

Summary
Mutations in RNA processing enzymes are increasingly linked to human disease. Telomerase RNA and related noncoding RNAs require 3’-end-processing steps including oligoadenylation. Germline mutations in poly(A)ribonuclease (PARN) cause accumulation of extended human telomerase RNA (hTR) species and precipitate dyskeratosis congenita and pulmonary fibrosis. Here, we develop nascent RNAend-Seq to quantitatively measure processing rates of RNA precursors. We find that mature hTR derives from extended precursors and with delayed kinetics compared to related RNAs. In PARN-mutant cells, telomerase levels diminish because hTR maturation kinetically stalls, leaving unprocessed precursors to be degraded. Loss of poly(A)polymerase PAPD5 in PARN-mutant cells accelerates hTR maturation and restores normal hTR processing, indicating that oligoadenylation and deadenylation set the rate of hTR maturation. These data reveal a feedforward circuit that slows RNA maturation, limits steady-state RNA levels and buffers transcriptional noise. Similar alterations in RNA processing rates may broadly contribute to mechanisms of RNA-based human disease.
Graphical Abstract
ETOC
Mutations in poly(A)ribonuclease (PARN) cause accumulation of telomerase RNA component (hTR) precursors and underlie a subset of Dyskeratosis Congenita. Using nascent RNAend-Seq to measure maturation rates of ncRNA precursors, Roake et al show that posttranscriptional oligoadenylation by PAPD5 and deadenylation by PARN set hTR maturation rate and determine telomerase levels.
Introduction
Abnormal RNA processing represents an emerging cause of tissue failure, developmental disorders and neurodegenerative disease (Burghes and Beattie, 2009). Putative defects in mRNA splicing or small noncoding RNA metabolism have been linked to diverse diseases. Mutations in telomerase pathway genes cause a stem cell disorder, dyskeratosis congenita (DC), associated with widespread tissue defects (Kirwan and Dokal, 2008). Telomerase is a ribonucleoprotein complex whose catalytic core includes telomerase reverse transcriptase TERT and an RNA component hTR that templates addition of telomeric repeats to chromosome ends (Pfeiffer and Lingner, 2013). hTR biogenesis occurs through a series of complex steps and is disrupted in patients with DC or familial idiopathic pulmonary fibrosis (IPF), a lethal and progressive interstitial lung disease (Stuart et al., 2015; Vulliamy et al., 2001). For telomerase and small noncoding RNAs more broadly the specific steps in RNA processing are challenging to reveal in vivo. Mechanisms governing precise formation of hTR and related H/ACA RNAs remain unclear; specifically, understanding how transcription termination occurs, the role of post-transcriptional modifications, and the method of trimming precursor RNAs to generate precise 3’ ends.
Despite the importance of hTR in scaffolding the telomerase enzyme and supporting telomerase activity, the biogenesis of this noncoding RNA is incompletely understood. Closely related vertebrate small Cajal body RNAs (scaRNAs) and small nucleolar RNAs (snoRNAs) are encoded in introns and liberated from the intron lariat structure before being trimmed to their mature forms (Matera et al., 2007). As an exception, hTR and a small subset of snoRNAs are transcribed by RNA polII from dedicated promoters (Feng et al., 1995; Lestrade and Weber, 2006). hTR transcripts in mammalian cells show a diversity of 3’ ends at steady state which were revealed by 3’ Rapid Amplification of cDNA Ends (3’ RACE) with approximately one-third of hTR molecules possessing genomically templated tails extending beyond the annotated 451 nt end (Goldfarb and Cech, 2013). A majority of extended molecules possess short untemplated oligo-A tails. The poly(A)polymerase (PAP) PAPD5 (TRF4-2) is the major poly(A)polymerase for snoRNAs in vertebrates (Berndt et al., 2012) and contributes to oligoadenylation and regulation of hTR levels (Nguyen et al., 2015; Shukla et al., 2016; Tseng et al., 2015).
The one-third of hTR molecules with 3’ ends extending beyond position 451 may either be precursors to the mature RNA or non-productive transcripts destined for destruction. The discovery that mutations in the exonuclease poly(A)ribonuclease (PARN) can cause DC and IPF through improper post-transcriptional processing of hTR supported the former model and suggested PARN may trim the extended hTR precursor (Moon et al., 2015; Stuart et al., 2015; Tummala et al., 2015). Knockdown of PARN in cell lines results in under-accumulation of hTR with a concomitant loss of telomerase activity. In addition, PARN-mutant iPS cells derived from DC patients show telomere shortening (Moon et al., 2015). However, PARN enzyme exhibits preferential nuclease activity towards poly(A) substrates in vitro, and PARN-mutant cells continue to generate mature hTR (Henriksson et al., 2010; Shukla et al., 2016; Wu et al., 2005). These observations indicate PARN contributes to processing, but it remains to be determined whether its action is restricted to oligo-adenylated residues or extends to trimming genomically encoded tails.
Processing of hTR transcripts is critical because a subset of hTR molecules become targets for the nuclear RNA surveillance pathway mediated by the RNA exosome. The exosome is recruited to targets by the human homolog of the yeast TRAMP complex containing PAPD5, Air2 homolog ZCCHC7, and MTR4 (LaCava et al., 2005). Knockdown of exosome or exosomal targeting components in human cell lines causes increased hTR levels (Tseng et al., 2015). Knockdown of exosome components can rescue hTR destabilization associated with loss of dyskerin binding or with PARN depletion, suggesting the exosome removes hTR transcripts that are improperly assembled or processed (Shukla et al., 2016). These data support a model in which processing of hTR competes with degradation.
Vertebrate telomerase RNAs (TERs) belong to a scaRNA family containing a box H/ACA motif, a two-hairpin structure connected by a single stranded hinge and bordered on the 3’ end by a single stranded tail containing the sequence ACA (Kiss et al., 2006; Mitchell et al., 1999). H/ACA box-containing scaRNAs and snoRNAs serve as guides for psuedouridylation of rRNA and small nuclear RNAs (snRNAs), respectively (Matera et al., 2007). The dyskerin-NOP10- NHP2 complex binds the H/ACA box, stabilizes the RNAs and executes pseudouridylation on targets of the small RNA guides. Incorporation of the H/ACA sequence in vertebrate TERs serves an important role in telomerase RNA biogenesis as mutations in the H/ACA box of hTR or mutations in components of the dyskerin complex cause hTR loss and DC (Fu and Collins, 2003; Mitchell et al., 1999). A 3 nt sequence in the terminal loop of the hTR H/ACA domain termed the CAB box binds to TCAB1 which regulates both catalytic activity of telomerase and directs trafficking of the enzyme to Cajal bodies and to telomeres (Chen et al., 2018; Venteicher et al., 2009; Zhong et al., 2011).
Noncoding RNAs (ncRNAs) serve important roles in the cell and recently a number of diseases have been linked to impaired ncRNA processing. Pontocerebellar hypoplasia (PCH) is a heterogeneous set of related diseases characterized by neurodevelopment defects (MIM # PS607596). Many PCH genes are required for RNA processing, including components of the RNA exosome and the RNA exonuclease TOE1 (Lardelli et al., 2017; Wan et al., 2012). Mutations in snRNA assembly factor SMN cause spinal muscular atrophy (SMA), a progressive loss of motor neurons accounting for the leading genetic cause of infant deaths (Burghes and Beattie, 2009). Understanding molecular mechanisms underlying this diverse set of diseases requires an understanding of how RNA is metabolized in the cell and consequences of perturbing these processes.
To understand how hTR and snoRNAs are processed in vivo, we developed a means for quantitatively measuring maturation of RNA precursors using nascent RNA sequencing. This approach defined the hTR precursor and revealed that defects in hTR maturation kinetics rather than a block in hTR processing underlie telomerase loss in PARN-mutant DC.
