

Abstract
The Bromodomain and Extra-Terminal domain family (BET) family of proteins are involved in the regulation of gene transcription, and their dysregulation is implicated in several diseases including cancer. BET proteins contain two tandem bromodomains (BD1 and BD2) that independently recognize acetylated-lysine residues and appear to have distinct biological roles. We compared several published co-crystal structures and found five positions near the substrate binding pocket that vary between BET bromodomains. One position located in the ZA loop has unique properties. In BRD2-4, this residue is glutamine in BD1 and lysine in BD2; in BRDT, this residue is arginine in BD1 and asparagine in BD2. Using molecular modeling, we identified differences in the water-mediated network at this position between bromodomains. Molecular dynamics simulations helped rationalize the observed bromodomain selectivity for exemplar BET inhibitors and a congeneric series of tetrahydroquinolines (THQ) that differed by a single heteroatom near the ZA channel. The 2-furan SJ830599, the most BD2-selective THQ analog, did not disrupt the water-mediated networks in either domain, but was electrostatically-repulsed by the specific arrangement of the W5 water dipole in BD1. Our work underscores the value of exploring water-mediated interactions to study ligand binding, and highlights the difficulty of optimizing polar interactions due to high desolvation penalties. Finally, we suggest further modifications to THQ-based BET inhibitors that would increase BD2-selectivity in BRD2-4, while minimizing affinity for one or both bromodomains of BRDT.
1. Introduction
Covalent post-translational modification (PTMs) of histone proteins via methylation, de-methylation, and acetylation affects a number of cellular processes including gene expression, DNA damage response, cell-cycle control, and differentiation1,2. In particular, lysine acetylation has been detected in over 1750 proteins, many of which are components of large macromolecular complexes that drive chromatin remodeling or maintenance3. Histone lysine acetylation is often a hallmark of transcriptionally active genes4,5, and deregulation of histone acetylation patterns can drive the aberrant expression of oncogenes. Bromodomains are evolutionary conserved protein interaction modules that exclusively recognize acetylation motifs6. A total of 61 bromodomains have been identified in the human genome, and these can be classified into eight families based on similarities in sequence and three-dimensional structure.
Members of the Bromodomain and Extra-Terminal domain family (BET) family of proteins (BRD2, BRD3, BRD4, and BRDT) contain two tandem bromodomains (the N-terminal, BD1, and C-terminal, BD2) that independently recognize acetylated-lysine (KAc) residues in nucleosomal histone tails, facilitating the recruitment of transcriptional machinery. Because BETs are involved in the regulation of many genes, the therapeutic potential of BET inhibitors (BETi) is likely to span a wide range of therapeutic indications7-10. While three members of this family (BRD2, BRD3, and BRD4) are universally expressed, the fourth member, BRDT/BRD6, is expressed only in male germ cells and appears to be critical for spermatogenesis11. Disruption of BRDT may offer therapeutic benefit as a male contraceptive; however, this reproductive effect may be unwanted or deleterious in certain patient populations such as developing children.
Baud et al used a ‘bump-and-whole’ chemical genetic approach to show that inhibition of BRD4-BD1, but not BD2, was sufficient to dislodge BRD4 protein from chromatin12. Other work has shown that knockout of BRDT-BD1 alone was sufficient to impair male germ cell differentiation13. Isothermal titration calorimetry studies of BRD4-BD1 and BRD4-BD2 bound to various acetylated lysine sequences revealed distinct substrate specificities: BD1 preferred histone H3 sequences, whereas BD2 had higher affinity for histone H4 sequences and acetylated lysine peptides derived from cyclin T1, a member of the P-TEFb complex that stimulates transcriptional elongation by RNA polymerase II14. Indeed, comparative analysis of the two bromodomains of BRD4 indicate that, although both share similar acetylated lysine binding pockets, the presence of more basic and less acidic residues in the ZA loop of BD1 leads to differences in the electrostatic surface potentials between bromodomains that could contribute to selective recognition of acetylated lysine motifs based on their extended sequence composition14. Furthermore, higher order structure may also contribute to the differential binding of acetylated lysine peptides. BRD2-BD1, for example, has been shown to dimerize in a manner that creates a binding pocket for lysine at the dimer interface which increases selectivity for histone H4 tails that are acetylated at lysine 12 and hypo-acetylated at lysine 815. In summary, BD1 and BD2 bromodomains appear to have distinct biological roles, and this supports the rationale to pursue BET bromodomain-selective inhibitors.
Most BETi reported thus far show excellent selectivity vs. other bromodomain families, but are either pan-BET active with little selectivity between BD1 and BD2, such as (+)-JQ116 (1) (Figure 1A), or their BD selectivity has not yet been reported. Notable exceptions include RVX-208 (2), a quinazolinone based BET inhibitor, reported to show 15–80-fold selectivity for BD2 over BD117; GSK1324726A (3), a tetrahydroquinoline (THQ) reported to show 5-8 fold selectivity for BD28; olinone, a tetrahydro-pyrido-indol reported to show moderate affinity for the BD1 domains of BRD2, 3 and 4, (Kd ~3.4 μM activity on BD1 according to ITC while exhibiting no binding to BD2)18; and MS436, a diazobenzene derivative, reported to potently inhibit BRD4-BD1 (Ki=30-50 nM) over BRD4-BD2 (Ki = 340 nM)19.
Figure 1. Exemplar BETi and comparative analysis of BET bromodomains.

(A) Exemplar BETi explored in this study. (B) Sequence alignment of BET BD1 and BD2 domains. Strictly conserved residues are highlighted in yellow. Conserved binding pocket residues are annotated with * and variable residues are annotated with #. (C) Structural model of BRD2-BD1 highlighting strictly conserved substrate binding pocket residues and other structural landmarks. A co-crystal pose of the 1 is shown for reference. (D), Similar to (C), but highlighting variable residues near the substrate binding pocket.
The three-dimensional structures of BET bromodomains share a conserved left-handed helix-bundle containing four α helices, termed αZ, αA, αB, and αC6,20,21. The first two alpha helices, αZ and αA, are connected by a long ZA loop and the latter two, αB and αC, are connected by a short BC loop. The ZA and BC loops flank the KAc recognition site and are the most variable regions between BET bromodomains, consistent with their role in discriminating KAc in the context of different substrate peptide sequences. A portion of the ZA loop forms a valley called the ‘ZA channel’, whereas elsewhere, residues from both the ZA and BC loops form a hydrophobic pocket called the tryptophan-proline-phenylalanine (WPF) shelf22 (Figures 1B-C). The KAc-binding pocket, ZA and BC loops, and WPF shelf make significant contributions to the affinity and selectivity of BETi. In co-crystal structures of BRD2-BD2 and BRD4-BD1 bound to 2, H433 on the BC loop of BRD2-BD2 makes favorable ligand interactions, whereas its counterpart in BRD4-BD1, D144, is detrimental to binding17. Compound 3 makes strong hydrophobic interactions with histidine and tryptophan to achieve BD2 selectivity8. In contrast, BD1-selective olinone makes favorable polar interactions with D144 and sterically clashes with H437 of BRD4-BD218. MS436 appears to take advantage of interactions with the ZA channel and BC loop to achieve BD1-selectivity19. Together, these findings indicate that subtle sequence variations in the BC and ZA loops can be exploited to achieve BD1- or BD2-selective BETi.
