

Abstract
Cyclization is a common strategy to confer proteolytic resistance to peptide scaffolds. Thus, cyclic peptides have been the focus of extensive bioengineering efforts. Ribosomally synthesized and posttranslationally-modified peptides (RiPPs) are a superfamily of peptidic natural products that often contain macrocycles. In the RiPP family of lanthipeptides, macrocyclization is accomplished through formation of thioether crosslinks between cysteines and dehydrated serines/threonines. The recent production of lanthipeptide libraries and development of methods to display lanthipeptides on yeast or phage highlights their potential for bioengineering and synthetic biology. In this regard, the prochlorosins are especially promising as the corresponding class II lanthipeptide synthetase ProcM matures numerous precursor peptides with diverse core peptide sequences. To facilitate future bioengineering projects, one of its native substrates, ProcA2.8, was subjected in this study to in-depth mutational analysis to test the limitations of ProcM-mediated cyclization. Alanine scan mutagenesis was performed on all residues within the two rings, and multiple prolines were introduced at various positions. Moreover, mutation, deletion, and insertion of residues in the region linking the two lanthionine rings was tested. Additional residues were also introduced or deleted from either ring, and inversion of ring forming residues was attempted to generate diastereomers. The findings were used for epitope grafting of the RGD integrin binding epitope within prochlorosin 2.8, resulting in a low nanomolar affinity binder of the αvβ3 integrin that was more stable towards proteolysis and displayed higher affinity than the linear counterpart.
Keywords: RiPP, lanthipeptide, prochlorosin, substrate scope, epitope grafting, integrin
Graphical Abstract:
INTRODUCTION
Lanthipeptides belong to the natural product superfamily of ribosomally synthesized and posttranslationally-modified peptides (RiPPs).1 All RiPPs share a similar biosynthetic logic: After translation of a genetically encoded precursor peptide, modification enzymes process the precursor into the mature natural product. Typically, these precursor peptides can be subdivided into an N-terminal leader and a C-terminal core peptide. While the leader region mediates the interaction with the processing enzymes, the core peptide is where modifications are introduced. Eventually, the leader peptide is removed and the mature compound is released.
In this manner, nature accomplishes impressive chemical and structural diversity despite the fact that the precursor can only be assembled from amino acids incorporated by the ribosome. Diversity in RiPP scaffolds ranges from small organic molecules (e.g. PQQ,2,3 mycofactocins4,5) to large and heavily modified peptide scaffolds (e.g. proteusins6). The range of biological functions of RiPPs is just as impressive, including roles as bacterial cofactors, signaling molecules, antimicrobial agents, antiviral compounds, anticancer agents, and receptor antagonists.1–5,7–13
A feature that is intrinsic to many RiPP systems is the high promiscuity of their biosynthetic machineries.7,10,14–29 With exception of residues involved in the posttranslational modifications, amino acid substitutions in the core region are generally tolerated well. Additionally, the corresponding processing enzymes allow access to complicated peptide topologies that are not readily accessible by synthetic means.1,6–8,30 These features make this superfamily of peptidic natural products interesting for synthetic biology and bioengineering applications. Examples can be found in the form of epitope grafting studies (using e.g. cyclotides,11,12,31 lasso peptides,10 or θ-defensins32,33), generation and screening of peptide libraries (utilizing e.g. cyclotides,11,12,31 lasso peptides,34 and lanthipeptides17), as well as yeast- and phage-display techniques employing lanthipeptides.19,28
For lanthipeptides, the defining structural feature is the presence of thioether bonds.1,7 These are generated in two steps. First, the side chain of a Ser or Thr is dehydrated to yield a dehydroalanine (Dha) or dehydrobutyrine (Dhb), respectively (Figure 1a). Subsequently, the Cys thiol group can attack the unsaturated double bond in a 1,4-conjugate addition to yield a cyclic enolate that is protonated to the corresponding lanthionine or methyllanthionine (Figure 1b). In some cases, these enolate intermediates can undergo a second nucleophilic attack on another Dha residue to generate a labionin moiety (Figure 1b).35–37
Figure 1.
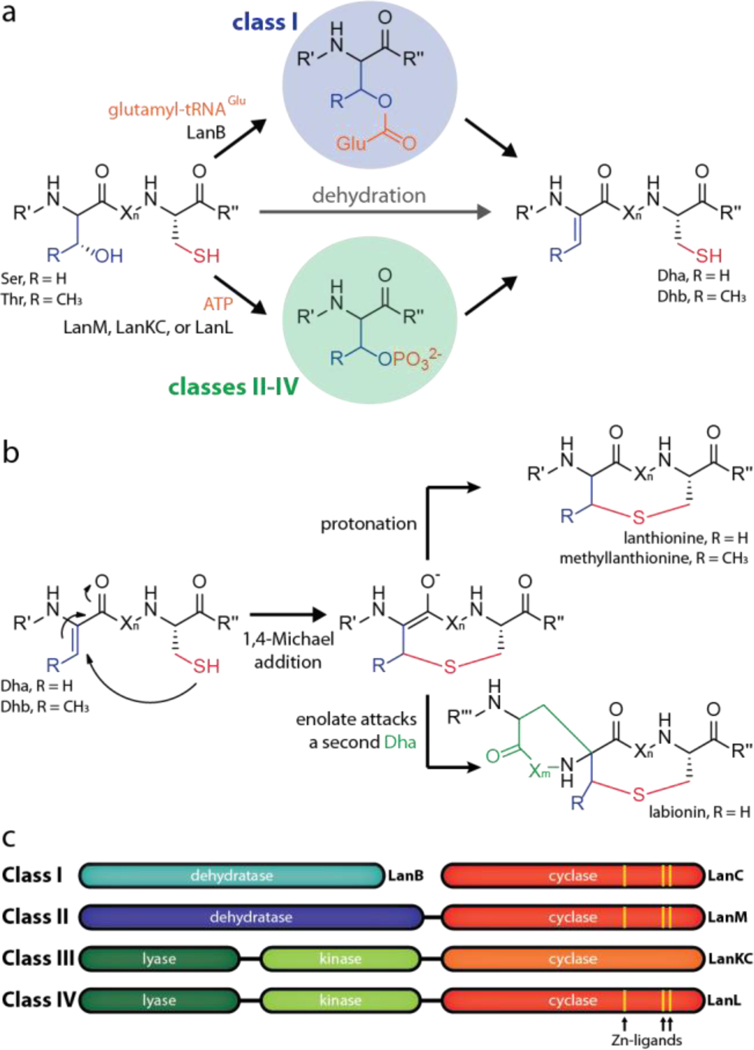
Lanthipeptide biosynthesis starts with (a) dehydration of Ser/Thr residues, which is followed by subsequent (b) cyclization through nucleophilic attack of Cys thiol groups. (c) Overview of the four classes of lanthipeptide processing enzymes.