Results
Identification of RNA precursors using nascent RNAend-Seq
All small noncoding RNAs (ncRNAs) require specific mechanisms to ensure proper formation of 3’ ends. For small ncRNAs, development of 3’ RACE sequencing enabled a complete description of these extended RNA species in cells at steady-state (Goldfarb and Cech, 2013). However, determining the relationship between extended species and the mature RNA, or revealing the dynamics of stepwise conversion of elongated species to processed RNA has not been possible. Furthermore, extended species are likely a mixture of transcripts destined to mature and transcripts that will be discarded through RNA surveillance (Tseng et al., 2015).
We analyzed hTR molecules in steady-state (SS) using 3’ RACE sequencing in a collection of human cell lines. Sequencing of SS-RNA in HeLa cervical carcinoma cells, human embryonic stem cells (hES), and UMUC3 bladder carcinoma cells indicated that hTR is a heterogeneous pool of molecules (Figure 1A), consistent with previous findings (Goldfarb and Cech, 2013). The majority of transcripts are 451 nts long and a subset have short genomically templated tails, often with additional untemplated adenosines (Figure 1A). Two general hypotheses could explain this pattern: (1) extended molecules are degradation intermediates or (2) extended species are precursors of the mature 451 nt species. To identify precursor molecules of RNA in an unbiased fashion, we developed a technique for sequencing nascent RNA ends. Nascent RNAend-Seq combines the nascent RNA labeling technique applied to mRNAs (Radle et al., 2013) with 3’ RACE (Goldfarb and Cech, 2013) and analyzed using high throughput sequencing (Figure 1B). Nascent RNA was labeled by treating cultured cells with nucleotide analogue 4-thiouridine (4SU) (Figure 1B). We estimated labeling efficiency by streptavidin (SA) dot blot to achieve sufficient labeling of nascent RNA while minimizing 4SU exposure (Figure S1A). With one-hour 4SU labeling, we recovered 1.7% of input RNA as nascent compared to 0.04% in untreated cells, indicating a very low non-nascent RNA contamination (Figure 1C). To understand how effectively this protocol discriminates between nascent RNA and SS-RNA, we performed qRT-PCR for well-known RNA precursors including the 47S rRNA precursor and intron-containing beta2-microglobulin (B2M) hnRNA. We found these short-lived RNAs were>1000 fold enriched in nascent RNA compared to SS-RNA (Figure 1D). To determine whether we could isolate small ncRNA precursors, we analyzed U2 snRNA, which is transcribed by RNA polII and cleaved near a cis element termed the 3’ box (Hernandez, 1985). The U2 precursor is trimmed to the mature 3’ snRNA end by exonucleases including TOE1 (Lardelli et al., 2017). Analysis of U2 snRNA using nascent RNAend-Seq revealed enrichment in nascent RNA of a 10 nt extended precursor (Figure 1E). These results show nascent RNAend-seq enriches for short-lived RNA precursors.
Figure 1: Identification of RNA precursors using nascent RNAend-Seq.

A. 3’ RACE-Seq of hTR in human embryonic stem (hES), HeLa, and UMUC3 cells. hTR transcripts either end at the annotated end of 451 nts or shorter (brown), are >451 nts with genomically templated tails (grey), or are >451 nts with untemplated oligo-A tails (blue). hTR Reads≥200k
B. Nascent RNAend-Seq to enrich nascent RNA tails. 4SU labeled RNA from pulsed cells is conjugated to biotin (Biotin-HPDP, B). Biotin-conjugated nascent RNA is captured with streptavidin (SA) beads and separated from steady state (SS) RNA. Nascent RNA is eluted with DTT, ligated with a 3’ ssDNA adapter, reverse transcribed (RT) and amplified with a gene-specific primer (GSP)
C. RNA bioanalyzer shows 1.7% of 4SU labeled input RNA is recovered vs. 0.04% of 4SU-unlabeled RNA.
D. qRT-PCR for RNA precursors including 47S rRNA and beta-2-microglobulin (B2M) in nascent RNA (blue) and steady state RNA (black). 47S rRNA primers span 5’ externally transcribed spacer (ETS) and B2M primers span an intron-exon junction. Data normalized to exonic GAPDH primers. Error bars represent S.E.M.
E. Sequencing of nascent U2 3’ end with the proportion of reads having a given tail length. Height of the line at templated tail length=0 indicates the proportion of reads with no tail; height of the line at templated tail length=10 indicated the proportion of reads with a tail≤10 nucleotides. Structure of the U2 snRNA transcription unit shown with annotated U2 3’ end graphed at 0 nts and 3’ box at 17 nts. Nascent RNAend-Seq results for U2 nascent (blue line) and steady state (black line) RNA.
hTR is transcribed as an extended molecule that accumulates in PARN-KO cells
Unlike nearly all vertebrate snoRNAs and scaRNAs, hTR is expressed from its own RNA PolII-promoter and the mechanism for its transcriptional termination as well as the length of its transcriptional unit remain undefined (Feng et al., 1995). To gain additional insights into the nature of the extended oligoadenylated hTR species evident in SS-RNA, we applied nascent RNAend-Seq to hTR transcripts and to related snoRNAs U17a, U68, U3, and U8 (Figure 2A)(Figure S2B,C). In nascent RNA, extended hTR species represented 75% of species in HeLa cells labeled for 1 hour and 72% of species in hES labeled for 4 hours, with a maximum templated tail length of 51 nts (Figure S2A). For U8 snoRNA we were able to detect abundant transcripts>100 nt in length, suggesting nascent RNAend-Seq can detect longer precursors (Figure S2A). In addition, many nascent hTR molecules were oligoadenylated (36% in HeLa, 37% in hES). In contrast, SS hTR molecules from the same cells were predominately comprised of the 451 nt mature species (extended RNAs HeLa 19%, hES 15%). The abundance of oligoadenylated hTR molecules was reduced in SS (HeLa 15%, hES 12%), reflecting the elevated representation of extended molecules in the nascent fraction (Figure S2D). These findings show the hTR locus produces an extended precursor that is rapidly oligoadenylated.
Figure 2: hTR is transcribed as an extended molecule that accumulates in PARN-KO cells.

A. Nascent RNAend-Seq of hTR in HeLa and hES cells with tail profiles for nascent (N) and steady-state (SS) transcripts. Cells are pulsed for 1 hour (1H) or 4 hours (4H). Grey shading indicates 95% CI intervals from 2 biologic replicates. Percent of hTR reads composed of oligo-A transcripts plotted for nascent (N) and steady state (SS) HeLa and hES hTR transcripts. Error bars are S.E.M. hTR Reads≥12k.
B. Nascent RNAend-Seq of H/ACA snoRNAs U17a and U68 with tail profiles for nascent and steady-state transcripts. Gray shading indicates 95% CI derived from 2 biologic replicates. Diagram of U17a and U68 intronic loci shows distance to nearest exon. Percent of snoRNA reads composed of oligo-A transcripts is plotted for nascent (N) and steady state (SS) U68 and U17a transcripts. U68 reads≥5k, U17a reads≥7k
C. Western blot (WB) of PARN CRISPR-KO clones and rescued cells. Parental (WT) cells and PARN-KO clones (4,2,6) and PARN-KO clone 4 transduced with lentiviral Flag-GFP (FL-GFP) or Flag-PARN (FL-PARN).
D. Telomere Repeat Amplification Protocol (TRAP) in parental (WT) and PARN-KO (clones 2,4,6) HeLa extract as well as TCAB1-KO extract. Each sample has 1x and 4x dilutions. Buffer indicates no extract, +RNAse indicates RNAse A added to WT extract. Presence of internal control (I.C) indicates successful PCR. TRAP activity relative to WT is quantified (right) and represents the results of 3 experiments. Error bars are S.E.M.
E. qRT-PCR of hTR relative to parental (WT) and normalized to GAPDH in PARN-KO cells (clones 2,4,6) and clone 4 with either GFP or PARN expressed. Error bars are S.E.M.