In this study, we analyzed the variable binding pocket residues of BET proteins by comparing several available co-crystal structures, and identified one putative hotspot residue (V4) in the ZA loop with unique properties. In BRD2-4, this residue is glutamine in BD1 and lysine in BD2; in BRDT, this residue is arginine in BD1 and asparagine in BD2. The physiochemical difference in residues at this position suggests the potential to design both bromodomain- and BET- selective inhibitors. For example, glutamine presents both a hydrogen bond acceptor and donor, whereas lysine presents only hydrogen bond donors and is positively charged. In BRDT, these differences are reversed in BD1 and BD2, though arginine and asparagine are less flexible and may be unable to form the same type of interactions. We used molecular dynamics (MD) simulations with several reported chemotypes bound to both bromodomains to interrogate the behavior of this region. Our simulations identified a water-mediated interaction network between protein and ligand molecules. We then applied this analysis to a series of congeneric THQ analogs that varied at the ZA channel to rationalize their observed bromodomain selectivity. Our work indicates that the V4 position can be exploited for bromodomain selectivity through water-mediated interactions, and suggests further modifications to increase BD2-selectivity while minimizing affinity for one or both bromodomains of BRDT.
2. Results
2.1. Comparative analysis of BD1 and BD2 BET bromodomains
We compared structures from 81 BD1 and 15 BD2 BET bromodomains (Table 1). While a high percentage of sequence identity (65 to 90%) was found across BET-BD1s, and across BET-BD2s, only 35 to 45% sequence identity was observed between BD1 and BD2 domains. However, the substrate binding pockets exhibited 100% sequence conservation in all eight BET-BDs (highlighted in Figure 1B). The carbonyl of the acetyl group on KAc (or the corresponding moiety on BETi) interacted directly with a conserved asparagine (N156) and tyrosine (Y113) residue through direct and water-mediated interactions14. Four highly conserved water molecules formed the base of the binding pocket, and were vital for proper active site architecture. Most BETi made hydrophobic interactions with the WPF shelf (W97, P98, and F99) and two leucine residues, L108 and L110 (residue numbering as defined in BRD2-BD1) (Figure 1C).
Table 1.
Experimental PDB structures used in comparative structural analysis.
| BRD2-BD1 | 2YDW, 2YEK, 1X0J, 4A9E, 4A9F, 4A9H, 4A9I, 4A9J, 4A9M, 4A9N, 4A9O, 4AKN, 4ALG, 4ALH, 4UYF, 4UYH |
| BRD2-BD2 | 3ONI, 2DVV, 2E3K, 4MR5, 4MR6, 4J1P, 4QEU, 4QEW, 4UYG, 5BT5, 4QEV |
| BRD3-BD1 | 3S91 |
| BRD3-BD2 | 3S92 |
| BRD4-BD1 | 3MXF, 4BJX, 2YEL, 3P50, 3SVF, 3SVG, 3U5J, 3U5K, 3ZYU, 4A9L, 4BW1, 4BW2, 4BW3, 4BW4, 4C66, 4C67, 4DON, 4F3I, 4HXL, 4HXM, 4HXR, 4HXS, 4LRG, 4MEN, 4MEO, 4CL9, 4CLB, 4NUC, 4NUD, 4NUE, 4O7A, 4O7B, 4O7C, 4O7E, 4O7F, 4O70, 4O71, 4O72, 4O74, 4O75, 4O76, 4O77, 4O78, 4PCE, 4PCI, 4PS5, 4QB3, 4QR3, 4QR4, 4QR5, 4QZS, 4UIX, 4UIY, 4UIZ, 4UYD, 4WIV, 4XY9, 4XYA, 4ZIQ, 4Z1S, 5A85, 5BT4 |
| BRD4-BD2 | 2YEM, 4Z93, |
| BRDT-BD1 | 4FLP, 4KCX |
| BRDT-BD2 | 2WP1 |
Structural alignment followed by inspection of residues immediately adjacent to the substrate binding pocket identified five variable positions between BD1 and BD2: two were present on the BC-loop and three were located in the ZA-loop region (Figure 1D, ‘V1-V5’). A gatekeeper-like residue at the end of BC loop was conserved as isoleucine in all BD1 and valine in all BD2 (V1). Most of the BETi co-crystal structures in our study showed similar interaction patterns with this region, although we speculate that the one carbon difference between isoleucine and valine could be exploited better by introducing subtle steric clashes. The second variant in the BC loop was an aspartate residue in BD1 and a histidine in BD2 (V2). As noted earlier, this region has been successfully exploited by several BETi to achieve bromodomain selectivity. On the ZA loop, a lysine residue in BD1 was replaced by alanine in BD2 (V3). The diazobenzene-based BETi appear to take advantage of this position to achieve BD1 selectivity19. Position V5 was unique in that it does not vary between bromodomains in BRD3, and was different in BRD2 vs. BRD4/BRDT. This might be an interesting region to explore for selectivity between BET proteins; however, it is the farthest away from the peptide substrate binding pocket and may be difficult to exploit without substantially increasing inhibitor size. Interestingly, position V4 in the ZA channel had a glutamine in BRD2-4 BD1, a lysine in BRD2-4 BD2, an arginine in BRDT-BD1, and an asparagine in BRDT-BD2. In BRD2-4 BD1, the glutamine side chain adopted either an ‘in’ or ‘out’ conformation (Figure 2A). In all BRD2-4 BD2s examined, the lysine side chain was in a solvent-exposed, ‘out’ conformation (Figure 2B). In the two BRDT-BD1 structures examined, the arginine adopted a solvent-exposed ‘out’ conformation; in the one BRDT-BD2 in our study, the asparagine also adopted an ‘out’ conformation (Figures 2C-D).
Figure 2. Structural variation at V4 in the ZA channel.

(A) BD1 glutamine can adopt an ‘in’ (cyan and green) or an ‘out’ conformation (magenta). (B) BD2 lysine adopts an ‘out’ orientation in all structures examined in this study. (C) In BRDT, the residues types are swapped between BD1 and BD2. Positively-charged arginine in BDRT-BD1 adopts an ‘out’ conformation, and (D) neutral amide asparagine in BRDT-BD2 also adopts an ‘out’ conformation. The four conserved bound waters present in all BET structures are depicted as red spheres, and 1 (yellow sticks) is shown for reference. Cluster analysis of bound waters indicates the presence of W5 in BD1 (E) and W5 and W6 in BD2 (F).
We aligned available crystal structures of both bromodomains to identify differences in water-mediated protein-ligand interactions near the BET substrate pocket and ZA channel (Figures 2E-F). As expected, four water molecules (W1 to W4) were conserved in all structures and formed well-defined hydrogen bond networks in BD1 and BD2. For example, W4 bridged the main chain carbonyl of Q101 (BD1)/K374 (BD2) and the main chain carbonyl of P98 (BD1)/P371 (BD2) in all structures. However, our analysis identified two more water clusters – W5 and W6. W5 was present in structures of both bromodomains and formed a hydrogen bond with the backbone carbonyl of P102 (BD1) or P375 (BD2). When Q101 adopts the ‘in’ conformation, the amide side chain acts as a hydrogen bond donor to W5 and the backbone carbonyl of W97. In BD2s, the corresponding residue, K374, adopted a solvent exposed ‘out’ conformation. Here, W5 usually picked up additional hydrogen bonds with neighboring water molecules. W6 was found primarily in BD2 structures and appeared to occupy the void created near W370 when the side chain of K374 adopts the ‘out’ conformation. W6 hydrogen bonded to the backbone amide of K374, and interestingly, appeared to mimic the role of the ‘in’ orientation of the Q101 side chain by forming a hydrogen bond with the backbone carbonyl of W370.
