

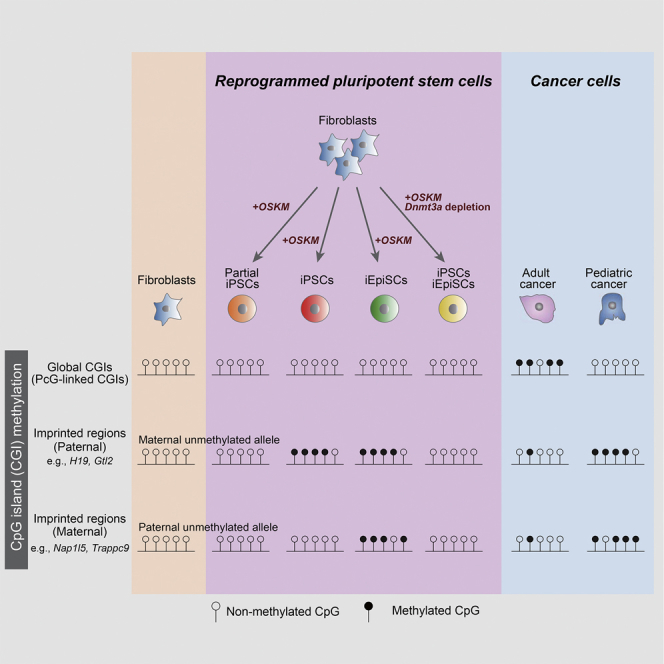
Summary
CpG islands (CGIs) including those at imprinting control regions (ICRs) are protected from de novo methylation in somatic cells. However, many cancers often exhibit CGI hypermethylation, implying that the machinery is impaired in cancer cells. Here, we conducted a comprehensive analysis of CGI methylation during somatic cell reprogramming. Although most CGIs remain hypomethylated, a small subset of CGIs, particularly at several ICRs, was often de novo methylated in reprogrammed pluripotent stem cells (PSCs). Such de novo ICR methylation was linked with the silencing of reprogramming factors, which occurs at a late stage of reprogramming. The ICR-preferred CGI hypermethylation was similarly observed in human PSCs. Mechanistically, ablation of Dnmt3a prevented PSCs from de novo ICR methylation. Notably, the ICR-preferred CGI hypermethylation was observed in pediatric cancers, while adult cancers exhibit genome-wide CGI hypermethylation. These results may have important implications in the pathogenesis of pediatric cancers and the application of PSCs.
Keywords: pluripotent stem cells, naive and primed pluripotency, reprogramming, pediatric cancers, DNA methylation, genomic imprinting, CpG islands, Dnmt3a
Graphical Abstract
Highlights
-
•
Several ICRs are de novo methylated in reprogrammed PSCs
-
•
De novo ICR methylation in iPSCs is linked with transgene silencing
-
•
Depletion of Dnmt3a prevents reprogrammed PSCs from de novo ICR methylation
-
•
Pediatric cancers exhibit reprogrammed PSC-like aberration in CGI methylation
In this article, Yamada, Yamamoto and colleagues show that Dnmt3a-mediated DNA methylation occurs at imprinting loci during reprogramming into naive and primed PSCs. Similar de novo ICR methylation is detected in pediatric cancers, which raises the possibility that reprogramming is involved in development of childhood cancers.
Introduction
Pluripotent stem cells (PSCs) have self-renewing activity and are capable of differentiating into various types of cells, making them invaluable tools for regenerative medicine and disease modeling (Stadtfeld and Hochedlinger, 2010, Yamanaka, 2012). In mice there are two types of pluripotent states, naive and primed. Mouse embryonic stem cells (ESCs) have naive pluripotency and are derived from inner cell mass (ICM) of a blastocyst, while mouse epiblast stem cells (EpiSCs) have primed pluripotency and are derived from post-implantation epiblast (Nichols and Smith, 2009). Naive and primed PSCs display distinct transcriptional and epigenetic profiles with different developmental potential (Nichols and Smith, 2009). Notably, naive and primed PSCs can be established from somatic cells by the enforced expression of defined transcription factors, such as Oct4, Sox2, Klf4, and c-Myc (OSKM), under appropriate culture conditions (induced pluripotent stem cells [iPSCs] and induced epiblast stem cells [iEpiSCs], respectively) (Han et al., 2011, Takahashi and Yamanaka, 2006). Although a number of studies have revealed that iPSCs and iEpiSCs display shared molecular characteristics with ESCs and EpiSCs, respectively (Choi et al., 2015, Han et al., 2011, Maherali et al., 2008, Mikkelsen et al., 2008), whether induced naive and primed PSCs faithfully recapitulate in vivo pluripotency is unknown.
Genomic imprinting is an essential epigenetic mechanism that controls the monoallelic expression of genes and is mediated by gamete-derived allele specific DNA methylation (Ferguson-Smith, 2011). Imprints are established exclusively in the male or female germline through de novo DNA methylation at imprinting control regions (ICRs) (Bourc'his et al., 2001, Kaneda et al., 2004). Established ICR methylation together with the concomitant unmethylated state at the other allele is strictly maintained in somatic cells throughout life (Ferguson-Smith, 2011). Previous studies demonstrated that Dnmt1, a maintenance DNA methyltransferase, together with Uhrf1 is responsible for the preservation of ICR methylation (Branco et al., 2008). In contrast, it is not fully understood how unmethylated allele at ICRs are maintained in the unmethylated state. It is known that CpG islands (CGIs) are generally protected from de novo methylation in somatic cells. Considering that ICRs often consist of CpG-rich regions including CGIs, protection from de novo ICR methylation could be mediated through mechanisms whereby CGIs are protected from de novo DNA methylation. Notably, cancer cells often display abnormal DNA hypermethylation at both CGIs and ICRs (Sharma et al., 2010), indicating that the machinery for avoiding de novo CGI methylation is impaired in cancer cells.
The dysregulation of imprinted genes is implicated in developmental defects and tumorigenesis (Kato et al., 1999, Steenman et al., 1994). Indeed, it has been reported that the dysregulation of imprints compromises the developmental potential of PSCs (Choi et al., 2017b, Yagi et al., 2017a). In addition, CGI hypermethylation in cancer cells are often detectable in tumor-suppressor genes with concomitant transcriptional silencing, which supports the notion that de novo CGI methylation plays a role in cancer development. It is therefore important to evaluate the stability of CGI/ICR methylation in PSCs. Several studies have previously examined the status of imprints in mouse and human iPSCs (Bar et al., 2017, Johannesson et al., 2014, Ma et al., 2014, Nazor et al., 2012, Pick et al., 2009). Differentially methylated regions (DMRs) at the Dlk1-Dio3 gene cluster are often hypermethylated in mouse iPSCs, which is linked with impaired developmental potential (Stadtfeld et al., 2010). A large-scale analysis of allele-specific RNA sequencing (RNA-seq) data revealed that primed human iPSCs display a higher incidence of biallelic expression of imprinted genes (Bar et al., 2017). However, the genome-wide stability of CGI methylation during the reprogramming process of naive and primed pluripotency remains to be fully elucidated.