F. 3’ RACE-Seq of hTR in parental (black), PARN-KO+GFP (green), PARN-KO+PARN cells (grey). Percent of oligo-A hTR reads are plotted. hTR Reads≥160k
G. W.B. of HeLa or PARN-KO clone 4 HeLa cells stably transduced with FL-GFP or FL-PARN cDNA.
H. qRT-PCR of hTR relative to parental (WT+GFP) and normalized to GAPDH in PARN-KO cells with GFP, PARN WT or PARN-mutant cDNA stably expressed.
I. qRT-PCR of extended hTR relative to parental (WT+GFP) and normalized to total hTR in PARN-KO cells with GFP, PARN-WT or PARN-mutant cDNA stably expressed. Position of reverse primers is shown
To ask whether similar precursors exist for other H/ACA RNAs, we sequenced nascent 3’ ends of U17a and U68, two intron-encoded H/ACA snoRNAs. The mature 3’ ends of U17a and U68 are 556 and 197 nts distant from the nearest 3’ splice site, respectively, defining the maximum length of their precursors (Figure 2B). For both U17a and U68, a high proportion of nascent molecules were extended (mean extended 46% U17a, 77% U68). Both U17a and U68 nascent transcripts were oligoadenylated (mean oligo-A 5% U17a, 20% U68) (Figure 2B). We did not capture the full length of the intronic sequences in nascent RNA, suggesting initial processing may be rapid.
PARN is mutated in telomere diseases and PARN depletion results in an accumulation of extended oligoadenylated hTR molecules (Boyraz et al., 2016; Moon et al., 2015). To compare the pattern of hTR species in nascent RNA with those in PARN-deficient cells, we generated PARN knockout HeLa cells clones using CRISPR/Cas9 and stably expressed either GFP or PARN in a PARN-KO clone (Figure 2C). Consistent with the role of PARN in telomere disease, PARN-KO cells showed a 53% reduction in telomerase activity compared to WT cells (Figure 2D). Telomerase activity in PARN-KO cells was higher than in TCAB1-KO cells (83% reduced), in which hTR conformation is perturbed by loss of TCAB1 binding (Chen et al., 2018) (Figure 2D). PARN-KO cells exhibited a reduced SS level of hTR by northern blot or by qRT-PCR, and reduced hTR levels were rescued by expression of PARN cDNA (Figure 2E, Figure S2E). In PARN-KO cells, extended hTR molecules were markedly increased (42% PARN-KO+GFP vs. 12% in PARN-KO+PARN) and oligoadenylated species were elevated (36% PARN-KO+GFP vs. 11% in PARN-KO+PARN), consistent with reports from PARN knockdown cells (Moon et al., 2015; Tseng et al., 2015) (Figure 2F). Therefore, the SS hTR profile from the PARN-KO cells represents an intermediate case between nascent hTR molecules and SS hTR molecules in a PARN-WT cell (42% extended in WT PARN-KO vs. 75% in WT nascent vs. 19% in WT SS) suggesting hTR maturation is impaired in the absence of PARN.
We asked whether disease-linked mutations in PARN were deficient in supporting hTR maturation. We cloned 3 PARN mutants from families with pulmonary fibrosis (Q177X, R251Efs*14, K421R) as well as a PARN mutant predicted to have no catalytic activity (H377A) (Stuart et al., 2015; Wu et al., 2005). Transiently transfected cDNA encoding PARN-Q177X and PARN-R251Efs*14 generated truncated proteins, whereas PARN-K421R and PARN-H377A yielded full-length protein (Figure S2F, G). Stable expression of PARN-mutant cDNAs in PARN-KO cells revealed two full-length mutants (K421R and H377A) accumulated at levels near to endogenous PARN, while PARN-Q177X did not accumulate and PARN-R251Efs*14 accumulated at reduced levels (Figure 2G). Expression of WT PARN in PARN-KO cells, but not expression of 3 patient mutants, rescued hTR levels (Figure 2H). Interestingly, PARN H377A expression caused partial rescue of hTR levels (85% of WT levels in PARN-KO+H337A vs. 44% in PARN-KO+GFP) (Figure 2H). To test whether this mutant could support maturation of extended hTR molecules, we designed a qRT-PCR strategy that utilizes a reverse primer annealed downstream of the mature hTR 3’ end to specifically amplify extended but not mature hTR transcripts. PARN-KO cells showed a 3-5 fold increase of extended hTR in this assay (Figure 2I). Expression of WT PARN cDNA suppressed the increase in extended hTR, but expression of either the 3 patient PARN-mutants or PARN-H377A failed to decrease the levels of extended hTR (Figure 2I). We conclude that the 3 PARN-mutant proteins are deficient in promoting hTR maturation and accumulation. The PARN catalytically dead variant is also deficient in promoting hTR maturation; however, hTR accumulation is partially rescued by PARN-H377A, suggesting this variant may protect extended hTR from degradation.
Extended nascent hTR molecules convert to the mature species with slow kinetics
Extended hTR species in nascent RNA could represent hTR precursors or unproductive hTR transcripts targeted by RNA surveillance. Nuclear RNA surveillance acts on hTR and may destroy some hTR transcripts (Tseng et al., 2015). To distinguish these possibilities, we designed a pulse-chase experiment where labeled nascent RNA molecules are followed over time and analyzed using nascent RNAend-Seq (Figure 3A). To characterize RNA maturation in chased fractions, we assayed total nascent RNA using RNA bioanalyzer. In early RNA fractions, high molecular weight bands representing hnRNAs and rRNA precursors are present but diminish over the 24 hr chase (Figure 3B). Nascent RNAend-Seq showed only 24% of labeled hTR transcripts were mature at t=0 hrs, labeled RNAs became increasingly mature during the chase (67% t=8 hrs; 96% mature t=24 hrs) (Figure 3C, Table S3). Analyses of these samples using gel-based 3’ RACE gave similar results (Figure S3A). Concomitant with maturation, the extent of oligoadenylation diminished during the chase. We repeated these analyses for snRNA U2 and snoRNAs U3 and U17a and found similar patterns of extended molecules evolving into mature molecules (Figure S3B). During this process, overall levels of hTR were maintained, indicating the extended species primarily undergo maturation rather than degradation (Figure 3D). These data demonstrate extended oligoadenylated hTR molecules are precursors for mature hTR-451.
Figure 3: Extended nascent hTR molecules convert to the mature species with slow kinetics.

A. HeLa cells are pulsed for 1 hr with 4SU and chased with uridine-containing media for 0, 5, 8, 12 or 24 hrs.
B. RNA bioanalyzer gel of total nascent RNA isolated from (A). Sizes of 28S rRNA, 18S rRNA are marked
C. Nascent RNAend-Seq of hTR in pulse-chase RNA. Data is derived from≥2 independent sequencing experiments. Gray shading is 95% CI derived from 2 biologic replicates. Percent of oligo-A reads from the same cohorts are graphed. hTR Reads≥15k
D. qRT-PCR of hTR in chased RNA fractions normalized to a spike-in IVT luciferase RNA to control for immunoprecipitation efficiency and to either U1 or U3. Error bars represent S.E.M.
E. ncRNA molecules greater than annotated length are defined as precursors, those equal to or shorter than annotated length are defined as mature, and precursor fraction is plotted vs. time since pulse induction in hours (t0=1 hr, t5=6 hr, t8=9 hr, t12= 13 hr, t24=25 hr). For each time point, at least two biologic replicates are plotted. 3 time points plotted for U17a, U3, and U68 while 5 time points plotted for U2 and hTR. Precursor percentage at time of transcription is assumed to be 100% and the fraction is assumed to approach 0%. Exponential decay curves are fitted to the data using the equation Precursor(time)=Precursor(initial)e−k(time) or P(t) = P(0)e−kt.
F. Half-lives of precursors are calculated from curves fitted in (E) following the equation . Ind=independent locus. Precursor % at steady state represents the percent that the precursor comprises in steady-state RNA.