In BRDT, the residue type is switched at V4: BD1 has a positively-charged arginine and BD2 has a neutral amide. Moreover, asparagine is shorter than glutamine by one atom and is not long enough to hydrogen bond with W97 in the ‘in’ orientation. Thus, variation in the physicochemical characteristics of the residues and the water-mediated interactions present at V4 suggests the potential to design inhibitors that selectively target BD1 or BD2 in BRD2-4, while potentially reducing affinity to one or both BRDT bromodomains.
2.2. ZA channel interaction patterns of three exemplar BETi
To study how existing BETi interact with the ZA channel, we performed ligand interaction analysis (LIA) using 20ns MD simulations with three exemplar BETi: 1, 2 and the dual PLK/BETi BI-2536 (4)23-25. In addition to quantifying the propensity to form direct protein-ligand interactions such as hydrogen bonds, ionic bridges, hydrophobic contacts, pi-pi, and pi-cation effects, LIA also quantifies the extent of water bridges – hydrogen-bonded protein-ligand interactions mediated by a water molecule.
The majority of interactions for 1 were observed near the KAc binding pocket and the BC loop region for both bromodomains (Figures 3A-B). Conserved W1 made a water-mediated interaction between Y113 (BD1)/Y386 (BD2) and one of the nitrogen atoms on the ligand triazole. N156 (BD1)/N429 (BD2) directly interacted with the adjacent triazole nitrogen atom, and was observed to form water-mediate interactions with the ester group of 1. In addition to hydrophobic contacts, L108/L381 formed water-mediated interactions with the ligand ester carbonyl, though at low occupancy. At BD1 V2, the D160 side chain formed a water-mediated hydrogen bond with the ester carbonyl. In BD2, the corresponding residue, H433, formed a similar water-mediated interaction at lower occupancy and a direct hydrogen bond to the epsilon nitrogen of H433. Interestingly, several BRD2-BD2 co-crystal structures with 1 and analogs (PDB IDs: 4QEW and 4QEV) revealed that the ester carbonyl orientation was crucial for directing the face of H433 towards the tryptophan from the WPF shelf. When abutting the WPF shelf, a hydrophobic pocket is formed between the histidine and tryptophan residues and this has been exploited by several reported BETi. Our simulations showed that, in the absence of such a directing hydrogen bond interaction, the side chain fluctuated between closed and open conformations. Notably, 1 made no direct or water-mediated interactions with the ZA channel.
Figure 3. Interaction patterns of exemplar BETi bound to BRD2-BD1 and BRD2-BD2.
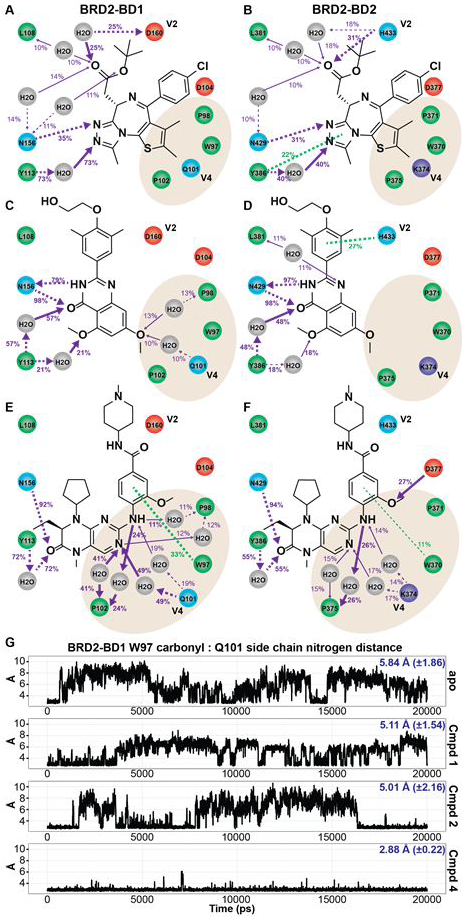
Ligand interaction analysis based on MD simulations of 1 (A,B), 2 (C,D), and 4 (E,F). Polar, hydrophobic, and charged residues are blue, green, and red circles, respectively. Purple and green arrows represent polar and pi-stacking interactions, respectively. Atoms receiving arrows are hydrogen bond acceptors. Solid and dotted lines indicate interactions with backbone and side chain, respectively. Percent values report the occupancy of the interaction, with 100% meaning the interaction was present in every snapshot of the MD simulation. Occupancies ≤10% and non-polar interactions are suppressed in the figures for clarity. For emphasis, arrow widths are larger for interactions with occupancy >20%. (G) Trajectory analysis of the distance between the backbone carbonyl of W97 and the Q101 side chain nitrogen in BRD2-BD1 MD simulations of the apo structure and exemplar BETi. Average distance and standard deviation reported in blue.
Like 1, 2 also formed water-mediated interactions with conserved W1 (Figures 3C-D). N156/N429 acted as both a hydrogen bond donor and acceptor in a bi-dentate interaction with the quinazolin-4(3H)-one scaffold. The stacking interaction between the phenyl group of the ligand and the V2 histidine imidazole ring was specific to BD2 and could account for the selectivity of this compound. In both bromodomains, 2 formed water-mediated interactions with ZA channel residues Q101/K374 and P98/P371, albeit with low occupancy (interactions were at single-digit occupancy in BD2 and were suppressed from the figure for clarity).
In contrast to 1 and 2, 4 did not substantially interact with V2 in the BC loop, and instead made significant direct and water-mediated interactions with the ZA channel (Figures 3E-F). In fact, several kinase inhibitors may take advantage of water-mediated interactions in this region25. In BD1, the pyrimidine and amine nitrogen atoms on 4 made water-mediated interactions with Q101 and P102 at 49% and 41% occupancy; in BD2, the pyrimidine interactions with K374 and P375 were less prevalent at 17% and 15%, respectively. LIA reports average behavior from the MD simulation and does not distinguish between individual water molecules. Detailed inspection of available crystal structures of 4 bound to BD1 (PDB IDs: 4OGI and 4O74) indicated that in all structures, Q101 adopted an ‘in’ conformation, whereby the Q101 amide side chain and ligand pyrimidine nitrogen form hydrogen bonds with W5 (Figure S1). This orientation pulled the water away from the backbone carbonyl of P102. Thus, rather than stabilizing the W5 orientation described in Figure 2E, 4 re-oriented the water-mediated network to accommodate the hydrogen bond requirements of the pyrimidine nitrogen. While the extent of the water-mediated interactions for 4 was less in BD2, the net effect of the re-orientation of the water-mediated network in BD1 may be energetically neutral or even unfavorable. Indeed, BD1-selectivity for 4 was <2-fold as assessed by thermal shift23.
Figure 4. Analysis of water-mediated interactions in the ZA channel for 3 bound to BRD2-BD1 and BRD2-BD2.

(A) Representative water-mediated interactions from different chains in BRD2-BD1 (4UYF) and BRD2-BD2 (4UYG). Hydrogen bonds are depicted as dotted lines. (B) Trajectory analysis of the distance between the backbone carbonyl of W97 and the Q101 side chain nitrogen following MD simulations of chains A,B, and C from 4UYF. Average distance and standard deviation reported in blue. Ligand interaction analysis of 3 bound to (C) BRD2-BD1 and (D) BRD2-BD2. Legend is identical to that reported in Figure 3.