Here we conducted comprehensive methylation analysis for CGIs and ICRs to understand the epigenetic stability in naive and primed PSCs. Given that ICR methylation is highly affected by culture conditions and gender in mouse PSCs (Choi et al., 2017a, Pasque et al., 2018, Yagi et al., 2017a, Yagi et al., 2017b), in this study we focus on the male reprogramming process under conventional serum-containing culture conditions. PSCs derived from cells with (129X1/SvJ × MSM/Ms) F1 genetic background allowed us to investigate allele-specific DNA methylation at ICRs by single-nucleotide polymorphisms (SNPs) (Takada et al., 2013, Yagi et al., 2017a). This effort revealed aberrant methylation at several ICRs during the reprogramming of somatic cells into naive and primed pluripotency. Furthermore, our data unveiled similar epigenetic aberrations in pediatric cancers with iPSCs, providing an unappreciated link between reprogramming and childhood cancer development.
Results
Generation of Naive and Primed Mouse PSCs in which Parental Alleles Are Distinguishable
To elucidate the stability of CGI methylation during reprogramming into naive and primed PSCs, we generated iPSCs and iEpiSCs from male mouse embryonic fibroblasts (MEFs) with piggyBac (PB) vector containing a doxycycline (Dox)-inducible polycistronic transgene encoding OSKM (Kim et al., 2016) (Figures 1A–1C, S1A, and S1B). For their control, we established male ESCs and EpiSCs derived from embryonic day 3.5 (E3.5) blastocyst and E6.5 post-implantation epiblast, respectively (Figures 1A and 1B). These cell lines were derived from (129X1/SvJ × MSM/Ms) F1 MEFs or embryos in which the parental alleles are distinguishable by a large number of SNPs. Early-passage PSCs (passage 3 [p3] to p4) were used to analyze gene expression and DNA methylation in this study. RNA-seq analysis confirmed that the iPSCs and iEpiSCs expressed general pluripotency-associated genes at levels comparable with those of control ESCs and EpiSCs (Figure S1C). iPSCs expressed naive pluripotency-associated genes and iEpiSCs expressed primed pluripotency-associated genes (Figures S1D and S1E). The established iPSC and iEpiSC clones exhibited silencing of transgenes even in the presence of Dox, except for iPSCs 9, 21, and 37, which continuously expressed mCherry, indicating transgene expression (Figures 1C and S1F). The silencing of transgenes is critical for achieving complete reprogramming to a stable pluripotent state (Jaenisch and Young, 2008). Consistent with this, iPSC 9 displayed distinct global transcriptional and methylome patterns compared with other ESC/iPSC clones and exhibited a decreased expression level of naive pluripotency-associated genes, which reflects the partial reprogramming state (Figures 1D and 1E). Consistent with this, there were DMRs in partially reprogrammed (partial) iPSC 9 compared with control iPSCs and ESCs (Figure S1G). Naive PSC lines and primed PSC lines were separately clustered in both transcriptome and DNA methylation profiling (Figures 1D and 1E). Overall, the established naive and primed PSC clones by somatic cell reprogramming harbored shared molecular signatures with naive and primed control PSC lines, which were directly derived from embryos.
Figure 1.

Establishment of Naive and Primed Mouse PSCs and CGI Methylation Analysis
(A) Schematic diagram of the experimental design. Generation of naive and primed PSCs derived directly from embryos or from somatic cells by reprogramming. Parental alleles can be distinguished by SNPs in (129X1/SvJ × MSM/Ms) F1 cells.
(B) Representative images of naive and primed PSCs (ESCs, EpiSCs, iPSCs, iEpiSCs). Scale bars, 100 μm.
(C) An image of mouse partial iPSCs. The mCherry signal represents the expression of the OSKM transgene. The transgene is not silenced in iPSC 9. Scale bars, 100 μm.
(D) Principal component (PC1 and PC2) analysis of transcriptional profiles by RNA-seq.
(E) Hierarchical clustering analysis of the global DNA methylation status by MethylC-seq.
(F) Box plot of DNA methylation levels at all CGIs in MEFs, ESCs, EpiSCs, iPSCs, and iEpiSCs. Solid lines in each box indicate the median. The bottom and top of the boxes are lower and upper quartiles, respectively. Whiskers extend to ±1.5 interquartile range (IQR).
(G) Venn diagram of CGIs with increased DNA methylation in PSCs compared with MEFs (DNA methylation difference >0.2). Number in parentheses indicates the number of CGIs linked to ICR. Note that ICR-linked CGIs are enriched in reprogrammed PSC-specific methylated CGIs. ∗p < 0.05, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 (Fisher's test).
(H) Box plot of DNA methylation levels at unmethylated alleles in ICRs in MEFs, ESCs, EpiSCs, iPSCs, and iEpiSCs. Solid lines in each box indicate the median. The bottom and top of the boxes are lower and upper quartiles, respectively. Whiskers extend to ±1.5 IQR.
(I) DNA methylation levels at unmethylated alleles in paternal and maternal ICRs in MEFs, ESCs, EpiSCs, iPSCs, and iEpiSCs.
De Novo DNA Methylation at ICRs in Reprogrammed PSCs
We next investigated genome-wide CGI methylation patterns by conducting target-captured MethylC sequencing (MethylC-seq) analysis of various PSC lines. We first confirmed that MethylC-seq analysis is suitable for the comprehensive analysis of CGI and ICR methylation since the probes capture 94.2% CGIs among all mouse CGIs (21,648 out of 22,948 CGIs). In fact, MethylC-seq analysis had higher sequencing coverage for CGIs and ICRs than whole-genome bisulfite sequencing (WGBS), with comparable numbers of sequencing reads (Figure S2A).
MethylC-seq analysis revealed that most CGIs remained hypomethylated in all PSC lines examined (Figure 1F). However, a small subset of CGIs exhibited increased methylation in reprogrammed PSCs compared with MEFs, which is the origin of the reprogrammed cells (DNA methylation difference >0.2) (Figure 1G). The majority of CGIs with increased methylation in iPSCs and iEpiSCs were similarly methylated in ESCs and EpiSCs, respectively (Figure 1G), which suggests that methylation at these CGIs is cell-type-related methylation in PSCs. Notably, we also observed iPSC/iEpiSC-specific CGI methylation (Figure 1G), suggesting that such CGI methylation is associated with the reprogramming. The reprogramming-associated CGI methylation was observed in various genetic elements (Figure S2B). Of particular interest, CGIs linked to ICRs (n = 27) were significantly enriched within CGIs with reprogramming-associated methylation in most reprogrammed PSC clones (p < 0.05 in iPSC 1, p < 0.001 in iPSC 7, p = 0.05466 in iPSC 13, p < 0.0001 in iEpiSC B1, and p < 0.0001 in iEpiSC F1, Fisher's test) (Figure 1G). Indeed, 9 out of 27 ICR-linked CGIs exhibited methylation in iEpiSC F1, and 8 of the 9 ICR-linked CGIs were not methylated in embryo-derived EpiSCs (Figure 1G), indicating that ICR methylation is closely related to reprogramming. The comprehensive allele-specific analysis for ICR methylation further confirmed that unmethylated alleles at ICRs were frequently de novo methylated in reprogrammed PSCs, a feature especially pronounced in iEpiSCs (Figure 1H). The unmethylated alleles of paternally imprinted ICRs (H19 DMR and Dlk1-Gtl2 DMR) were heavily methylated in both iPSCs and iEpiSCs, whereas maternally imprinted ICRs (e.g., Nap1l5 DMR, Kcnq1ot1 DMR, Trappc9 DMR) were often de novo methylated in iEpiSCs (Figure 1I).