To understand the kinetics of RNA precursor maturation, we plotted percentage of precursor molecules vs. time. Pulse-chase data for hTR as well as U2 snRNA, U17a H/ACA snoRNA, U68 H/ACA snoRNA, and U3 C/D snoRNA were analyzed. For each RNA, conversion of precursor to mature species followed exponential decay kinetics, characterized by diminishing maturation rates at lower concentrations of substrate (Figure 3E). First order-kinetics are consistent with processing of the precursor substrates by one or more enzymes. We extracted half-lives (t1/2) for maturation of each RNA from the fitted curves. Maturation t1/2 for hTR was greater than for U17a H/ACA snoRNA and U3 C/D snoRNA (4.94 hr, 0.92 hr and 0.60 hr), indicating hTR matures more slowly than conventional snoRNAs. Maturation t1/2 for U2 snRNA was closer to that of hTR (3.19 hr) (Figure 3E, F), reflecting the multi-step processing of snRNA transcripts (Patel and Bellini, 2008). To compare our hTR kinetic values to similar calculations, we observed maturation t1/2 is related to decay t1/2 of mature RNA and to the proportion of precursor in the SS-RNA pool. Using decay rates from label-free experiments in the literature we calculate maturation t1/2 of hTR to be within 3-8 hrs which encompasses our measured t1/2 of 4.94 hours (Figure S3C). These data reveal extended oligoadenylated species represent precursors of mature hTR-451 and maturation of extended precursors occurs slowly compared to related small non-coding RNAs.
RNA processing enzymes PARN and PAPD5 define the rate of hTR precursor maturation
PARN is proposed to both deadenylate oligo-A containing precursors and to trim templated residues to generate mature hTR-451 (Tseng et al., 2015). However, our results showing diminished but persistent expression of mature hTR-451 suggest PARN is not essential for hTR processing. Instead, we hypothesized reduced hTR in PARN-mutant cells could reflect a reduction in hTR maturation rate. To answer this question, we performed pulse-chase studies with 4SU in PARN-KO cells and analyzed newly synthesized RNA by nascent RNAend-Seq. In PARN-KO cells, mature hTR-451 was reduced in nascent RNA compared with WT cells (6% at t=0 hr; 85% at t=24 hr)(Figure S5A), reflecting a striking delay in maturation kinetics (t1/2= 12.1 hr)(Figure 4D, E). To interrogate the reduced total levels of hTR in PARN-KO cells, we measured hTR levels in pulse-chase fractions and found that while PARN-KO cells and WT cells had similar hTR levels at time 0, by time 24 hrs the PARN-KO cells showed an 84% decrease in levels compared to WT cells (Figure 4F). These data show PARN-KO cells continue to produce mature hTR, but at a markedly slowed rate.
Figure 4: RNA processing enzymes PARN and PAPD5 define the rate of hTR precursor maturation.

A. Western blot for PARN and PAPD5 showing CRISPR-Cas9 KO of PAPD5, PARN, or PAPD5 and PARN (DKO). Unused KO clones are omitted. Asterisk (*) indicates background band
B. qRT-PCR of hTR relative to parental cells in parental, PARN-KO, PAPD5-KO, and DKO cells normalized to GAPDH. Error bars represent S.E.M.
C. 3’ RACE-Seq of steady-state hTR in indicated genotypes. Proportion of oligo-A molecules at steady-state for the indicated genotypes. hTR Reads≥20k
D. Exponential decay curves are fitted to the pulse chase data as in Figure 3F. For each time point, two biologic replicates are plotted. hTR Reads per time point≥13k.
E. Half-life measurements for maturation rate are calculated from curves in (D) using method from Figure 3G.
F. qRT-PCR of hTR relative to parental cells at indicated time points normalized to IVT luciferase spike-in.
G. Percent of hTR transcripts with the indicated oligo-A tail length is plotted for indicated genotypes for molecules at t0, t8, and t24. Error bars represent the S.E.M. from two biologic replicates. hTR Oligo-A reads: Parental≥1k, PARN-KO≥2.4k, PAPD5-KO≥0.3k, DKO≥1.3k.
PAPD5 is proposed to antagonize PARN by adding untemplated adenosine residues to hTR and other RNAs (Berndt et al., 2012). To understand the role of PAPD5 in hTR processing, we generated PAPD5-KO and double PARN/PAPD5-KO HeLa cells (DKO) (Figure 4A). Consistent with the role of PAPD5 in oligoadenylation of hTR, loss of PAPD5 caused an 80% reduction in oligoadenylated hTR reads by sequencing (Figure S4C). Loss of PAPD5 alone led to elevated hTR levels and increased telomerase activity compared with WT cells (Figure 4A, B, S5D). Stable overexpression of PAPD5 in HeLa cells or PAPD5 KO HeLa cells suppressed hTR levels (Figure S4A, B). In the absence of PAPD5, mature hTR-451 represented a greater percentage of hTR reads compared with WT cells (PAPD5-KO 95% vs. WT 81% of reads)(Figure 4C). Pulse-chase analysis revealed that maturation of extended precursors was accelerated in PAPD5-KO cells (maturation t1/2=2.83 vs. 5.07 hr) (Figure S5A)(Figure 4D, E). Associated with rapid maturation, oligoadenylation of hTR precursors was reduced ∼5-fold (Figure 4C). PAPD5 functions to slow hTR maturation by oligoadenylation of hTR precursors, and in the absence of these modifications hTR processing proceeds more rapidly leading to a net increase in mature hTR.
Given PAPD5 enzyme slows hTR processing, we wondered whether loss of PAPD5 could correct the kinetic defect in hTR maturation in PARN-mutant cells. In DKO cells, the percentage of hTR extended precursor molecules was reduced to WT levels (Figure 4C). To assess kinetics of maturation, we performed pulse-chase studies in DKO cells. Loss of PAPD5 in DKO cells rescued the immature profile of nascent RNA in PARN-KO cells (Figure S5A) and corrected the kinetic defect in hTR maturation in PARN-KO cells (maturation t1/2 4.23 vs.12.1 hrs). Maturation of hTR in DKO cells lacking both PAPD5 and PARN was effectively equivalent to hTR in WT cells (95% CI WT 3.2-7.4 vs. DKO cells 3.5-5.0 hrs)(Figure 4D, E). Loss of PAPD5 in DKO cells rescued all the central defects in the telomerase pathway, including hTR levels, telomerase activity, and telomere length (Figure 4A, S4D, S4F). In the presence of TCAB1, hTR localizes to the Cajal body, a nuclear structure marked by coilin (Zhong et al., 2011). In PARN-KO cells, hTR Cajal body foci were severely diminished. However, in DKO cells localization of hTR was fully restored (Figure S4E). These data reveal PAPD5 and PARN act in opposition to define the rate of hTR precursor maturation. PARN accelerates the maturation rate of the hTR precursor, while PAPD5 slows this process. In the absence of both enzymes, hTR processing proceeds at a normal rate enabling effective production of mature hTR-451 and unperturbed levels and function of telomerase (see Discussion). These data suggest PAPD5 and PARN set the rate of hTR maturation by oligoadenylating and deadenylating the hTR precursor.
Oligoadenylation marks hTR precursors for delayed processing
Given their biochemical activities, we hypothesized that PAPD5 and PARN compete to determine the extent of 3’ oligoadenylation of hTR precursor RNAs. Under steady-state conditions oligoadenylation was elevated in PARN-KO cells, diminished in PAPD5-KO cells and restored to a level similar to that of WT cells in DKO cells (WT 15%, PARN-KO 36%, PAPD5-KO 3%, DKO 11%)(Figure 4C). To understand the relationship between the length of each A tail on extended RNA precursors over time, we labeled RNA with 4SU and isolated samples after chases of 0, 8 or 24 hours. In WT cells, the earliest nascent RNA molecules show very short A-tails (mean 1.8 nt). The distribution of A-tails increases in length at t=8 hrs and at t=24, as nascent molecules are processed to mature hTR-451 and become less abundant (Figure 4G). In PARN-KO cells, initial patterns of A-tail length are comparable, but A-tail lengths elongate at t=8 and t=24 hrs. At later time points elongation of A-tails is strongly inhibited in PAPD5-KO cells (Figure 4G). In DKO cells, the pattern and kinetics of oligoadenylation were similar to those of WT cells. In each genetic background, we note that overall A-tail length is inversely proportional to the rate of precursor maturation. Consistent with this model, loss of PAPD5 and PARN had no effect on maturation of snRNA U2 that is not subject to adenylation (Figure S5B). For snoRNAs PARN-KO differentially affected maturation depending upon extent of oligoadenylation. Maturation of U68 but not U17a was slowed with PARN loss (maturation t1/2 3.5 vs. 1.1 hrs), reflecting higher levels of oligoadenylation of U68 precursors compared to U17a (20% U68 vs. 5% U17a) (Figure S5B, Figure 2B). These data show A-tail length is the critical molecular determinant that dictates rates of precursor maturation.