To further explore how BETi interact with Q101 in the BD1 ZA channel, we plotted the distance between the amine nitrogen on the Q101 side chain and the backbone carbonyl from W97 as a function of time in the MD simulation (Figure 3G). This metric oscillated between 2.9Å to 8Å in the apo, 1, and 2 simulations, consistent with a lack of significant interaction with the ZA channel. In contrast, the metric rarely deviated from 2.9Å when 4 was bound, indicative of strong stabilization of the Q101 ‘in’ conformation. However, as noted in the analysis above, stabilization of the ‘in’ conformation does not necessarily imply favorable energetic interaction with W5, and consequently, may not result in BD1-selectivity.
2.3. THQs induce distinct water-mediated networks in the ZA channel of BD1 and BD2
Given our observations on the potential to achieve bromodomain selectivity through water-mediated interactions in the ZA channel, we sought to identify a chemically-tractable scaffold to rationally design inhibitors that could be selective for either bromodomain. The tetrahydroisoquinoline (THQ) scaffold in 3 appeared to be ideal for such an experiment. Co-crystal structures of 3 bound to BRD2-BD1 (PDB ID: 4UYF) and BRD2-BD2 (PDB ID: 4UYG) have been reported8. We performed protonation and hydrogen bond network optimization on each chain present in those structures in order to better examine water-mediated protein-ligand interactions (Figure 4A).
Q101 adopted three distinct conformations in the 3 BRD2-BD1 co-crystal structure. In chain A, Q101 was solvent exposed and a water molecule was recruited to the W6 position to form a hydrogen bond with the backbone carbonyl of W97 (orange circle). An additional water molecule was observed to occupy the position of the Q101 amide carbonyl when oriented in the ‘in’ conformation (purple circle). W5 hydrogen bonded to the backbone carbonyl of P102, as expected, and also formed a hydrogen bond to a water that has been recruited by the side chain amine of K107. In chain B, Q101 adopted the ‘in’ conformation, trapping W5 in a hydrogen-bond network with the backbone carbonyl of P102 and a bound water that was also stabilized by the backbone amine of V103. In chain C, Q101 adopted a conformation that is intermediate to that observed in chain A and chain B: the side chain amine swings out and was solvent exposed, while the side chain carbonyl was pointed inward and forms a hydrogen bond with W6. While W6 still formed a hydrogen bond with the backbone carbonyl of W97, the water molecule was re-oriented and forms a hydrogen bond with W5. This in turn breaks W5’s interaction with the backbone carbonyl of P102, but enabled it to maintain its interaction with the water bound to the backbone amide of V103.
In contrast to the variable water-mediated networks observed in BD1, these interactions in BD2 appeared to be more constant. W5 and W6 were present in most chains. W6 formed hydrogen bonds with the backbone amide of K374 and the backbone carbonyl of W370 (orange circle). W5 formed hydrogen bonds with either the backbone carbonyl of P375 or K374, and often picked up an additional interaction with a water bound to the backbone amine of V376. In reality, the interactions reported in Figure 4A reflect snapshots of hydrogen-bond networks that are dynamic. Nevertheless, it is apparent that the water-mediated interactions observed in the Q101 ‘in’ orientation were significantly different from those present in BD2. If the ‘in’ orientation reflects the preferred conformation of the side chain when a THQ scaffold is bound, then it is conceivable that differences in the hydrogen-bond network at this location could be exploited to achieve bromodomain selectivity.
To investigate this hypothesis, we performed a series of molecular dynamics simulations to probe the energetics of the Q101 side chain in the presence of 3 (Figure 4B). Chain A began with Q101 extended completely into solvent with the Q101 side chain nitrogen – W97 carbonyl distance metric averaging approximately 6 Å. However, within 5 ns, the complex converged to a local minimum with a distance metric of around 4 Å. Inspection of the simulation trajectory indicated that the Q101 has swung inward towards ligand but appeared to be unable to overcome a local energy barrier to achieve the ‘in’ orientation. We subjected the initial structure to accelerated MD (‘aMD’) – a modeling technique that enables more efficient conformational sampling by lowering energy barriers26. This simulation quickly converged to the ‘in’ conformation in the first hundred picoseconds, and remained in that conformation before the entire complex dissociated at around 10 ns. Chain B, already in the ‘in’ conformation, remained in that orientation throughout the length of the simulation. Two additional independent simulations showed identical behavior. Chain C began with Q101 in an intermediate state between ‘in’ and ‘out’: however, within 2-3 ns, the structure converged to the ‘in’ conformation. Two additional independent simulations showed similar convergence to the ‘in’ orientation within 2-18 ns. These experiments strongly suggest that BRD2-BD1 prefers Q101 in the ‘in’ conformation when bound to the THQ scaffold of 3.
Next, we performed LIA on the co-crystal structures of 3 bound to BD1 and BD2 (Figures 4C-D). The ligand made no water-mediated interactions with the ZA channel in either bromodomain. K107 (BD1) and K374 (BD2) made ionic interactions with the carboxylic acid moiety. As mentioned earlier, the hydrophobic interaction between the benzoic acid moiety on 3 and the pocket formed by H433 and the WPF shelf appears to drive BD2-selectivity for this compound – a mechanism used by other BETi to achieve bromodomain selectivity.
Finally, we swapped the glutamine and lysine residues at V4 in BRD2-BD1 and BRD2-BD2, respectively, to explore the influence of that residue on W5 and W6 water occupancy (Table S2). W5 occupancy was defined as the percent of MD snapshots where the carbonyl from P102 (BD1) or P375 (BD2) acted as a hydrogen bond acceptor to water. W6 occupancy was defined similarly for the carbonyl of W97 (BD1) or W370 (BD2). The point mutations did not destabilize either bromodomain: average root mean squared fluctuation per residue across all simulations was in the range 0.70Å to 1.06Å. In all simulations, W5 remained close to full occupancy (86% to 139%), consistent with our earlier observation that W5 is present in both bromodomains. In apo-BD1, W6 occupancy was 67% compared to 26% when 3 was bound, and this is consistent with our observation that the Q101 side chain is mobile in the absence of ligand and consequently, the V4 site is more accessible to solvent. However, when 3 is bound to the Q101K BD1 mutant, W6 occupancy was 88% and 89% (two independent simulations) – a significant increase compared to the wild-type structure with 3 bound that supports our earlier finding that the BD2 lysine was solvent exposed and this increased solvent accessibility at V4. In contrast, the K374Q BD2 mutant had decreased W6 occupancy, 23% and 29%, compared to 54% observed in the wild-type BD2 structure with 3 bound. Moreover, the average distance between the W370 carbonyl and the side chain amide nitrogen of the K374Q mutant was 2.90Å and 2.93Å, compared to 2.88Å for the analogous distance in wild-type BD1. In summary, these point mutation experiments suggest that the water-mediated network at the V4 position in BD1 and BD2 is largely dependent on a single residue – Q101 in BD1 and K374 in BD2.
2.4. Exploiting water-mediated interactions in the ZA channel for bromodomain selectivity
Our analysis suggests that while 3 stabilizes the ‘in’ conformation in BRD2-BD1, the ligand presents no hydrogen bond donors or acceptors to interact with W5, and instead appears to have little interaction with the ZA channel. We hypothesized that replacement of the benzoic acid substituent on 3 with an appropriately-oriented hetero-aromatic group could either increase BD1-selectivity by strengthening interactions with W5 and Q101, or increase BD2-selectivity by disrupting the BD1 W5 water-mediated network and/or picking up an additional water mediated interaction with BD2 W6, which does not exist in BD1 when Q101 is ‘in’.