De Novo DNA Methylation at Paternal ICRs during Reprogramming
We next generated heatmaps and dot plots showing methylation levels and allelic methylation patterns at ICRs (Figures 2A and 2B). We confirmed that control MEFs retain monoallelic ICR methylation patterns. Consistent with this, the total ICR methylation level in MEFs exhibited approximately 50% as in vivo tissues (Figures 2A, 2B, and S2C), except for partial gain of methylation at a part of Zac1 DMR and Dlk1-Gtl2 DMR (Figures 2A, 2B, and S2D). In mice, there are three paternal imprinted loci (Dlk1-Gtl2 DMR, H19 DMR, and Rasgrf1 DMR). De novo methylation at Dlk1-Gtl2 DMR was observed not only in iPSC clones but also in iEpiSC clones, but the loci remained unmethylated in both ESCs and EpiSCs (Figures 2A and 2B). Consistent with this observation, Meg3 and Rian, which are regulated by DNA methylation at Dlk1-Dio3 loci, were repressed in iPSCs and iEpiSCs (Figure S2E). H19 DMR also acquired de novo methylation in reprogrammed PSCs, which was further confirmed with multiple independent PSC clones (Figures 2A, 2B, and S3A). The unmethylated allele of H19 DMR was similarly methylated in ESCs to some extent, but not in EpiSCs, indicating that de novo methylation at H19 DMR takes place during both the reprogramming and maintenance of naive PSCs (Figures 2A, 2B, and S3A). We next examined the DNA methylation status at Rasgrf1 DMR, which was not captured by MethylC-seq analysis. De novo methylation at the unmethylated allele of Rasgrf1 DMR was observed exclusively in iEpiSCs (Figure S3B), providing additional evidence that paternally imprinted DMRs are epigenetically unstable during reprogramming.
Figure 2.

DNA Methylation of ICRs during Reprogramming to Naive and Primed Pluripotency in Mice
(A) Heatmap for DNA methylation levels and allelic balance at ICRs in MEFs, ESCs, EpiSCs, iPSCs, and iEpiSCs. The heatmap depicts the methylation status at CpG sites in which parental alleles have been distinguished. CpG methylation levels and allelic balance for the methylation are shown for each CpG site. Color scale is shown for DNA methylation levels and allelic balance.
(B) CpG methylation at representative ICRs of MEFs, ESCs, EpiSCs, iPSCs, and iEpiSCs. Each black dot represents a methylation percentage for each CpG site. Red and blue dots indicate methylation levels at maternal 129 allele and paternal MSM allele, respectively.
De Novo ICR Methylation Is Linked with Silencing of Exogenous Reprogramming Factors
We found that partial iPSC 9 harbors a smaller number of methylated CGIs compared with iPSCs (p < 0.0001, Fisher's test) (Figure 1G). Notably, iPSC 9 tended to preserve the monoallelic methylation pattern at ICRs (Figures 1H, 2A, 2B and S3A), which raised the possibility that aberrant ICR de novo methylation occurred at a late stage of the reprogramming. To examine this possibility, we extended the culture of iPSC 9 (Figure 3A). We found that a subset of iPSC 9 cells turned into mCherry-negative cells during the prolonged culture. Consistent with the fact that the silencing of reprogramming factors is a critical event for complete reprogramming, mCherry-negative iPSC 9 cells expressed higher levels of naive pluripotency genes (Figure S3C), suggesting that a subset of partial iPSCs converted into fully reprogrammed iPSCs. Most notably, mCherry-negative iPSC 9 cells acquired de novo methylation at the unmethylated allele of H19 DMR and Gtl2 DMR, while the allele remained unmethylated in mCherry-positive cells (Figure 3B). This transgene silencing-linked de novo ICR methylation was similarly observed in partial iPSC clones 21 and 37 (Figure 3B). These data indicate that de novo DMR methylation at H19 and Gtl2 is coupled with the silencing of exogenous reprogramming factors.
Figure 3.

De Novo ICR Methylation Occurs at the Late Stage of Somatic Cell Reprogramming
(A) Schematic diagram of the experimental design. Partial iPSC 9 cells were passaged three times, and mCherry-negative/-positive cells (p7) were sorted by fluorescence-activated cell sorting for expression analysis and DNA methylation analysis. Successful sorting was confirmed after expansion of sorted cells. Scale bars, 100 μm.
(B) DNA methylation analysis at H19 DMR and Gtl2 DMR in mCherry-positive and mCherry-negative iPSCs (clones 9, 21, 37) by conventional bisulfite sequencing. Open circles represent unmethylated CpGs and closed circles represent methylated CpGs. Crosses indicate undermined methylation status.
(C) DNA methylation at ICRs in preimplantation embryos. Note that ICMs exhibit reduced methylation levels at Gnas_1A ICR, while methylation levels are not altered at H19 or Impact ICRs. WGBS data of ICMs were obtained from GEO: GSE84236. MethylC-seq data of MEFs from our previous study (Yagi et al., 2017a) were used (GEO: GSE84165).
(D) Allelic expression analysis of Igf2 in MEFs and iPSCs. iPSCs exhibit biallelic expression of Igf2, which is consistent with biallelic methylation at H19 DMR in these cells. Red and blue indicate maternal and paternal alleles, respectively. Numbers in the pie chart display numbers of the subcloned allele.
(E) Establishment of iPSC-derived secondary MEFs by blastocyst injection. iPSC-derived MEFs were selected by neomycin treatment for 7 days.
(F) DNA methylation analysis of H19 DMR in iPSC-derived MEFs by conventional bisulfite sequencing.
(G) Allelic expression analysis of Igf2 in iPSC-derived MEFs. Biallelic expression of Igf2 is detectable in iPSC-derived MEFs. Red and blue indicate maternal and paternal alleles, respectively. Numbers in the pie chart display numbers of the subcloned allele.
De novo DNA Methylation at a Subset of Maternal ICRs in Primed PSCs
In contrast to the monoallelic DNA methylation pattern at maternally imprinted loci in iPSCs, iEpiSCs exhibited biallelic ICR methylation at a subset of maternal ICRs (e.g., Nap1l5, Trappc9), whereas embryo-derived EpiSCs showed only a modest increase of methylation at these ICRs (Figures 1I, 2A, and 2B). Hypermethylation at Nap1l5 DMR was confirmed with multiple iEpiSC clones (Figure S3D). We also found that Gnas1A ICR is hypomethylated in naive PSCs (both ESCs and iPSCs) but maintained in primed PSCs (both EpiSCs and iEpiSCs) (Figure 2A). Given that Gnas1A ICR is hypomethylated in ICM of preimplantation embryos (Figure 3C), the decreased Gnas1A ICR methylation in naive PSCs may reflect the reduced methylation in ICM. Apart from primary ICRs, secondary DMRs (sDMRs) acquire parent-of-origin-dependent DNA methylation patterns after implantation. We found that naive PSCs exhibit reduced methylation levels at Nespas sDMR and Cdkn1c sDMR, whereas primed PSCs retain monoallelic methylation (Figure S4A), presumably reflecting the DNA methylation status in the in vivo counterpart. Collectively, these results suggest that the epigenetic integrity of imprinted DMRs in PSCs is variable and depends on the imprinted loci and two types of pluripotent states (naive and primed).
