

Summary
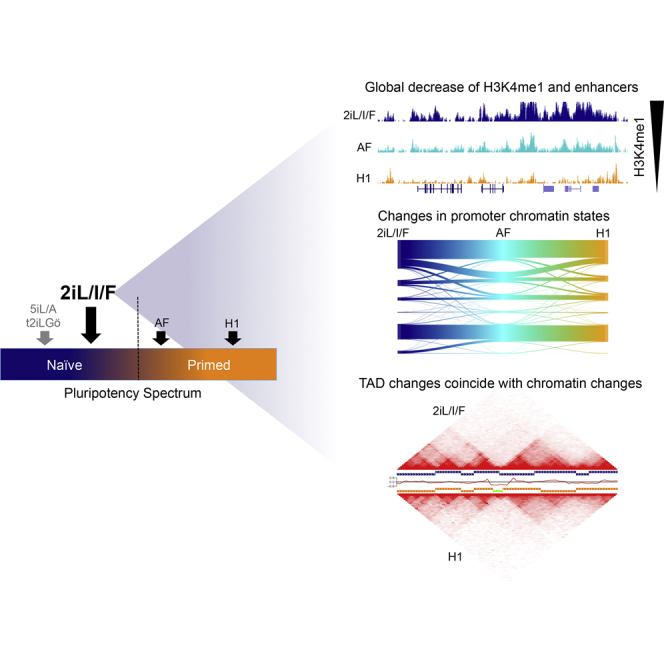
During mammalian embryogenesis, changes in morphology and gene expression are concurrent with epigenomic reprogramming. Using human embryonic stem cells representing the preimplantation blastocyst (naive) and postimplantation epiblast (primed), our data in 2iL/I/F naive cells demonstrate that a substantial portion of known human enhancers are premarked by H3K4me1, providing an enhanced open chromatin state in naive pluripotency. The 2iL/I/F enhancer repertoire occupies 9% of the genome, three times that of primed cells, and can exist in broad chromatin domains over 50 kb. Enhancer chromatin states are largely poised. Seventy-seven percent of 2iL/I/F enhancers are decommissioned in a stepwise manner as cells become primed. While primed topologically associating domains are largely unaltered upon differentiation, naive 2iL/I/F domains expand across primed boundaries, affecting three-dimensional genome architecture. Differential topologically associating domain edges coincide with 2iL/I/F H3K4me1 enrichment. Our results suggest that naive-derived 2iL/I/F cells have a unique chromatin landscape, which may reflect early embryogenesis.
Keywords: enhancers, 3D genome architecture, naive hESCs, embryogenesis
Graphical Abstract
Highlights
-
•
2iL/I/F hESCs are useful for studying naive-to-primed epigenomic transitions
-
•
H3K4me1 is expanded across the genome of naive 2iL/I/F hESCs
-
•
The majority of naive 2iL/I/F enhancers are decommissioned upon priming
-
•
Differential topologically associating domains have H3K4me1 at their boundaries
Human ESCs grown in 2iL/I/F are useful for studying the transition from naive to primed pluripotency. Battle et al. show that the naive epigenome is globally enhanced for H3K4me1. Many of the H3K4me1 regions are putative enhancers and are lost, or decommissioned, during the transition to primed. Differential topologically associating domains (TADs) exist between states and contain H3K4me1 enrichment at TAD boundaries.
Introduction
Dynamic changes in the epigenome are associated with morphological and gene expression changes during early embryogenesis. Soon after fertilization DNA methylation is actively removed from the paternal genome, passively lost from the maternal genome, and regained in the postimplantation epiblast (Guo et al., 2014). In addition to resetting the DNA methylome, the early embryonic epigenome maintains an open chromatin structure as repressive heterochromatin is gained later over the course of development, lineage commitment, and differentiation (Ahmed et al., 2010, Liu et al., 2004, Sarmento et al., 2004). These changes in histone modification correlate with the hypothesis that a more open chromatin structure is a key aspect of pluripotency and allows embryonic cells to respond to a broad array of developmental signaling cues (Hawkins et al., 2010, Meshorer et al., 2006).
Pre- and postimplantation pluripotent embryonic stem cells (ESCs) provide a system to model epigenomic reprogramming during early embryogenesis and to study changes in pluripotency. In the past few years, several groups described the first set of naive human ESCs (hESCs), whereby primed hESCs or human pluripotent stem cells were induced, or reset, to the naive state (Chan et al., 2013, Gafni et al., 2013, Hanna et al., 2010, Takashima et al., 2014, Theunissen et al., 2014, Valamehr et al., 2014, Ware et al., 2014). Additionally new hESC lines were derived, each under a different naive growth condition (Gafni et al., 2013, Guo et al., 2016, Theunissen et al., 2014, Ware et al., 2014; for review see Ware, 2016). Similar to mouse, naive hESCs exhibit DNA hypomethylation and two active X chromosomes (Gafni et al., 2013, Theunissen et al., 2016, Ware et al., 2014), hallmarks of the preimplantation state.
Given the differences between early human and mouse embryogenesis (Blakeley et al., 2015, Rossant, 2015), naive-derived hESC lines provide an opportunity to study changes that are reflective of early human development and pluripotency. To better our understanding of epigenomic reprogramming as hESCs transition from naive to primed, we present data from whole-transcriptome RNA sequencing (RNA-seq), chromatin immunoprecipitation sequencing (ChIP-seq) for five histone modifications, and topologically associating domains (TADs) from in situ DNaseI Hi-C for the naive-derived Elf1 line (Ware et al., 2014) grown in 2i + leukemia inhibitory factor (LIF) + insulin-like growth factor 1 (IGF1) + fibroblast growth factor (FGF) (2iL/I/F). Elf1 cells grown in this culture condition were previously shown to be naive based on gene expression, but in a later stage of development compared with 5iL/A and t2iL + Gö cells, and are more similar to mouse ESCs (mESCs) (Figure 1A) (Moody et al., 2017). We include data from cells that are exiting or transitioning out of the naive state (activin + FGF) and compared our results with data from primed H1 hESCs (Dixon et al., 2012, Hawkins et al., 2010). Extensive chromatin remodeling occurs at promoters and enhancer elements as cells transition from naive to primed. Our analysis reveals that 2iL/I/F hESCs have a more open chromatin structure due to large expansions of H3K4me1 and H3K27ac in the genome. Seventy-seven percent of 2iL/I/F enhancers are decommissioned in the primed state. TADs are largely stable between pluripotent states, but our data reveal limited 2iL/I/F-specific shifts in TAD boundaries. Overall, these data provide an extensive view of the epigenome and three-dimensional (3D) genome for hESC states and a model for epigenomic reprogramming during early human embryogenesis.
Figure 1.

Overview of Chromatin States
(A) Schematic of where 2iL/I/F and other ESCs lie on the pluripotency spectrum. Dashed line represents transition from naive to primed. Adapted from Moody et al. (2017).
(B) Global view of chromatin structure for 2iL/I/F (navy), transitioning (TR; cyan) and primed (orange) hESCs. These colors are used throughout all figures. UCSC Genome Browser images of TBX3 and CDX2 gene loci showing enrichment of H3K4me1 (RPKM range 1–20), H3K27ac (RPKM range 1–20), and H3K27me3 (RPKM range 1–30) in 2iL/I/F, transitioning and primed cells.
