA1 region (Figure 5A). Acute slices were prepared 2–3 weeks following injection, and expression was verified by fluorescent reporters engineered into the constructs (STAR Methods; Figures 5B and S5A–S5D). Whole-cell voltage clamp recordings of AMPA receptor EPSCs were made from primary subicular neurons visually confirmed to be uninfected by either virus (Figure 5C). After establishing baseline EPSC amplitude, we exposed the slices to blue light pulses for 30 min. We observed a robust reduction in EPSC amplitude coincident with the onset of blue light exposure (Figures 5D–5G). Slices from animals infected with vPA-BoNT but not exposed to blue light or uninfected slices that were exposed to blue light showed a mild run-down (10% ± 6%) over the same time period. Neither fragment expressed on its own was sufficient to disrupt neurotransmission (Figures 5D, 5E, and 5H). Nearly identical results were obtained when we used SspBmilli or SspBmicro as the iLID dimerizer (Figure 5G).
Figure 5. vPA-BoNT Inhibits Excitatory Neurotransmission in Hippocampal Projections to the Subiculum.
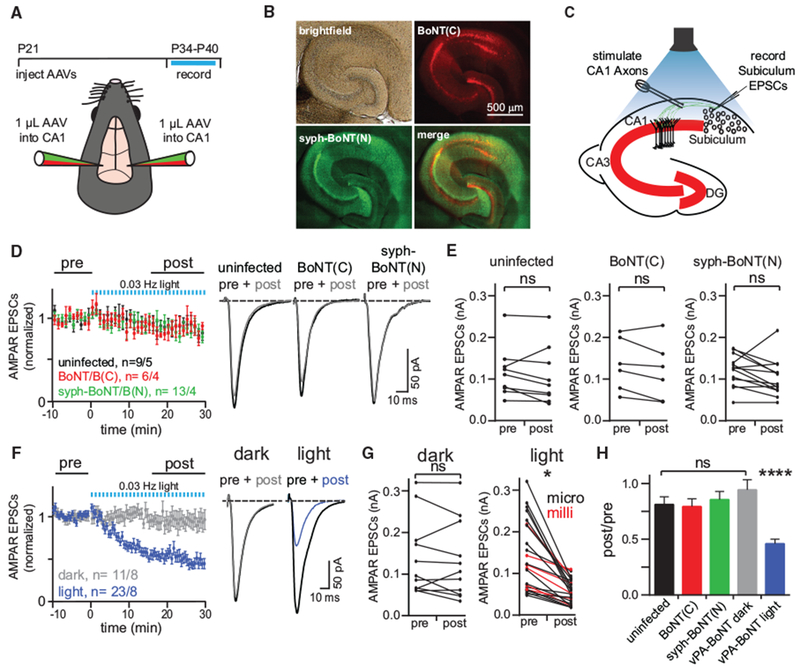
(A) Top: timeline of the experiment. Bottom: schematic of the viral injection. Two AAVs encoding vPA-BoNT N- and C-terminal fragments were bilaterally co-injected into the hippocampus.
(B) Representative image displaying expression of vPA-BoNT in the hippocampus. Bright-field, red (mCherry-IRES-SspBmicro-BoNT(C)), green (syph-GFP-BoNT(N)-iLID), and merged channel images are displayed.
(C) Schematic of ex vivo recordings in acute hippocampal slices. Hippocampal CA1 axons were electrically stimulated to evoke AMPAR-mediated EPSCs in uninfected subicular pyramidal cells.
(D) N- and C-terminal vPA-BoNT fragments expressed alone do not affect neurotransmission. Summary of evoked responses from slices prepared from uninfected (black) orsingly infected animals(red, C-terminal fragment; green N-terminal fragment). Slices were illuminated after 10 min dark baseline with 473-nm light for 30 s every min for 30 min. Right: representative traces of averaged responses: pre, 10-min baseline average; post, 15- to 30-min average. n refers to number of cells and number of animals; error bars, SEM.
(E) Paired plots showing EPSC amplitudes recorded from individual cells pre- and post-light for uninfected (left), mCh-IRES-SspBmicro-BoNT(C)-infected (center), and syphGFP-BoNT(N)-iLID-infected (right) animals.
(F) Summary of evoked responses from slices prepared from animals infected with AAVs encoding both fragments of vPA-BoNT. Slices were either maintained in darkness (gray, n = 11 cells from 8 animals) or illuminated after 10 min dark baseline with 473-nm light for 30 s every minute for 30 min (blue, n = 23 cells from 8 animals). Right: representative traces of averaged responses (pre, 10-min baseline average; post, 15-to 30-min average) for slices maintained in darkness (left traces) or treated with light (right traces).
(G) Paired plots of EPSC amplitudes averaged over the first 10 min (pre) and last 15 min (post) for individual dark-treated (left) and light-treated (right) cells. Similar light-evoked reductions in EPSC amplitudes were obtained using vPA-BoNTmilli (red) and/or vPA-BoNTmicro (black). *p < 0.05, paired Student’s t test.
(H) Summary of the ratio of EPSC amplitudes measured before and after light exposure (or for slices maintained in darkness for the same time period) and for each condition in (D) and (F). Error bars, SEM; ****p < 0.0001, one-way ANOVA.
Although vPA-BoNT robustly inhibited neurotransmission in this circuit, it was not completely eliminated. Residual neurotransmission could arise from incomplete block of vesicular release. It is also likely that a significant fraction of stimulated presynaptic inputs do not express vPA-BoNT. To discriminate between these possibilities, we estimated presynaptic release probability (Pr) using a paired-pulse paradigm. We reasoned that, if neurotransmission was efficiently blocked in vPA-BoNT neurons but uninfected neurons contributed to residual neurotransmission, then Pr would not change when measured before and after light exposure. Alternatively, if most stimulated inputs expressed PA-BoNT but were only partially blocked, then we would observe decreased Pr following light exposure. We observed no significant change in Pr from baseline levels following 30 min of blue light exposure (Figures S5E and S5F) even though the evoked EPSC amplitude was reduced approximately 2-fold. Importantly, expression of vPA-BoNT did not influence Pr on its own compared with uninfected controls, indicating that vPA-BoNT did not affect baseline neurotransmission prior to light exposure (Figure S5G). These observations are consistent with robust impairment of neurotransmission in vPA-BoNT-expressing neurons, with a significant fraction of residual transmission arising from presynaptic input from uninfected neuronal projections from other brain areas or unbalanced expression of the two vPA-BoNT components in a subset of CA1 neurons. Indeed, based on the fluorescent reporters expressed from the two vPA-BoNT AAVs, we determined that 29% ± 5% of inputs expressing one of the vPA-BoNT components did not express detectable quantities of the other component (Figure S5D).
To test whether vPA-BoNT could be used for controlling neurotransmission over longer-ranging projections, we co-injected the two AAVs encoding the vPA-BoNT fragments into the motor cortex (Figures 6A and 6B). Medium spiny neurons (MSNs) of the dorsal striatum receive strong excitatory input from the sensorimotor cortex (Gerfen, 1992). To examine whether cortically evoked glutamate transmission to striatal MSNs could be regulated by vPA-BoNT, acute dorsal striatal slices were cut three weeks (21–25 days) after viral injection, and cortical glutamatergic EPSCs were evoked in MSNs with a bipolar stimulating electrode placed in the cortex at the border of the corpus callosum (Figure 6C). As above, we found that photoactivation of vPA-BoNT in the dorsal striatum (470-nm light, 1-s flash, 10 pulses at 0.5 Hz) robustly inhibited glutamatergic EPSCs in MSNs (Figures 6D–6F). The same light regime had no effect on uninfected control slices (Figures 6D–6F). Together, these results confirm that vPA-BoNT/B can be used to acutely disrupt excitatory neurotransmission in intact circuits.
Figure 6. vPA-BoNT Inhibits Corticostriatal Excitatory Transmission.
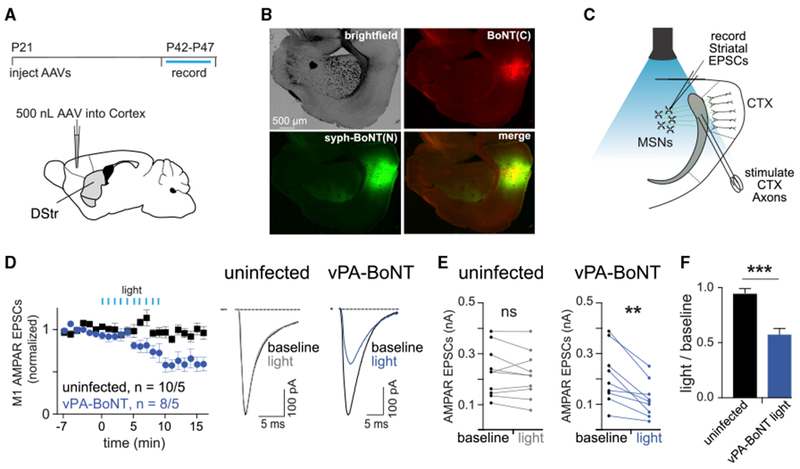
(A) Top: timeline of the experiment. Bottom: schematic of the viral injection. Two AAVs encoding vPA-BoNT N- and C-terminal fragments were bilaterally co-injected into the M1 motor cortex.