Inheritance of De Novo ICR Methylation and Biallelic Expression of Imprinted Genes after Differentiation
We next examined whether an altered DNA methylation status at ICRs affects the allelic expression pattern of imprinted genes. Among maternally imprinted genes, expressed genes retained monoallelic expression in iEpiSC lines (Figure S4B), which is consistent with our methylome data showing that the corresponding ICRs were stably methylated/unmethylated in reprogrammed PSC lines (Figure 2A). We noticed that maternally imprinted genes that acquire de novo ICR methylation (e.g., Nap1l5, Airn) showed lower or no detectable expression levels in EpiSCs (data not shown). These results may suggest that imprinted genes with lower expression levels are targets of de novo methylation at maternally imprinted ICRs in iEpiSCs.
It is well known that paternally imprinted H19 DMR regulates the monoallelic expression of Igf2 from the paternal allele (Steenman et al., 1994). Consistent with our observation that H19 DMR is biallelically methylated in reprogrammed PSCs, Igf2 was expressed from both paternal and maternal alleles in iPSC clones (Figure 3D). A previous report suggests that ICR methylation is not recovered in somatic cell lineages once it is lost in PSCs (Holm et al., 2005). Therefore, we next investigated whether the acquired de novo ICR methylation in PSCs is inherited after differentiation. For this purpose, we injected iPSCs, which harbor de novo methylation at H19 DMR, into blastocyst and established iPSC-derived secondary MEFs by neomycin selection (Figure 3E). Notably, aberrant DNA methylation patterns at H19 DMR and biallelic expression of Igf2 were detected in differentiated MEFs (Figures 3F and 3G), indicating that aberrant de novo ICR methylation was sustained even after the differentiation of PSCs. This result is consistent with the silencing of H19 in ESC nuclei being sustained in differentiated cells after nuclear cloning (Humpherys et al., 2001).
Variable ICR Methylation Status in Human PSCs
To investigate any overlap of aberrant CGI methylation patterns between mouse and human PSCs, we next analyzed the genome-wide CGI methylation status in human primed PSC lines (Nishizawa et al., 2016). Consistent with our results in mouse primed iEpiSCs, most CGIs were hypomethylated in human iPSCs (Figure 4A). However, a subset of imprinted loci was aberrantly methylated in human iPSCs (hypermethylated DMR, n = 10 loci; hypomethylated DMR, n = 5 loci), whereas cells of origin for human iPSCs exhibited normal methylation levels (approximately 50%) at the imprinted DMRs (Figures 4B–4D). The aberrant DMR methylation was not associated with the method of reprogramming (Figure 4B). Notably, paternally imprinted genes such as H19, MEG3, and ZDBF2 were frequently hypermethylated in multiple human PSCs, which was similarly observed in mouse PSCs (Figure 4E). These observations are consistent with a recent study demonstrating that the biallelic expression of imprinted genes is observed more frequently at paternally imprinted genes than maternally imprinted genes in human iPSCs (Bar et al., 2017). As observed in mouse iEpiSCs, several maternally methylated DMRs (e.g., TRAPPC9, SNRPN, RB1, and PEG3) were hypermethylated in human PSCs (Figure 4E). Conversely, a subset of maternally methylated DMRs (FAM50B and GNAS) were hypomethylated in human PSCs (Figure 4D). Importantly, FAM50B and GNAS are hypomethylated in human preimplantation embryos (Hanna et al., 2016), which supports the notion that hypomethylation at imprinted DMRs in PSCs may reflect the decreased methylation level of preimplantation embryos.
Figure 4.

Variable ICR Methylation Aberrations in Human PSCs
(A) Box plot of DNA methylation levels at all CGIs in human somatic cells and PSCs. Solid lines in each box indicate the median. The bottom and top of the boxes are lower and upper quartiles, respectively. Whiskers extend to ±1.5 IQR. Infinium 450K data of human somatic cells and PSCs were obtained from GEO: GSE60821 and GSE60923.
(B) Heatmap for DNA methylation levels at imprinted DMRs in human iPSCs (hiPSCs), human ESCs (hESCs), and various somatic cells of origin for hiPSCs. Color scale is shown for DNA methylation levels. Infinium 450K data of 35 hiPSC lines (20 male lines and 15 female lines), 4 hESCs, and 16 somatic cells were obtained from GEO: GSE60821 and GSE60923. Names of the hPSC lines are shown at the right of the panel. Colors depict the methods of reprogramming. HDF, human dermal fibroblasts; CB, cord blood cells; PBMN, peripheral blood mononuclear cells; DP, dental pulp cells.
(C) List of 36 human imprinted DMRs shown in (A) (Court et al., 2014). Origin (maternal or paternal allele) and timing (germline or somatic) of methylation are shown for each DMR. IG-DMR (17) is not considered in further analyses because the probes of Infinium 450K are not designed at IG-DMR.
(D) Difference of median methylation levels at the 35 imprinted DMRs between hiPSCs and somatic cells. The median methylation levels of the 35 iPSC lines and 16 somatic cells in (B) are compared. Hypermethylated DMRs (false discovery rate [FDR] <0.01, median methylation in iPSCs >60%) and hypomethylated DMRs (FDR <0.01, median methylation in iPSCs <40%) are shown in pink and blue, respectively.
(E) Box plots of DNA methylation levels at representative paternal and maternal imprinted DMRs in human iPSCs. Solid lines in each box indicate the median. The bottom and top of the boxes are lower and upper quartiles, respectively. Whiskers extend to ±1.5 IQR. Each color in the boxes represents a cell of origin. ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001 (Mann-Whitney U test).