To explore this hypothesis, we applied our modeling framework to three hetero-aromatic substituted THQs: 2-furan SJ830599 (5), 2-thiophene SJ830629 (6), and 2-pyrrole SJ852844 (7)27. These molecules differed by a single atom that was expected to be within close proximity to the W5 and W6 waters in the ZA channel, and therefore, enabled study of the role of water-mediated interactions at that position. Compound 5 showed modest BD2-selectivity in a TR-FRET assay (3.3-fold selective), compared to 6 (2.1-fold selective) and 7 (equipotent) (Table 2). Docking models of the three analogs bound to BRD2-BD1 and BRD2-BRD2 confirmed that the variable heteroatom position is oriented toward W5 in BD1 and W5 and W6 in BD2 (Figures 5A-C). Interestingly, the docked pose of 5 in BD1 was displaced away from the ZA channel relative to the pose in BD2 (Figures 5D-E), presumably because of the electrostatic clash between the lone pairs on W5 and the lone pairs on the furan oxygen. In BD2, the lone pairs on W5 and W6 were directed away from the furan oxygen. In contrast, 7 appeared to make stable hydrogen bonds to waters in both bromodomains. Consequently, docking predicted 5 to be the most BD2 selective (2.1-fold) compared to 1.5- and 1.4-fold selectivity for 6 and 7, respectively (Table 2).
Table 2.
TR-FRET and Docking affinities for THQ analogs bound to BRD2 bromodomains
| Model | BD1 pIC50 |
BD2 pIC50 |
TR- FRET BD2/BD1 |
Dock BD1 pKd |
Dock BD2 pKd |
Dock BD2/BD1 |
|---|---|---|---|---|---|---|
| 5 | 5.69 | 6.21 | 3.3 | 7.24 | 7.56 | 2.1 |
| 6 | 5.75 | 6.08 | 2.1 | 7.46 | 7.65 | 1.5 |
| 7 | 5.85 | 5.85 | 1.0 | 7.40 | 7.56 | 1.4 |
Figure 5. Docking studies of the THQ analogs in BRD2-BD1 and BRD2-BD2.

Alignment of BD1 and BD2 docked poses of (A) 2-furan 5, (B) 2-pyrole 6, and (C) 2-thiophene 7. W5 and W6 waters (blue) and key residues are shown for reference. (D-E) Same as (A), but with 3 superimposed for reference (yellow sticks) for (D) BRD2-BD1 and (E) BRD2-BD2.
Though estimated binding affinities obtained from docking correctly rank ordered the THQ analogs according to BD2-selectivity, the magnitude of the selectivity was less while the affinity for either domain was more than observed in the TR-FRET assay (Table 2). We applied LIA on the docked poses of the three THQ analogs to better understand water-mediated interactions. The 2-furan oxygen atom is a weak hydrogen bond acceptor, and a modest degree of water-mediated hydrogen bond formation was observed in these MD simulations for 5 with W5 and P102 in BD1 and P375 in BD2 (Figures 6A-B). Compound 6 behaved similarly to the benzoic acid substitution on 3 and did not interact with the ZA channel (Figures 6C-D). Both 5 and 6 stabilized the ‘in’ conformation of Q101 in BRD2-BD1 (Figure 6G). In contrast, 7 directed a strong hydrogen bond donor into the ZA channel, and consequently, significantly disrupted the water-mediated network at that position (Figures 6E-F). Indeed, we observed that the stable ‘in’ conformation of Q101 was lost after 10 ns of simulation (Figure 6G) when the pyrrole nitrogen induced a partial dislocation of the Q101 side chain to compete for water binding and eventually re-organized the water-mediated network at that site. Strong hydrogen bond donors and acceptors also have strong desolvation penalties, and these groups can negatively impact binding affinity unless they are reasonably satisfied with new bonds to the target. Thus, our models suggested that the lack of bromodomain selectivity for 7 was similar to the rationale applied to 4: by disrupting the existing water-mediated network to form strong interaction with bound waters in the ZA channel, the net contribution to the free energy of binding may be zero or negative if geometries are less than ideal and the desolvation penalty is not repaid. On the other hand, the 2-furan 5 appeared to preserve the water-mediated networks in both domains, and differential electrostatic interaction with water (repulsive in BD1 and/or neutral or favorable in BD2) accounted for the modest BD2 selectivity of 5.
Figure 6. Analysis of water-mediated interactions in the ZA channel for THQ analogs targeting V4 in the ZA channel.
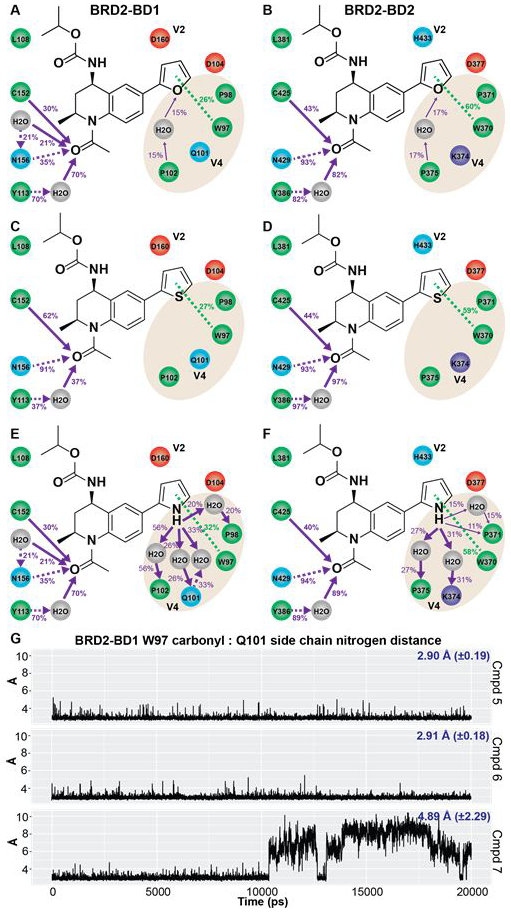
Ligand interaction analysis of 2-furan 5 (A,B), 2-thiophene 6 (C,D), and 2-pyrrole 7 (E,F) docked to the co-crystal structures of 3 bound to BRD2-BD1 and BRD2-BD2. Legend is identical to that reported in Figure 3. (G) Trajectory analysis of the distance between the backbone carbonyl of W97 and the Q101 side chain nitrogen in BRD2-BD1 MD simulations for each THQ analog. Average distance and standard deviation reported in blue.
3. Discussion
Though several potent BETi have been reported in recent years, few exhibit both high potency and selectively between the N- and C-terminal BD1 and BD2 bromodomains. This lack of selectivity has hindered study of the specific biological role of each bromodomain, and may account for some of the off-target effects and/or safety concerns observed from this class of molecules. Indeed, the male contraceptive effect associated with BETi therapy are likely the result of inhibition of testes-specific BRDT.
Comparative analysis of BET bromodomains using sequence and structural alignment identified five variable regions. Of the two variable regions in the BC-loop, the aspartate in BD1 and histidine in BD2 at V2 have been exploited by reported BETi to achieve bromodomain selectivity. In the ZA channel, we focused on V4 because it differentiated BD1 and BD2 among BRD2-4, while also offering the possibility to reduce affinity to one or both bromodomains of BRDT. Small molecules which access the ZA channel can induce an ‘in’ orientation of Q101 in BD1: this induces a water-mediated network that is different from that observed in BD2.