(B) Representative image displaying expression of vPA-BoNT in the cortex. Bright-field, red (mCherry-IRES-SspBmicro-BoNT(C)), green (syph-GFP-BoNT(N)-iLID), and merged channel images are displayed.
(C) Schematic of ex vivo recordings in acute striatal slices. Cortical axons were electrically stimulated at the boundary of the cortex and corpus callosum to evoke AMPAR-mediated EPSCs in medium spiny neurons in the striatum.
(D) Summary of evoked responses from slices prepared from wild-type (WT) control uninfected animals (black) or animals infected with AAVs encoding both fragments of vPA-BoNT (blue). Slices were illuminated after 6 min dark baseline with 470-nm light (10-s exposure each minute, 85 μW/mm2) (blue). Right: representative traces of averaged responses (baseline, 7-min baseline average; light, 10- to 15-min post flash average) for uninfected slices (left traces) or vPA-BoNT-expressing slices (right traces).
(E) Paired plots of EPSC amplitudes from medium spiny neurons (MSNs) averaged over the first 7 min (baseline) and 10–15 min after light (light) from uninfected (left) and vPA-BoNT-expressing slices. **p < 0.01, paired Student’s t test.
(F) Summary of the ratio of EPSC amplitudes measured before and after light exposure (light-treated value divided by baseline value) for WT control uninfected slices (gray) or slices infected with AAVs encoding both fragments of vPA-BoNT (blue). Error bars, SEM; ***p < 0.001, Student’s t test.
PA-BoNT Can Be Used to Disrupt Coordinated Movement in C. elegans
To further examine the functionality of PA-BoNT, we tested the ability of the system to induce a behavioral phenotype in C. elegans. Prior studies have shown that blocking GABAergic motor neuron neurotransmission in C. elegans results in behavioral defects that include a “shrinker” locomotion phenotype (McIntire et al., 1993). Efficiency of neurotoxins in C. elegans is well established for tetanus toxin (TeNT) LC (Davis et al., 2008; Husson et al., 2012; Macosko et al., 2009) but has not been explored for BoNT/B. We thus first verified that BoNT/B can silence neuronal activity by expressing it in GABAergic neurons. More than 90% of transgenic animals expressing BoNT/B (codon-optimized either for yeast or mammalian expression) showed the shrinker phenotype compared with more than 95% for the positive control, TeNT (Figure S6A). Another GABA-dependent behavior is the defecation motor program, in which gut content is expelled by enteric muscle contractions (EMCs). Animals expressing BoNT or TeNT showed largely abolished EMCs and were constipated because the gut content could not be emptied (Figures S6B–S6D).
We next tested sPA-BoNT and vPA-BoNT in C. elegans. With sPA-BoNT, we fused an mCh tag to the N terminus of the BoNT(N)-iLID fragment and fused GFP to the N terminus of SspBmilli-BoNT(C, Y365A) to allow monitoring of protein expression and localization (Figure 7A). With vPA-BoNT, we fused the GFP-SspBmilli-BoNT(C, Y365A) fragment to the C terminus of synaptogyrin (SNG-1), encoding a tetraspan vesicle protein homologous to syph (Abraham et al., 2006), and coexpressed it along with mCh-BoNT(N)-iLID in a single bi-cistronic construct (Figure 7A). In both cases, the two fragments were expressed pan-neuronally using the Psng-1 promoter (Abraham et al., 2006). For sPA-BoNT, expression could be observed diffusely in neuronal cell bodies and processes (Figure 7B). Expression of each vPA-BoNT fragment was detectable in neuronal processes in the ventral nerve cord, specifically in en passant synaptic specializations (Figure 7B).
Figure 7. Soluble and Synaptic Vesicle-Targeted PA-BoNT Affect Locomotion Behavior in C. elegans after Photoactivation.

(A) Schematic of sPA- and vPA-BoNT constructs targeted to and activated at the synaptic vesicle membrane.
(B) Expression of sPA-BoNT fragments (top row) or vPA-BoNT fragments (bottom row) with GFP or mCherry tags as indicated. From left to right: transmitted light differential interference contrast (DIC), GFP, mCherry, and merged fluorescence. Shown are head (top) and body regions (bottom). Square arrows indicate fluorescence in nerve ring (top) and neuronal processes and synaptic boutons (bottom). Arrowheads indicate neuronal cell bodies. Scale bars, 50 μm.
(C) Left: mean ± SEM swimming cycles of WT (N2) or transgenic animals expressing soluble or synaptic vesicle (SV)-targeted variants of PA-BoNT, as indicated, without or with pulsed illumination (blue bars, 30 s every min for 60 min, 85 μW/mm2). n = 61–176 animals, assayed in n = 2-3 experiments, as indicated for each strain and condition. Right: mean ± SEM for statistical comparisons of baseline swimming cycles, measured prior to light exposure for all groups tested. Statistical analysis: unpaired t test with Bonferroni correction, ***p < 0.00025.
(D) Normalized data from (C), with data normalized to t = 0 time point.
(E) Group data of animals analyzed in (C) and (D) at the 75-min time point and after 24 h recovery in the dark. Statistical analysis: paired or unpaired t test with Bonferroni correction, *p < 0.0125, **p < 0.0025, ***p < 0.00025.
We assessed the performance of sPA-BoNT and vPA-BoNT using a swimming behavioral assay that requires function of cholinergic motor neurons. In liquid medium, C. elegans thrashes with a c-shaped body posture for ~100 full cycles per minute in the wild-type. We imaged 61–176 animals simultaneously (accumulated over n = 2–3 experiments) with a multi-worm tracker (Swierczek et al., 2011) every 15 min for 75 min in the dark or with pulsed blue light illumination for 1 h and automatically analyzed the thrashes. We first compared the thrashing rates of animals expressing sPA-BoNT and vPA-BoNT with control animals (not expressing the toxin) prior to light exposure to assess potential dark background activity of the toxins. We did observe a modest but significant reduction in baseline thrashing rate in PA-BoNT-expressing animals, which was more pronounced with sPA-BoNT compared with vPA-BoNT (Figure 7C). Importantly, when maintained in the dark, N2 wild-type animals as well as animals expressing sPA-BoNT or vPA-BoNT swam for the entire duration of the experiment with only a slightly reduced thrashing rate after 60 or 75 min because of phases of spontaneous quiescence (Figures 7C and 7D). When the animals were illuminated with blue light, PA-BoNT animals exhibited significantly reduced thrashing rates compared with non-illuminated PA-BoNT animals (to about 50% of the thrashes of non-illuminated animals for vPA-BoNT) after 60 and 75 min (Figures 7C–7E). The kinetics of onset of the behavioral effect was slower than the neurotransmission suppression we observed in mammalian experiments, possibly because of the differences in temperature (~25°C for C. elegans studies and 32°C-35°C for mammalian studies). We also note that animals that were actively swimming were more susceptible to light effects than animals foraging on a bacterial lawn (data not shown). This may point to a use dependence of the toxin because of a need to effectively “cycle” available VAMP molecules through exo- and endocytosing synaptic vesicles to make them accessible to the toxin (Dittman and Kaplan, 2006).
Finally, we assessed recovery of the light-induced behavioral deficit by maintaining the animals in the dark for a 24-h recovery period following light treatment and reassessing their swimming locomotion. The light-treated samples showed significant recovery to control levels after 24 h (Figures 7D and 7E). Combined, these results indicate that PA-BoNT can be used in vivo to conditionally and reversibly suppress a coordinated movement behavior.