Dnmt3a Contributes to De Novo ICR Methylation during Reprogramming in Mice and Humans
During germ cell development, imprinted DMR is de novo methylated by Dnmt3a in conjunction with Dnmt3l (Bourc'his et al., 2001, Kaneda et al., 2004). A previous study revealed that Dnmt3a but not Dnmt3l is responsible for the increased methylation at Dlk1-Dio3 imprinted loci during reprogramming (Stadtfeld et al., 2012). We therefore asked whether aberrant hypermethylation at other ICRs during reprogramming is also dependent on Dnmt3a activity. To test this hypothesis, we analyzed iPSCs derived from C57/BL6 MEFs lacking Dnmt3a with lentivirus-mediated Dox-inducible OKSM (Stadtfeld et al., 2012) for ICR methylation. We observed hypermethylation at H19 DMR in control iPSCs, affirming that ICR hypermethylation occurred irrespective of the mouse genetic background or the PB system. Notably, Dnmt3a-deficient iPSCs displayed decreased DNA methylation levels at H19 DMR compared with control cells (Figure 5A). We further established iEpiSCs from Dnmt3a KO MEFs and examined ICR methylation (Figure S4C). Dnmt3a knockout (KO) iEpiSCs exhibited reduced methylation levels at multiple ICRs compared with control Dnmt3a wild-type (WT) iEpiSCs (Figures 5B–5D). Of note, methylation levels at multiple ICRs were close to 50% in Dnmt3a-KO iEpiSCs (Figures 5A–5D). These findings suggest that Dnmt3a plays a dominant role in de novo ICR methylation, although our study does not exclude the possibility of the contribution of Dnmt3b. Collectively, Dnmt3a contributes to aberrant de novo DNA methylation at ICRs during somatic cell reprogramming in mice.
Figure 5.

Dnmt3a Mediates Reprogramming-Associated De Novo ICR Methylation in Mice and Humans
(A) DNA methylation status at H19 DMR by conventional bisulfite sequencing in Dnmt3a control (2lox) and KO iPSCs.
(B) DNA methylation status at Nap1l5 DMR and H19 DMR by conventional bisulfite sequencing in Dnmt3a wild-type (WT) and null (KO) iEpiSCs.
(C) DNA methylation status at paternally methylated DMRs in Dnmt3a wild-type (WT) and null (KO) iEpiSCs. Each bar indicates a CpG site, and bar height represents methylation percentage (0%–100%) by MethylC-seq.
(D) DNA methylation status at maternally methylated DMRs in Dnmt3a wild-type (WT) and null (KO) iEpiSCs. Each bar indicates a CpG site, and bar height represents methylation percentage (0%–100%) by MethylC-seq.
(E) DNA methylation status at MEG3 DMR and IGF2 DMR2 by conventional bisulfite sequencing in hiPSCs established with DNMT3A short hairpin RNA (shRNA) and control shRNA treatment.
We further tested whether the suppression of DNMT3A can prevent human iPSCs from hypermethylation at imprinted DMRs during reprogramming. We generated human iPSCs from human peripheral blood mononuclear cells by inducing reprogramming factors with simultaneous knockdown of DNMT3A (Figures S4D and S4E). Of note, DNA methylation levels at MEG3 DMR and IGF2 DMR2 were reduced in DNMT3A KO human iPSCs compared with control iPSCs (Figure 5E), indicating that DNMT3A is responsible for hypermethylation at imprinted DMRs during reprogramming in both mice and humans.
Increased Methylation Levels at a Subset of Imprinted DMRs in Pediatric Cancers
Increased methylation levels at imprinted DMRs have been observed in a subset of cancers. Particularly, H19 DMR hypermethylation and the concomitant biallelic expression of IGF2 are frequently detectable in Wilms' tumors, the most common human pediatric kidney cancer (Hubertus et al., 2011, Steenman et al., 1994). We previously demonstrated that premature termination of in vivo reprogramming in mice leads to the development of cancers (Ohnishi et al., 2014). Notably, reprogramming-associated kidney cancers resembled Wilms' tumor and often harbored imprinting aberrations, including H19 DMR hypermethylation (Ohnishi et al., 2014). These results may provide a link between reprogramming-associated ICR methylation and epigenetic aberrations in pediatric cancers. Therefore, to investigate a possible association of aberrant DNA methylation between Wilms' tumors and iPSCs, we performed comprehensive DMR methylation analysis in Wilms' tumors as well as normal kidney tissues and renal cell carcinomas (RCCs), a representative of adult kidney cancer, using public datasets. Wilms' tumors often exhibited increased methylation levels at imprinted DMRs not only at H19 DMR but also other DMRs, whereas normal kidney samples and RCCs displayed relatively stable DMR methylation levels (Figures 6A and 6B). Increased DMR methylation levels at imprinted loci including H19 DMR were also detectable in neuroblastomas, another type of pediatric cancer (Figure S4F). Together, these results suggest that de novo methylation at particular imprinted DMRs is a shared aberration in reprogrammed PSCs and pediatric cancers.
Figure 6.

Pediatric Cancers Exhibit Hypermethylation at ICRs but Not at Global CGIs
(A) Heatmap for DNA methylation levels at imprinted DMRs in human normal kidney samples, renal cell carcinomas (RCC), and Wilms' tumors. Color scale is shown for DNA methylation levels. Infinium 450K data of normal kidney tissues and Wilms' tumors were obtained from GEO: GSE59157. Those of RCCs were obtained from GEO: GSE70303.
(B) Box plots of DNA methylation levels at representative hypermethylated imprinted DMRs in Wilms' tumors. Solid lines in each box indicate the median. The bottom and top of the boxes are lower and upper quartiles, respectively. Whiskers extend to ±1.5 IQR. Note that H19 DMR and RB1 DMR are hypermethylated in Wilms' tumor but not in normal kidney or RCC. ∗∗∗∗p < 0.0001 (Mann-Whitney U test).
(C) Box plot of DNA methylation levels at all CGIs in normal tissues, pediatric cancers, adult cancers, somatic cells, and PSCs. Solid lines in each box indicate the median. The bottom and top of the boxes are lower and upper quartiles, respectively. Whiskers extend to ±1.5 IQR. Note that increased CGI methylation is observed in adult cancers but not in pediatric cancers. Data of somatic cells and PSCs are the same as in Figure 4A.
(D) Hierarchical clustering analysis based on the methylation status at all CGIs.
(E) Box plot of DNA methylation levels at CGIs linked to PcG target genes (Lee et al., 2006) in normal tissues, pediatric cancers, adult cancers, somatic cells, and PSCs. Solid lines in each box indicate the median. The bottom and top of the boxes are lower and upper quartiles, respectively. Whiskers extend to ±1.5 IQR.
Previous studies demonstrated that CGI hypermethylation, particularly at CGIs linked to polycomb (PcG) target genes, is a general feature of epigenetic abnormalities found in cancers (Feinberg et al., 2006, Schlesinger et al., 2007, Widschwendter et al., 2007). In contrast, we found that most CGIs, except for imprinted DMRs, remain hypomethylated in iPSCs, which suggests that aberrant CGI methylation patterns in cancers are distinct from those in iPSCs. Consistent with previous reports, we detected CGI hypermethylation in a wide variety of adult cancers and found it was more prominent at CGIs close to PcG target genes (Figures 6C–6E). Notably, however, the same analysis revealed that pediatric cancers as well as iPSCs displayed no evidence of hypermethylation at global CGIs or even at PcG target gene-linked CGIs (Figures 6C–6E). Collectively, our data demonstrated that pediatric cancers harbor similar patterns of aberrant CGI methylation with iPSCs and highlighted the distinct features of epigenetic abnormalities in pediatric cancers and adult cancers.