Lanthionines are differentiated into four different classes, which are defined by characteristics of the corresponding biosynthetic enzymes (Figure 1c). Class I lanthipeptides are produced through action of a glutamyl tRNA-dependent dehydratase (LanB) and a cyclase (LanC).7,38–40 In the other classes, both dehydration and cyclization are accomplished by a single lanthipeptide synthetase enzyme, which uses NTPs to phosphorylate the Ser/Thr hydroxy groups to activate them for the subsequent elimination reaction.35–37,41–45 Class II lanthipeptide synthetases (LanM) feature an N-terminal dehydratase and a C-terminal cyclization domain. In contrast, class III (LanKC) and class IV (LanL) enzymes consist of three domains; an N-terminal lyase, a central kinase, and a C-terminal cyclase domain.
The general applicability of lanthipeptides in the field of bioengineering and synthetic biology was shown in several studies. Phage-display was used to select for urokinase plasminogen activators and streptavidin ligands,28 as well as to optimize lipid II binding by nisin.19 Moreover, yeast-display was employed to screen for potent αvβ3 integrin receptor binders,19 and a library consisting of about two million lanthipeptides was successfully screened for inhibitors of the protein-protein interaction between the HIV p6 protein and the UEV domain of the human TSG101 protein.17
The prochlorosins (Pcns) originate from Prochlorococcus MIT 9313, which harbors a single gene encoding the class II lanthipeptide synthetase ProcM as well as 30 different genes encoding ProcA precursor peptides.7,44,46,47 The latter contain a highly conserved leader sequence of ~70 amino acids that is followed by short and highly variable core peptides that yield great structural diversity (e.g. Figure 2a). ProcM was observed to modify all 17 ProcA substrates tested thus far.7,44,46,47 As such, nature already demonstrated the vast structural and chemical diversity that can be accessed by this single enzyme.
Figure 2.
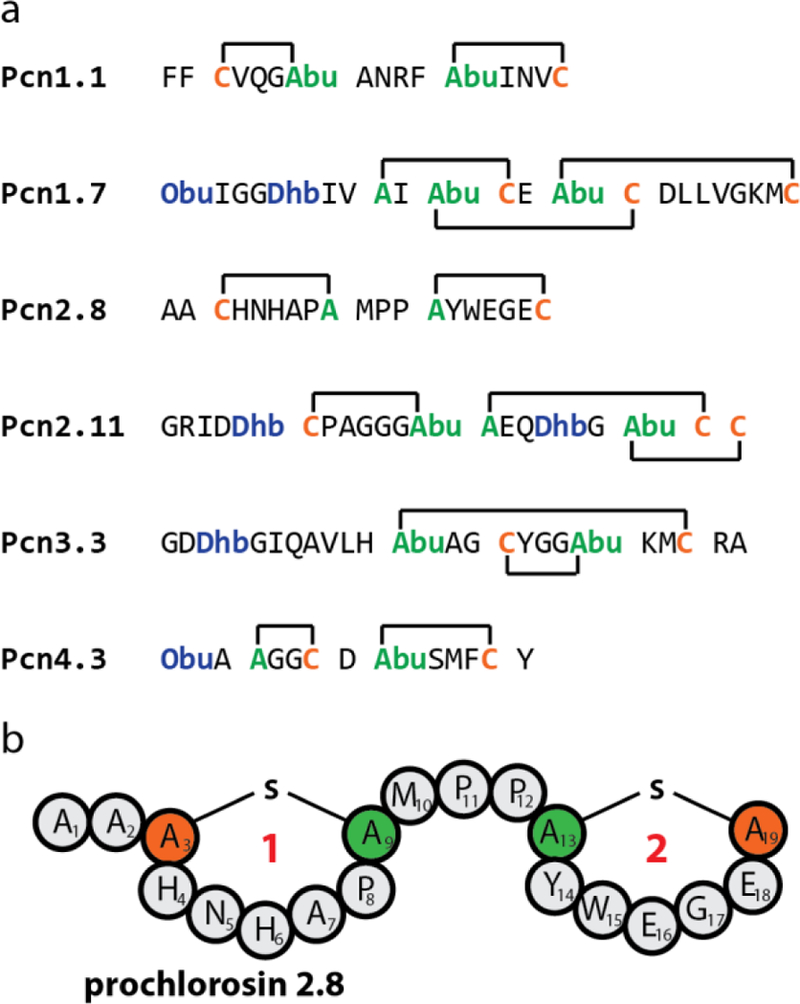
(a) Structures of six prochlorosins (Pcns).7,44 Cys residues are highlighted in orange and other residues involved in thioether macrocycles in green. Residues that are posttranslationally modified during maturation, but not involved in cyclization are shown in blue. (b) Schematic depiction of Pcn2.8. Abu = aminobutyric acid. Obu = 2-oxobutyryl.
While ProcA precursors were successfully employed to generate peptide libraries to screen for Pcn variants with desired activities, the limits of ProcM substrate tolerance have not been tested since a systematic mutational analysis of the ProcA precursor peptides has not been reported. Therefore, we choose the ProcA2.8 precursor peptide as a model system to investigate the effects of various substitutions in its amino acid sequence and of alterations of topology defining features on the ProcM-mediated maturation. We chose ProcA2.8, because heterologous production in Escherichia coli is already well-established,44,46,47 because this scaffold has been successfully used in previous bioengineering studies,17,28 and because the two adjacent seven-residue rings are an attractive peptide scaffold that may mimic two substrate binding loops of antibodies and offer potential application in epitope grafting efforts.
After assessing the flexibility of this scaffold, we tested the utility of Pcn2.8 for epitope grafting by rational design approaches. Therefore, we incorporated the RGD epitope at different positions in ProcA2.8, isolated the resulting variants, released the core peptides by proteolysis and after HPLC purification used them in αvβ3 integrin binding studies to assess their affinity to the receptor. We could thereby show that one of the tested RGD variants has a binding affinity to αvβ3 in the low nanomolar range.
RESULTS AND DISCUSSION
Assessing the tolerance of ProcM towards mutations in ProcA2.8
As basis for our investigations, we used a His6-tagged ProcA2.8(G−1K) variant, which is expressed and processed as efficiently as wild type (WT) ProcA2.8, but has the advantage that the core peptide can be released by treatment with LysC endoproteinase.44 In this study, all ProcA2.8 variants were co-expressed with ProcM in E. coli, purified via affinity chromatography using nickel nitrilotriacetic acid (NiNTA) resin and then treated with LysC. The dehydration state of the peptides was assessed by mass spectrometry (MS) (Supporting Information Figures S1–S38). As the lanthionine cyclization is a mass-neutral process, reactions with N-ethylmaleimide (NEM) were also performed. Under assay conditions, this compound is a thiol specific electrophile and thus reports on the number of free cysteines in a peptide and in turn on the cyclization state of the peptide (Supporting Information Figures S1–S38). Finally, the topology of the formed lanthionine rings was assessed by collision-induced fragmentation (CID) of the isolated peptides (Supporting Information Figures S39–S76). The results of these experiments are summarized in Table 1.
Table 1.