(C) The number of ChIP-seq peaks called by MACS with FDR cutoff ≤0.05.
(D) The percentage of genome covered by each histone modification.
(E) Cartoon showing different categories of promoter states.
(F) Violin plots showing the distribution of RPKM values of NNGs of active, poised, and bivalent promoter peaks in each cell type. p values for pairwise comparisons are computed using two tailed t tests with pooled SD. p values are adjusted with Benjamini-Hochberg method. ∗∗∗p < 0.001.
(G) Sankey plot of primed bivalent gene promoters and their origins from the 2iL/I/F state.
(H) Significance level of GO terms from bivalently marked gene promoters.
Results
Gene Expression in 2iL/I/F hESCs
It is currently accepted that pluripotency exists as a spectrum (Wu and Izpisua Belmonte, 2015, Zimmerlin et al., 2017), and 2iL/I/F cells are useful for studying the naive-to-primed transition (Figure 1A). As additional support of their position on the naive spectrum, we tested the presence of naive-specific cell-surface markers previously identified by Collier et al. (2017) using fluorescence-activated cell sorting (FACS). We found that the majority of 2iL/I/F cells expressed naive cell-surface markers CD77 and CD75 (Figures S1A and S1B). We also performed reduced representation bisulfite sequencing (RRBS) to measure the global DNA methylation level in 2iL/I/F cells. 2iL/I/F cells are more methylated than cells grown in the naive 5iL/A condition but hypomethylated compared with primed cells (Figure S1C). 2iL/I/F cells also exist in a metabolic state similar to preimplantation embryos, unlike the glycolytic state of primed cells (Sperber et al., 2015, Zhou et al., 2012). Altogether, this indicates that 2iL/I/F cells have characteristics that are reflective of preimplantation development and in vitro naive states. We then performed strand-specific, whole-transcriptome RNA-seq in replicate on Elf1 2iL/I/F, Elf1 transitioning (activin + FGF; referred to as TR) and H1 primed (mTeSR) cells of equal cell numbers (Figures S1D–S1F). We identified differentially expressed genes (DEGs) in a pairwise manner (Figures S1G and S1H). The largest number of DEGs was observed between 2iL/I/F and primed hESCs (Figure S1H and Table S1), signifying just how distinct these cellular states are. Highlighted in Figures S1G and S1H are several genes known to be upregulated in the human preimplantation epiblast (Blakeley et al., 2015, Yan et al., 2013) and other key genes of interest.
We identified gene ontology (GO) categories and KEGG pathways for 2iL/I/F DEGs, which were significantly enriched for embryo development and pluripotency signaling pathways along with other pathways important during preimplantation development (Figures S1I and S1J). In particular, genes in the transforming growth factor β (TGF-β) pathway were found to be upregulated in 2iL/I/F cells, including LEFTY1, SMAD3, and NODAL. The TGF-β pathway was shown to be important for maintenance of NANOG in the human epiblast, whereas inhibition of this pathway has insignificant effects on mouse embryos (Blakeley et al., 2015). The PI3K-AKT signaling pathway was also enriched, and is known to promote ESC self-renewal through inhibition of the ERK signaling pathway (Chen et al., 2012). The WNT signaling pathway was enriched for naive upregulated genes including WNT8A, WNT5B, and TCF7 (Sperber et al., 2015). A number of terms associated with embryonic development and morphogenesis were enriched for 2iL/I/F upregulated genes.
We identified cell-type-specific genes in the different hESC stages by applying a cutoff of an RPKM (reads per kilobase per million) value greater than or equal to 2 in one cell type and less than 1 in the other two cell types (Figure S2A). Using this cutoff we determined 429 2iL/I/F-specific genes, 229 TR-specific genes, and 333 primed-specific genes. Compared with the primed states, 2iL/I/F-specific genes were enriched for GO terms associated with morphogenesis and pattern specification (Figure S2A). This is due, in part, to the many HOX genes that are uniquely expressed in 2iL/I/F hESCs and not in transitioning or primed cells. Primed cells were enriched for terms associated with extracellular communication and protein/histone demethylation.
A recent report showed that the transposable element (TE) transcriptome can be used as a state-specific signature in hESCs (Theunissen et al., 2016). 2iL/I/F and primed hESCs segregate when clustered on the top 1,000 highest expressed TEs (Figure S2B). We see that HERVK and LTR5 are upregulated in 2iL/I/F cells while HERVH and LTR7 are upregulated in primed. Lastly, we compared upregulated genes with human embryo RNA-seq data from Yan et al. (2013). We find that a similar percentage of upregulated genes from 2iL/I/F and primed are expressed in pre-zygotic genome activation stages, while 2iL/I/F hESCs share more upregulated genes with the post-zygotic genome activation (ZGA) embryo than primed (Figure S2C). This strengthens reports that Elf1 cells are a good representative model of the preimplantation stage of human development (Moody et al., 2017, Sperber et al., 2015, Ware et al., 2014).
Global Chromatin Features of 2iL/I/F hESCs
To assess global chromatin dynamics between the cellular states, we performed ChIP-seq on five histone modifications from 2iL/I/F and transitioning cells (Table S2), and used data previously generated in H1 hESCs for the primed state (Hawkins et al., 2010). These modifications include H3K4me3 for Pol II-bound promoters, H3K4me1 for enhancers, H3K27ac for active regions, H3K27me3 for Polycomb-repressed regions, and H3K9me3 for heterochromatin. All five modifications along with ChIP inputs were sequenced in duplicates for both Elf1 2iL/I/F and Elf1 transitioning cells for a total of >270 million and >213 million sequencing reads, respectively (Table S2).
We inspected genes with known expression differences during early embryogenesis through the blastocyst/epiblast stage to ensure our chromatin maps reflect known changes during differentiation from naive to primed. TBX3 was shown to be expressed in naive ESCs and human epiblasts (Blakeley et al., 2015). The TBX3 locus exhibits high levels of H3K4me1 and H3K27ac in naive hESCs, a reduction of H3K27ac in the transitioning state, followed by a reduction of H3K4me1 and a gain of H3K27me3 in primed hESCs (Figure 1B). KLF2, which was shown not to be expressed in human naive cells (Blakeley et al., 2015), lacks the H3K27ac modification in all three hESC stages (Figure S3A). CDX2 has active histone modifications in naive hESCs but transitions to a loss of acetylation and gain of H3K27me3 in primed hESCs (Figure 1B). CDX2 protein has been shown to be present after blastocyst formation in human embryos and overlaps OCT4 protein in preimplantation embryos (Niakan and Eggan, 2013). Expansion of H3K27me3 domains are also shown at the HOXA locus as hESCs move from naive to primed (Figure S3B). Next, we asked whether these trends observed at specific loci held true genome wide.