Discussion
In the present study, we conducted comprehensive methylation analysis for CGIs to elucidate the epigenetic stability during somatic cell reprogramming into naive and primed PSCs. This analysis unveiled that most CGIs are precluded from being methylated, but CGIs linked to ICRs preferentially undergo de novo methylation during reprogramming. Consistent with this, paternally methylated ICRs often gained aberrant de novo methylation in both iPSCs and iEpiSCs. A subset of maternally methylated ICRs (e.g., Nap1l5 and Trappc9) was also hypermethylated in primed iEpiSCs. We also show that human iPSCs exhibit aberrant CGI methylation at several imprinted DMRs. Notably, the biallelic expression or silencing of imprinted genes in reprogrammed PSCs are sustained in PSC-derived differentiated cells. Since the proper establishment and maintenance of genomic imprints, particularly at H19 DMR (Kono et al., 2004), are important for normal development in mammals, our results may have important implications in various applications of PSCs, including regenerative medicine, drug screening, and the study of early developmental biology.
Interestingly, de novo ICR methylation was not prominent in partial iPSCs, suggesting that the aberrant ICR methylation in PSCs was not related to incomplete reprogramming. Notably, de novo ICR methylation occurred in accordance with the silencing of reprogramming factors. A previous study demonstrated that de novo DNA methylation plays a role in PB silencing (Troyanovsky et al., 2016). It is thus possible that the same machinery is involved in de novo ICR methylation and PB silencing. Nevertheless, considering that aberrant DMR hypermethylation is detectable in human iPSCs established by various methods, we propose that the de novo methylation at particular ICRs is a general phenomenon during somatic cell reprogramming. However, it should be noted that there exists clonal variation in ICR methylation patterns. The clonal variation may reflect the stochastic nature of the de novo methylation.
Although we demonstrated that Dnmt3a contributes to the ICR methylation in both mice and humans, the precise mechanism for de novo ICR methylation remains unclear. A previous study demonstrated that unmethylated alleles of imprinted genes are marked with both H3K4me2/3 and H3K27me3 and exhibit monoallelic bivalent chromatin when they are transcriptionally inactive (Maupetit-Mehouas et al., 2016). Of note, a recent study demonstrated that Dnmt3a preferentially binds to bivalent regions (Manzo et al., 2017). Collectively, it is possible that bivalent modifications at the unmethylated allele of imprinted genes might be a target of Dnmt3a binding, thus acting as a possible cause of de novo methylation during reprogramming.
We found that CGIs of multiple ICRs are aberrantly methylated in both human PSCs and pediatric cancers. However, we also identified that adult cancers exhibit distinct patterns of CGI hypermethylation from iPSCs; adult cancers display genome-wide global CGI hypermethylation while iPSCs exhibit ICR-preferred CGI hypermethylation. Notably, in sharp contrast to adult cancers, pediatric cancers did not show global CGI hypermethylation but exhibited ICR-preferred CGI hypermethylation, indicating that pediatric cancers harbor shared aberrant epigenetic signatures with human PSCs. A recent study demonstrated that CGI hypermethylation in adult cancers is associated with activated fibroblast growth factor (FGF) signaling (Smith et al., 2017). The fact that pediatric cancers infrequently harbor genetic aberrations in components of the FGF pathway might explain the absence of global CGI in pediatric cancers. Collectively, these findings highlighted the unique patterns of epigenetic aberrations in pediatric cancers, which exhibit differences from adult cancers but similarities with iPSCs. Considering that the premature termination of in vivo reprogramming causes pediatric cancer-like tumors in mice (Ohnishi et al., 2014) and that pediatric cancers harbor PSC-like transcriptional signatures (Terada et al., 2019), these results raised the possibility that some aspects of reprogramming to PSCs may drive the development of pediatric cancers with concomitant aberrations in ICR methylation.
To conclude, our findings about de novo CGI methylation may provide important insights into the faithful recapitulation of in vivo pluripotent cells in vitro. Our findings may also underscore the significant relevance of reprogramming-associated epigenetic aberrations in the development of pediatric cancers.
Experimental Procedures
Detailed descriptions of experimental procedures can be found in Supplemental Information.
Establishment and Culture of Male ESCs and EpiSCs
Zygotes with an (129X1/SvJ × MSM/Ms) F1 genetic background were obtained by in vitro fertilization (IVF). ESCs established in a previous study were used in this study (Yagi et al., 2017a). Epiblasts were divided from the extraembryonic regions and transferred into culture plates to derive EpiSCs.
Generation and Culture of Male iPSCs and iEpiSCs
Dox-inducible PB vector containing tetO-Oct4-Sox2-Klf4-cMyc-IRES-mCherry-EF1-rtTA-IRES-Neo (PB-OSKM) was used for reprogramming. After transfection, cultured medium was switched to ESC medium containing 2 μg/mL Dox (Sigma) for iPSC derivation and iEpiSC medium for iEpiSC derivation.
Generation of iPSCs and iEpiSCs from Dnmt3a-Deficient MEFs
Dnmt3a WT and KO MEFs were obtained by crossing Dnmt3a hetero KO (B6; 129S4-Dnmt3a <tm1Enl>) mice (Okano et al., 1999). Dnmt3a WT and KO iEpiSCs were generated by Dox-inducible PB-OSKM. Dnmt3a WT and KO iPSCs were generated in a previous study (Stadtfeld et al., 2012).
Animals
All experiments using animals were performed under the ethical guidelines of Kyoto University, University of Tokyo, and Kumamoto University. MSM/Ms were obtained from RIKEN Bio Resource Center (Takada et al., 2013, Takada et al., 2015).
Generation and Culture of hiPSCs
For generation of human iPSCs (hiPSCs), human peripheral blood mononuclear cells (Cellular Technology) were cultured. After transduction of the plasmid mixture (pCXLE-hOCT3/4-shp53-F, pCXLE-hUL, pCXWB-EBNA1, and pCXLE-hSK encoding short hairpin RNA for DNMT3A), the cells were seeded on a 6-well plate coated with iMatrix-511 (Takara) and cultured until hiPSC colony formation. The selected hiPSCs were expanded in StemFit AK02N (Takara).
Library Preparation
Library preparation was performed with SureSelect Mouse Methyl-Seq Reagent Kit (Agilent Technologies). DNA was bisulfite-treated using the EZ DNA Methylation-Gold Kit. The libraries were then sequenced on HiSeq 2500 (2 × 100-bp or 2 × 101-bp paired-end reads, Illumina). RNA-seq libraries were generated using the Truseq Stranded mRNA LT sample prep kit (Illumina). RNA-seq libraries were sequenced on NextSeq500 (75-bp single read, Illumina).
DNA Methylation Analyses
For allelic methylation analyses, the SNP data for MSM/Ms were obtained from NIG Mouse Genome Database (MSMv4HQ, http://molossinus.lab.nig.ac.jp/msmdb/index.jsp). The B6-derived and MSM/Ms-derived sequenced reads were determined based on the MSM/Ms SNP data. Previously described mouse CGIs (Illingworth et al., 2010) and mouse ICRs (Court et al., 2014, Tomizawa et al., 2011) were used for CGI and imprinting analyses, respectively. Infinium array data were obtained from publicly available datasets. Previously described human CGIs (Illingworth et al., 2010), human ICRs (Court et al., 2014, Tomizawa et al., 2011), and PcG target genes (Lee et al., 2006) were used for methylation analyses. The average methylation signals of each ICR were used for comparison among samples.