Overview of all His6-ProcA2.8(G−1K) variants co-expressed with ProcM in E. coli in this study. Observed dehydration states, number of NEM adducts, and ring topologies as determined by tandem MS experiments are shown. The amino acid sequences of the core peptides are presented in Supporting Information Table S1.
| Name | dehydration state | NEMs added | ring topology |
|---|---|---|---|
| WT | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
| H4A | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
| N5A | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
| H6A | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
| P8A | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
| M10A/P11A/P12A | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
| ΔP11 | −2 H2O | 0 | Cys3-Ser9, Ser12-Cys18 |
| ΔP11P12 | −2 H2O | 0 | Cys3-Ser9, Ser11-Cys17 |
| linker +1 aa | −2 H2O | 0 | Cys3-Ser9, Ser14-Cys20 |
| linker +2 aa | (unmodified/−1)a/−2 H2O | 0 | Cys3-Ser9, Ser15-Cys21 |
| Y14A | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
| W15A | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
| E16A | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
| G17A | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
| E18A | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
| ring1 −1 aa (ΔH6) | −2 H2O | 0 | Cys3-Ser8, Ser12-Cys18 |
| ring1 −2 aa (ΔH6A7) | −2 H2O | 0 | Cys3-Ser7, Ser11-Cys17 |
| ring1 +1 aa | −2 H2O | 0 | Cys3-Ser10, Ser14-Cys20 |
| ring1 +2 aa | −2 H2O | 0 | Cys3-Ser11, Ser15-Cys21 |
| ring1 +3 aa | −2 H2O | 0 | Cys3-Ser12, Ser16-Cys22 |
| ring2 −1 aa (ΔW15) | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys18 |
| ring2 −2 aa (ΔW15E16) | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys17 |
| ring2 +1 aa | −1/−2 H2O | 1/0 | Cys3-Ser13 / Cys3-Ser9, Ser13-Cys20 |
| ring2 +2 aa | −1/−2 H2O | 1/0 | Cys3-Ser13 / Cys3-Ser9, Ser13-Cys21 |
| C3S/S9C | −2 H2O | 0 | Ser3-Cys9, Ser13-Cys19 |
| S13C/C19S | −1 H2O | 1 | Cys3-Ser9 |
| S13C/C19S-A | unmodified | 2 | none |
| S13C/C19S-AA | unmodified/ −1 H2O | 2/1 | none / Cys3-Ser9 |
| H4P | (−1)a/−2 H2O | (1) a/0 | Cys3-Ser9, Ser13-Cys19 |
| H6P | (−1) a/−2 H2O | (1) a/0 | Cys3-Ser9, Ser13-Cys19 |
| H4P/H6P | unmodified | 2 | none |
| Y14P/E16P | unmodified | 2 | none |
| Y14P/E18P | −1 H2O | 1 | n.d.b |
| E16P/E18P | −2 H2O | 0 | n.d.b |
| Y14P/E16P/E18P | −1 H2O | 1 | n.d.b |
| 5RGD | −1/−2 H2O | 1/0 | Ser13-Cys19 / Cys3-Ser9, Ser13-Cys19 |
| 15RGD | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
| 16RGD | −2 H2O | 0 | Cys3-Ser9, Ser13-Cys19 |
parentheses emphasize that only trace amounts of this species were observed
n.d. = not determined because of low yields
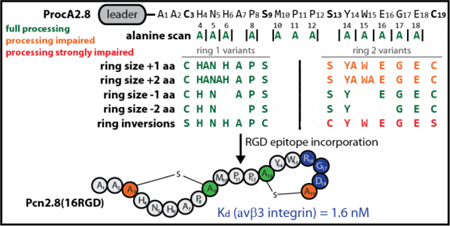
First, an Ala scan of residues within the rings was performed to see if any residue is important for processing. As expected based on the previously reported library studies (but which did not include Ala mutations),17 the ProcA2.8 scaffold showed high tolerance for Ala exchanges inside both ring 1 and ring 2. Next, variation of the Met10-Pro11-Pro12 linker region was investigated (Figure 2b) as we hypothesized that the conformational constraints conferred by the two prolines linking both rings could be important for preorganization of the core peptide and thus enzymatic processing. Exchange of these linking residues to three Ala residues did not affect correct maturation by ProcM, and neither did the deletion of a single or both prolines of the linker region. Thus, the secondary structural constraints imposed by the two consecutive Pro in the linker do not appear to be important for ProcM activity, and hence preorganization of the substrate conformation by this linker does not appear to be critical. Incorporation of additional residues in the linker region was tested as well by introduction of one or two additional Ala residues in between the two lanthionine ring forming sequences (designated linker+aa, Table 1). For both variants, complete maturation of the core peptide was observed, although small amounts of unmodified and only once cyclized side products were detected for double Ala insertion (Supporting Information Figure S10). These results demonstrate that ProcM can readily process ProcA2.8 variants with linker sizes of one to five residues.
We next tested whether ring size is important to ensure correct and complete modification. Being able to alter the ring sizes could be interesting for future bioengineering projects, as it would allow the screening of a larger structural space. As shown in Table 1, ring 1 tolerates both Ala insertions and deletions well, whereas ring 2 does so only for deletions (Supporting Information Figures S54–S64); addition of one or two Ala residues to the sequence forming ring 2 yielded a mixture of fully processed core peptide with the expected ring topology and core peptide that was only dehydrated and cyclized once. Interestingly, tandem MS data suggest that in the latter case an unusual ring is formed between Cys3 and Ser13 (Supporting Information Figures S62 and S64).
Previous studies have shown that ring 2 is the first ring to be formed during processing of ProcA2.8 by ProcM.48 It seems that if this ring is not formed correctly in the ring expansion variants, formation of ring 1 is also perturbed. We therefore hypothesize that formation of ring 2 is important for formation of ring 1, presumably by preorganizing the structure to facilitate cyclization. These findings also suggest that extensions of the sequence leading to what would be an expanded ring 2 slow down its formation such that Cys3 now can attack Dha13 instead of Dha9. This provides a dead-end once-cyclized product. The precise reasons for this outcome are not known, but these findings do inform on the types of cyclization patterns that are accessible from the ProcA2.8 sequence. To further support this notion, we attempted to increase the size of ring 1 even further by inserting three amino acids at once. If indeed the correct formation ring 2 is essential for overall maturation, a further expansion of ring 1 is likely to be tolerated. In agreement with this hypothesis, full modification of the core peptide and formation of WT ring topology was observed (Supporting Information Figure S58).
We also investigated the scope of the ProcM-catalyzed formation of cyclic peptides with respect to stereochemistry. In native Pcn2.8, stereoselective protonation of the enolate formed upon conjugate addition of Cys3 and Cys19 to Dha9 and Dha13, respectively, results in D stereochemistry.47 Thus, Pcn2.8 has L stereochemistry at positions 3 and 19 originating from Cys, and D stereochemistry at positions 9 and 13, originating from Ser. Inverting the positions of the lanthionine forming residues of ring 1 in the variant ProcA2.8(C3S/S9C) would potentially result in a diastereomer of Pcn2.8 having L stereochemistry at position 9 and D stereochemistry at position 3 (see Figure 3a).
Figure 3.