DISCUSSION
In this work, we describe the development of a first-in-class optogenetic tool for regulating neurotransmission, vPA-BoNT. We demonstrate its utility in suppressing neurotransmitter release in cultured neurons and slices and in vivo. Since the original recognition that botulinum and tetanus neurotoxins cleave SNARE proteins involved in neurotransmitter release, such toxins have been widely adopted as tools to disrupt synaptic function (Blasi et al., 1993; Schiavo et al., 1992). Although genetic expression of the catalytic LC of these toxins affords targeted silencing within specific populations of neurons, these reagents lack spatial and temporal control. Given these limitations, botulinum and tetanus toxin reagents have been overshadowed by light-activated opsin-based tools, which offer unparalleled spatiotemporal precision. Opsin-based tools are powerful for short-term (milliseconds to seconds) neural silencing applications, but it is increasingly recognized that there are limitations for their use in longer-term (minutes to hours) silencing experiments (Wiegert et al., 2017). For long-term applications, a variety of useful chemical-genetic approaches have been developed (Alexander et al., 2009; Armbruster et al., 2007; Lechner et al., 2002; Lerchner et al., 2007; Magnus et al., 2011; Slimko et al., 2002; Wulff et al., 2007). Although many of these methods have seen widespread use, they can be difficult to activate with spatial and temporal precision and, to a similar extent, within all targeted neurons because they are actuated by small molecules whose precise local delivery in the brain can be difficult to control (Wiegert et al., 2017). Chemical-genetic approaches can also have off-target effects that can affect neuronal physiology in unexpected ways (Gomez et al., 2017; Saloman et al., 2016).
Approaches for long-term silencing that can be activated with greater spatial precision will complement existing opto- and chemogenetic approaches. Efforts in this direction have been made using chromophore-assisted light inactivation (CALI) approaches to disrupt neurotransmitter release (Lin et al., 2013; Marek and Davis, 2002). However, damaging radicals generated from CALI methods can have off-target effects on cellular physiology. For example, activation of a singlet oxygen generator in presynaptic terminals led to an unexpected increase in spontaneous neurotransmission (Lin et al., 2013). Furthermore, these approaches require relatively long-term (tens of seconds to minutes) constant light exposure to generate damaging radicals, which can be impractical when targeting a large region using scanning-based light delivery methods common in many confocal and multi-photon microscopes. PA-BoNT overcomes these limitations because it acts through a defined mechanism (cleavage of VAMP proteins) and does not require constant illumination. Another advantage is that PA-BoNT activity can be monitored using a commonly available antibody (used in this study) that allows discrimination of uncleaved VAMP2 from VAMP2 BoNT/B cleavage products. Thus, post hoc immunohistochemistry could be used to precisely calibrate toxin activity under different illumination conditions and to define the anatomic region of activation. Together, these features make PA-BoNT a unique and powerful silencing approach that fills a substantial gap in the current optogenetic toolkit for spatially restricted, long-term silencing.
Although PA-BoNT has unique advantages, we also acknowledge current limitations. First, the onset of synaptic inactivation using PA-BoNT is orders of magnitude slower than pump- and channel-based optogenetic tools for neural silencing. For many experimental scenarios that require long-term silencing, however, the speed of onset is less of a concern, as evidenced by the widespread use of chemogenetic tools that activate with similarly slow (minutes) kinetics. Second, persistent synaptic inhibition comes at the cost of rapid reversibility. Because BoNT/B cleaves VAMP proteins, recovery of synaptic transmission depends either on synthesis of new VAMP proteins (if the entire neuron was illuminated) or on lateral trafficking of uncleaved VAMP proteins to the inactivated region from non-illuminated synaptic sites or the cell body, which takes hours. A third potential limitation is that the current configuration requires target neurons to be co-infected with two viruses, decreasing the efficacy of silencing. A bicistronic version with the fragments separated by a 2A peptide led to high dark activity (Figure S1H), whereas use of an IRES element retained robust light-dark differences but is currently too large for AAV packaging (Figure S1F). On the other hand, a two-component system could be advantageous for manipulating neural subtypes using intersectional approaches or for manipulating specific projections by introducing one of the fragments in a retrograde trafficking virus injected at the target site (Tervo et al., 2016). Finally, the current iteration of PA-BoNT has modest but significant dark background activity. For example, we observed an ~25% reduction in VAMP2 levels in PA-BoNT-expressing cultured hippocampal neurons prior to light exposure. Surprisingly, this level of background activity did not appear to impair synaptic function, at least as measured by FM dye loading-unloading, optical quantal analysis, and electrophysiology. Thus, some degree of background activity may be tolerated. However, in the C. elegans studies, we did observe a modest but significant impairment in baseline swimming rates in PA-BoNT-expressing animals compared with controls. This could be due to higher expression levels of the toxin or an increased sensitivity of vesicle fusion to VAMP2 levels at cholinergic synapses in worms.
With additional engineering, we expect to reduce the dark background activity of the toxin and improve onset kinetics, potency, and organelle specificity. We have already demonstrated that simply targeting PA-BoNT to synaptic vesicles greatly enhances its ability to silence neurotransmitter release and, surprisingly, reduces dark background activity. Indeed, this strategy could allow potent and selective disruption of different secretory molecules released from the same cell. For example, many neurons can release more than one neurotransmitter from distinct vesicle pools, but the functional significance of co-transmission has been difficult to unravel (Vaaga et al., 2014). In principle, PA-BoNT could be targeted to selectively disrupt one class of neurotransmitter. In addition to neurotransmitter release, the mechanism of regulated secretion targeted by BoNT is important for many other activities. PA-BoNT could thus also be used to conditionally disrupt secretion of diverse biomolecules from neuroendocrine cells, pancreatic cells, immune cells, glia, and other cell types. In addition to systems-level applications, this tool will also be generally useful for advancing our understanding of the molecular mechanisms of vesicular fusion. The ability to rapidly disrupt SNARE proteins will help elucidate the machinery responsible for priming, docking, and fusion of secretory vesicles in diverse cell types whose fusion mechanisms remain obscure or controversial.
Finally, because the botulinum toxin protein family is structurally conserved, these engineering efforts for BoNT/B will be broadly applicable to related toxins with distinct substrate specificities, including other BoNT serotypes. In addition to engineering conditional versions of toxins that act on different endogenous substrates, coevolution of orthogonal protease-substrate pairs could lead to novel light-dependent protease systems that would expand our ability to precisely manipulate cellular systems in space and time.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Matthew J. Kennedy, matthew.kennedy@ucdenver.edu.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293 cells were obtained from ATCC and maintained and propogated under standard conditions (10% FBS in DMEM, 37°C, 5% CO2).
Vertebrate animals
All animal procedures were carried out in accordance with a protocol approved by the University of Colorado Denver Institutional Animal Care and Use Committee. All dissociated cultures were prepared from Sprague-Dawley rats. Timed pregnant dams (typically embryonic day 16) were obtained from Charles River Laboratories and housed under standard conditions. This study used dissociated cultures from both male and female pups. C57BL/6 mice were used for testing PA-BoNT in acute brain slices. Mice were obtained from Jackson Laboratories. Both male and female mice were used for these studies.
C. elegans
C. elegans (wild-type Bristol N2) were obtained from the Caenorhabditis Genetics Center (University of Minnesota). C. elegans were cultivated at 20°C on standard nematode growth medium (NGM) and fed with Escherichia coli OP50-1 bacteria according to standard methods (Brenner, 1974).
METHOD DETAILS
Cloning and Mutagenesis
Full sequences of constructs used in this manuscript and oligo details are provided in Table S2. To generate CRY2- and CIBN-fused N- and C-terminal BoNT/B LC fragments, we first replaced CRY2 and CIBN in mCh-IRES-CRY2-CreN and mCh-IRES-CIBN-CreC (Kennedy et al., 2010) with NLS-deleted versions at XhoI and XmaI sites. CRY2(ΔNLS) was PCR-amplified using oligos 744F/1702R, and CIBN(ΔNLS) was amplified using oligos 748F/694R, followed by 748F/1702R. Next, the Cre fragments were replaced at Not I and XmaI sites by BoNT/B-LC N-terminal or C-terminal fragments to produce pQL6/pQL16 (80/81 split); pQL7/pQL17 (146-147 split) pQL8/pQL15 (329/330 split); pQL11/pQL13 (211/212 split); pQL19/pQL14 (254/255 split). The BoNT/B-LC plasmid used as a template to amplify the N- and C-terminal fragments was a gift from Dr. Thomas Binz. For the full-length positive control (pQL24, mCh-IRES-BoNT/B), BoNT/B was amplified (1740F/1704R) and cloned into mCh-IRES-CRY2(ΔNLS)-CreN between Sac I and XmaI sites. To generate the EGFP-VAMP-GST reporter (pQL47), pEGFP-VAMP3 (Galli et al., 1998) (Addgene 42310) was PCR-amplified (oligos 1794F/1795R) to add a linker between VAMP3 and Not I site. GST was PCR-amplified (oligos 1811F/1812R) to add Not I and Xba I sites, and inserted after the linker. Mutations in BoNT/B-LC and iLID were introduced by one-step Phusion mutagenesis using protocols from New England Biolabs. Briefly, non-overlapping oligos containing desired mutations (Table S2) were used to amplify the entire plasmid using Phusion High-Fidelity DNA Polymerase (NEB), then the PCR products were treated by T4 Polynucleotide Kinase (NEB, M0201S) in the presence of ATP at 37°C for 30 min before self-ligation using T4 Quick Ligase (NEB).