Author Contributions
M.Y. and Y.Y. designed and conceived the study and wrote the manuscript. M.Y., T.U., Y.S., M.S., and K.A. generated the cell lines, performed the experiments, and generated the MethylC-seq and RNA-seq libraries. A.T. performed the blastocyst injections. K.O. established the human iPSCs with short hairpin RNA. K.W. provided the OSKM piggyBac vector. K.H. provided Dnmt3a 2flox and null iPSCs. M.K., S.O., and T.Y. analyzed the RNA-seq, MethylC-seq, and Infinium 450K data.
Acknowledgments
We are grateful to K. Nakabayashi for providing details on human imprinted DMRs. Y.Y. was supported in part by cancer research grant P-CREATE, Japan Agency for Medical Research and Development (AMED); SICORP, AMED; JSPS KAKENHI 18H04026; the Takeda Science Foundation; and the Naito Foundation. Y.Y. and T.Y. were supported by AMED-CREST under grant numbers JP18gm1110004 (Y.Y and T.Y.) and JP18gm0610017 (T.Y.); and Core Center for iPS Cell Research, Research Center Network for Realization of Regenerative Medicine, AMED. T.Y. was supported by the iPS Cell Research Fund. M.Y. was supported by JSPS KAKENHI 15J05792. The ASHBi is supported by the World Premier International Research Center Initiative, MEXT, Japan.
Published: May 2, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2019.04.008.
Contributor Information
Takuya Yamamoto, Email: takuya@cira.kyoto-u.ac.jp.
Yasuhiro Yamada, Email: yasu@ims.u-tokyo.ac.jp.
Accession Numbers
The accession numbers for the MethylC-seq and RNA-seq data reported in this paper are GEO: GSE111173 and GSE84165.
Supplemental Information
References
- Bar S., Schachter M., Eldar-Geva T., Benvenisty N. Large-scale analysis of loss of imprinting in human pluripotent stem cells. Cell Rep. 2017;19:957–968. doi: 10.1016/j.celrep.2017.04.020. [DOI] [PubMed] [Google Scholar]
- Bourc'his D., Xu G.L., Lin C.S., Bollman B., Bestor T.H. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- Branco M.R., Oda M., Reik W. Safeguarding parental identity: Dnmt1 maintains imprints during epigenetic reprogramming in early embryogenesis. Genes Dev. 2008;22:1567–1571. doi: 10.1101/gad.1690508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Clement K., Huebner A.J., Webster J., Rose C.M., Brumbaugh J., Walsh R.M., Lee S., Savol A., Etchegaray J.P. DUSP9 modulates DNA hypomethylation in female mouse pluripotent stem cells. Cell Stem Cell. 2017;20:706–719.e7. doi: 10.1016/j.stem.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Huebner A.J., Clement K., Walsh R.M., Savol A., Lin K., Gu H., Di Stefano B., Brumbaugh J., Kim S.Y. Prolonged Mek1/2 suppression impairs the developmental potential of embryonic stem cells. Nature. 2017;548:219–223. doi: 10.1038/nature23274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Lee S., Mallard W., Clement K., Tagliazucchi G.M., Lim H., Choi I.Y., Ferrari F., Tsankov A.M., Pop R. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat. Biotechnol. 2015;33:1173–1181. doi: 10.1038/nbt.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court F., Tayama C., Romanelli V., Martin-Trujillo A., Iglesias-Platas I., Okamura K., Sugahara N., Simon C., Moore H., Harness J.V. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014;24:554–569. doi: 10.1101/gr.164913.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A.P., Ohlsson R., Henikoff S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- Ferguson-Smith A.C. Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011;12:565–575. doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- Han D.W., Greber B., Wu G., Tapia N., Arauzo-Bravo M.J., Ko K., Bernemann C., Stehling M., Scholer H.R. Direct reprogramming of fibroblasts into epiblast stem cells. Nat. Cell Biol. 2011;13:66–71. doi: 10.1038/ncb2136. [DOI] [PubMed] [Google Scholar]
- Hanna C.W., Penaherrera M.S., Saadeh H., Andrews S., McFadden D.E., Kelsey G., Robinson W.P. Pervasive polymorphic imprinted methylation in the human placenta. Genome Res. 2016;26:756–767. doi: 10.1101/gr.196139.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm T.M., Jackson-Grusby L., Brambrink T., Yamada Y., Rideout W.M., 3rd, Jaenisch R. Global loss of imprinting leads to widespread tumorigenesis in adult mice. Cancer Cell. 2005;8:275–285. doi: 10.1016/j.ccr.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Hubertus J., Lacher M., Rottenkolber M., Muller-Hocker J., Berger M., Stehr M., von Schweinitz D., Kappler R. Altered expression of imprinted genes in Wilms tumors. Oncol. Rep. 2011;25:817–823. doi: 10.3892/or.2010.1113. [DOI] [PubMed] [Google Scholar]
- Humpherys D., Eggan K., Akutsu H., Hochedlinger K., Rideout W.M., 3rd, Biniszkiewicz D., Yanagimachi R., Jaenisch R. Epigenetic instability in ES cells and cloned mice. Science. 2001;293:95–97. doi: 10.1126/science.1061402. [DOI] [PubMed] [Google Scholar]
- Illingworth R.S., Gruenewald-Schneider U., Webb S., Kerr A.R., James K.D., Turner D.J., Smith C., Harrison D.J., Andrews R., Bird A.P. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010;6:e1001134. doi: 10.1371/journal.pgen.1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R., Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–582. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannesson B., Sagi I., Gore A., Paull D., Yamada M., Golan-Lev T., Li Z., LeDuc C., Shen Y., Stern S. Comparable frequencies of coding mutations and loss of imprinting in human pluripotent cells derived by nuclear transfer and defined factors. Cell Stem Cell. 2014;15:634–642. doi: 10.1016/j.stem.2014.10.002. [DOI] [PubMed] [Google Scholar]
- Kaneda M., Okano M., Hata K., Sado T., Tsujimoto N., Li E., Sasaki H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- Kato Y., Rideout W.M., 3rd, Hilton K., Barton S.C., Tsunoda Y., Surani M.A. Developmental potential of mouse primordial germ cells. Development. 1999;126:1823–1832. doi: 10.1242/dev.126.9.1823. [DOI] [PubMed] [Google Scholar]
- Kim S.I., Oceguera-Yanez F., Sakurai C., Nakagawa M., Yamanaka S., Woltjen K. Inducible transgene expression in human iPS cells using versatile all-in-one piggybac transposons. Methods Mol. Biol. 2016;1357:111–131. doi: 10.1007/7651_2015_251. [DOI] [PubMed] [Google Scholar]
- Kono T., Obata Y., Wu Q.L., Niwa K., Ono Y., Yamamoto Y., Park E.S., Seo J.S., Ogawa H. Birth of parthenogenetic mice that can develop to adulthood. Nature. 2004;428:860–864. doi: 10.1038/nature02402. [DOI] [PubMed] [Google Scholar]