Comparisons across cell types reveal a genome-wide depletion of repressive histone modifications in 2iL/I/F cells (Figures 1C and 1D). H3K27me3 repressed regions are more abundant and broader in primed than in 2iL/I/F cells, covering ∼1.4% of the genome in primed cells compared with 0.5% in 2iL/I/F (Figures 1D and S3C), which we previously showed is linked to metabolic differences between the cell states (Sperber et al., 2015). H3K9me3 heterochromatin regions, which are sparse in primed cells (Hawkins et al., 2010), are further depleted in transitioning and 2iL/I/F cells (Figures 1C, 1D, and S3D; Table S3). There is a notable abundance of H3K4me1 regions in 2iL/I/F hESCs (Figure 1C and Table S3). Over 9% of the 2iL/I/F genome is marked by H3K4me1, 3-fold more than primed cells and 1.7-fold more than transitioning cells (Figure 1D and Table S3). Monomethylation is present in larger domains, reaching sizes of over 30 kb in transitioning cells and over 50 kb in 2iL/I/F cells (Figure S3E). Acetylation is also more enriched in 2iL/I/F cells with 3-fold more peaks than primed, which is similar to H3K27ac in naive 5iL/A cells where it also covers ∼4% of the genome (Ji et al., 2016). Similar to H3K4me1, broad H3K27ac domains can reach over 50 kb (Figures 1C, 1D, and S3F; Table S3). We found H3K4me3 to be the most stable mark although cell-specific peaks exist (Figures 1C, 1D, and S3G). The trends for histone modifications also hold true for a second primed line, H9 (Figure S3H).
Promoter Transitions as hESCs Exit the Naive State
We investigated how DEGs were reflected through promoter chromatin states using >19,000 GENCODE defined autosomal protein coding genes. Over 12,000 promoters are marked with H3K4me3 (Figure S4A). We divided promoters into six categories: (1) active—H3K4me3 and H3K27ac; (2) poised—H3K4me3 only; (3) bivalent—H3K4me3 and H3K27me3; (4) H3K27ac—H3K27ac only; (5) H3K27me3—H3K27me3 only; and (6) unmarked—lacking all three modifications (Figures 1E and S4B). Although the largest percentages of gene promoters remain static as either active or unmarked across all three stages, many promoters change chromatin state (Figure S4C), which exemplifies the dynamic nature of the epigenome. To illustrate that chromatin patterns coincide with general trends of expression, we plotted the RPKM values of genes with active, poised, and bivalent promoters. As expected, genes with active promoters had overall higher expression levels than genes with promoters in the other two categories (Figure 1F).
We observed an increase in bivalent gene promoters from 2iL/I/F to primed cells (1,097 versus 2,674), and determined from which epigenetic states the primed bivalent promoters arose. Roughly 60% of primed bivalent promoters are bivalent in transitioning cells, and of those, their promoter states are split between active (42%), bivalent (32%), and poised (20%) in 2iL/I/F hESCs (Figure 1G). Of the ∼7% of 2iL/I/F active gene promoters that become bivalent in transitioning cells, these genes were enriched for GO terms such as morphogenesis and WNT signaling, and included genes such as HOXA1, HOXA4, HOXD8, and ZEB1. 2iL/I/F bivalent genes fall into categories involving GO terms for synaptic transmission, ion transport, and neuron differentiation (Figure 1H). Thus, it appears that the neural lineage is one of the first lineages to be bivalently marked in 2iL/I/F cells and further suggests that naive hESCs may be an excellent model for further investigation of the establishment of Polycomb-repressive regions in the early epigenome.
Enhancers in 2iL/I/F Cells
Enhancer elements are cis-acting regulatory sequences that control gene expression via interaction with transcription factors and promoters. We defined enhancers as H3K4me1 peaks lacking overlap with H3K4me3 (Table S3) (Heintzman et al., 2007, Heintzman et al., 2009). Investigation of the enhancer landscape across hESC states revealed that 2iL/I/F cells harbor the most cell-type-specific enhancers (>47k; Figures 2A and 2B), while transitioning and primed cells had roughly the same number of unique enhancers at ∼17k and ∼14k, respectively (Figures 2B, S5A, and S5B). Sixty-four percent of transitioning enhancers and 55% of primed enhancers are marked in the 2iL/I/F state (Figures S5A and S5B). We asked whether the expansion of 2iL/I/F H3K4me1 was random or occurred at known regulatory elements. Using DNase I hypersensitive sites (DHS) data from 177 ENCODE cells (ENCODE Project Consortium, 2012), we found 25%–30% of the H3K4me1-marked genome (enhancer-verse) to be hypersensitive in each of our pluripotent cell types (Figure 2C). Of the 177 cell and tissue types, fetal tissues had the largest collection of DHS overlapping 2iL/I/F enhancers (Figure 2D and Table S4). Additionally, over 92% of the enhancer regions (quantified by number of bases) covered by 2iL/I/F H3K4me1 peaks are utilized as enhancers in 127 Roadmap Epigenome Project cell types, as indicated by H3K4me1 (Figures S5C and S5D). Single-cell RNA-seq data from early human embryogenesis (Yan et al., 2013) indicate that 92% of annotated transcription factors (Zhang et al., 2015) are expressed by the late blastocyst stage (Figure S5E). Their expression provides a plausible means for aiding the localization of H3K4me1 to known enhancers.
Figure 2.

2iL/I/F Enhancer Repertoire
(A) Venn diagram of 2iL/I/F (navy) enhancers overlapped with transitioning (cyan) and primed (orange) enhancers.
(B) Heatmap of H3K4me1 normalized ChIP-seq signal centered at cell type-specific enhancers in a 3-kb window.
(C) Percentage of hESC H3K4me1 genomic space, i.e., number of base pairs, occupied by ENCODE DNase hypersensitive sites (DHS) from 177 cell types.
(D) Number of ENCODE DHS from 177 cell types overlapping with 2iL/I/F and primed H3K4me1 enhancers.
(E) Percentage of active (H3K4me1 + H3K27ac) and poised (H3K4me1 only or H3K4me1 + H3K27me3) enhancer states in each cell type.
Enhancer elements can exist in distinct chromatin states, or classes, that indicate whether they are active or poised (Figures 2E and S5F) (Creyghton et al., 2010, Hawkins et al., 2011, Rada-Iglesias et al., 2011). We characterized differences in the classes of enhancers in each hESC state. We defined active enhancers as regions having H3K4me1 and H3K27ac and poised enhancers as regions with either H3K4me1 only or H3K4me1 and H3K27me3. In all three stages of pluripotency, the majority of enhancers are in the H3K4me1 only poised state (67%, 84%, and 73% in 2iL/I/F, transitioning, and primed cells, respectively; Figure 2E). There is an increase of H3K27me3 containing poised enhancers moving from 2iL/I/F to primed (1%–4%; Figure 2E), which correlates with the increase of H3K27me3.
Our comparative analysis of enhancers indicates that both active and H3K4me1 only poised enhancers are largely decommissioned as 2iL/I/F hESCs transition to the primed state (Figure 3A). When assessing overlapping H3K4me1 peaks across hESCs, we see that the chromatin-marked genomic space of 2iL/I/F enhancers is greatly reduced in primed cells (Figures 3A and 3B). This is exemplified at the H19 locus, where broad enhancers are shorter and broken up in the transitioning state, and most have disappeared by primed state (Figure 3A). This process happens in a stepwise manner, as is evidenced by the loss of acetylation as cells exit the 2iL/I/F state followed by the gradual loss H3K4me1. We observed this globally and at shared 2iL/I/F enhancer regions (Figures 1B, 1C, 3A, and 3B). This introduces an alternative view of development compared with previous studies showing that poised enhancers gain acetylation following differentiation and were often enriched near genes that became activated later in development (Creyghton et al., 2010, Hawkins et al., 2011, Rada-Iglesias et al., 2011). By using 2iL/I/F hESCs as a model, we can infer that not only is H3K4me1 likely maintaining open chromatin to aid in the pluripotency phenotype, but that a substantial fraction of enhancers in the human genome are premarked early during embryogenesis and subsequently decommissioned during priming.