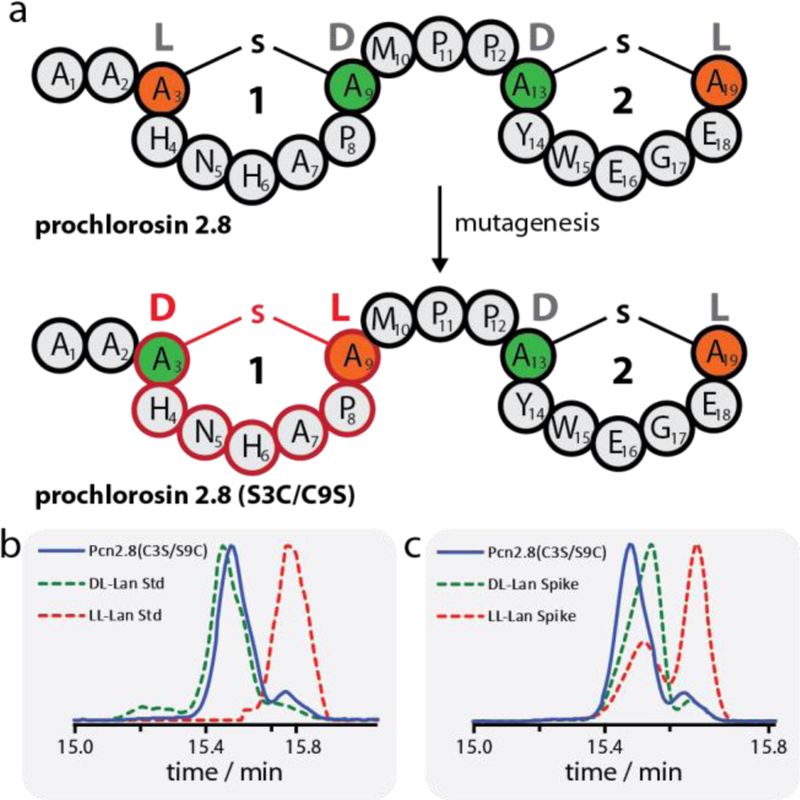
(a) Schematic representation of WT Pcn2.8 and Pcn2.8(S3C/C9S) demonstrating how mutagenesis can be used to invert the stereochemistry of ring 1 and thus yield a diastereomer of the WT lanthipeptide. Orange residues derive from Cys, green residues derive from Ser. (b-c) GC-MS analysis of the lanthionine bis-amino acids in Pcn2.8(C3S/S9C). Shown in (b) is the sample trace (blue) overlayed with traces of the DL-lanthionine (green dashed) and LL-lanthionine (red dashed) standards. Shown in (c) are the traces of the sample (blue) and sample spiked with either the DL-lanthionine (green dashed) or LL-lanthionine (red dashed) standard. The experiments clearly show that the lanthionine bis-amino acids of Pcn2.8(C3S/S9C) have DL stereochemistry. The acid hydrolysis of the peptide results in a small amount of epimerization as previously reported.50
Co-expression of this ProcA2.8 variant with ProcM resulted in the desired bicyclic analog of Pcn2.8 with the same ring topology as WT. The resulting Pcn2.8(C3S/S9C) peptide was hydrolyzed to the constituent amino acids, which in turn were derivatized and then analyzed by GC-MS with a chiral stationary phase alongside DL- and LL-lanthionine standards49 treated the same way (Figure 3b-c). These experiments clearly show that both lanthionines in Pcn2.8(C3S/S9C) have DL-stereochemistry. Thus, as intended, this exchange of Cys and Ser residues indeed allows access to diastereomers of Pcn2.8, thereby extending the scope of cyclic peptides that can be made with ProcM. Again, this has interesting implications for bioengineering studies, as the generation of libraries from both diastereomers of ring 1 expands the chemical space that can be accessed and thus increases the likelihood of finding more potent binders or inhibitors in future screens.
In contrast, the swap of the lanthionine forming residues of ring 2 in the variant ProcA2.8(S13C/C19S) was not successful. Co-expression with ProcM yielded a singly dehydrated and cyclized precursor peptide, with only the Cys3-Ser9 thioether connection formed (Supporting Information Figures S26 and S66). We suspected that placement of a Ser at the C-terminus interfered with the dehydration by ProcM since no lanthipeptides are known with C-terminal Ser/Thr residues that are dehydrated.
Therefore, two new ProcA2.8 variants were generated, both featuring the S13C/C19S exchanges and addition of one or two Ala residues at the C-terminus. Surprisingly, processing was poor for these variants, with ProcA2.8(S13C/C19S-A) completely unmodified (Supporting Information Figures S27 and S67), whereas the ProcM-product of ProcA2.8(S13C/C19S-AA) was isolated as a mixture of unmodified precursor and peptide with a ring between Cys3 and Ser9 (Supporting Information Figures S28, S68, and S69). These data once again show that ring 2 is less amenable to variation, and this observation may be related to this ring usually being formed first. These findings also support the notion that correct formation of native ring 2 might facilitate formation of ring 1, which therefore is more tolerant of changes.
Next, we investigated whether ProcM would tolerate substrates with increasingly constrained backbones by introduction of non-native Pro residues to rings 1 and 2. This approach was taken to test the limits of the machinery. Additional Pro residues were incorporated at positions 4, 6, 14, 16, or 18 (Supporting Information Figures S29–35 and S70–72). In these variants, ring 1 or 2 would contain two or three Pro residues. Once more, ring 1 exhibited a higher tolerance than ring 2 for these changes. Variants with two Pro in ring 1 (ProcA2.8(H4P) and ProcA2.8(H6P)) were processed nearly completely with a small amount of singly dehydrated product. In contrast, only one ring 2 variant containing two Pro was dehydrated and cyclized twice (ProcA2.8(E16P/E18P)), whereas ProcA2.8(Y14P/E18P) was only dehydrated and cyclized once and ProcA2.8(Y14P/E16P) was not modified at all. The presence of three Pro in sequences leading to ring 1 (ProcA2.8(H4P/H6P) or ring 2 (ProcA2.8(Y14P/E16P/E18P)) resulted in unmodified or only once dehydrated and cyclized peptide, respectively. Thus, whereas substitutions of residues in either ring 1 or 2 with Ala, Asn, Asp, His, Ile, Leu, Phe, Tyr and Val were all tolerated by ProcM in this and previous studies,16 substitution with Pro is not accepted as well showing that ProcM tolerance is not unlimited. In turn, these observations also suggest that the folding energy landscape that results in formation of well-defined ring topologies is determined by the sequence of the substrates.