Dyskerin and the Box/HACA element confer RNA end-formation and regulation by PARN
Previous studies found hTR biogenesis is not reliant on 3’ cis sequences distal to mature hTR-451, making it unclear what elements guide 3’ end-formation (Fu and Collins, 2003). Based on the critical requirement for dyskerin and its binding element, the H/ACA domain, in stability of H/ACA RNAs (Theimer et al., 2007), we considered that the dyskerin core complex and the H/ACA element contributed to RNA end formation. Because the H/ACA domain of hTR is essential for its accumulation, it is difficult to study H/ACA element deletion. To overcome this limitation, we tested whether the H/ACA domain was sufficient to confer upon a heterologous RNA the characteristics of an H/ACA RNA end. We studied U1 snRNA and two chimeric RNAs comprising U1 fused to the hTR H/ACA domain either at the far 3’ end of the chimera (U1-H/ACA) or slightly internal, leaving a tail of U1 sequences at the 3’ end (U1-H/ACA+6) (Figure 5A). We expressed these chimeric RNAs in HeLa cells under control of the U1 promoter together with U1 3’ sequences. To distinguish between transfected molecules and endogenous U1, we incorporated a 5’ RNA affinity (RAT) tag enabling detection by northern blot (NB) using a RAT tag probe (Hernandez, 1985; Hogg and Collins, 2007; Ishikawa et al., 2014) (Figure 5A). RAT-U1 was detected by NB as a 230 nt species (Figure 5B lane 1). Addition of the H/ACA element to U1 resulted in the formation of an elongated RNA, consistent with incorporation of the H/ACA element into a mature chimeric RNA (Figure 5B lane 2-3). In addition to this RAT-U1-H/ACA RNA species, a similarly abundant RAT-U1 RNA was detected from the RAT-U1-H/ACA construct (lane 3). These results indicate addition of the H/ACA element confers formation of a stable 3’ end, but H/ACA-dependent end-formation competes with U1 termination mechanisms. Interruption of 3’ U1 terminal sequences in RAT-U1-H/ACA+6 prevented formation of a stable RAT-U1 species (lane 2) consistent with a requirement for contiguous 3’ elements in U1. The H/ACA element can confer formation of a stable extended 3’ terminus when fused to the U1 snRNA.
Figure 5: Dyskerin and the Box/HACA element confer RNA end formation and regulation by PARN.

A. hTR H/ACA is grafted to the 3’ end of U1 snRNA with U1 distal sequence elements (DSE), proximal sequence elements (PSE) and the U1 3’ box. H/ACA is added at the 3’ end (U1-H/ACA) or internal (U1-H/ACA+6 nt). A RAT tag recognized by a northern blot (N.B.) probe is added in the U1 5’ region. Expected sizes indicated.
B. N.B. of chimeric RNA constructs transfected into HeLa cells. RAT-U1 indicates the band formed by termination at the U1 3’ end. U1-H/ACA indicates the band formed by termination at the chimeric U1-H/ACA end. Per lane, % of chimeric reads with untemplated A or U nucleotides (% Oligo-A, % Oligo-U) from total reads>80k. Relative U1-H/ACA levels are normalized to U4 and quantified from 2 biologic replicates. Error bars represent S.E.M.
C. N.B. of chimeric RNA constructs transiently transfected into HeLa or PARN-KO cells. Relative U1-H/ACA levels are normalized to U4 and quantified from 4 biologic replicates.
D. 3’ RACE-Seq of RAT-U1-H/ACA in cells treated with siCTRL (black), treated with siDKC1 (red), or PARN-KO cells with siCTRL (blue), with total reads>80k. The 3’ downstream sequence of the U1-H/ACA chimera is indicated on the x-axis. Gray shading is 95% CI derived from 2 sequencing replicates.
E. Percent of Oligo-A tails with the indicated lengths. Total Oligo-A reads>2.5k. Error bars represent S.E.M. Blue asterisks indicate oligoA tails with significantly more reads in PARN-KO cell lines (**p<0.01, * p<0.05). Black asterisks indicate oligoA tails with significantly more reads in parental cell lines.
To determine whether the H/ACA element is sufficient for yielding extended oligoadenylated precursors, we performed 3’ RACE-Seq on RNAs expressed from the U1-hTR chimeras. Sequencing revealed the H/ACA domain produced a heterogenous 3’ end consisting of mature species and extended species, reminiscent of endogenous hTR 3’ ends (Figure 1A, Figure 5D). The H/ACA domain caused a 5-fold (RAT-U1-H/ACA+6) or 10-fold (RAT-U1-H/ACA) increase in oligoadenylation over RAT-U1, indicating the H/ACA domain is sufficient to confer precursor oligoadenylation (Figure 5B). During hTR biogenesis, binding of the dyskerin core complex to the H/ACA domain is essential for retaining hTR RNA. To test whether dyskerin is required for the formation of the chimeric H/ACA ends, we expressed chimeric constructs along with siCTRL or siDKC1. For both RAT-U1-H/ACA and RAT-U1-H/ACA+6, knockdown of dyskerin resulted in loss of chimeric H/ACA species, but retention of RAT-U1 (lanes 4-6, Figure S6A). Sequencing of residual RAT-U1-H/ACA transcripts in dyskerin knockdown cells showed a decreased fraction of transcripts terminating precisely at the H/ACA domain end (12% in siDKC1 vs. 44% in siCTRL cells), suggesting dyskerin defines the end of H/ACA RNAs (Figure 5D). Residual U1-H/ACA transcripts in siDKC1 cells had reduced oligoadenylation (3% in siDKC1 vs. 9% in siCTRL cells), suggesting the binding of dyskerin to the H/ACA element is required for oligoadenylation.
To understand if the U1-H/ACA chimeras are under control of PARN, we repeated experiments in PARN-KO cells. Loss of PARN caused a 70% reduction in the accumulation of chimeric U1-H/ACA RNAs but not of co-transfected luciferase cDNA transcripts, consistent with a role for PARN in regulating maturation of H/ACA RNAs (Figure 5C, S6B). Loss of PARN caused increases in extended transcripts (43% in PARN KO vs. 34% in WT cells) (Figure 5D). Although PARN-KO did cause an increase in mean A-tail length (6.3 in PARN-KO vs. 5.2 nts in WT cells) PARN-KO did not cause a large increase in proportion of oligo-A transcripts (10% PARN-KO vs. 9% WT cells) (Figure 5C, D). Instead, PARN-KO caused an increase in the proportion of longer oligo-A tails on U1-H/ACA compared to WT cells, with PARN-KO cells having more transcripts with oligo-A tails longer than 6 nts (Figure 5E).
These data show the end-formation and processing characteristics of hTR are conferred by the 3’ H/ACA cis element and mediated by the dyskerin core complex that recognizes this RNA domain in trans. Together, this modular ribonucleoprotein subcomplex dictates RNA end-formation, including formation of oligoadenylated and extended RNA precursor intermediates, and subsequent regulation by PARN.
Discussion
A feed-forward pathway regulates maturation kinetics of human telomerase RNA
Defects in RNA metabolism are increasingly being linked to human diseases, including developmental disorders, tissue failure, neurodegeneration and cancer. In most cases, the precise disease-causing defect is not understood. We developed nascent RNA sequencing technology to determine the mechanisms underlying RNA maturation. This approach allows the identification of RNA precursors, their relationship with the mature RNA and a quantitative assessment of maturation kinetics.