Figure 3.

2iL/I/F Enhancers Are Decommissioned but Active in Other Cell Types
(A) UCSC Genome Browser image illustrating loss of H3K27ac, followed by loss of H3K4me1 at the H19 locus as cells move from 2iL/I/F (navy), to transitioning (cyan), to primed (orange); RPKM range 1–20 for each track. This region also contains a broad enhancer domain in 2iL/I/F hESCs. Enhancer peak calls are represented as bars above the H3K4me1 track.
(B) Number of bases under peaks overlapping shared (all three cell types) 2iL/I/F enhancers. H3K27ac is reduced greatly in the transitioning genome, then there is a great reduction in H3K4me1.
(C) Heatmaps of H3K4me1 and H3K27ac normalized ChIP-seq signal at broad enhancer regions (≥5 kb).
(D) Histograms of average H3K4me1 and H3K27ac normalized ChIP-seq signal at all broad enhancers or broad H3K27ac domains.
(E) Violin plots showing the distribution of RPKM values of nearest neighboring genes of all enhancers, broad enhancers (H3K4me1 ≥5 kb), or active broad enhancers (H3K4me1 ≥5 kb overlapping H3K27ac ≥5 kb) in each cell type. p values for pairwise comparisons are computed using two-tailed t tests with pooled SD. p values are adjusted with Benjamini-Hochberg method. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Broad Enhancer Domains in the 2iL/I/F Epigenome
Super (Whyte et al., 2013) and stretch (Parker et al., 2013) enhancers, which are largely based on H3K27ac, were originally identified in primed ESCs. These regions were shown to upregulate nearby and cell-specific genes and were stronger than conventional enhancers. We asked to what degree these regions were present in 2iL/I/F hESCs. To identify both broad H3K4me1 and H3K27ac domains, we identified regions ≥5 kb in all cell types (Figure 3C). The H3K4me1 broad enhancers are almost 20-fold more abundant in the 2iL/I/F epigenome compared with the primed hESC stage (7,412 in 2iL/I/F hESCs compared with 371 in primed) with an average size of 8.1 kb compared with 6.1 kb in primed (Figures S5G and S5H). The number of broad enhancers steadily declines as hESCs transition from 2iL/I/F to primed. We observed the same trend with H3K27ac broad domains (2,330 in 2iL/I/F compared with 803 in primed), although the number of broad H3K27ac domains in 2iL/I/F cells is 3-fold less than the number of H3K4me1 broad enhancers (Figure S5G). For perspective, we looked for broad H3K4me3 peaks, and found them to be limited across the different hESC stages (Figure S5G).
Next, we determined whether H3K4me1 broad enhancers and H3K27ac broad domains occupy the same genomic space. The average number of bases contained within the overlap of broad H3K4me1 and H3K27ac domains is over 70% of the average length of each domain (Figure S5H). Over 78% of broad H3K27ac domains in 2iL/I/F cells are found within H3K4me1 broad enhancers (Figure S5I). In the 2iL/I/F and primed states 87% and 71% of H3K4me1 broad enhancers, respectively, contained some overlap with H3K27ac, indicating that they are active enhancers (Figures 3D and S5I). The average ChIP-seq signal for H3K4me1 is high at H3K27ac broad domains in all cells except primed hESCs (Figure 3D). The active state of broad enhancers is supported by the distribution of expression values of nearest neighboring genes (NNGs) (Figure 3E). Only in the primed state are there more broad H3K27ac domains than H3K4me1 domains, and the difference in the expression distribution of NNGs at broad enhancers versus active broad enhancers in primed cells was the only comparison not found to be significant (Figure 3E). This may explain why H3K27ac was originally associated with “super/stretch” enhancers, as most broad H3K4me1 domains are found at broad H3K27ac domains and broad H3K27ac domains are more abundant. However for naive pluripotency the frequent occurrence of H3K4me1 and H3K27ac broad domains, and the observation that broad H3K27ac domains lie within broad H3K4me1 enhancers, provides an additional means of giving the genome its “open structure.”
2iL/I/F hESCs Enhancers in Different Growth Conditions
To determine whether the expansion of H3K4me1 in the 2iL/I/F epigenome was indicative of the naive state and independent of a single growth condition or cell line, we grew three lines in 4i (2i + p38 kinase inhibitor + JNK inhibitor) + LIF + IGF1 + FGF (4iL/I/F [Sperber et al., 2015]): Elf1, H1 reset to naive, and the naive-derived LIS1 line (Gafni et al., 2013) (Figure S6A). The LIS1 line grew slightly better in 4iL/I/F compared with the original growth conditions. To determine whether growth conditions or genetic background had an effect on the enhancer landscape, we compared the enhancer profiles from H3K4me1 ChIP-seq data across cell types and conditions. Overall, all naive cells have a similar enhancer profile (Figure 4A). Cells grown in 4iL/I/F exhibit a stronger enhancer signal at Elf1 2iL/I/F-specific enhancers (Figures 4B and 4C), and less enrichment at primed- and transitioning-specific enhancers (Figures 4B and S6B). Principal component analysis (PCA) of gene expression data shows a clear separation between naive and primed cells (Figure S6C). PCA of H3K4me1 signal reveals that all lines grown in 4iL/I/F are largely indistinguishable, and most similar to 2iL/I/F (Figure 4D). Our analysis suggests that 2iL/I/F enhancers do not vary greatly in 4iL/I/F conditions, although 2iL/I/F hESCs have some distinct H3K4me1 features. The acquired expansion upon resetting primed H1 cells to 4iL/I/F may suggest that this epigenetic feature is necessary for maintenance in the naive state. Further experiments will be needed to confirm this hypothesis. Additionally, we find that one-third of H3K27ac peaks in 5iL/A reset hESCs(Ji et al., 2016) are also present in 2iL/I/F hESCs (Figure S6D), highlighting the epigenetic changes that must occur as cells transition through the spectrum of pluripotency, eventually becoming primed.
Figure 4.

2iL/I/F Enhancers from Various 2iL/I/F Culture Conditions
ChIP-seq of naive cells grown in different culture conditions including Elf1 2iL/I/F (navy), Elf1 4iL/I/F (purple), Elf1 AF (transitioning; cyan), primed H1 mTeSR (orange), H1 4iL/I/F (red), and LIS1 4iL/I/F (green).
(A) H3K4me1 enrichment in different growth conditions at DNMT3L and SOX2 loci.
(B) Average ChIP-seq signal at 2iL/I/F-specific enhancers (top panel) and primed-specific enhancers (bottom panel).
(C) Heatmap of H3K4me1 ChIP-seq signal at 2iL/I/F-specific enhancers, defined as not in TR or primed. Scale is RPKM.