Protease susceptibility and epitope grafting
In addition to assessing the promiscuity of ProcM for exchanges in ProcA2.8 on a single residue level, we tested whether this scaffold can be used for rational incorporation of bioactive peptide epitopes. Cyclic peptides are attractive scaffolds in this regard, as macrocyclic structures often significantly increase proteolytic stability. To probe if this holds true for the Pcn2.8 lanthipeptide, we assayed both the lanthipeptide and the unmodified core sequence against a panel of proteases that are able to cleave at several positions of the primary structure: elastase, chymotrypsin, GluC, and proteinase K (Supporting Information Figure S77a). Whereas the linear peptide is completely degraded by all of the tested proteases, cyclized Pcn2.8 is completely resistant against elastase, chymotrypsin, and GluC. However, proteinase K was able to cleave inside of one of the lanthionine rings, which led to addition of one water molecule. Nonetheless, these assays clearly show that Pcn2.8 has superior proteolytic stability over a linear analog and therefore is a suitable candidate scaffold.
To investigate if Pcn2.8 is able to efficiently present a peptide epitope to a binding partner, we tested incorporation of the RGD integrin binding epitope. This peptide epitope not only has a fitting size for introduction in the Pcn2.8 lanthionine rings, but integrin binding of the products can be assessed by well-established assays and potent αvβ3 integrin binders can inhibit angiogenesis and hence are of interest for generation of anti-tumor agents.10,19,33,51–55 Previous studies have demonstrated that the RGD motif could be incorporated successfully in the third ring of lacticin 481 (Figure 4a–c), a ring consisting of nine amino acids.19 In this work, we grafted the RGD peptide epitope into the smaller rings of Pcn2.8 (Figure 4d–f) to assess whether this would still be tolerated and potentially would lead to tighter integrin-binding.
Figure 4.
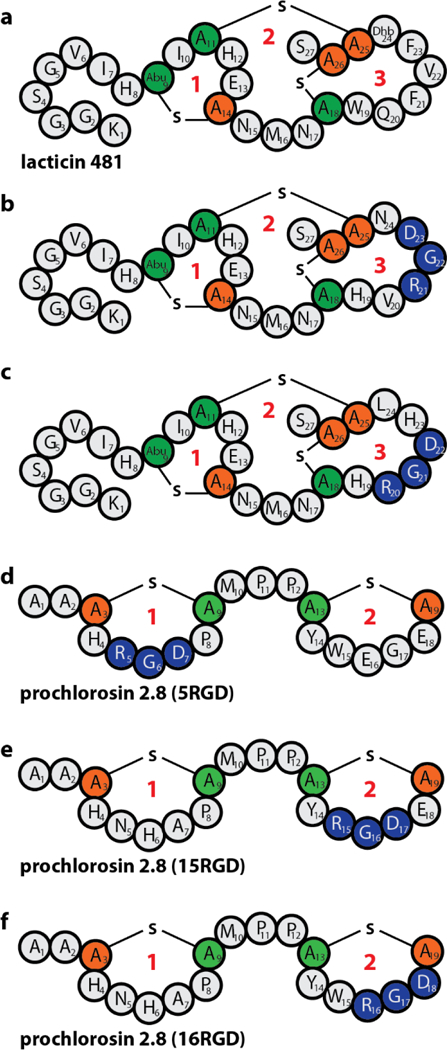
Schematic depiction of (a) lacticin 481, (b-c) the two best αvβ3 binders identified by yeast-display screening using lacticin 481 as a scaffold19 (Ki(b) = 2.5 ± 0.6 nM, Ki(c) = 3.0 ± 1.0 nM), and (d-f) the Pcn2.8 RGD variants generated in this study. Cys residues are in orange and other residues involved in thioether macrocycles in green. Ring number designations are in red. Positions where the RGD epitope was introduced are shown in blue. Abu = Aminobutyric acid.
The motif was introduced centrally into ring 1 (ProcA2.8(5RGD)) and ring 2 (ProcA2.8(15RGD)). In addition, another variant (ProcA2.8(16RGD)) was generated to take advantage of the fact that the Glu16 Gly17 Glu18 sequence in ring 2 only needs one major (E16R) and one conserved (E18D) exchange for introduction of the RGD motif. All variants were isolated in the fully modified state, although for ProcA2.8(5RGD), the majority of the isolated peptide carried a single ring between Ser13 and Cys19, which was separated from the desired, fully modified core peptide. To accomplish this, the mixture of twice- and singly-cyclized peptide was treated with trypsin. The constrained conformation of Arg5 inside a lanthionine ring makes it resistant against proteolysis by trypsin, while the linear peptide chain around Arg5 in the singly-cyclized species allows for fast hydrolysis (Supporting Information Figure S77b). This observation again emphasizes the utility of lanthionine macrocycles for stabilizing peptide scaffolds and thus making them promising leads for drug development.56,57
After isolation and HPLC purification, all three RGD variants were tested in a fluorescence polarization (FP) competition assay with fixed concentrations of αvβ3 integrin and a fluorescently labelled, previously reported55 cyclic RGD peptide (Figure 5). While Pcn2.8(5RGD) turned out to be a poor (Ki > 2 μM) and Pcn2.8(15RGD) a moderate (Ki = 350 ± 80 nM) αvβ3 integrin binder, Pcn2.8(16RGD) exhibited low nanomolar affinity (Ki = 1.6 ± 0.3 nM), similar to the best binders derived from lacticin 481 by yeast-display screening.19 These results demonstrate that the Pcn2.8 scaffold can present a small bioactive peptide epitope to its binding partner, but also highlight that predicting the optimal position for conferring the desired bioactivity is difficult. To assess if the cyclic structure improved or decreased integrin binding affinity, we produced a linear analog of Pcn2.8(16RGD) and employed it in the FP competition assay (Supporting Information Figure S78b). As expected due to the presence of the RGD epitope, binding was observed as well for this peptide, although it displayed roughly an order of magnitude (Ki = 18 ± 3 nM) less affinity than the lanthipeptide variant. To evaluate if cyclization improved proteolytic stability, we treated all RGD lanthipeptides and the linear analog of Pcn2.8(16RGD) with trypsin. Although the fully matured versions of Pcn2.8(5RGD) and Pcn2.8(15RGD) were completely resistant against trypsin (Supporting Information Figure S77b), a portion of the twice cyclized Pcn2.8(16RGD) was hydrolyzed inside ring 2 (Supporting Information Figure S77c). However, the linear Pcn2.8(16RGD) core peptide was completely degraded under identical assay conditions (Supporting Information Figure S77c), which demonstrates that although Pcn2.8(16RGD) is eventually hydrolyzed by trypsin, it still exhibits both an improved proteolytic stability as well as a higher binding affinity to the target integrin. Taken together, the results of these experiments show that Pcn2.8 is a promising scaffold for epitope grafting.
Figure 5.