To generate the iLID and SspB-fused N- and C-terminal BoNT/B LC constructs, we first replaced CRY2 in pQL7 with BoNT(1-146) (oligos 2009F/2010R) at Xho I and Not I sites. Next, to generate mCh-IRES-BoNT(1-146)-iLID (pQL155), iLID (amplified from Venus-iLID-Mito, Addgene 60413, oligos 2011F/2013R then 2012F/2013R) was inserted between Not I and XmaI sites. Similarly for mCh-IRES-BoNT(147-441, N157A)-SspBnano (pQL162), CRY2 in pQL7 was first replaced with BoNT(147-441, N157A) (amplified using oligos 2014F/ 2015R) at Xho I and BspEI sites. Subsequently, SspBnano (PCR-amplified from tgRFPt-SspB-WT, Addgene 60415, oligos 2016F/2017R) was inserted between BspEI and XmaI sites. For mCh-IRES-BoNT(147-441, N157A)-iLID (pQL176), SspBnano in pQL162 was removed between EcoRI and XmaI sites, then replaced by iLID (PCR-amplified using oligos 2053F/2013R). To generate pQL170, pQL173, pQL174, and pQL184 containing SspB-BoNT(1-146) and SspB-BoNT(147-441) configurations, SspBnano, SspBmicro (SspB with R73Q, Addgene 60416), or SspBmilli (SspB with A58V/R73Q, gift from Dr. Brian Kuhlman) was PCR-amplified using oligos 2054F and 2055R/1702R and cloned into pQL7, pQL17, pQL54, or pQL56 between Xho I and BspEI sites. The V416I mutation was introduced using Phusion site-directed mutagenesis. To generate the bicistronic split construct (pQL253, Figure S1F), mCh-BoNT/B(N)-iLID-IRES-SspB(A58V/R73Q)-BoNT/B(147-441, Y365A), BoNT(N)-iLID was cloned into pQL185 at BsrGI and KpnI sites.
All AAV plasmids were propagated in Stbl3 E. coli (Invitrogen). To generate pQL262 (pAAV-hSYN-mCh-IRES-BoNT(1-146)-iLIDV416I), mCh-IRES-BoNT(1-146)-iLIDV416I was PCR-amplified from pQL193 (oligos 2167F/2169R) to add BglII and EcoRI sites, and then cloned into pAAV-hSYN-mRuby (derived from pAAV1-hSYN-mGFP-2A-syph-mRuby, a gift from Dr. Robert Malenka, by inserting mRuby into AAV-hSYN at EcoRI and BamHI sites, oligos mRubyF/mRubyR) between BamHI (removed) and EcoRI sites. Similarly, to generate pQL269 [pAAV-hSYN-mCh-IRES-SspBmicro-BoNT(147-441, Y365A)] and pQL261 [pAAV-hSYN-mCh-IRES-SspBmilli-BoNT(147-441, Y365A)], mCh-IRES-SspBmicro-BoNT(147-441, Y365A) and mCh-IRES-SspBmilli-BoNT(147-441, Y365A) were amplified from pQL173 and pQL185 (oligos 2167F/2168R) and inserted into pAAV-hSYN-mRuby in the same way. To generate syph-tagged versions (pQL280, pAAV-hSYN-syph-EGFP-HA-SspBmilli-BoNT(147-441, Y365A); pQL281, pAAV-hSYN-syph-EGFP-myc-BoNT(1-146)-iLIDV416I), syph-EGFP was PCR-amplified using 2216F/2217R or 2216F/2218R to add a NheI site, SalII site and HA or myc tag, then cloned into pQL261 or pQL262 between NheI and SalII. The combination of constructs designated sPA-BoNTmicro consists of (pQL269 + pQL262), sPA-BoNTmilli is (pQL261 + pQL262), vPA-BoNTmicro consists of (pQL269 + pQL281), and sPA-BoNTmilli consists of (pQL261 + pQL281).
C. elegans constructs
For studies in C. elegans, the following plasmids were constructed: for pMSM07 [unc-47p::eGFP::ScBoNT/B] the S. cerevisiae codon-optimized version of full-length botulinum neurotoxin B (ScBoNT/B, Genscript) (full sequence provided in Table S2) was PCR-amplified (primers oMSM33/34) and recombined via Gibson-assembly reaction with the fragments unc-47p, generated by PCR amplification (oMSM31/32), eGFP and backbone of pPD95.79 (A. Fire, Addgene plasmid #1496), generated from pPD95.79 by restriction digest (XmaI, SalI, BsmI). The same reaction was performed with the mammalian codon-optimized TeNT (Eisel et al., 1986) PCR-amplified with (oMSM35/36) to generate pMSM09 [unc-47p::eGFP::TeNT]. For pMSM08 [unc-47p::eGFP:: MmBoNT/B] the Mus musculus (mammalian) codon-optimized synthetic gene was PCR-amplified (oMSM37/38) and processed via restriction digest (XmaI, SpeI) for subcloning into pMSM07 (XmaI, NheI). For the soluble PaBoNT the two plasmids pMSM10 [sng-1p::mCh::ScBoNT/B(1-146)::iLID(V416I)] and pMSM11 [sng-1p::eGFP::SspBmilli::ScBoNT/B(147-441, Y365A) were generated via Gibson-assembly reaction of pPD95.79 backbone (XmaI, SalI, BsmI) and sng-1p (PCR-amplified with oMS18/19) with either worm codon-optimized mCh (PCR-amplified with oMS25/26), ScBoNT/B(1-146) (PCR-amplified with oMS27/28 from pQL221) and iLID(V416I) (PCR-amplified with oMS29/30 from pQL193), or with worm-optimized eGFP (XmaI, SalI, BsmI restriction digest of pPD95.79) and SspBmilli-ScBoNT/B(147-441, Y365A) (PCR-amplified with oMS20/21 from pQL221). For the vesicular PaBoNT both parts of PaBoNT were combined into one plasmid pMSM14 [sng-1p::sng-1::eGFP::SspBmilli::ScBoNT/B(147-441, Y365A)::SL2::mCh::ScBoNT/B(1-146)::iLID(V416I)] by ligation of pMSM12 [sng-1p::sng-1::eGFP::SspBmilli::ScBoNT/B(147-441, Y365A)] backbone (AvrII, XmaI) with mCh-ScBoNT/B(1-146)-iLID(V416I) from pMSM10 (XmaI, Bsu36I) and the trans-splicing site SL2, PCR-amplified with oMS23/24 and restriction digested (AvrII, Bsu36I). All plasmids were validated by restriction digest and sequencing.
Characterization of split BoNT/B in HEK293T cells
HEK293T cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS at 37°C with 5% CO2. To test split constructs, 1 μg of the GFP-VAMP-GST cleavage reporter and each BoNT/B fragment were transfected into HEK293T cells on 12-well plates using standard calcium phosphate transfection methods. Cells were wrapped in aluminum foil after transfection and kept in dark for 24 h before blue light treatment (461 nm delivered from a custom-built LED array, 15.6 mW/cm2). For CRY2-CIBN systems, 2 s pulses were delivered every 3 min; for iLID-SspB systems, 1 s pulses were delivered every 30 s, unless noted otherwise. Dark samples were kept in the dark throughout the experiment. Cells were harvested after 4-5 h of light treatment, unless specified otherwise (28-29 h post transfection). For harvest, cells were washed in 1× PBS, collected, and lysed in 2× Laemmli sample buffer with boiling. Proteins were separated by electrophoresis on an SDS-PAGE gel and transferred to nitrocellulose membranes, followed by probing with primary (anti-GFP, Sigma G1544; anti-mCh, Thermo Fisher Scientific PA5-34974) and secondary (goat anti-rabbit IR-Dye 800CW, LiCOR, 926-32211) antibodies. An Odyssey FC Imager (Li-COR) was used to visualize labeled immunoblots. HEK293T cell culture experiments were repeated as noted on figure and table legends, 2-6 independent experiments for all main figures, 1-6 independent experiments for supplementary figures. No data were excluded; no experiments were blinded or randomized. Statistical significance was determined using a two-tailed unpaired Student’s t test.