(D) PCA of top 500 10-kb bins of H3K4me1 with largest variance.
3D Genome Architecture in 2iL/I/F hESCs
Genome architecture is an important component of gene regulation. TADs identified in primed hESCs proved to be surprisingly stable upon differentiation to distinct cell types despite diverse changes to chromatin structure (Dixon et al., 2015). However, domain-scale 3D genome architecture is still missing for the naive state. To characterize TADs in hESCs at the naive spectrum of pluripotency, we generated deeply sequenced in situ DNase Hi-C maps (Deng et al., 2015) in Elf1 2iL/I/F (Figure S7A), which exhibited characteristic reductions in contact frequency as a function of linear distance between two loci (Figure 5A). We processed raw Hi-C read pairs produced from H1 primed hESCs (Dixon et al., 2012, Dixon et al., 2015) and compared the architectural features identified in each cell type at 40-kb resolution. A total of 6,119 TADs were identified in 2iL/I/F hESCs and 5,822 TADs in primed hESCs (Figure S7B), consistent with previous observations in primed hESCs (Shin et al., 2016). We defined boundaries as regions between two adjacent TADs and found that 7.3% and 6.2% of boundaries were greater than 40 kb in 2iL/I/F and primed cells, respectively (Figure S7C). To provide confidence in our TAD calls, we calculated insulation scores at each Hi-C bin (Crane et al., 2015, Giorgetti et al., 2016). The insulation score represents how insulated each bin is from TAD boundaries. It is expected that TAD boundaries occur at the valleys/minima of insulation scores, and TAD centers occur at the peaks/maxima. We found that boundary insulation scores were significantly different from TAD center scores (Figure 5B), providing high confidence to our defined TAD regions.
Figure 5.

3D Genome Architecture in 2iL/I/F hESCs
(A) Hi-C contact heatmap of chromosome 3 in 2iL/I/F cells at 500-kb resolution.
(B) Box plots of the insulation scores at all TAD centers and boundaries for both 2iL/I/F and primed cells. p values are computed using individual Wilcoxon signed-rank tests.
(C) Global size distributions of TADs within 2iL/I/F and primed cells (left panel) and size differences of overlapping TADs (40-kb bin resolution) in 2iL/I/F and primed cells (right panel).
(D) Differential heatmap of 2iL/I/F minus primed Hi-C. Negative (red) values indicate a stronger bin signal in primed cells relative to 2iL/I/F cells.
(E) Naive CTCF ChIP-seq signal from Ji et al. (2016) centered at 2iL/I/F TAD boundaries.
(F) Number of TADs or differential TADs with cohesin ChIA-PETs within 80 kb of boundary.
Overall, TAD size distributions are similar (Figure 5C, first panel), with means of 420 kb in 2iL/I/F and 444 kb in primed. We observed 2,024 TADs whose genomic coordinates are identical at 40-kb resolution while the remaining overlapping TADs differ by at least 40 kb (Figure 5C, second panel). We could detect differences in TAD boundaries as illustrated by 2iL/I/F-specific boundaries exhibiting an enrichment of primed Hi-C signal (Figure 5D). The enrichment of primed signal at 2iL/I/F-specific boundaries confirms that this is not a boundary in primed cells. We asked whether the higher number of 2iL/I/F Elf1 TADs may be due to technical differences of our in situ data. We found that some H1 TADs were split into two or more Elf1 TADs, which accounts for an “extra” 427 Elf1 TADs (Figure S7D). The average overlap between 2iL/I/F and primed TADs is 319 kb, suggesting that while some difference exist in TAD structure, the overall TAD structure remains intact between the 2iL/I/F and primed states (Figure S7E), To confirm that our in situ DNaseI Hi-C data are accurately capturing the 3D structure of the naive genome, we utilized cohesin chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) and CTCF ChIP-seq data from primed and reset naive hESCs, which previously revealed changes in looping structures (Ji et al., 2016). Most TADs have a CTCF binding site near their boundaries. We asked whether the reset naive CTCF ChIP-seq signal was also enriched at our 2iL/I/F-derived hESC TAD boundaries. Indeed, we found the CTCF signal to be enriched near our 2iL/I/F boundaries and that this enrichment was present in both our 2iL/I/F and primed hESCs (Figure 5E). This may have been expected, as Ji et al. (2016) found that 80% of CTCF binding sites were shared between their reset naive and primed hESC lines.
We also determined whether the cohesin ChIA-PET data could additionally validate our naive TAD boundary calls, since it was shown to recapitulate Hi-C TADs in H1 primed hESCs (Ji et al., 2016). An overlap analysis with cohesin PETs yielded 1,363 2iL/I/F and 1,818 primed TADs with at least one PET, from the respective cell type, whose termini are located within 40 kb of each boundary of a given TAD (Figure 5F). This corresponds to 22% of our 2iL/I/F TADs having a naive PET and 31% of primed TADs having a primed PET within 40 kb of the TAD boundary. We examined whether any of the PETs were localize at differential TAD boundaries, focusing on those boundaries in 2iL/I/F that are 80 kb or greater from the boundary in primed. Of 1,363 PETs near a 2iL/I/F boundary, 529 (39%) are near a 2iL/I/F differential TAD (Figure 5F). Although the authors note that their ChIA-PET data were undersaturated, these data confirm some of the structural differences observed in the 2iL/I/F 3D genome.
We investigated whether there was a relationship between higher-order chromatin structure at differential TAD boundaries and changes in chromatin modifications. We observe a significant enrichment for H3K4me1 and H3K27ac across differential TAD boundaries in 2iL/I/F relative to random (2iL/I/F H3K4me1 and H3K27ac p < 5 × 10−5), and a similar enrichment for primed H3K27me3 (p < 1 × 10−4) (Figure 6A). A clear example illustrating these differences in TAD and chromatin structure is the HOXA cluster, where a broad boundary spans the HOXA cluster in primed hESCs and is enriched for H3K27me3 (Figure 6B). The 2iL/I/F TAD to the left of the primed boundary is extended across the cluster and marked by H3K4me1 and H3K27ac. As mentioned previously, HOXA genes are expressed in 2iL/I/F cells. We asked whether there was a significant difference in the TAD structure around the HOXA locus between 2iL/I/F and primed cells. To do this we calculated the differential insulation scores by comparing the 2iL/I/F and primed insulation scores. The differential insulation score represents the differential TAD structure between two conditions. We observed that there was a noticeable drop in the differential insulation score at the HOXA locus, confirming that the TAD structure at the HOXA locus is different between 2iL/I/F and primed cells (Figure 6B).
Figure 6.

Changes in 3D Genome Architecture in 2iL/I/F hESCs
(A) Enrichment of ChIP peaks for histone marks H3K4me1, H3K27ac, H3K27me3, and H3K4me3 at overhanging TAD regions. The p value was calculated from a permutation test (N = 10,000 permutations) of randomized genomic regions with centromeres and telomeres masked.
(B) Interaction matrices of region of chr7 containing HOXA locus. Between matrices, horizontal bars with a vertical offset represent an individual TAD, 2iL/I/F in navy and primed in orange. The green bar indicates a primed boundary region >40 kb. Track of differential insulation score of 2iL/I/F versus primed cells around the HOXA locus nested in between TAD calls. ChIP-seq signal (RPKM) scaled from 0 to 20 for H3K4me1 and H3K27ac, scaled 0 to 30 for H3K27me3, 2iL/I/F in navy and primed in orange.