We studied a series of THQ analogs that differed in bromodomain selectivity and were predicted to differentially engage the water-mediated network in the ZA channel. Docking and molecular dynamics suggested that the 2-furan 5, the most BD2-selective analog, did not disrupt the water-mediated networks in either domain, but was electrostatically-repulsed by the specific arrangement of the W5 water dipole in BD1. On the other hand, though docking suggested that the 2-pyrrole 7 made favorable interactions with bound water in both domains, molecular dynamics indicated significant perturbation of both networks that essentially cancelled out any benefit, and this finding could explain the observed lack of bromodomain selectivity. While the magnitude of selectivity is modest and does not meet current guidelines for a selective chemical probe28, 5 supports the hypothesis that exploiting a specific water-mediated interaction in the ZA channel can lead to bromodomain selectivity.
Moreover, our work suggests weak hydrogen bond donors and acceptors may be useful for taking advantage of water mediated interactions compared to functional groups which interact more strongly, simply because the latter may be more likely to re-arrange, as opposed to stabilize, the water-mediated network. Indeed, improving binding affinity by increasing enthalpic contributions via hydrogen bonds is often difficult to achieve not only because such interactions are subject to precise geometric constraints, but also because the penalty associated with the desolvation of polar groups is often high29. For example, it was recently shown in a series of thrombin inhibitors that increasing the basicity of a key hydrogen bond acceptor on a ligand paradoxically led to reduced binding affinity due to an increase in the desolvation penalty30. Therefore, a more sophisticated treatment of water, such as the use of molecular dynamics simulation with explicit water molecules, is critical to understanding the determinants of ligand-protein binding.
Finally, while the aspartate/histidine variance is present in the BD2 domains of all BET proteins, the V4 position is different in BRDT compared to BRD2-4. Simultaneously targeting the V2 and V4 positions could increase BD2 potency in BRD2-4, while reducing potency on both bromodomains of BRDT. Thus, our work provides a road map to develop inhibitors that retain potency on BRD2-4, the bromodomains implicated in a number of diseases, while limiting the male contraceptive effect associated with BRDT inhibition.
4. Experimental Methods
Ligand preparation for docking simulations
The 2D structures of docked compounds (5-7) were prepared using the Ligprep31 module in Schrodinger suite31. Ligprep accepts molecules in 2D format, and converts them to 3D. The Epik sub-module of Ligprep was used to generate tautomers and possible ionization states for each molecule. Each ligand was manually inspected to ensure that the correct tautomer state and ionization state at physiologically-relevant pH (7.4) was present. Low energy ring conformations were generated by utilizing the ring_conf sub-module. Output structures from Ligprep were considered as input for multiple conformation generation using the MacroModel-MTLM (mixed torsional/low-mode) method32. MacroModel-MTLM combines a Monte Carlo method of exploring torsional space (that efficiently locates widely separated minima on a potential energy surface) with a low-mode conformational search method that searches along energetically “soft” degrees of freedom. The OPLS_2005 force field was used, and energy minimization was performed for 500 steps using the TNCG method. The energy window for acceptable structures was set to 21 kJ/mol (default value). Conformations of the same molecule within 0.5 Å RMSD were culled. A maximum of 500 steps was allowed for Monte Carlo sampling, and a maximum of 50 steps was allowed for low-mode searching. A maximum of 25 conformers per molecule were accepted.
Molecular docking
The Glide v5.533-34 molecular docking module (Schrodinger, 2012) in SP-mode was used to generate unbiased binding modes for THQ analogs 5-7 in the BRD2-BD1 and BRD2-BD2 bromodomains. GW841819X bound to the BRD2-BD1 structure (PDB ID: 2YDW, resolution: 1.9 Å) and 1 bound to BRD2-BD2 (PDB ID: 3ONI, resolution: 1.61 Å) were processed initially using the Protein Preparation Wizard in the Maestro suite from Schrodinger to add the correct protonation state and bond orders to hetero atoms of protein residues, water molecules, and the bound ligands. These processed complexes were used to generate the pre-computed grid. Four conserved water molecules present in both crystal structures were also included in this step. The docking protocol started with systematic conformational expansion of the ligand, followed by placement in the receptor site. Minimization of the ligand in the field of the receptor was then carried out using the OPLS-AA force field with the default distance-dependent dielectric. The lowest energy poses were then subjected to a Monte Carlo procedure that sampled nearby torsional minima. Posed were ranked using GlideScore, a modified version of the ChemScore function that includes terms for steric clashes and buried polar groups. The default van der Waal’s scaling was used (1.0 for the receptor and 0.8 for the ligand). Next, the docked poses for THQ analogs 5-7 generated above were further refined by aligning to their respective bromodomain using the crystal structures of 3 bound to BRD2-BD1 (PDB ID: 4UYF) and BRD2-BD2 (PDB ID: 4UYG). All crystallographic waters were preserved and hydrogen-bond networks were optimized as described below (see section “Hydrogen-bond network analysis). The ligand pose was refined using the ‘Dock’ module in the MOE application (v2016.08, Chemical Computing Group) as follows: ‘Placement’ was set to ‘None’ and ‘Refinement’ was set to ‘Induced Fit’ using the ‘GBVI/WSA dG’ scoring function35 and default parameters.
Molecular Dynamics simulations
Initial binding orientations of bound ligands were obtained from co-crystal structures with BET family members. When no co-crystal structures were available, the superimposition method or docking was used to extract ligand conformation. Details on initial complex structures used for simulations are shown in Table S1. Force field parameters for ligands were created with the Antechamber program36 from the Amber10 package using the General Amber Force Field (GAFF) and AM1-BCC37-38 partial charges. All molecular dynamics simulations were performed using the Amber12 molecular dynamics package39 and the Amber99SB40 force field. Energy minimization and MD was carried out with PMEMD MD. A total of four minimization steps were executed prior to the heating step. For minimization, restraints employed a harmonic force constant of 100.0 kcal/(mol•Å2). First, each ligand molecule was minimized with restraints applied to all protein heavy atoms. Next, the total system (protein and ligand) was minimized with no restraints. Each minimized system was then inserted in a water box of TIP3P water, which extended at least 10 Å away from any given protein atom, and neutralized by adding counter ions. Solvent molecules were then minimized while restraining all protein and ligand heavy atoms. Finally all restraints were removed and the total system (protein, ligand, and solvent) was minimized. Each energy minimization procedure used the steepest descent method for the first 3000 steps and then conjugated gradient method for the subsequent 2000 steps. After energy minimization, the system was slowly heated from 0 to 300.0 K over 100 ps in the NPT ensemble under 1 atm pressure to equilibrate the solvent. A harmonic restraint weight of 10.0 kcal/(mol•Å2) was applied to all heavy atoms in this first 100 ps. An additional MD equilibration of 100 ps was performed with a decreased restraint weight of 1.0 kcal/(mol•Å2), followed by a final equilibration lasting 100 ps with no restraints. Production MD simulations of 20 ns each were carried out without any restraint at a temperature of 300.0 K and a pressure of 1 atm. Unless otherwise noted, all MD simulations used a time step of 2 fs, periodic boundary conditions were employed, and all electrostatic interactions were calculated using the particle mesh Ewald (PME) method41. A 10.0 Å cutoff was used to calculate the direct space sum of PME, and bond lengths involving bonds to hydrogen atoms were constrained using the SHAKE algorithm42. The coordinates were stored every 2 ps for each production MD run, and these snapshots were used for RMSD and water analysis. Ligand RMSD was measured by utilizing the Amber analyses tool PTRAJ. Ligand RMSD values were computed by aligning the protein backbone from MD snapshots and using the “nofit” option for ligand molecule. The Accelerated Molecular Dynamics (aMD) protocol implemented in Amber was used to reduce the height of local barriers, allowing the trajectories to evolve much faster. A boost was applied specifically to torsion angles (iamd=3). Other required input parameters such as average total potential energy threshold (EthreshP), inverse strength boost factor for the total potential energy (alphaP), average dihedral energy threshold (EthreshD), and inverse strength boost factor for the dihedral energy (alphaD) were calculated based on unbiased simulation output by following equations specified in the Amber user manual39.