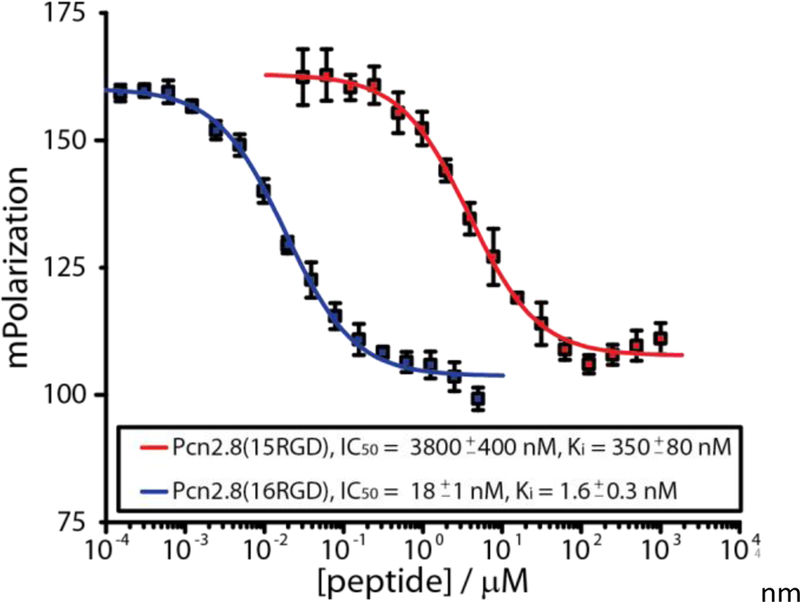
FP competition experiments with Pcn2.8(15RGD) and Pcn2.8(16RGD). The curve for Pcn2.8(5RGD) could not be completed due to poor binding to the αvβ3 integrin of this compound (Supporting Information Figure S78a).
The different affinities of the three different Pcn2.8 RGD lanthipeptide variants towards the αvβ3 integrin is hard to explain without structural data. Factors than can contribute or interfere with target binding include contributions of the neighboring residues to binding, backbone conformation, stereochemistry at the ring juncture, and accessibility of the interacting residues. As such, the observation that Pcn2.8(16RGD) is both the highest affinity binder and the only RGD peptide that is susceptible to trypsin might imply that the Arg16 residue adopts an exposed conformation that facilitates accessibility for both integrin binding as well as trypsin cleavage.
CONCLUSION
In summary, our mutational analysis of ProcA2.8 revealed both the potential and the limits of what can be done with this peptide scaffold (Figure 6). Generally, single residue exchanges inside and between the rings are tolerated well and ring 1 seems to be highly amenable to alterations of its ring size. Ring 1 also accepts inversion of the positions of the ring forming residues, providing access to a diastereomer of Pcn2.8. Conversely, ring 2 only accepts reductions in size, while ring expansions and inversion of the ring forming residues are not tolerated. Introduction of additional Pro residues into either ring is accepted by ProcM but only up to a certain point. Ring 1 again shows a higher tolerance than ring 2 for these kinds of alterations. Our findings seem to indicate that in general the ring formed first during processing is more sensitive towards exchanges than the subsequently formed one. Because of the high intrinsic variation amongst ProcA core peptides, it is hard to predict how well these results can be transferred to other members of the Pcn family, although it is likely that they are similarly receptive to simple exchanges of residues not involved in ring formation. Investigation of the tolerance of these other prochlorosin scaffolds is currently underway.
Figure 6.
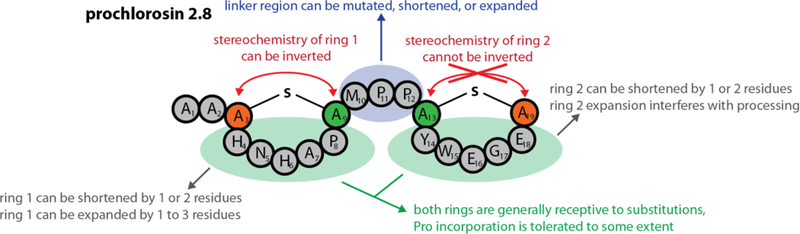
Schematic summary of our findings highlighting which changes in ProcA2.8 are tolerated well by ProcM and lead to a fully dehydrated and cyclized variant of Pcn2.8, and what changes are not tolerated by the biosynthetic enzyme and thus interfere with processing.
The incorporation of the RGD epitope highlights the potential of Pcn2.8 for epitope grafting approaches, but also shows that these require trial-and-error exercises to find an optimal binder. Recent reports19,28 of a yeast-display system for the lacticin 481 lanthipeptide and phage-display systems for nisin and Pcn lanthipeptides are promising starting points for generating a screening platform to help streamline both epitope grafting and library generation efforts for development of lanthipeptides with new biological activities. The improved protease stability and/or binding affinity of these cyclic compounds compared to the linear counterparts further illustrates their potential.
The general implications of our experiments for future bioengineering efforts using Pcn2.8 as basis for library generation are that ring 1 diastereomers should also be considered in screenings as well as libraries starting with altered ring sizes compared to the WT scaffold. Moreover, the high tolerance of ring-internal residues for substitutions might allow development of bifunctional Pcn derivatives, where each ring carries a different peptide epitope and therefore allows binding to multiple targets. Hence, the insights garnered in this study can be of use for planning and executing future engineering studies using Pcns.
METHODS
Bacterial Strains and Materials.
For mutagenesis, E. coli DH10b cells were used, while all expressions were carried out in E. coli BL21(DE3). Oligonucleotide primers were synthesized by Integrated DNA Technologies and the correct sequences of all mutant plasmids were confirmed by sequencing performed by ACGT, Inc. Phusion DNA polymerase and LysC were purchased from New England Biolabs. Micro Bio-Spin Chromatography Columns were bought from BioRad. His60 Ni Superflow Resin was obtained from Clontech. Trypsin was purchased from Worthington Biochemical Corporation. Millipore C18 ZipTips and NHS-fluorescein were obtained from Thermo Fisher Scientific. The c(RGDyK) peptide was bought from AnaSpec Inc.
Mutagenesis.
The plasmid his6-procA2.8(G−1K)[MCS1]:procM[MCS2] pRSF was used as template for generating the mutant library via site-directed ligase-independent mutagenesis (SLIM) using standard protocols.58,59 Oligonucleotide primers used are listed in Supporting Information Table S2.
Small Scale Expression and Isolation of ProcA2.8 Variants.
All media used in this study contained 50 μg/mL kanamycin as selection marker. For small scale His6-ProcA2.8 co-expression with ProcM, aliquots of 100 mL of lysogeny broth (LB) medium were inoculated 1:100 with a 37 °C LB overnight culture. Cells were grown at 37 °C until the optical density at 600 nm (OD600) reached 0.4. At this point, the culture flasks were moved into another shaker at 18 °C and incubated for another hour before expression was induced at OD600 0.5–0.7 by addition of 20 μL of a 0.5 M stock solution (final concentration 0.1 mM) of isopropyl β-D-1-thiogalactopyranoside (IPTG).
After induction, cultures were grown overnight at 18 °C. On the next day, 15 mL aliquots of each culture were pelleted by centrifugation and cells were resuspended in 1 mL of guanidinium chloride containing lysis buffer (500 mM NaCl, 20 mM phosphate, 6 M guanidinium chloride, 0.5 mM imidazole, pH 7.5) and lysed at room temperature (RT) by sonication employing a Vibra Cell sonicator (Sonics & Materials) with a microtip at the following settings: 40% amplitude, 30 s total sonication time alternating between 2 s on- and 2 s off-pulse.