(C) Example of a 2iL/I/F-specific A compartment relative to primed. PC1 scale from −60 to 60.
(D) Heatmap of PC1 values at 2iL/I/F- and primed-specific A compartments. PC1 values at randomized compartments are displayed underneath. “N” and “P” denote 2iL/I/F and primed, respectively.
(E) Box plot of transposable element expression (RPKM) overlapping A to B compartment switches. “A to B” and “B to A” are 2iL/I/F to primed directions. Stable are compartments that do not switch. p values are computed using two-sample t test with one-sided alternative. ∗∗∗p < 2.2 × 10−16.
Finally, to compare the spatial organization of chromatin within the nuclei of 2iL/I/F and primed hESCs, we partitioned the genome into active and inactive (A/B) compartments by performing a PCA of each intrachromosomal contact matrix (Dixon et al., 2015, Lieberman-Aiden et al., 2009). Compartments identified using the first principal component (PC1) ranged in size from 40 kb to over 49 Mb in both cell types, with means of 3.6 Mb in 2iL/I/F cells and 3.4 Mb in primed. An overwhelming majority of compartments are static, with only 23 switching from being active in 2iL/I/F cells to inactive in primed (A to B), and 124 switching from being active in primed cells to inactive in 2iL/I/F (B to A; Figures 6C and 6D). While there is enrichment of primed-specific active compartments, a previous study showed that inactive B subcompartments are largely devoid of histone modifications, including H3K27me3 and H3K9me3 (Rao et al., 2014). It is therefore likely that the primed-specific active compartments are driven by the lack of repressive modifications in 2iL/I/F hESCs (alternatively, these are 2iL/I/F-specific inactive B compartments). Additionally, cell-specific active compartments are enriched for TE expression relative to stable compartments (Figure 6E), and gene expression to a lesser extent (Figure S7F).
Discussion
mESCs derived from the preimplantation blastocyst and grown in 2iL, as compared with the traditional serum and Lif, have been described as the ground state of pluripotency (Nichols and Smith, 2009, Silva and Smith, 2008). Recent gene expression studies of 2iL mESCs illustrated that their transcriptional and network analysis profiles reflect that of preimplantation blastocysts (Boroviak et al., 2014, Boroviak et al., 2015). Our comparison of single-cell RNA-seq from the developing human blastocyst (Blakeley et al., 2015) to 2iL/I/F and primed hESCs demonstrated an enrichment of key developmental genes and pathways, including WNT and TGF-β, in 2iL/I/F hESCs. In addition, we described the enrichment of hundreds of 2iL/I/F-specific genes that are expressed during early embryogenesis. Coupling transcriptome data with evidence that that the genome of naive hESCs are also hypomethylated, naive versus primed hESCs can be used to model epigenomic reprogramming that occurs as cells shift between these developmental states.
The early mouse embryo undergoes dramatic chromatin remodeling. Immunofluorescence studies in mouse embryos showed that during the first few hours after fertilization, H3K4me1 levels in the paternal genome increase to match maternal levels, while histone 3 lysine 9 dimethylation (H3K9me2) is rapidly removed from the maternal genome (Lepikhov and Walter, 2004, Sarmento et al., 2004, van der Heijden et al., 2005). This is accompanied by hyperacetylation of histones prior to zygotic genome activation (Adenot et al., 1997, Wiekowski et al., 1997). The presence of H3K4 methylation inhibits interaction of DNMT3L with DNA, thus potentially contributing to the hypomethylated epigenome (Ooi et al., 2007). Based on equivalent cells in mouse, naive and primed hESCs are thought to be reflective of early human embryogenesis, capturing the preimplantation and postimplantation states, respectively (Nichols and Smith, 2009). We observe novel features of the epigenome transitioning from naive to primed. We hypothesize that widespread deposition of H3K4me1 and histone acetylation are part of the mechanism to reset the zygotic genome along with known global DNA demethylation, and that this aids to open chromatin structure for, or in response to, genome activation.
These dramatic chromatin changes during embryogenesis are likely further altered upon implantation. For example, recent ChIP-seq results for H3K27ac in the mouse embryo and serum-maintained mESCs confirmed an enrichment of H3K27ac genome-wide post-ZGA followed by a decline in mESCs (Dahl et al., 2016). Enhancer decommissioning requires LSD1 activity (Whyte et al., 2012), which is inhibited by acetylation (Forneris et al., 2005, Lee et al., 2006). Deacetylation must, therefore, precede the removal of H3K4me1 by LSD1. This stepwise decommissioning was observed as cells exited the naive state, lending support to our hypothesis that enhancer premarking is a likely component of epigenetic reprogramming during embryogenesis.
Lastly, changes in chromatin modifications are reflected in changes in 3D genome architecture. Although primed TADs are largely unchanged upon hESC differentiation (Dixon et al., 2015), most 2iL/I/F TAD boundaries are unique to this pluripotent state, subsets of which are validated by recent ChIA-PET data. Our findings suggest that TAD structures are still formalizing prior to implantation, i.e., while overall TAD architecture is in place naive boundaries are expanded or contracted relative to primed cells. This was also recently observed in the mouse embryo, where 80% of inner cell mass (ICM) TAD boundaries are also present in mESCs, as the other TADs did not reach a mESC or somatic cell-like state until later development (Du et al., 2017). We, therefore, suspect a large fraction of 2iL/I/F TADs may also be reflected in the naive human ICM. Collectively, these characteristics are likely to shape naive pluripotency and provide new insights on epigenetic reprogramming through this model of development.
Experimental Procedures
Human Embryonic Stem Cell Culture
All human ESC culture conditions were as previously described (Sperber et al., 2015), with modifications described in Supplemental Experimental Procedures. Base media were supplemented with specific inhibitors for each growth condition.
Genome-wide Assays, Library Construction, and Sequencing
ChIP-seq was performed as previously described (Hawkins et al., 2013). Raw sequence reads were obtained from the Roadmap Epigenome Project (Hawkins et al., 2010). Replicates of aligned files were merged prior to peak calling. RNA-seq libraries, performed in duplicate, were constructed using the Scriptseq RNA-seq Library Preparation Kit. All Libraries were sequenced single-end 75 cycles on Illumina NextSeq or single-end 50 cycles on HiSeq 2500.
Computational Analysis
Details of computational analysis are provided in Supplemental Experimental Procedures. Sequencing raw reads were trimmed for low quality and adapters using TrimGalore!, and mapped to human genome (hg19) using Bowtie2 (ChIP-seq) (Langmead and Salzberg, 2012) and TopHat2 (RNA-seq) (Kim et al., 2013). Duplicates were merged. Transcript quantification was performed by Cufflinks (Trapnell et al., 2010) using GENCODE gene annotation release 19 as reference annotation. ChIP-seq peaks were called using MACS v1.4 using the --nomodel mode.