Neuronal cell culture
Primary hippocampal neurons were prepared from neonatal Sprague-Dawley rats. Hippocampi were dissected from the brains of postnatal day 0–2 rats and dissociated by papain digestion. Neurons were plated at 150,000 cells/well in MEM, 10% FBS (Hyclone) containing penicillin/streptomycin on poly-d-lysine–coated 18 mm coverslips. After 1 d the media was replaced with Neurobasal-A supplemented with B27 (Invitrogen) and GlutaMAX (ThermoFischer). The neurons were then fed with Neurobasal-A, B27, and mitotic inhibitors (uridine + fluoro-deoxyuridine [Ur+FdUr]) by replacing half the media on day 5 or day 6 and then weekly. Neurons were maintained at 37°C in a humidified incubator at 5% CO2. Neurons were transfected with 0.75 μg of each split construct using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s recommendations and allowed to express for 48-72 h.
Live cell imaging
Live cell imaging of dissociated neurons was carried out at 34°C on an Olympus IX71 equipped with a spinning disc scan head (Yokogawa). Excitation illumination was delivered from an acousto-optic tunable filter (AOTF) controlled laser launch (Andor). Images were acquired using a 60× Plan Apochromat 1.4 NA objective, and collected on a 1024×1024 pixel Andor iXon EM-CCD camera. Data acquisition and analysis were performed with Metamorph (Molecular Devices) and ImageJ software.
Measurement of endogenous VAMP2 cleavage in neurons
Cultured hippocampal neurons were transfected with PA-BoNT or full-length BoNT/B or mCh alone and allowed to express for 48 h in the dark. Cells were fixed in the dark or following exposure to blue light (4.5 mW/cm2), permeabilized with 0.1% Triton X-100 and blocked with 5% BSA. Cells were incubated with a primary antibody against VAMP2 (Synaptic Systems, 104211) that does not recognize VAMP2 following cleavage by BoNT/B, followed by goat-anti-Mouse Alexa Fluor 647 secondary antibody (Invitrogen, A-32728). The amount of VAMP2 staining in presynaptic boutons was compared to neighboring untransfected neurons and normalized to positive (full-length BoNT/B) and negative (mCh alone) controls. Synaptic boutons were selected for analysis using the mCh (cell fill) or GFP channel (when syph-GFP was co-expressed), blinded to the VAMP2 signal in the far red (Alexa647) channel.
Ca2+ imaging and analysis
To image quantal Ca2+ transients (QCTs), neurons transfected with jRGECO1a and infected with AAVs expressing PA-BoNT were incubated in an artificial cerebro-spinal fluid (ASCF) solution containing (in mM): 130 NaCl, 5 KCl, 10 HEPES, 30 glucose, 2.5 CaCl2, 0.03 glycine and 0.002 tetrodotoxin (Tocris) (pH 7.4). Single z-plane images of a portion of the dendritic arbor were acquired at 7 Hz for 1 min to record baseline QCTs. Cells were then either exposed to 488 nm light every two min or kept in the dark. The same z plane was then imaged again to record QCTs post-treatment.
To measure the frequency and amplitude of QCTs, regions of interest (ROIs) were drawn around 12 clearly resolved spines per cell in the baseline movie. The ROIs were saved and the same synapses were analyzed in the post-treatment movies. The mean background-subtracted jRGECO1a fluorescence within each ROI was measured. A baseline of 10 frames was established and each frame was compared to that baseline. A threshold of a 40% increase in fluorescence over baseline was established to remove small variations in fluorescence. Event frequency and average peak amplitudes were compared between baseline and time points after blue light-treatment.
Local vPA-BoNT activation and Ca2+ imaging was performed in a similar manner, except a digital micromirror array was used to illuminate a sub-region of the dendritic arbor with a 450 nm LED to activate the toxin in presynaptic terminals. Neurons infected with AAVs expressing vPA-BoNT and sparsely transfected with jRGECO1a were imaged for 1 min at baseline and then either kept in the dark or exposed to locally delivered blue light for 5 s every 2 min for 30 min and then jRGECO was imaged again. Spontaneous postsynaptic Ca2+ transients were measured from synapses within the illuminated region and compared to synapses outside the illuminated region on the same neurons. Controls included uninfected neurons exposed to the same level of blue light using the digital micromirror array and infected neurons not exposed to blue light. Analysis was not blinded for these experiments.
Electrophysiology in primary cultured neurons
Dissociated hippocampal neurons infected with AAVs expressing PA-BoNT were either kept in the dark or exposed to at least 1 h of blue light (1 s pulse every 2 min, 4.5 mW/cm2). Whole cell voltage clamp recordings were carried out from dissociated hippocampal neurons (DIV17-19) bathed in (mM): 10 HEPES, 130 NaCl, 5 KCl, 30 D-glucose, 2 CaCl2 and 1 MgCl2 supplemented with 1 μM tetrodotoxin and 30 μM bicuculline (Tocris). Intracellular solution contained (in mM): 130 cesium methanesulfonate, 3 Na2ATP, 0.5 Na3GTP, 0.5 EGTA, 10 phosphocreatine, 5 MgCl2, 2.5 NaCl, 10 HEPES (290-300 mOsm). The pH was adjusted to 7.25 with CsOH. Data were collected using a multiclamp 700b amplifier and digitized using a National Instruments DAQ board at 10 KHz and filtered at 2 KHz (single pole Bessel filter) and collected with WinLTP software (University of Bristol). Data were analyzed using WinLTP (University of Bristol), the NeuroMatic package in IGOR Pro (WaveMetrics) and Mini Analysis software (Synaptosoft). mEPSC analysis was carried out blinded to the experimental condition (e.g., dark versus light treated).
FM dye loading-unloading experiments
For initial screening of toxin efficacy, dissociated hippocampal neurons were transfected with PA-BoNT or full-length BoNT/B or mCh and allowed to express for 48 h. Neurons were either kept in the dark or exposed to blue light (1 s blue light every 2 min, 4.5 mW/cm2) for indicated times, then surface membrane was saturated with FM1-43FX(5 μM) in ACSF containing 10 μM NBQX and 100 μM APV. Cells were exposed to 50 mM KCl (in the presence of NBQX/APV) for 1 min to induce exocytosis then returned to baseline ACSF containing FM1-43FX for 5 min to allow for compensatory endocytosis and dye uptake. Cells were washed to remove excess dye and remaining surface dye was quenched with 1 mM Advasep7. Cells were washed and fixed with 4% paraformaldehyde and imaged. To quantify FM dye uptake, fluorescence within presynaptic boutons of transfected cells was measured and compared to dye uptake of neighboring untransfected cells in the same imaging fields. Values were then normalized between positive (full-length BoNT/B) and negative (mCh alone) controls. For vPA-BoNT FM dye unloading experiments we used the red shifted FM4-64 so we could image syph-GFP-BoNT-iLID in the green channel to identify terminals from vPA-BoNT expressing neurons. Prior to illumination, FM4-64 was loaded as above. Cells were then exposed to blue light using 488 nm illumination through the microscope objective for 2 s every 1.5 min for a total of 25 min, or kept in darkness as a control. We then triggered dye release by rapidly perfusing the cells with isosmotic ACSF containing 50 mM K+ while imaging the FM4-64 channel at 2Hz. The rate of dye release was quantified at terminals containing vPA-BoNT and compared to the same number of randomly selected neighboring, untransfected terminals in the same imaging fields.
Production of AAVs for primary culture and in vivo injection
AAV-DJ expressing PA-BoNT constructs were generated as previously described (Aoto et al., 2013). Briefly, HEK293T cells were co-transfected with the AAV vector along with helper plasmids (pDJ and pHelper) using calcium phosphate transfection. 72 h post-transfection cells were harvested, lysed, and purified over an iodixanol gradient column (2 h at 63,500 rpm in a Beckman Type80Ti rotor). Virus was dialyzed to remove excess iodixanol and aliquoted and stored at −80°C until use.
Stereotactic viral injection, hippocampus
P21 C57BL6J male and female mice were anesthetized with an intraperitoneal injection of 2,2,2-Tribromoethanol (250 mg/kg) then head fixed to a stereotactic frame (KOPF). An incision was made in the scalp with sterilized scissors, and small holes (~0.5 mm diameter) were drilled into the skull using a handheld dental drill. Viral solutions containing either AAV1-hSYN-mCh-IRES-SspBmicro/milli-BoNT/B(C),AAV1-hSYN-syph-GFP-BoNT/B(N)-iLIDV416I, or a premixed solution of both, were injected into each hemisphere with a pulled glass micropipette. Using a syringe pump (World Precision Instruments), a total volume of 0.8-1.0 μL was delivered into intermediate CA1 at an infusion rate of 14 μL/h at the following coordinates: AP: −3.2, M/L: +/− 3.45 (relative to Bregma), and D/V: −2.5 (relative to pia). The micropipette was held in place for 5 min after injection to prevent backflow of virus, then slowly retracted. Correct localization and expression of viral infection was verified post hoc by presence of mCh and/or GFP.