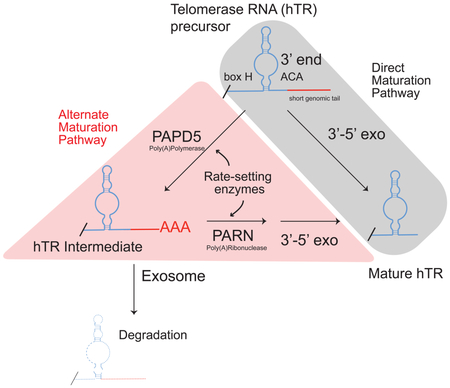
Our nascent RNAend-Seq data show hTR is processed through a non-linear pathway, whereby extended precursors are both directly processed to mature hTR and indirectly processed to mature hTR after a cycle of oligoadenylation by PAPD5 and deadenylation by PARN (Figure 6A, WT cells). This pathway represents feed-forward regulation where an input (transcription of hTR) feeds into two processes (maturation and oligoadenylation) that both contribute to formation of mature hTR species. This type of circuit is commonly found in transcriptional networks, where a transcription factor X activates a second transcription factor Y, and both jointly regulate a gene Z (Mangan et al., 2003). Compared to direct regulation of gene Z by X (or direct production of mature hTR from extended hTR) feed-forward regulation is inherently time-delayed and buffered against noisy transcriptional inputs (Mangan et al., 2003).
Figure 6: A feed-forward pathway regulating hTR maturation is determined by the H/ACA domain.

A. A feed-forward pathway governs maturation of human telomerase RNA (Wild-type cells): hTR extended precursors mature directly or are fed into a side pathway and oligoadenylated by PAPD5. Oligoadenylated precursors may be deadenylated by PARN to form mature hTR, or destroyed by the RNA exosome. Final trimming of hTR precursor is achieved through a 3’-5’ RNA exonuclease.
(PARN-KO cells): In the absence of PARN, oligoadenylated intermediates cannot robustly convert to mature hTR. There is a relative increase in oligoadenylated hTR precursors, and a loss of mature hTR.
(PAPD5-KO cells): In the absence of PAPD5, fewer hTR precursors are fed into the cycle of adenylation and deadenylation, leading to rapid maturation of hTR molecules.
B. The hTR H/ACA domain and dyskerin constitute a 3’ end formation module. The H/ACA domain in the presence of dyskerin promotes transcriptional termination and oligoadenylation of an extended precursor.
An important junction in this pathway is gated by disease associated poly(A)ribonuclease PARN which deadenylates extended hTR precursors to prevent their destruction and return them to the maturing hTR pool (Figure 6A, PARN-KO). In the absence of PARN, one arm of the circuit is broken and adenylated precursors accumulate before being degraded by the RNA exosome. Our data support a role for PARN in deadenylation of adenylated precursors but are inconsistent with a direct role for PARN in trimming templated extensions from hTR precursors. These data are consistent with biochemical studies showing a 10-100 fold greater activity of PARN on oligo-A substrates compared to other RNA sequences (Henriksson et al., 2010). However, a recent study found recombinant PARN could digest genomically encoded 3’ sequences in vitro (Tseng et al., 2018). In our studies in living cells, maturation of hTR persists in the absence of PARN albeit at a reduced rate. Furthermore, maturation of hTR is fully normalized in cells lacking both PARN and PAPD5, indicating that PARN is dispensable for trimming 3’ genomically encoded tails. Rather than trimming these 3’ genomic sequences, PARN removes oligo-A residues from these intermediates thereby allowing adenylated intermediates to be completely processed to the mature hTR species. Based on these observations, we propose that extended precursors mature through the action of an as yet undefined 3’-5’ exonuclease.
The poly(A)polymerase PAPD5 oligoadenylates hTR precursors, preventing them from immediate maturation and causing them to enter into the pool of oligoadenylated precursors that is susceptible to destruction (Figure 6A, PAPD5-KO cells). Loss of PAPD5 impairs the cycle of adenylation/deadenylation, favoring direct maturation of hTR precursors rather than degradation (Figure 6A, PAPD5 KO cells). This direct pathway of maturation occurs more rapidly than in cells with PAPD5 present and results in greater accumulation of hTR transcripts. Because the oligo-A mark inhibits maturation, removal of PAPD5 causes complete rescue in PARN KO cells either by CRISPR-KO or shRNA mediated knockdown (unpublished data). Therefore, PARN and PAPD5 set the rate of hTR precursor maturation by gating the oligoadenylation and deadenylation steps in an hTR processing circuit.
Limiting and controlling levels of mature telomerase through RNA processing
Why does the maturation pathway of hTR include a regulatory cycle of adenylation and deadenylation? In human development and adult homeostasis, telomere lengths are tightly controlled to ensure sufficient regenerative potential during life while preventing the development of cancer. This balancing act is accomplished in part through regulation of telomerase, including transcriptional control of TERT, regulation of enzyme activity, and recruitment to telomeres. In addition to these steps, controlling hTR levels is critical for maintaining telomeres in an appropriate range. Modest germline reductions in hTR levels cause telomere syndromes (Vulliamy et al., 2001) and somatic amplification of hTR is common in human cancers (www.cbioportal.org). Transcriptional noise represents another challenge to maintaining consistent expression of RNAs over time and from cell-to-cell. This feed-forward circuit both limits the steady-state concentration of hTR while buffering against perturbations in transcription.
Remarkably, these varied characteristics of hTR processing are mediated simply by the presence of the H/ACA cis sequences encoded within hTR. Adding the H/ACA element from hTR to a heterologous snRNA conferred each of the properties associated with hTR maturation, including formation of a defined 3’ end to the mature chimeric RNA, recruitment of polyA polymerase activity and sensitivity to PARN (Figure 6B). The H/ACA element acts by recruiting the dyskerin core complex, which promotes transcriptional termination downstream of the H/ACA element. In steps that remain to be understood, the dyskerin-bound RNP helps to recruit polyA polymerases, PARN and additional 3’-5’ exonuclease activities to complete processing. It is notable that telomerase RNAs from single cell eukaryotes lack an H/ACA element and that telomerase RNA acquired an H/ACA element during evolution to complex invertebrates and vertebrates. The need for fine-tuned regulation of telomerase in complex multicellular organisms may explain the incorporation of the H/ACA element into telomerase RNA during evolution.
Altered RNA maturation kinetics: an emerging disease mechanism
Precursor accumulation is observed in other human diseases involving small non-coding RNAs, suggesting that kinetic defects in RNA maturation represents an emerging disease mechanism. Defects in RNA maturation may contribute to disease by: (1) causing degradation of stalled precursors and decreased amounts of steady state RNA, (2) accumulation of precursor RNA with impaired function, or (3) both of the above. Investigation of these processes using similar nascent RNA isolation approaches as the ones introduced here may lead to both better understanding of the regulatory circuits that dictate processing of ncRNAs and strategies for treating diseases by correcting kinetic defects in RNA processing. In this study, we show that perturbing the relative activities of two enzymes, PARN and PAPD5, can accelerate or decelerate hTR maturation in a predictable, tunable way. Manipulating levels of hTR in this manner has important therapeutic implications for treating telomere syndromes and pulmonary fibrosis.