After centrifugation (20 min at 15,700×g), the cleared lysates were transferred to fresh microreaction tubes containing 100 μL of NiNTA resin that was equilibrated with the same lysis buffer and samples were incubated for 30 min at RT while shaking lightly. Afterwards, the resin-lysate mixtures were transferred in a stepwise manner to micro bio-spin chromatography columns (BioRad) and centrifuged at 270×g until all liquid had passed through. For the first wash, the resin was resuspended inside the columns with 800 μL of guanidinium chloride containing wash buffer (500 mM NaCl, 20 mM phosphate, 4 M guanidinium chloride, 30 mM imidazole, pH 7.5) by carefully pipetting up and down. Buffer was then removed by centrifugation at 270×g and this step was repeated with wash buffer without guanidinium chloride (300 mM NaCl, 20 mM phosphate, 30 mM imidazole, pH 7.5). Then, the columns were placed into fresh microreaction tubes and the resin was resuspended inside the columns with 500 μL of elution buffer (300 mM NaCl, 20 mM phosphate, 500 mM imidazole, 1 mM tris(2-carboxyethyl)phosphine, pH 7.5) and incubated at RT for 5 min before it was spun at 270×g until all eluate had passed through. Elution fractions were stored at −20 °C until used again.
MS analysis, NEM labeling and Tandem MS.
For mass spectrometric (MS) analysis of the dehydration states of the ProcA2.8 peptides, the leader peptides were removed by treatment with LysC. An aliquot of 100 μL of an elution fraction (see above) was mixed with 2.5 μL of a LysC stock solution (0.1 mg/mL) and incubated overnight at 37 °C. For direct MS analysis, 10 μL of the LysC reactions were purified by using C18 ZipTips according to the manufacturer’s protocol and samples were eluted into 3 μL of 80% MeCN. Dehydration states were determined by matrix-assisted laser desorption/ionization-time-of-flight MS (MALDI-TOF-MS) on an UltrafleXtreme MALDI TOFTOF (Bruker Daltonics) after samples were prepared using sinapinic acid as matrix (Supporting Information Figures S1–S38).
For analyzing the cyclization state of a peptide, labeling reactions with N-ethylmaleimide (NEM) were performed, which under assay conditions is an electrophile selective for free thiol groups. To accomplish NEM-labeling, 20 μL of the LysC-treated elution fractions were mixed with 1 μL of a tris(2-carboxyethyl)phosphine (TCEP) stock solution (20 mM in dH2O; to ensure reduction of all free thiols) and 1 μL of an NEM stock solution (250 mM in EtOH) and incubated for 15 min at RT. Afterwards, samples were purified by use of C18 ZipTips as described above and analyzed via MALDI-TOF-MS using sinapinic acid as matrix for sample preparation (Supporting Information Figures S1–S38). Each addition of an NEM molecule to a Cys thiol increases the monoisotopic mass of a peptide by 125.05 Da.
Tandem MS of the core peptides was performed to assess the topology of the lanthionine rings. For this, 10 μL of the LysC treated elution fractions were used for high-resolution liquid chromatography-MS (LC-MS) employing an Acquity Ultraperformance LC system (Waters) connected to a SYNAPT-MS instrument (Waters) with a C18 column (Phenomenex, Jupiter 5 μm C18 300 Å, 150×1 mm). A flow rate of 0.15 mL/min at a column temperature of 35 °C was used for LC with the following gradient of solvents A (0.1% formic acid in H2O) and B (0.1% formic acid in MeCN): Holding 3% B for 3 min, followed by a linear increase from 3% to 97% B in 12 min and then holding 97% B for 3 min. The doubly charged core peptide ions were selected for fragmentation by collision-induced dissociation (CID) at a ramping-cone voltage setting of 25–30 kV. The obtained fragmentation data were analyzed with the Mass Lynx software (Waters) using the TOF transform method for deconvolution of the spectra. Masses were calibrated to the exact mass of the fragmented parent ion. Identification of the observed fragments and the intensity of each signal allowed determination of the ring topology (Supporting Information Figures S39–S76). A comparison between the tandem MS spectra of WT cyclized Pcn2.8 −2 H2O (Supporting Information Figure S39a) and WT unmodified Pcn2.8 (Supporting Information Figure S39b) shows that linear and cyclized topologies can be readily distinguished: The linear peptide yields high intensity signals for many of the predicted b- and y-fragments, whereas the only high intensity peaks observed for the cyclized peptide results from bond breakages outside of the lanthionine rings. For the cyclized peptides, sometimes also low-intensity signals relating to b-/y-fragments of dehydrated, but not cyclized peptides were observed. These likely result from the presence of minor species of dehydrated, non-cyclized or only partially cyclized peptides in our isolated peptide samples and were annotated in the spectra for the sake of thoroughness. This notion is supported by the observation that the NEM Cys alkylation assays with cyclized peptides (e.g. WT ProcA2.8, Supporting Information Figure S1a) sometimes show some low intensity ions consistent with NEM adducts, while the cyclized main product is unaffected by NEM treatment. This explains why the intensity of the corresponding b-/y-fragment ions is so low, despite the fact that some of them (e.g. b7) are the major species detected in tandem MS of linear ProcA2.8 core peptide (Supporting Information Figure S39b). These minor side product ions were not detected in a recent study,17 as fragmentation was performed with MALDI-TOF-MS LIFT mode instead of CID, which results in a much lower overall abundance of fragment ions and makes these peaks harder to detect (Supporting Information Figure S79).
For WT Pcn2.8 −2 H2O and Pcn2.8(C3S/S9C) −2 H2O, the triply charged ion of a LysC fragment of the leader peptide is so close to the mass of the doubly charged ions of the twice dehydrated core peptides that the instrument cannot discriminate between them for CID selection and fragments both at once. Therefore, the elution fractions of both full length peptides were digested with trypsin, which would not yield this particular leader peptide fragment. For trypsin proteolysis, 100 μL of an elution fraction was mixed with 2.2 μL CaCl2 (0.1 M in H2O) and 10 μL of a trypsin stock solution (3 mg/mL in 50 mM Tris, 1 mM CaCl2 at pH 7.6) and incubated overnight at RT. Of these digests, 10 μL were applied to LC-MS and the doubly charged core peptide ions were fragmented as described above.
Hydrolysis, Derivatization and GC-MS analysis of Pcn2.8(C3S/S9C).
Purified Pcn2.8(C3S/S9C) (2 mg) was dissolved in 6 M DCl in D2O (3 mL) and transferred to a glass pressure tube. The sample was heated to 110 °C for 18 h, then cooled to RT before removing solvent under reduced pressure. Acetyl chloride (1.5 mL) was added dropwise to 5 mL of MeOH in an ice-water bath. This solution (3 mL) was added to the hydrolyzed sample residue and the mixture was heated to 110 °C for 1 h with reflux. The solution was allowed to cool before removing solvent by rotary evaporation. The residue was dissolved in 3 mL of dichloromethane and the solution cooled in an ice-water bath. Pentafluoropropionic anhydride (1 mL) was added to the reaction vessel and the mixture was then heated to 110 °C for 1 h with reflux. The reaction vessel was cooled to RT and the sample was dried under a gentle stream of nitrogen. The resulting residue was dissolved in 0.1 mL of MeOH and transferred to a clean vial and stored at −20 °C prior to analysis.