Stereotactic viral injection, cortex
Male and female C57BL6J mice were injected at postnatal day 21 under isofluorane anesthesia. 250 nL of pAAV-hSYN-mCh-IRES-SspBmicro-BoNT/B(C,Y365A) and 250 nL of pAAV-hSYN-syphGFP-BoNT/B(N)-iLIDV416I were injected into one hemisphere with the following coordinates (relative to bregma): AP +1.1mm, ML +1.45mm, DV −1.0 mm. Virus was injected using a pulled pipette using a Nanoject III (Drummond Scientific). Mice were allowed to recover for at least 3 weeks. Correct localization and expression of viral infection was verified post hoc by presence of mCh and/or GFP.
Electrophysiology in acute slices, hippocampus
At P34-P40, animals were deeply anesthetized with isoflurane and decapitated. Brains were rapidly dissected and 300 μm horizontal slices were sectioned with a vibratome (Leica VT1200) in ice cold, oxygenated solution containing (in mM) 85 NaCl, 75 sucrose, 25 D-glucose, 24 NaHCO3, 4 MgCl2, 2.5 KCl, 1.3 NaH2PO4, and 0.5 CaCl2. Slices were then allowed to recover for 30 min in oxygenated ACSF at 31.5°C containing (in mM) 126 NaCl, 26.2 NaHCO3, 11 D-Glucose, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4-7H2O, and 1 NaH2PO4 before resting at room temperature for at least 1 h. Slices were superfused in ACSF containing 100 μM picrotoxin and 50 μM D-AP5. Subicular pyramidal neurons were visually identified with an Olympus BX51W microscope with a 40× dipping objective collected on a Hamamatsu ORCA-Flash 4.0 V3 digital camera using an IR bandpass filter. Cells were patched in whole cell configuration using glass pipettes pulled to a resistance of 3-5 mΩ and filled with an internal solution containing (in mM) 117 Cs-methanesulfonate, 15 CsCl, 10 HEPES, 10 Phosphocreatine, 10 TEA, 8 NaCl, 4 Mg-ATP, 1 MgCl2, 0.5 GTP, and 0.2 EGTA. AMPAR-mediated EPSCs were evoked by electrically stimulating CA1 axon efferents within the alveus/stratum oriens at the border of CA1 and subiculum at 0.1 Hz with a home-made Nichrome electrode. Stimulus intensity was adjusted to evoke 50-300 pA AMPAR-mediated EPSCs and baseline was acquired for 10 min before photoactivation of split toxins using 473 nm blue light. Slices were illuminated with blue light pulses for 30 min (30 s every min). Release probability was assessed before and after the 40 min recording (10 min baseline + 30 min light treatment) by measurements of paired pulse ratios at inter-stimulus intervals of 33 ms. Slices were then fixed in 4% PFA then mounted for posthoc imaging to validate expression of each split toxin. All experiments were performed using a Multiclamp 700B amplifier and a Digidata 1440 or 1550B digitizer. Recordings were collected using a 2 kHz lowpass filter and digitized at 10 kHz. All slice preparations and baseline recordings were performed in the dark using red LED illumination and under infrared optics to prevent inadvertent photoactivation of PA-BoNT. Recordings were discarded if series resistance changed by more than 10% during the recording period. Dark and light-treated slice recordings were interleaved and analysis was carried out blind with respect to the treatment.
Electrophysiology in acute slices; striatum
At 42-47, animals were deeply anesthetized with isoflurane and decapitated under dim red right light. Brains were rapidly dissected and 240 μm coronal slices were sectioned with a vibratome (Leica VT1200s) in ice cold, oxygenated solution containing (in mM) 75 NaCl, 2.5 KCl, 6 MgCl2, 0.1 CaCl2, 1.2 NaH2PO4, 25 NaHCO3, 2.5 D-glucose, 50 sucrose and bubbled with 95% O2 and 5% CO2. Slices were incubated for 1 h at 32°C in ACSF containing (in mM): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 21.4 NaHCO3, 11.1 D-glucose and 10 μM MK-801, and bubbled with 95% O2 and 5% CO2. Slices were then transferred into a recording chamber and perfused with ACSF (33 ± 2°C) containing picrotoxin (100 μM) at a rate of 2 mL/min. MSNs were visualized using a BX51WI microscope (Olympus) with an infrared LED (Thorlabs). Recordings were made using a Axopatch 200B amplifier (Molecular Devices) and using Axograph × (Version 1.6.9) at 10 kHz and filtered to 5 kHz. Pipettes (~5-6MΩ) for whole-cell recordings from MSNs contained (in mM): 135 D-gluconic acid (K), 10 HEPES(K), 0.1 CaCl2, 2 MgCl2, 10 BAPTA. All internal solution also contained 0.1 mg/mL GTP, 1 mg/mL ATP, and 1.5 mg/mL phosphocreatine (pH 7.35, 275 mOsm). MSNs were held at −60 mV. No series resistance compensation was applied and recordings were discarded if series resistance was over 15 MU. AMPA EPSCs were evoked (50-300 μA, 0.1 ms, every 60 s) with a tungsten concentric bipolar stimulating electrode placed at the boundary between motor cortex layer 6 and the corpus callosum. EPSCs were acquired for 7 min before photoactivation of split toxins using 470 nm blue light (Thorlabs). Slices were illuminated with a train blue light pulses for 10 min (1 s flash, 2 Hz, 10 pulses) once per minute. All experiments were performed under dim conditions using red light. Dark controls and light-treated recordings were interleaved.
C. elegans studies
All C. elegans strains were cultivated at 20°C on standard nematode growth medium (NGM) and fed with Escherichia coli OP50-1 bacteria according to standard methods (Brenner, 1974). The following strains were generated: ZX2489: zxEx1152[unc-47p::ScBoNTB; myo-2p::mCherry], ZX2490: zxEx1153[unc-47p::MmBoNTB; myo-2p::mCherry], ZX2491: zxEx1154[unc-47p::TeNT; pmyo-2::mCherry], ZX2495: zxEx1158[sng-1p::mCherry::BoNTB(1-146)::iLID(V416I), sng-1p::eGFP::SspBmilli:: BoNTB(147-441, Y365A); elt-2p::NLS-eGFP], ZX2496: zxEx1159[sng-1p::sng-1::eGFP::SspBmilli::BoNTB(147-441, Y365A):: SL2::mCherry::BoNTB(1-146)::iLID(V416I); myo-2p::mCherry]. Transgenic C. elegans were obtained by microinjection of DNA into the gonads of nematodes by standard procedures using Bristol-N2 genetic background (Mello et al., 1991). For ZX2489 and ZX2490 50 ng μL−1 of either pMSM07 or pMSM08 and for ZX2491 5 ng μL−1 of pMSM09 were microinjected using 2.5 ng μL−1 myo-2p::mCherry as co-injection marker. For ZX2495 5 ng μL−1 of both pMSM10 and pMSM11 were microinjected using 30 ng μL−1 elt-2p::NLS-eGFP as co-injection marker. For ZX2496 50 ng μL−1 of pMSM14 was microinjected using 2.5 ng μL−1 of myo-2p::mCherry as co-injection marker.
Expression of BoNT/B and PaBoNT fragments in neurons of C. elegans was analyzed via fluorescence microcopy on an Axio Observer.Z1 (Zeiss) microscope equipped with a 40× oil immersion objective (Zeiss EC-Plan 40× /1.3 oil) and a 100 W HBO mercury lamp, using eGFP (F36-525) and mCh (F41-007, both AHF Analysetechnik) excitation/emission filter sets. Images were captured using the Evolve 512 Delta (Photometrics) camera together with μManager software.