STAR Methods
Cell Culture, RNAi, Viral Transductions and CRISPR knockout generation
HeLa S3 and 293T cells were cultured in DMEM containing 10% fetal bovine serum at 37 deg C, 5% CO2. Lipofectamine 2000 (ThermoFisher 11668019) was used for all cDNA transfection experiments. All siRNAs were siGENOME pools purchased from Dharmacon/Thermo and used at 50 nM. Cells were assayed 48 hr after transfection. Passage 29 human WA01 H1 hESC were grown in mTeSR1 (Stem Cell 05850) feeder free system on matrigel (Corning 356230) coated plates. CRISPR KO cells were generated by transfection of HeLa cells with px458 plasmid with Cas9 T2A GFP and containing guide RNAs to the indicated locus. Sequences of guide RNAs can be found in Table S1. GFP positive cells were single cell cloned in 96 well plates and clones were screened for gene KO by western blot. For transduction of Flag-PARN and Flag-GFP, 293T cells were transfected with pCDH T2A GFP and packaging constructs; 48 hours later, viral supernatant was collected. Target HeLa cells were transduced in the presence of 5 ug/ml polybrene and FACS sorted for GFP expression. For western blotting, cells were lysed in NP40 buffer (25 mM HEPE-KOH, 150 mM KCl, 1.5 mM MgCl2, 0.5% NP40, 10% Glycerol) supplemented with protease inhibitors (Sigma 11836153001). PARN antibody is a rabbit polyclonal from Bethyl Laboratories (A303-562A). PAPD5 antibody is a rabbit polyclonal from Atlas antibodies (HPA042968).
Nascent RNAend-Seq and steady-state 3’ RACE-Seq
Cells were pulsed with 1 hr 4SU 250 uM or 4 hr 4SU 50 uM. For chase experiments, 4SU was removed, the cells were washed in PBS, and 2.5 mM uridine containing media was added. Cells were harvested and cell pellets resuspended in Trizol. RNA was extracted using standard Trizol protocol. 100 ug RNA was biotinylated with Biotin-HPDP (Pierce 21341) at 0.5 mg/mL in 40% DMF and 10 mM Tris pH 7.4, 1 mM EDTA, for 1.5 hours at room temperature. In vitro transcribed luciferase RNA transcribed in the presence of 4SU was spiked into the biotinylation mixture for a final concentration of .01 ng/uL. RNA was extracted using isopropanol/ethanol precipitation. Biotinylated RNA was immunoprecipitated from total RNA using μMacs Streptavidin Kit (Miltenyi 130-074-101). The depleted fraction was recovered using isopropanol/ethanol precipitation. Biotinylated RNA was released from beads using 100 mM DTT and cleaned using RNeasy MinElute Kit (Qiagen 74204). Half of eluted RNA or 600 ng of depleted RNA was ligated to 5 uM of 5’ adenylated, 3’ blocked adapter (Universal miRNA cloning linker, NEB S1315S) with 250 units of T4 RNA ligase truncated KQ (NEB M0373S), 25% PEG 8000, and 1 uL RnaseOUT (ThermoFisher 10777019) in a 20 uL reaction at 25 degrees for 16 hours. Ligated RNA was cleaned up with RNA clean and concentrator columns (Clontech 740955.50) and DNAse treatment, cDNA was synthesized with universal primer and Superscript III (ThermoFisher 18080093). Amplification was carried out with Phusion (New England Biosystems M0530) and primer sets universal/TERCR1. PCR products were directly run on an 8% PAGE gel and visualized with SYBR Gold (ThermoFisher S-11494), or subject to AMPure XP beads (Beckman Coulter A63881) for PCR cleanup and library preparation. Libraries were prepped using Kapa Hyperprep Kit (Kapa KK8504), quantified with Qubit and bioanalyzer, and run on Illumina miSeq at the Stanford Functional Genomics Facility. Reads were paired using fastq-join tool at Galaxy (usegalaxy.org). Reads were binned into the various forms of hTR using custom python scripts (cmroake.people.stanford.edu/links-python-scripts) and the number of reads in each bin was normalized to total hTR reads. Primer sequences can be found in Table S1.
qRT-PCR
RNA was extracted by Trizol. ∼100ng of RNA was used for qRT-PCR using Brilliant II SYBR Green QRT-PCR 1-Step Master Mix (Stratagene, 600825). Primers used for qPCR are shown in Table S1.
Telomere Repeat Amplification Protocol and Telomere Restriction Fragment Analysis
To measure telomerase activity, a two-step TRAP procedure was performed according to (Kim and Wu, 1997). Briefly, cell extracts were incubated with telomeric primers (Table S1) for a 30 min initial extension step at 30°C in a PCR machine , followed by 5 min of inactivation at 72°C.1 μl of the extended reaction was subjected to PCR amplification (24 cycle of 30 s at 94°C, followed by 30 s at 59°C) in presence of 32P end-la beled telomeric primers purified using a micro-spin G-25 column (GE healthcare, 27-5325-01). The PCR reactions were resolved by 9% polyacrylamide gel electrophoresis at room temperature, and the gel was exposed to a phosphor-imager and scanned by a Typhoon scanner. To measure telomere lengths by telomere restriction fragment analysis, cells were harvested and digested with Proteinase K at 6 ug/mL overnight. DNA was extracted by the phenol-chloroform method and digested overnight with Hinfl and RsaI before electrophoresis and southern blotting with end labelled (CCCTAA)4 oligonucleotide probe (Table S1).
Northern Blotting
Indicated U1 chimera was cloned with a RAT tag as described in Hogg et al. RNA 2007. Sequences of oligos for NB probes can be found in Table S1. Whole cell total RNA was extracted using Trizol reagent, and quantified using nanodrop. ∼4 μg of total RNA was mixed 1:1 (v/v) with formamide-EDTA loading buffer (95% deionized formamide, 0.025% [w/v] of Bromophenol blue and Xylene Cyanol FF, 5mM of EDTA pH8.0), boiled at 95 °C for 5 min, and immediately chilled on ice. The RNA sample was then resolved by 6% Urea-polyacrylamide gel (6% 19:1 acrylamide:bis, 7M Urea, 0.6x TBE) at room temperature under constant 60V, and transfer to Hybond-XL (GE healthcare, RPN2020S) in cold room under 400A. The post-transferring gel was stained by ethidium bromide to visualize the residual ribosomal RNA signal for loading control. The membrane is air-dried, UV crosslinked, and blocked in a hybridization tube containing Ultrahyb (Ambion, AM8670) at 42°C for 1 hour. The oligonucleotide probes were end-labeled by 32p-gamma ATP using T4 polynucleotide kinase, and purified through a G-50 column. Labeled probes with a minimum of ∼2 million CPM were boiled, snap-chilled, combined with the Ultrahyb blocking buffer, and hybridize overnight at 42°C. The membrane was then washed twice by 2x SSC/0.1% SDS, once each with 1x SSC/0.1% SDS, and 0.1X SSC/0.1% SDS, before proceeding to autoradiography using a phosphor imager, which was scanned by a Typhoon imager after overnight exposure.
Immunofluorescence and RNA FISH
All immunofluorescence was carried out on cells seeded on coverslips. Cells were fixed with 10% formalin and permeabilized with 0.1% Triton X-100 in PBS. Coverslips were incubated with primary antibody to coilin (Santa Cruz SC-32860) in 1% BSA for 1 hr at room temperature. Coverslips were washed 3x with PBS and incubated with secondary Alexa Flour conjugated antibodies (Jackson Immunoresearch) in 1% BSA for 45 minutes at room temperature. Coverslips were washed 3x in PBS and then dehydrated for 10 min in 70% ethanol in preparation for RNA FISH. RNA FISH was done using labeled oligonucleotide probes (Biosearch, Table S1). Ethanol was removed and RNA FISH probes (Quasar 570 conjugated, 75 nM working concentration) in RNA FISH hybridization buffer (2x SSC, 10% Formamide, and 10% dextran sulfate) was immediately added. Coverslips were incubated at 37°C overnight, then washed twice with 2x SSC, 10% Formamide at 37°C for 30 min each. Coverslips were mounted in 2x SSC. For each genotype > 50 nuclei were scored for the number of hTR foci at Cajal bodies.
Quantification and Statistical Analysis
Error bars presented represent S.E.M; p values were calculated by Student’s t test; p < 0.05 was defined as significant. Error bars, S.E.M, and significance of the IF data were calculated by GraphPad Prism Software. Statistical details can be found in figures, and in the Results section.
Data and Software Availability
The nascent RNAend-Seq data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GEO: GSE117054 For Raw data used to construct the Figures, please refer to data depository hosted by Mendeley via the following URL: https://doi:10.17632/f7ckbdfrgf.1
Supplementary Material
Highlights.