Author Contributions
R.D.H. and C.B.W. conceived the study. S.L.B., C.B.W., and R.D.H. planned the study. S.L.B. and R.D.H. wrote the manuscript. S.L.B. performed ChIP-seq, RNA-seq, RRBS, analyzed ChIP-seq and RRBS data, and contributed to analyses related to RNA-seq and Hi-C. N.D.J. analyzed RNA-seq data and performed related analyses, and contributed to Hi-C analysis. R.N.A. performed Hi-C and analyzed data. J.H. performed all cell culture with guidance from C.B.W., H.R.-B., J.M., and F.N.A. contributed to ChIP-seq experiments. E.G.O. performed FACS experiments and analysis. J.A.Z. contributed to Hi-C analysis.
Acknowledgments
The authors thank Dr. Gurkan Yardimci (PhD, Department of Genome Sciences, University of Washington, Seattle, WA, USA) for communication and advice on Hi-C analysis. R.D.H. is supported by funds from a John H. Tietze Award from the Tietze Foundation, the NIH/NIAMS (R01AR065952), NIH/NIDDK (R01DK103667), and the United States-Israel Binational Science Foundation (2013027). S.L.B. was supported by a Ford Foundation Fellowship Dissertation award administered by the National Academies of Sciences, Engineering, and Medicine and by the Molecular Medicine Training grant (T32GM095421). E.G.O. was supported by the Big Data for Genomics and Neuroscience training grant (T32LM012419).
Published: May 2, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2019.04.004.
Accession Numbers
All data are available from NCBI GEO accession number GEO: GSE90680.
Supplemental Information
References
- Adenot P.G., Mercier Y., Renard J.P., Thompson E.M. Differential H4 acetylation of paternal and maternal chromatin precedes DNA replication and differential transcriptional activity in pronuclei of 1-cell mouse embryos. Development. 1997;124:4615–4625. doi: 10.1242/dev.124.22.4615. [DOI] [PubMed] [Google Scholar]
- Ahmed K., Dehghani H., Rugg-Gunn P., Fussner E., Rossant J., Bazett-Jones D.P. Global chromatin architecture reflects pluripotency and lineage commitment in the early mouse embryo. PLoS One. 2010;5:e10531. doi: 10.1371/journal.pone.0010531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeley P., Fogarty N.M., del Valle I., Wamaitha S.E., Hu T.X., Elder K., Snell P., Christie L., Robson P., Niakan K.K. Defining the three cell lineages of the human blastocyst by single-cell RNA-seq. Development. 2015;142:3151–3165. doi: 10.1242/dev.123547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T., Loos R., Bertone P., Smith A., Nichols J. The ability of inner-cell-mass cells to self-renew as embryonic stem cells is acquired following epiblast specification. Nat. Cell Biol. 2014;16:516–528. doi: 10.1038/ncb2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T., Loos R., Lombard P., Okahara J., Behr R., Sasaki E., Nichols J., Smith A., Bertone P. Lineage-specific profiling delineates the emergence and progression of naive pluripotency in mammalian embryogenesis. Dev. Cell. 2015;35:366–382. doi: 10.1016/j.devcel.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan Y.S., Goke J., Ng J.H., Lu X., Gonzales K.A., Tan C.P., Tng W.Q., Hong Z.Z., Lim Y.S., Ng H.H. Induction of a human pluripotent state with distinct regulatory circuitry that resembles preimplantation epiblast. Cell Stem Cell. 2013;13:663–675. doi: 10.1016/j.stem.2013.11.015. [DOI] [PubMed] [Google Scholar]
- Chen Y.-G., Li Z., Wang X.-F. Where PI3K/Akt meets smads: the crosstalk determines human embryonic stem cell fate. Cell Stem Cell. 2012;10:231–232. doi: 10.1016/j.stem.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier A.J., Panula S.P., Schell J.P., Chovanec P., Plaza Reyes A., Petropoulos S., Corcoran A.E., Walker R., Douagi I., Lanner F. Comprehensive cell surface protein profiling identifies specific markers of human naive and primed pluripotent states. Cell Stem Cell. 2017;20:874–890.e7. doi: 10.1016/j.stem.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane E., Bian Q., McCord R.P., Lajoie B.R., Wheeler B.S., Ralston E.J., Uzawa S., Dekker J., Meyer B.J. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 2015;523:240–244. doi: 10.1038/nature14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U S A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl J.A., Jung I., Aanes H., Greggains G.D., Manaf A., Lerdrup M., Li G., Kuan S., Li B., Lee A.Y. Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature. 2016;537:548–552. doi: 10.1038/nature19360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X., Ma W., Ramani V., Hill A., Yang F., Ay F., Berletch J.B., Blau C.A., Shendure J., Duan Z. Bipartite structure of the inactive mouse X chromosome. Genome Biol. 2015;16:152. doi: 10.1186/s13059-015-0728-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y., Ye Z., Kim A., Rajagopal N., Xie W. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z., Zheng H., Huang B., Ma R., Wu J., Zhang X., He J., Xiang Y., Wang Q., Li Y. Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature. 2017;547:232–235. doi: 10.1038/nature23263. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forneris F., Binda C., Vanoni M.A., Battaglioli E., Mattevi A. Human histone demethylase LSD1 reads the histone code. J. Biol. Chem. 2005;280:41360–41365. doi: 10.1074/jbc.M509549200. [DOI] [PubMed] [Google Scholar]
- Gafni O., Weinberger L., Mansour A.A., Manor Y.S., Chomsky E., Ben-Yosef D., Kalma Y., Viukov S., Maza I., Zviran A. Derivation of novel human ground state naive pluripotent stem cells. Nature. 2013;504:282–286. doi: 10.1038/nature12745. [DOI] [PubMed] [Google Scholar]
- Giorgetti L., Lajoie B.R., Carter A.C., Attia M., Zhan Y., Xu J., Chen C.J., Kaplan N., Chang H.Y., Heard E. Structural organization of the inactive X chromosome in the mouse. Nature. 2016;535:575–579. doi: 10.1038/nature18589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H., Zhu P., Yan L., Li R., Hu B., Lian Y., Yan J., Ren X., Lin S., Li J. The DNA methylation landscape of human early embryos. Nature. 2014;511:606–610. doi: 10.1038/nature13544. [DOI] [PubMed] [Google Scholar]
- Guo G., von Meyenn F., Santos F., Chen Y., Reik W., Bertone P., Smith A., Nichols J. Naive pluripotent stem cells derived directly from isolated cells of the human inner cell mass. Stem Cell Reports. 2016;6:437–446. doi: 10.1016/j.stemcr.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J., Cheng A.W., Saha K., Kim J., Lengner C.J., Soldner F., Cassady J.P., Muffat J., Carey B.W., Jaenisch R. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc. Natl. Acad. Sci. U S A. 2010;107:9222–9227. doi: 10.1073/pnas.1004584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins R.D., Hon G.C., Lee L.K., Ngo Q., Lister R., Pelizzola M., Edsall L.E., Kuan S., Luu Y., Klugman S. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins R.D., Hon G.C., Yang C., Antosiewicz-Bourget J.E., Lee L.K., Ngo Q.M., Klugman S., Ching K.A., Edsall L.E., Ye Z. Dynamic chromatin states in human ES cells reveal potential regulatory sequences and genes involved in pluripotency. Cell Res. 2011;21:1393–1409. doi: 10.1038/cr.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins R.D., Larjo A., Tripathi S.K., Wagner U., Luu Y., Lonnberg T., Raghav S.K., Lee L.K., Lund R., Ren B. Global chromatin state analysis reveals lineage-specific enhancers during the initiation of human T helper 1 and T helper 2 cell polarization. Immunity. 