- Lee T.I., Jenner R.G., Boyer L.A., Guenther M.G., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H., Morey R., O'Neil R.C., He Y., Daughtry B., Schultz M.D., Hariharan M., Nery J.R., Castanon R., Sabatini K. Abnormalities in human pluripotent cells due to reprogramming mechanisms. Nature. 2014;511:177–183. doi: 10.1038/nature13551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N., Ahfeldt T., Rigamonti A., Utikal J., Cowan C., Hochedlinger K. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell. 2008;3:340–345. doi: 10.1016/j.stem.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzo M., Wirz J., Ambrosi C., Villasenor R., Roschitzki B., Baubec T. Isoform-specific localization of DNMT3A regulates DNA methylation fidelity at bivalent CpG islands. EMBO J. 2017;36:3421–3434. doi: 10.15252/embj.201797038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maupetit-Mehouas S., Montibus B., Nury D., Tayama C., Wassef M., Kota S.K., Fogli A., Cerqueira Campos F., Hata K., Feil R. Imprinting control regions (ICRs) are marked by mono-allelic bivalent chromatin when transcriptionally inactive. Nucleic Acids Res. 2016;44:621–635. doi: 10.1093/nar/gkv960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen T.S., Hanna J., Zhang X., Ku M., Wernig M., Schorderet P., Bernstein B.E., Jaenisch R., Lander E.S., Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazor K.L., Altun G., Lynch C., Tran H., Harness J.V., Slavin I., Garitaonandia I., Muller F.J., Wang Y.C., Boscolo F.S. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell. 2012;10:620–634. doi: 10.1016/j.stem.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J., Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4:487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Nishizawa M., Chonabayashi K., Nomura M., Tanaka A., Nakamura M., Inagaki A., Nishikawa M., Takei I., Oishi A., Tanabe K. Epigenetic variation between human induced pluripotent stem cell lines is an indicator of differentiation capacity. Cell Stem Cell. 2016;19:341–354. doi: 10.1016/j.stem.2016.06.019. [DOI] [PubMed] [Google Scholar]
- Ohnishi K., Semi K., Yamamoto T., Shimizu M., Tanaka A., Mitsunaga K., Okita K., Osafune K., Arioka Y., Maeda T. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell. 2014;156:663–677. doi: 10.1016/j.cell.2014.01.005. [DOI] [PubMed] [Google Scholar]
- Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Pasque V., Karnik R., Chronis C., Petrella P., Langerman J., Bonora G., Song J., Vanheer L., Sadhu Dimashkie A., Meissner A. X chromosome dosage influences DNA methylation dynamics during reprogramming to mouse iPSCs. Stem Cell Reports. 2018;10:1537–1550. doi: 10.1016/j.stemcr.2018.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick M., Stelzer Y., Bar-Nur O., Mayshar Y., Eden A., Benvenisty N. Clone- and gene-specific aberrations of parental imprinting in human induced pluripotent stem cells. Stem Cells. 2009;27:2686–2690. doi: 10.1002/stem.205. [DOI] [PubMed] [Google Scholar]
- Schlesinger Y., Straussman R., Keshet I., Farkash S., Hecht M., Zimmerman J., Eden E., Yakhini Z., Ben-Shushan E., Reubinoff B.E. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007;39:232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- Sharma S., Kelly T.K., Jones P.A. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z.D., Shi J., Gu H., Donaghey J., Clement K., Cacchiarelli D., Gnirke A., Michor F., Meissner A. Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature. 2017;549:543–547. doi: 10.1038/nature23891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M., Apostolou E., Akutsu H., Fukuda A., Follett P., Natesan S., Kono T., Shioda T., Hochedlinger K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010;465:175–181. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M., Apostolou E., Ferrari F., Choi J., Walsh R.M., Chen T., Ooi S.S., Kim S.Y., Bestor T.H., Shioda T. Ascorbic acid prevents loss of Dlk1-Dio3 imprinting and facilitates generation of all-iPS cell mice from terminally differentiated B cells. Nat. Genet. 2012;44:398–405. doi: 10.1038/ng.1110. S391-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M., Hochedlinger K. Induced pluripotency: history, mechanisms, and applications. Genes Dev. 2010;24:2239–2263. doi: 10.1101/gad.1963910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenman M.J., Rainier S., Dobry C.J., Grundy P., Horon I.L., Feinberg A.P. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms' tumour. Nat. Genet. 1994;7:433–439. doi: 10.1038/ng0794-433. [DOI] [PubMed] [Google Scholar]
- Takada T., Ebata T., Noguchi H., Keane T.M., Adams D.J., Narita T., Shin I.T., Fujisawa H., Toyoda A., Abe K. The ancestor of extant Japanese fancy mice contributed to the mosaic genomes of classical inbred strains. Genome Res. 2013;23:1329–1338. doi: 10.1101/gr.156497.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada T., Yoshiki A., Obata Y., Yamazaki Y., Shiroishi T. NIG_MoG: a mouse genome navigator for exploring intersubspecific genetic polymorphisms. Mamm. Genome. 2015;26:331–337. doi: 10.1007/s00335-015-9569-8. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Terada Y., Jo N., Arakawa Y., Sakakura M., Yamada Y., Ukai T., Kabata M., Mitsunaga K., Mineharu Y., Ohta S. Human pluripotent stem cell-derived tumor model uncovers the embryonic stem cell signature as a key driver in atypical teratoid/rhabdoid tumor. Cell Rep. 2019;26:2608–2621.e6. doi: 10.1016/j.celrep.2019.02.009. [DOI] [PubMed] [Google Scholar]
- Tomizawa S., Kobayashi H., Watanabe T., Andrews S., Hata K., Kelsey G., Sasaki H. Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development. 2011;138:811–820. doi: 10.1242/dev.061416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanovsky B., Bitko V., Pastukh V., Fouty B., Solodushko V. The functionality of minimal piggybac transposons in mammalian cells. Mol. Ther. Nucleic Acids. 2016;5:e369. doi: 10.1038/mtna.2016.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widschwendter M., Fiegl H., Egle D., Mueller-Holzner E., Spizzo G., Marth C., Weisenberger D.J., Campan M., Young J., Jacobs I. Epigenetic stem cell signature in cancer. Nat. Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- Yagi M., Kishigami S., Tanaka A., Semi K., Mizutani E., Wakayama S., Wakayama T., Yamamoto T., Yamada Y. Derivation of ground-state female ES cells maintaining gamete-derived DNA methylation. Nature. 2017;548:224–227. doi: 10.1038/nature23286. [DOI] [PubMed] [Google Scholar]
- Yagi M., Yamanaka S., Yamada Y. Epigenetic foundations of pluripotent stem cells that recapitulate in vivo pluripotency. Lab. Invest. 2017;97:1133–1141. doi: 10.1038/labinvest.2017.87. [DOI] [PubMed] [Google Scholar]
- Yamanaka S. Induced pluripotent stem cells: past, present, and future. Cell Stem Cell. 2012;10:678–684. doi: 10.1016/j.stem.2012.05.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.