Identification of water molecules and ligand interaction analysis (LIA)
The Amber tool PTRAJ was used to identify water molecules close to the bound ligand as follows. For each snapshot of the molecular dynamics simulation, the “center” command was used to re-center the coordinates to the center of mass of the protein. The “autoimage” command was then used to re-image the periodic box, and the 100 water molecules closest to the ligand were selected for each snapshot using the “closest” command. The processed snapshots were saved as a new trajectory using the “trajout” command without box information using the “nobox” option. Note that each snapshot in the trajectory may have contained different water molecules. But for each snapshot, the 100 water molecules closest to the bound ligand were kept. Processed trajectory files were converted to a Desmond molecular software readable format (.cms). The Simulation Interactions Diagram method incorporated in the Desmond module43 of Schrodinger was used to perform ligand interaction analysis (LIA). This analysis provides a statistical description of the interactions present during a molecular dynamics simulation. For water-mediated interactions, the interaction occupancy was calculated as the percent of frames in the trajectory where any water molecule present made that interaction. Thus, it is possible the more than one water may contribute to a water-mediated interaction during the course of a simulation.
Hydrogen-bond network analysis
The Protonate 3D module44 in the MOE application (v2016.08, Chemical Computing Group) was used to assign ionization states and to position hydrogens (rotamers and tautomers). The program optimizes the titration free energy of all titratable groups in the context of an all-atom model of the macromolecular structure (including ligands and solvent). The Generalized Born/Volume Integral45 electrostatics model is used for longer range interactions and solvation effects. After the system composed of protein and ligand is optimized, incidental water molecules are usually oriented one by one. For this study, the ‘precise’ flag was set for water molecules in the ZA channel: this treats the water molecules as part of the main calculation where geometric orientation is optimized in the context of all other system atoms. While substantially increasing computation time, the ‘precise’ flag enables better determination of water-mediated protein-ligand interactions. Only hydrogen bonds exceeding 0.5 kcal/mol as calculated by MOE are reported as bona fide interactions.
Generation of point-mutations at V4
The mutagenesis tool incorporated in Pymol46 was used to mutate Q101 in BRD2-BD1 (PDB ID: 4UYF) to lysine, and K374 in BRD2-BD2 (PDB ID: 4UYG) to glutamine. The Apo forms for this study were generated by removing the co-crystal ligand coordinates from the two PDB files above. MD simulations were performed as described above, and water molecules were re-imaged as was done for Desmond analysis. VMD was used to track waters in these re-imaged simulation trajectories and water occupancies were calculated over the 10,000 snapshots collected for each 20 ns simulation using the HBONDS plugin47.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported in part by an NIH Cancer Center Core grant CA21765, and by the American Lebanese Syrian Associated Charities (ALSAC).
Abbreviations Used
- BET
Bromodomain and Extra-Terminal
- BETi
Bromodomain inhibitors
- KAc
acetylated-lysine
- BD1
bromodomain 1
- BD2
bromodomain 2
- PTM
post-translational modification
- THQ
tetrahydroquinoline
- MD
molecular dynamics
- LIA
ligand interaction analysis
- aMD
accelerated molecular dynamics
- GAFF
General Amber Force Field
- AMBER
Assisted Model Building with Energy Refinement
- PMEMD
Particle Mesh Ewald Molecular Dynamics
References
- (1).Pedersen MT; Helin K Histone demethylases in development and disease. Trends Cell Biol. 2010, 20, 662–671. [DOI] [PubMed] [Google Scholar]
- (2).Turner BM Defining an epigenetic code. Nat. Cell Biol. 2007, 9, 2–6. [DOI] [PubMed] [Google Scholar]
- (3).Kouzarides T Acetylation: a regulatory modification to rival phosphorylation? EMBO. J 2000, 19, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Marushige K Activation of chromatin by acetylation of histone side chains, Proc. Natl. Acad. Sci. U. S. A 1976, 73, 3937–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Marmorstein R Protein modules that manipulate histone tails for chromatin regulation. Nat. Rev. Mol. Cell Biol 2001, 2, 422–432. [DOI] [PubMed] [Google Scholar]
- (6).Filippakopoulos P; Picaud S; Mangos M; Keates T; Lambert JP; Barsyte-Lovejoy D; Felletar I; Volkmer R; Muller S; Pawson T; Gingras AC; Arrowsmith CH; Knapp S Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Muller S; Filippakopoulos P; Knapp. S Bromodomains as therapeutic targets. Expert Rev. Mol. Med 2011, 13, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gosmini R; Nguyen VL; Toum J; Simon C; Brusq JM; Krysa G; Mirguet O; Riou-Eymard AM; Boursier EV; Trottet L; Bamborough P; Clark H; Chung CW; Cutler L; Demont EH; Kaur R; Lewis AJ; Schilling MB; Soden PE; Taylor S; Walker AL; Walker MD; Prinjha RK; Nicodeme E The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J. Med. Chem 2014, 57, 8111–8131. [DOI] [PubMed] [Google Scholar]
- (9).Brand M; Measures AR; Wilson BG; Cortopassi WA; Alexander R; Hoss M; Hewings DS; Rooney TP; Paton RS; Conway SJ Small molecule inhibitors of bromodomain-acetyl-lysine interactions. ACS Chem. Biol. 2015, 10, 22–39. [DOI] [PubMed] [Google Scholar]
- (10).Mirguet O; Gosmini R; Toum J; Clement CA; Barnathan M; Brusq JM; Mordaunt JE; Grimes RM; Crowe M; Pineau O; Ajakane M; Daugan A; Jeffrey P; Cutler L; Haynes AC; Smithers NN; Chung CW; Bamborough P; Uings IJ; Lewis A; Witherington J; Parr N; Prinjha RK; Nicodeme E Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem 2013, 56, 7501–7515. [DOI] [PubMed] [Google Scholar]
- (11).Jones MH; Numata M; Shimane M Identification and characterization of BRDT: A testis-specific gene related to the bromodomain genes RING3 and Drosophila fsh. Genomics 1997, 45, 529–534. [DOI] [PubMed] [Google Scholar]
- (12).Baud MG; Lin-Shiao E; Cardote T; Tallant C; Pschibul A; Chan KH; Zengerle M; Garcia JR; Kwan TT; Ferguson FM; Ciulli A Chemical biology. A bump-and-hole approach to engineer controlled selectivity of BET bromodomain chemical probes, Science 2014, 346, 638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shang E; Nickerson HD; Wen D; Wang X; Wolgemuth DJ The first bromodomain of Brdt, a testis-specific member of the BET sub-family of double-bromodomain-containing proteins, is essential for male germ cell differentiation. Development 2007, 134, 3507–3515. [DOI] [PubMed] [Google Scholar]