For behavioral studies, transgenic strains were kept in dark on standard NGM plates (6 cm diameter, 8ml NGM) with OP50-1 bacteria at 20°C. For the swimming assay, L4 stage animals were selected for mCh fluorescence under a Leica MZ16F dissection scope using a red filter to block transmission light < 590 nm and transferred to freshly seeded NGM plates. After 12-16h in the dark 30-40 young adult animals were picked onto a plain NGM plate (3.5 cm diameter, 3 mL NGM) and ~500-700 μL M9 buffer was added to induce swimming behavior. The plate was placed onto a multiworm tracker platform (Swierczek et al., 2011) equipped with a high-resolution camera (Falcon 4M30, DALSA) and a transmission light source, which was also covered with the red filter. A LabVIEW-based custom software called “MS-Acqu” was used for fully automated video acquisition of 1 min length at different time points. The experiment was started when the animals accommodated to swimming for 15 min. After the first video the plate was placed under a custom-built ring of high power 470 nm blue light-emitting diodes (Alustar 3W 30°, ledxon), which was controlled by transistor-transistor logic (TTL) pulses from MS-Acqu to allow pulsed blue light illumination (85 μW mm−2 at 0.03 Hz) of up to three culture dishes in parallel. Dishes are iteratively placed on the platform to acquire videos every 15 min during 1 h light illumination and 15 min post stimulation. Afterward excess M9 buffer was removed with a pipette and a drop of OP50-1 culture suspension was added. Worm plates were allowed to dry for 1 h in the dark. After 24 h recovery in the dark at room temperature, another swimming assay was performed as described, but without photostimulation. Swimming cycles are automatically analyzed using the “wrMTrck” plugin for the image processing software ImageJ (Nussbaum-Krammer et al., 2015). Samples were blinded to strain identity, then subjected to automated analysis. Objects not identified as worms (e.g., debris) were manually removed. A customized macro was used to adjust the region-of-interest (ROI) and to batch-process all video files. Tracking was validated using the labeled video-files. Custom Java-scripts were used to summarize the data and perform the statistical analysis. Experiments were independently performed 2-3 times, depending on the transgenic strain, as described in figure legends.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification for imaging experiments was carried out on raw fluorescent images using ImageJ to measure pixel intensities. Background values (estimated by measuring pixel intensities in image regions with no detectable signal) were routinely subtracted. mEPSCs amplitude and frequency were manually measured using Mini Analysis software (synaptosoft). EPSC amplitudes were quantified using the Neuromatic package with Igor Pro (Wavemetrics). Statistical analysis was carried out using standard statistical packages in Microsoft Excel. “n” values for electrophysiology experiments represent the number of cells recorded from and are reported for each experiment in the corresponding figure legend. In imaging experiments, the definition of n values are specified in figure legends but typically represent the number of synapses from at least 6 different neurons. Statistical significance for experiments comparing two populations was determined using a two-tailed unpaired Student’s t test. In cases where the two populations represented paired measurements, a paired Student’s t test was used. For experiments comparing three or more populations, a oneway ANOVA with Bonferroni multiple comparison test was used. All statistical analyses were performed using Graphpad Prism (Graphpad Software). For C. elegans studies, statistical analysis used two-tailed unpaired t tests, with Bonferroni-corrected p values. Data are presented as mean ± SEM unless otherwise noted.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal anti-VAMP2 | Synaptic Systems | Cat# 104 211BT; RRID: AB_2619758 |
| Polyclonal anti-mCh | Thermo Fisher Scientific | Cat# PA5-34974; RRID: AB_2552323 |
| Goat anti-rabbit IRDye 800CW secondary | Li-Cor | Cat# 926-32211; RRID: AB_621833 |
| Polyclonal anti-GFP | Sigma-Aldrich | Cat# G1544; RRID: AB_439690 |
| Polyclonal anti-GFP | Thermo/Molecular Probes | Cat#A-11122; RRID: AB_221569 |
| Goat anti-mouse Alexa647 conjugated secondary antibody | Life Technologies/Thermo Fisher | Cat#A-32728; RRID: AB_2633277 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FM4-64 | Molecular Probes/Thermo Fisher | Cat #13320 |
| FM1-43FX (fixable version of FM1-43) | Molecular Probes/Thermo Fisher | Cat #F35355 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | RRID: CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Rat, Sprague Dawley | Charles River | Crl:CD(SD)BR; RRID: RGD_734476 |
| C. elegans wild type Bristol N2 | CGC | RRID: WB-STRAIN:N2_ancestral |
| Mouse, C57BL/6 | The Jackson Laboratory | JAX:000664; RRID: IMSR_JAX:000664 |
| Recombinant DNA | ||
| jRGECO1a | Dana et al., 2016 | Addgene #61563 |
| Cryptochrome2 | Kennedy et al., 2010 | Addgene #26866 |
| CIBN | Kennedy et al., 2010 | Addgene #26867 |
| SspBmicro | Guntas et al., 2015 | Addgene #60416 |
| iLID | Guntas et al., 2015 | Addgene #60413 |
| SspBwt/nano | Guntas et al., 2015 | Addgene #60415 |
| pEGFP-VAMP3 | Galli et al., 1998 | Addgene #42310 |
| BoNT/B LC (S. cerevisiae codon optimized) | This manuscript | N/A |
| BoNT/B LC (M. musculus codon optimized) | This manuscript | N/A |
| Software and Algorithms | ||
| MATLAB | MathWorks | https://www.mathworks.com/ |
| Igor Pro neuromatic package | Wavemetrics | http://www.neuromatic.thinkrandom.com |
| Mini analysis | Synaptosoft | http://www.synaptosoft.com |
| Andor IQ3 acquisition software package | Andor Technologies | https://andor.oxinst.com/products/iq-live-cell-imaging-software |
| Metamorph | Molecular Devices | https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metamorph-microscopy |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| WinLTP | University of Bristol | https://www.winltp.com/ |
Highlights.
Few tools allow inhibition of neural activity for long time periods with light
We engineered botulinum neurotoxin B so that it can be activated with blue light
Photoactivated botulinum toxin efficiently cleaves the SNARE protein VAMP2
We demonstrate utility in diverse systems, from mammalian brain slices to worms
ACKNOWLEDGMENTS
We thank Brian Kuhlman for providing the iLID constructs used in this study. This work was supported by grants from the NIH (R01GM100225 to C.L.T.; R01NS082271 and R01NS107554 to M.J.K.; R21EY026363 to C.L.T. and M.J.K.; 1UF1NS107710 to M.J.K., C.L.T., and C.P.F.; R01DA35821 and R01NS95809 to C.P.F.; and R00MH103531 to J.A.) and from the Deutsche Forschungsgemeinschaft (GO1011/12-1, SPP1926 - Next Generation Optogenetics, and CRC1080-B2 to A.G.). This work was also supported by funding from the Brain Research Foundation (to C.L.T. and M.J.K.).
Footnotes
DECLARATION OF INTERESTS
A provisional patent application has been filed related to this study.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures, two tables, and two videos and can be found with this article online at https://doi.org/10.1016/j.neuron.2019.01.002.
A video abstract is available at http://dx.doi.org/10.1016/j.neuron.2019.01.002#mmc6.