2013;38:1271–1284. doi: 10.1016/j.immuni.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X., Dadon D.B., Powell B.E., Fan Z.P., Borges-Rivera D., Shachar S., Weintraub A.S., Hnisz D., Pegoraro G., Lee T.I. 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell. 2016;18:262–275. doi: 10.1016/j.stem.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.G., Wynder C., Bochar D.A., Hakimi M.A., Cooch N., Shiekhattar R. Functional interplay between histone demethylase and deacetylase enzymes. Mol. Cell. Biol. 2006;26:6395–6402. doi: 10.1128/MCB.00723-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepikhov K., Walter J. Differential dynamics of histone H3 methylation at positions K4 and K9 in the mouse zygote. BMC Dev. Biol. 2004;4:12. doi: 10.1186/1471-213X-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Kim J.M., Aoki F. Regulation of histone H3 lysine 9 methylation in oocytes and early pre-implantation embryos. Development. 2004;131:2269–2280. doi: 10.1242/dev.01116. [DOI] [PubMed] [Google Scholar]
- Meshorer E., Yellajoshula D., George E., Scambler P.J., Brown D.T., Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell. 2006;10:105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody J.D., Levy S., Mathieu J., Xing Y., Kim W., Dong C., Tempel W., Robitaille A.M., Dang L.T., Ferreccio A. First critical repressive H3K27me3 marks in embryonic stem cells identified using designed protein inhibitor. Proc. Natl. Acad. Sci. U S A. 2017;114:10125–10130. doi: 10.1073/pnas.1706907114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niakan K.K., Eggan K. Analysis of human embryos from zygote to blastocyst reveals distinct gene expression patterns relative to the mouse. Dev. Biol. 2013;375:54–64. doi: 10.1016/j.ydbio.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Nichols J., Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4:487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Ooi S.K., Qiu C., Bernstein E., Li K., Jia D., Yang Z., Erdjument-Bromage H., Tempst P., Lin S.P., Allis C.D. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker S.C., Stitzel M.L., Taylor D.L., Orozco J.M., Erdos M.R., Akiyama J.A., van Bueren K.L., Chines P.S., Narisu N., Black B.L. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. U S A. 2013;110:17921–17926. doi: 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A., Bajpai R., Swigut T., Brugmann S.A., Flynn R.A., Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossant J. Mouse and human blastocyst-derived stem cells: vive les differences. Development. 2015;142:9–12. doi: 10.1242/dev.115451. [DOI] [PubMed] [Google Scholar]
- Sarmento O.F., Digilio L.C., Wang Y., Perlin J., Herr J.C., Allis C.D., Coonrod S.A. Dynamic alterations of specific histone modifications during early murine development. J. Cell Sci. 2004;117:4449–4459. doi: 10.1242/jcs.01328. [DOI] [PubMed] [Google Scholar]
- Shin H., Shi Y., Dai C., Tjong H., Gong K., Alber F., Zhou X.J. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2016;44:e70. doi: 10.1093/nar/gkv1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J., Smith A. Capturing pluripotency. Cell. 2008;132:532–536. doi: 10.1016/j.cell.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperber H., Mathieu J., Wang Y., Ferreccio A., Hesson J., Xu Z., Fischer K.A., Devi A., Detraux D., Gu H. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015;17:1523–1535. doi: 10.1038/ncb3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y., Guo G., Loos R., Nichols J., Ficz G., Krueger F., Oxley D., Santos F., Clarke J., Mansfield W. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158:1254–1269. doi: 10.1016/j.cell.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen T.W., Powell B.E., Wang H., Mitalipova M., Faddah D.A., Reddy J., Fan Z.P., Maetzel D., Ganz K., Shi L. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell. 2014;15:471–487. doi: 10.1016/j.stem.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen T.W., Friedli M., He Y., Planet E., O'Neil R.C., Markoulaki S., Pontis J., Wang H., Iouranova A., Imbeault M. Molecular criteria for defining the naive human pluripotent state. Cell Stem Cell. 2016;19:502–515. doi: 10.1016/j.stem.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valamehr B., Robinson M., Abujarour R., Rezner B., Vranceanu F., Le T., Medcalf A., Lee T.T., Fitch M., Robbins D. Platform for induction and maintenance of transgene-free hiPSCs resembling ground state pluripotent stem cells. Stem Cell Reports. 2014;2:366–381. doi: 10.1016/j.stemcr.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden G.W., Dieker J.W., Derijck A.A., Muller S., Berden J.H., Braat D.D., van der Vlag J., de Boer P. Asymmetry in histone H3 variants and lysine methylation between paternal and maternal chromatin of the early mouse zygote. Mech. Dev. 2005;122:1008–1022. doi: 10.1016/j.mod.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Ware C.B. Concise review: lessons from naïve human pluripotent cells. Stem Cells. 2016;35:35–41. doi: 10.1002/stem.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware C.B., Nelson A.M., Mecham B., Hesson J., Zhou W., Jonlin E.C., Jimenez-Caliani A.J., Deng X., Cavanaugh C., Cook S. Derivation of naive human embryonic stem cells. Proc. Natl. Acad. Sci. U S A. 2014;111:4484–4489. doi: 10.1073/pnas.1319738111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte W.A., Bilodeau S., Orlando D.A., Hoke H.A., Frampton G.M., Foster C.T., Cowley S.M., Young R.A. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482:221–225. doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiekowski M., Miranda M., Nothias J.Y., DePamphilis M.L. Changes in histone synthesis and modification at the beginning of mouse development correlate with the establishment of chromatin mediated repression of transcription. J. Cell Sci. 1997;110(Pt 10):1147–1158. doi: 10.1242/jcs.110.10.1147. [DOI] [PubMed] [Google Scholar]
- Wu J., Izpisua Belmonte J.C. Dynamic pluripotent stem cell states and their applications. Cell Stem Cell. 2015;17:509–525. doi: 10.1016/j.stem.2015.10.009. [DOI] [PubMed] [Google Scholar]
- Yan L., Yang M., Guo H., Yang L., Wu J., Li R., Liu P., Lian Y., Zheng X., Yan J. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013;20:1131–1139. doi: 10.1038/nsmb.2660. [DOI] [PubMed] [Google Scholar]
- Zhang H.M., Liu T., Liu C.J., Song S., Zhang X., Liu W., Jia H., Xue Y., Guo A.Y. AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 2015;43:D76–D81. doi: 10.1093/nar/gku887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Choi M., Margineantu D., Margaretha L., Hesson J., Cavanaugh C., Blau C.A., Horwitz M.S., Hockenbery D., Ware C. HIF1alpha induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012;31:2103–2116. doi: 10.1038/emboj.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerlin L., Park T.S., Zambidis E.T. Capturing human naive pluripotency in the embryo and in the dish. Stem Cells Dev. 2017;26:1141–1161. doi: 10.1089/scd.2017.0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.