Nascent RNAend-Seq reveals that mature hTR derives from a 3’ extended precursor
PAPD5 and PARN control rates of hTR maturation by oligoadenylation/deadenylation
The H/ACA domain determines RNA processing pathway choice
Defects in hTR maturation rate cause telomerase loss in PARN-mutant cells
Acknowledgements
We are grateful to members of the Artandi laboratory and to D. Herschlag, A. Straight, J. Sage and K. Chua for critical comments. We thank Lifeng Xu for the generous gift of UMUC3 cells. This work was supported by NIH grants AG056575 and CA197563 (to S.E.A). C.M.R. was supported by MSTP Training Grant GM007365 and by a Gerald J. Lieberman Fellowship. L.C. was supported by a Stanford Cancer Institute 2018 Fellowship Award.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berndt H, Harnisch C, Rammelt C, Stohr N, Zirkel A, Dohm JC, Himmelbauer H, Tavanez JP, Huttelmaier S, and Wahle E (2012). Maturation of mammalian H/ACA box snoRNAs: PAPD5-dependent adenylation and PARN-dependent trimming. RNA (New York, NY) 18, 958–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyraz B, Moon DH, Segal M, Muosieyiri MZ, Aykanat A, Tai AK, Cahan P, and Agarwal S (2016). Posttranscriptional manipulation of TERC reverses molecular hallmarks of telomere disease. J Clin Invest 126, 3377–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghes AH, and Beattie CE (2009). Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 10, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Roake CM, Freund A, Batista PJ, Tian S, Yin YA, Gajera CR, Lin S, Lee B, Pech MF, et al. (2018). An Activity Switch in Human Telomerase Based on RNA Conformation and Shaped by TCAB1. Cell 174, 218–230.e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP, Adams RR, Chang E, Allsopp RC, Yu J, et al. (1995). The RNA component of human telomerase. Science 269, 1236–1241. [DOI] [PubMed] [Google Scholar]
- Fu D, and Collins K (2003). Distinct biogenesis pathways for human telomerase RNA and H/ACA small nucleolar RNAs. Molecular cell 11, 1361–1372. [DOI] [PubMed] [Google Scholar]
- Goldfarb KC, and Cech TR (2013). 3' terminal diversity of MRP RNA and other human noncoding RNAs revealed by deep sequencing. BMC Mol Biol 14, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson N, Nilsson P, Wu M, Song H, and Virtanen A (2010). Recognition of adenosine residues by the active site of poly(A)-specific ribonuclease. J Biol Chem 285, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez N (1985). Formation of the 3' end of U1 snRNA is directed by a conserved sequence located downstream of the coding region. EMBO J 4, 1827–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg JR, and Collins K (2007). RNA-based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA 13, 868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Nobe Y, Izumikawa K, Yoshikawa H, Miyazawa N, Terukina G, Kurokawa N, Taoka M, Yamauchi Y, Nakayama H, et al. (2014). Identification of truncated forms of U1 snRNA reveals a novel RNA degradation pathway during snRNP biogenesis. Nucleic Acids Res 42, 2708–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirwan M, and Dokal I (2008). Dyskeratosis congenita: a genetic disorder of many faces. Clin Genet 73, 103–112. [DOI] [PubMed] [Google Scholar]
- Kiss T, Fayet E, Jady BE, Richard P, and Weber M (2006). Biogenesis and intranuclear trafficking of human box C/D and H/ACA RNPs. Cold Spring Harb Symp Quant Biol 71, 407–417. [DOI] [PubMed] [Google Scholar]
- LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, Jacquier A, and Tollervey D (2005). RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121, 713–724. [DOI] [PubMed] [Google Scholar]
- Lardelli RM, Schaffer AE, Eggens VR, Zaki MS, Grainger S, Sathe S, Van Nostrand EL, Schlachetzki Z, Rosti B, Akizu N, et al. (2017). Biallelic mutations in the 3' exonuclease TOE1 cause pontocerebellar hypoplasia and uncover a role in snRNA processing. Nat Genet 49, 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lestrade L, and Weber MJ (2006). snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res 34, D158–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan S, Zaslaver A, and Alon U (2003). The coherent feedforward loop serves as a sign- sensitive delay element in transcription networks. J Mol Biol 334, 197–204. [DOI] [PubMed] [Google Scholar]
- Matera AG, Terns RM, and Terns MP (2007). Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat Rev Mol Cell Biol 8, 209–220. [DOI] [PubMed] [Google Scholar]
- Mitchell JR, Cheng J, and Collins K (1999). A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3' end. Mol Cell Biol 19, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon DH, Segal M, Boyraz B, Guinan E, Hofmann I, Cahan P, Tai AK, and Agarwal S (2015). Poly(A)-specific ribonuclease (PARN) mediates 3'-end maturation of the telomerase RNA component. Nat Genet 47, 1482–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, Grenier St-Sauveur V, Bergeron D, Dupuis-Sandoval F, Scott MS, and Bachand F (2015). A Polyadenylation-Dependent 3' End Maturation Pathway Is Required for the Synthesis of the Human Telomerase RNA. Cell reports 13, 2244–2257. [DOI] [PubMed] [Google Scholar]
- Patel SB, and Bellini M (2008). The assembly of a spliceosomal small nuclear ribonucleoprotein particle. Nucleic Acids Res 36, 6482–6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer V, and Lingner J (2013). Replication of telomeres and the regulation of telomerase. Cold Spring Harb Perspect Biol 5, a010405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radle B, Rutkowski AJ, Ruzsics Z, Friedel CC, Koszinowski UH, and Dolken L (2013). Metabolic labeling of newly transcribed RNA for high resolution gene expression profiling of RNA synthesis, processing and decay in cell culture. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Schmidt JC, Goldfarb KC, Cech TR, and Parker R (2016). Inhibition of telomerase RNA decay rescues telomerase deficiency caused by dyskerin or PARN defects. Nat Struct Mol Biol 23, 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, Choi M, Dharwadkar P, Torres F, Girod CE, et al. (2015). Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet 47, 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theimer CA, Jady BE, Chim N, Richard P, Breece KE, Kiss T, and Feigon J (2007). Structural and functional characterization of human telomerase RNA processing and cajal body localization signals. Mol Cell 27, 869–881. [DOI] [PubMed] [Google Scholar]
- Tseng CK, Wang HF, Burns AM, Schroeder MR, Gaspari M, and Baumann P (2015). Human Telomerase RNA Processing and Quality Control. Cell reports 13, 2232–2243. [DOI] [PubMed] [Google Scholar]
- Tseng CK, Wang HF, Schroeder MR, and Baumann P (2018). The H/ACA complex disrupts triplex in hTR precursor to permit processing by RRP6 and PARN. Nat Commun 9, 5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummala H, Walne A, Collopy L, Cardoso S, de la Fuente J, Lawson S, Powell J, Cooper N, Foster A, Mohammed S, et al. (2015). Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. The Journal of clinical investigation 125, 2151–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, Terns MP, and Artandi SE (2009). A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323, 644–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, and Dokal I (2001). The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 413, 432–435. [DOI] [PubMed] [Google Scholar]
- Wan J, Yourshaw M, Mamsa H, Rudnik-Schoneborn S, Menezes MP, Hong JE, Leong DW, Senderek J, Salman MS, Chitayat D, et al. (2012). Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat Genet 44, 704–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Reuter M, Lilie H, Liu Y, Wahle E, and Song H (2005). Structural insight into poly(A) binding and catalytic mechanism of human PARN. EMBO J 24, 4082–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong F, Savage SA, Shkreli M, Giri N, Jessop L, Myers T, Chen R, Alter BP, and Artandi SE (2011). Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes and Development 25, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The nascent RNAend-Seq data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GEO: GSE117054 For Raw data used to construct the Figures, please refer to data depository hosted by Mendeley via the following URL: https://doi:10.17632/f7ckbdfrgf.1