REFERENCES
- Abraham C, Hutter H, Palfreyman MT, Spatkowski G, Weimer RM, Windoffer R, Jorgensen EM, and Leube RE (2006). Synaptic tetraspan vesicle membrane proteins are conserved but not needed for synaptogenesis and neuronal function in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 103, 8227–8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, et al. (2009). Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonsa H, Merricks EM, Codadu NK, Cunningham MO, Deisseroth K, Racca C, and Trevelyan AJ (2015). The contribution of raised intraneuronal chloride to epileptic network activity. J. Neurosci. 35, 7715–7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Martinelli DC, Malenka RC, Tabuchi K, and Südhof TC (2013). Presynaptic neurexin-3 alternative splicing trans-synaptically controls post-synaptic AMPA receptor trafficking. Cell 154, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, and Roth BL (2007). Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. USA 104, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi J, Chapman ER, Link E, Binz T, Yamasaki S, De Camilli P, Siidhof TC, Niemann H, and Jahn R (1993). Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 365, 160–163. [DOI] [PubMed] [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks VB, Curtis DR, and Eccles JC (1957). The action of tetanustoxin on the inhibition of motoneurones. J. Physiol. 135, 655–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow BY, Han X, Dobry AS, Qian X, Chuong AS, Li M, Henninger MA, Belfort GM, Lin Y, Monahan PE, and Boyden ES (2010). High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 463, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP, Tsegaye G, Holt GT, Hu A, Walpita D, et al. (2016). Sensitive red protein calcium indicators for imaging neural activity. eLife 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MW, Morton JJ, Carroll D, and Jorgensen EM (2008). Gene activation using FLP recombinase in C. elegans. PLoS Genet. 4, e1000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, and Kaplan JM (2006). Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proc. Natl. Acad. Sci. USA 103, 11399–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisel U, Jarausch W, Goretzki K, Henschen A, Engels J, Weller U, Hudel M, Habermann E, and Niemann H (1986). Tetanus toxin: primary structure, expression in E. coli, and homology with botulinum toxins. EMBO J. 5, 2495–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli T, Zahraoui A, Vaidyanathan VV, Raposo G, Tian JM, Karin M, Niemann H, and Louvard D (1998). A novel tetanus neurotoxin-insensitive vesicle-associated membrane protein in SNARE complexes of the apical plasma membrane of epithelial cells. Mol. Biol. Cell 9, 1437–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR (1992). The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 15, 133–139. [DOI] [PubMed] [Google Scholar]
- Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA, Ellis RJ, Richie CT, Harvey BK, Dannals RF, et al. (2017). Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 357, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntas G, Hallett RA, Zimmerman SP, Williams T, Yumerefendi H, Bear JE, and Kuhlman B (2015). Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc. Natl. Acad. Sci. USA 112, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husson SJ, Costa WS, Wabnig S, Stirman JN, Watson JD, Spencer WC, Akerboom J, Looger LL, Treinin M, Miller DM 3rd, et al. (2012). Optogenetic analysis of a nociceptor neuron and network reveals ion channels acting downstream of primary sensors. Curr. Biol. 22, 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson N, and Varshavsky A (1994). Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. USA 91, 10340–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, and Tucker CL (2010). Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 7, 973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JC, Cook MN, Carey MR, Shen C, Regehr WG, and Dymecki SM (2009). Linking genetically defined neuronsto behavior through a broadly applicable silencing allele. Neuron 63, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner HAE, Lein ES, and Callaway EM (2002). A genetic method for selective and quickly reversible silencing of Mammalian neurons. J. Neurosci. 22, 5287–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerchner W, Xiao C, Nashmi R, Slimko EM, van Trigt L, Lester HA, and Anderson DJ (2007). Reversible silencing of neuronal excitability in behaving mice by a genetically targeted, ivermectin-gated Cl- channel. Neuron 54, 35–49. [DOI] [PubMed] [Google Scholar]
- Lin JY, Sann SB, Zhou K, Nabavi S, Proulx CD, Malinow R, Jin Y, and Tsien RY (2013). Optogenetic inhibition of synaptic release with chromophore-assisted light inactivation (CALI). Neuron 79, 241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link E, Edelmann L, Chou JH, Binz T, Yamasaki S, Eisel U, Baumert M, Sudhof TC, Niemann H, and Jahn R (1992). Tetanus toxin action: inhibition of neurotransmitter release linked to synaptobrevin proteolysis. Biochem. Biophys. Res. Commun. 189, 1017–1023. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Pokala N, Feinberg EH, Chalasani SH, Butcher RA, Clardy J, and Bargmann CI (2009). A hub-and-spoke circuit drives pheromone attraction and social behaviour in C. elegans. Nature 458, 1171–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnus CJ, Lee PH, Atasoy D, Su HH, Looger LL, and Sternson SM (2011). Chemical and genetic engineering of selective ion channel-ligand interactions. Science 333, 1292–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek KW, and Davis GW (2002). Transgenically encoded protein photoinactivation (FlAsH-FALI): acute inactivation of synaptotagmin I. Neuron 36, 805–813. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Jorgensen E, and Horvitz HR (1993). Genes required for GABA function in Caenorhabditis elegans. Nature 364, 334–337. [DOI] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, and Ambros V (1991). Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrmann R, de Wit H, Verhage M, Neher E, and Sørensen JB (2010). Fast vesicle fusion in living cells requires at least three SNARE complexes. Science 330, 502–505. [DOI] [PubMed] [Google Scholar]
- Molgo J, Comella JX, Angaut-Petit D, Pecot-Dechavassine M, Tabti N, Faille L, Mallart A, and Thesleff S (1990). Presynaptic actions of botulinal neurotoxins at vertebrate neuromuscular junctions. J. Physiol. (Paris) 84, 152–166. [PubMed] [Google Scholar]
- Nakashiba T, Young JZ, McHugh TJ, Buhl DL, and Tonegawa S (2008). Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science 319, 1260–1264. [DOI] [PubMed] [Google Scholar]
- Nussbaum-Krammer CI, Neto MF, Brielmann RM, Pedersen JS, and Morimoto R.l. (2015). Investigating the spreading and toxicity of prion-like proteins using the metazoan model organism C. elegans. J. Vis. Exp. 52321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto H, Hanson PI, and Jahn R (1997). Assembly and disassembly of a ternary complex of synaptobrevin, syntaxin, and SNAP-25 in the membrane of synaptic vesicles. Proc. Natl. Acad. Sci. USA 94, 6197–6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondo JV, Kay L, Ellender TJ, and Akerman CJ (2012). Optogenetic silencing strategies differ in their effects on inhibitory synaptic transmission. Nat. Neurosci. 15, 1102–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, and Kavalali ET (2011). Differential regulation of spontaneous and evoked neurotransmitter release at central synapses. Curr. Opin. Neurobiol. 21, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saloman JL, Scheff NN, Snyder LM, Ross SE, Davis BM, and Gold MS (2016). Gi-DREADD expression in peripheral nerves produces ligand-dependent analgesia, as well as ligand-independent functional changes in sensory neurons. J. Neurosci. 36, 10769–10781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, and Montecucco C (1992). Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 359, 832–835. [DOI] [PubMed] [Google Scholar]
- Sinnen BL, Bowen AB, Gibson ES, and Kennedy MJ (2016). Local and use-dependent effects of β-amyloid oligomers on NMDA receptor function revealed by optical quantal analysis. J. Neurosci. 36, 11532–11543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slimko EM, McKinney S, Anderson DJ, Davidson N, and Lester HA (2002). Selective electrical silencing of mammalian neurons in vitro by the use of invertebrate ligand-gated chloride channels. J. Neurosci. 22, 7373–7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland D, Lin Y, Wagner E, Hope CM, Zayner J, Antoniou C, Sosnick TR, Weiss EL, and Glotzer M (2012). TULIPs: tunable, light-controlled interacting protein tags for cell biology. Nat. Methods 9, 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan S, and Eswaramoorthy S (2000). Structural analysis of the catalytic and binding sites of Clostridium botulinum neurotoxin B. Nat. Struct. Biol. 7, 693–699. [DOI] [PubMed] [Google Scholar]
- Sweeney ST, Broadie K, Keane J, Niemann H, and O’Kane CJ (1995). Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron 14, 341–351. [DOI] [PubMed] [Google Scholar]
- Swierczek NA, Giles AC, Rankin CH, and Kerr RA (2011). High-throughput behavioral analysis in C. elegans. Nat. Methods 8, 592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervo DGR, Hwang BY, Viswanathan S, Gaj T, Lavzin M, Ritola KD, Lindo S, Michael S, Kuleshova E, Ojala D, et al. (2016). A designer AAV variant permits efficient retrograde access to projection neurons. Neuron 92, 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaaga CE, Borisovska M, and Westbrook GL (2014). Dual-transmitter neurons: functional implications of co-release and co-transmission. Curr. Opin. Neurobiol. 29, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G, Holt MG, Bunt G, Riedel D, Wouters FS, and Jahn R (2010). One SNARE complex is sufficient for membrane fusion. Nat. Struct. Mol. Biol. 17, 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegert JS, Mahn M, Prigge M, Printz Y, and Yizhar O (2017). Silencing neurons: tools, applications, and experimental constraints. Neuron 95, 504–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff P, Goetz T, Leppä E, Linden A-M, Renzi M, Swinny JD, Vekovischeva OY, Sieghart W, Somogyi P, Korpi ER, et al. (2007). From synapse to behavior: rapid modulation of defined neuronal types with engineered GABAA receptors. Nat. Neurosci. 10, 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Wada N, Kitabatake Y, Watanabe D, Anzai M, Yokoyama M, Teranishi Y, and Nakanishi S (2003). Reversible suppression of glutamatergic neurotransmission of cerebellar granule cells in vivo by genetically manipulated expression of tetanus neurotoxin light chain. J. Neurosci. 23, 6759–6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CR, Power J, Barnea G, O’Donnell S, Brown HEV, Osborne J, Axel R, and Gogos JA (2004). Spontaneous neural activity is required for the establishment and maintenance of the olfactory sensory map. Neuron 42, 553–566. [DOI] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N, Wood PG, Bamberg E, Nagel G, Gottschalk A, and Deisseroth K (2007). Multimodal fast optical interrogation of neural circuitry. Nature 446, 633–639. [DOI] [PubMed] [Google Scholar]
- Zimmerman SP, Hallett RA, Bourke AM, Bear JE, Kennedy MJ, and Kuhlman B (2016). Tuning the binding affinities and reversion kinetics of a light inducible dimer allows control of transmembrane protein localization. Biochemistry 55, 5264–5271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.