- (14).Vollmuth F; Blankenfeldt W; Geyer M Structures of the Dual Bromodomains of the P-TEFb-activating Protein Brd4 at Atomic Resolution. J. Biol Chem. 2009, 284, 36547–36556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Umehara T; Nakamura Y; Jang MK; Nakano K; Tanaka A; Ozato K; Padmanabhan B; Yokoyama S Structural basis for acetylated histone H4 recognition by the human BRD2 bromodomain. J. Biol. Chem 2010, 285, 7610–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I; Philpott. M; Munro S; McKeown MR; Wang Y; Christie AL; West N; Cameron MJ; Schwartz B; Heightman TD; La Thangue N; French CA; Wiest O; Kung AL; Knapp S; Bradner JE Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Picaud S; Wells C; Felletar I; Brotherton D; Martin S; Savitsky P; Diez-Dacal B; Philpott M; Bountra C; Lingard H; Fedorov O; Muller S; Brennan PE; Knapp S; Filippakopoulos P RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gacias M; Gerona-Navarro G; Plotnikov AN; Zhang G; Zeng L; Kaur J; Moy G; Rusinova E; Rodriguez Y; Matikainen B; Vincek A; Joshua J; Casaccia P; Zhou MM Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem. Biol 2014, 21, 841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang G; Plotnikov AN; Rusinova E; Shen T; Morohashi K; Joshua J; Zeng L; Mujtaba S; Ohlmeyer M; Zhou MM Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J. Med. Chem. 2013, 56, 9251–9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hewings DS; Rooney TP; Jennings LE; Hay DA; Schofield CJ; Brennan PE; Knapp S; Conway SJ Progress in the development and application of small molecule inhibitors of bromodomain-acetyl-lysine interactions. J. Med. Chem. 2012, 55, 9393–9413. [DOI] [PubMed] [Google Scholar]
- (21).Moriniere J; Rousseaux S; Steuerwald U; Soler-Lopez M; Curtet S; Vitte AL; Govin J; Gaucher J; Sadoul K; Hart DJ; Krijgsveld J; Khochbin S; Muller CW; Petosa C Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature 2009, 461, 664–668. [DOI] [PubMed] [Google Scholar]
- (22).Chung CW; Dean AW; Woolven JM; Bamborough P Fragment-based discovery of bromodomain inhibitors part 1: inhibitor binding modes and implications for lead discovery. J. Med. Chem 2012, 55, 576–586. [DOI] [PubMed] [Google Scholar]
- (23).Ciceri P; Muller S; O’Mahony A; Fedorov O; Filippakopoulos P; Hunt JP; Lasater EA; Pallares G; Picaud S; Wells C; Martin S; Wodicka LM; Shah NP; Treiber DK; Knapp S Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol 2014, 10, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ember SW; Zhu JY; Olesen SH; Martin MP; Becker A; Berndt N; Georg GI; Schonbrunn E Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen L; Yap JL; Yoshioka M; Lanning ME; Fountain RN; Raje M; Scheenstra JA; Strovel JW; Fletcher S BRD4 Structure-Activity Relationships of Dual PLK1 Kinase/BRD4 Bromodomain Inhibitor BI-2536. ACS Med. Chem. Lett 2015, 6, 764–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hamelberg D; Mongan J; McCammon JA Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys 2004, 120, 11919–11929. [DOI] [PubMed] [Google Scholar]
- (27).Shadrick WR; Slavish P; Sergio CC; Waddellb B; Connelly M; Low JA; Tallant C; Young BM; Bharathama N; Knappc S; Boyd VA; Morfouace M; Roussel MF; Chen T; Lee RE; Guy RK; Shelat AA; Potter PM Exploiting a water network to achieve enthalpy-driven, bromodomain-selective BET inhibitors. Bioor. Med. Chem 2018, 26, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Arrowsmith CH; Audia JE; Austin C; Baell J; Bennett J; Blagg J; Bountra C; Brennan PE; Brown PJ; Bunnage ME et al. The promise and peril of chemical probes. Nature Chemical Biology 2015, 11, 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Freire E Do Enthalpy and Entropy Distinguish First in Class From Best in Class? Drug Discov. Today 2008, 13, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nasief NN; Said AM; Hangauer H Modulating hydrogen-bond basicity within the context of protein-ligand binding: A case study with thrombin inhibitors that reveals a dominating role for desolvation. Eur. J. Med. Chem.2017, 125, 975–991. [DOI] [PubMed] [Google Scholar]
- (31).Chen IJ; Foloppe N Drug-like bioactive structures and conformational coverage with the LigPrep/ConfGen suite: comparison to programs MOE and catalyst. J. Chem. Inf. Model 2010, 50, 822–839. [DOI] [PubMed] [Google Scholar]
- (32).Suite 2012: Maestro, version 9.2; Schrodinger, LLC: New York, 2012. [Google Scholar]
- (33).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (34).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- (35).Naïm M; Bhat S; Rankin KN; Dennis S; Chowdhury SF; Siddiqi I; Drabik P; Sulea T; Bayly CI; Jakalian A; Purisima EO Solvated interaction energy (SIE) for scoring protein-ligand binding affinities. 1. Exploring the parameter space. J Chem Inf Model. 2007. Jan-Feb;47(1):122–33. [DOI] [PubMed] [Google Scholar]
- (36).Wang J; Wang W; Kollman PA; Case DA Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [DOI] [PubMed] [Google Scholar]
- (37).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (38).Jakalian A; Bush BL; Jack DB; Bayly CI Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [DOI] [PubMed] [Google Scholar]
- (39).Case DA; Cheatham TE III; Simmerling CL; Wang J; Duke JE; Luo R; Crowley M; Walker RC; Zhang W; Merz KM; Wang B; Hayik S; Roitberg A; Seabra G; Kolossváry I; Wong KF; Paesani F; Vanicek J; Wu X; Brozell SR; Steinbrecher T; Gohlke H; Yang L; Tan C; Mongan J; Hornak V; Cui G; Mathews DH; Seetin MG; Sagui C; Babin V; Kollman PA AMBER 12, University of California, San Francisco, 2012. [Google Scholar]
- (40).Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Darden T; York D; Pedersen L Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (42).Lambrakos SG; Boris JP; Oran ES; Chandrasekhar I; Nagumo M A modified shake algorithm for maintaining rigid bonds in molecular dynamics simulations of large molecules. J. Comput. Phys. 1989, 85, 473–486. [Google Scholar]
- (43).Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2015 Maestro-Desmond Interoperability Tools, version 4.2, Schrödinger, New York, NY, 2015. [Google Scholar]
- (44).Labute P The generalized Born/volume integral implicit solvent model: estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem 2008, 29, 1693–1698. [DOI] [PubMed] [Google Scholar]
- (45).Labute P The Generalized Born / Volume Integral (GB/VI) Implicit Solvent Model: Estimation of the Free Energy of Hydration Using London Dispersion Instead of Atomic Surface Area. J. Comp. Chem 2008, 19, 1693–1698. [DOI] [PubMed] [Google Scholar]
- (46).The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. [Google Scholar]
- (47).Humphrey W,; Dalke A; Schulten K “VMD - Visual Molecular Dynamics”. J. Molec. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.