GC-MS analysis of derivatized samples was performed on an Agilent HP 6890N mass spectrometer equipped with a CP-Chirasil-L-Val (Agilent) fused silica column (25 m x 0.25 mm x 0.12 μm). Samples, dissolved in MeOH, were introduced to the instrument via splitless injection with a helium gas flow rate of 1.7 or 2.0 mL/min. Runs were held at 160 oC for 5 min, then raised to 190 °C at a rate of 3 °C/min then held at 190 °C for 5 min. The instrument was operated in selected-ion monitoring (SIM) mode, monitoring for 365 Da for derivatized lanthionine. Co-injection with known standards47,49 was used to determine the stereochemistry of Pcn2.8(C3S/S9C). Derivatized DL- and LL-lanthionine standards had been prepared according to previously reported procedures.49
Large Scale Isolation and Purification of ProcA2.8 Variants.
To simplify isolation, RGD mutants of the WT ProcA2.8 encoding gene lacking an additional G−1K mutation were generated. With the WT leader sequence, the core peptide can be released by treatment with the recently characterized LahT150 protease (prepared as recently described),60 which facilitates purification as only two peptides (leader and modified core peptide) are obtained compared to the numerous peptide fragments resulting from treatment with LysC or trypsin. For production of the ProcA2.8 RGD variants in large scale, culture flasks containing terrific broth (TB) medium with 2% glucose and 2 mM MgCl2 were inoculated with 37 °C LB overnight cultures (1:50; volume overnight culture: volume overexpression culture). The cultures were incubated at 37 °C until OD600 reached 1.2–1.5, then cooled to 22 °C and induced by addition of IPTG (0.5 mM final concentration). After overnight expression at 22 °C, the cells were harvested by centrifugation at 5,000×g for 10 min and resuspended in guanidinium chloride containing lysis buffer (6.0 M guanidine hydrochloride, 0.5 mM imidazole, 20 mM NaH2PO4, pH 7.5), using 30–50 mL of buffer for each liter of culture. Resuspended cells were stored at −80 °C until purification. Cell lysis was accomplished by freeze-thawing in the 6 M guanidinium buffer and lysates were cleared by centrifugation at 30,000×g for 30 min at 4 °C. Lysates were applied to NiNTA affinity chromatography using 2 mL of resin that had been equilibrated with 20 mL of the guanidinium chloride containing lysis buffer. After loading, the resin was washed with 20 mL of guanidinium chloride containing wash buffer (4.0 M guanidine hydrochloride, 20 mM NaH2PO4, 30 mM imidazole, 300 mM NaCl, pH 7.5), followed by another wash step using 20 mL of wash buffer without guanidinium chloride (20 mM NaH2PO4, 30 mM imidazole, 300 mM NaCl, pH 7.5). Finally, the His6-tagged peptides were eluted with 20 mL of elution buffer (20 mM NaH2PO4, 500 mM imidazole, 300 mM NaCl, pH 7.5). Core peptides were released via proteolysis through addition of 4 mL of 1 M Tris at pH 8, 16 mL of ddH2O and 50 μL of LahT stock solution (5 mg/mL), followed by incubation overnight at RT. Afterwards, the modified Pcn variants were purified by preparative HPLC using a Nexera HPLC system (Shimadzu) and a C18 column (Macherey-Nagel, VP 250/10 Nucleodur C18 HTec, 5 μM). For separation, solvents A (0.1% trifluoroacetic acid in water) and B (0.1% trifluoroacetic acid in MeCN) were used at a flow rate of 4 mL/min at RT employing a linear gradient from 2% to 100% B in 60 min. In this way, Pcn2.8(15RGD) −2 H2O was obtained with a yield of 2.7 mg/L culture, Pcn2.8(16RGD) −2 H2O with a yield of 2.0 mg/L, linear/unmodified Pcn2.8(16RGD) with a yield of 0.25 mg/L, and Pcn2.8(C3S S9C) −2 H2O with a yield of 0.5 mg/L. For Pcn2.8(5RGD) a mixture of −1 and −2 H2O peptides were isolated with a total yield of 1.9 mg/L culture, which eluted too close to each other for direct separation. Therefore, the once purified peptide mixture was treated with trypsin by dissolving 5.7 mg of peptide mixture in 1 mL of buffer (50 mM Tris, 1 mM CaCl2, pH 7.6) and adding 20 μL of a trypsin stock solution (3 mg/mL). Then, the digest was incubated ON at RT. Under these conditions, the peptide can only be cleaved after Arg5 when ring 1 has not been formed as otherwise this residue is not accessible for proteolysis. After another round of HPLC purification, Pcn2.8(5RGD) −2 H2O was obtained with a yield of 0.2 mg/L culture.
Fluorescence Polarization Competition Assays.
The concentration of Pcn2.8(5RGD), Pcn2.8(15RGD), and Pcn2.8(16RGD) was determined using calculated61 extinction coefficients at 205 nm of 86,280 M−1cm−1, 71,480 M−1cm−1, and 91,880 M−1cm−1 respectively. Fluorescence polarization competition assays were performed as previously described.19 Briefly, initial samples (160 μL) were prepared with 3 nM αvβ3 integrin, and 5 nM fluorescein-c(RGDyK) in integrin binding buffer (IBB) (25 mM TrisHCl, 150 mM NaCl, 2 mM CaCl2, 1 mM MgCl2, 1 mM MnCl2, 0.1% BSA, pH 8)55 with 200 μM, 1,000 μM, or 5 μM of Pcn2.8(5RGD), Pcn2.8(15RGD), or Pcn2.8(16RGD). Then, 15 2-fold serial dilutions of the initials samples into 3 nM αvβ3 integrin and 5 nM fluorescein-c(RGDyK) in IBB were prepared in a black 96-well plate and incubated at RT for 1 h. The fluorescence polarization was measured on a BioTek Synergy H4 Hybrid Reader plate reader with excitation at 485/20 nm and emission measured at 528/20 nm. Non-linear regression was performed in Origin using the following equation:
where A1 is the minimum polarization, A2 is the maximum polarization, logx0 is the log of the IC50, and x is the concentration of lanthipeptide. The data is shown in Supporting Information Tables S3a–d.
The inhibition constant (KI) was calculated with the following equation:
[L]tot is the concentration of fluorescein-c(RGDyK) and KD is the dissociation constant of fluorescein-c(RGDyK) (0.5 ± 0.1 nM19).
As the linear Pcn2.8(16RGD) peptide contains two free Cys residues, the assay had to be slightly altered to ensure presence of only the reduced, linear species and avoid formation of the oxidized, disulfide bridged peptide. Initially, a peptide stock solution was prepared that contained ~75 μM of linear Pcn2.8(16RGD) in dH2O with 1 mM TCEP as reducing agent. This sample was incubated for 10 min at 50 °C to ensure full reduction of the thiol groups. Peptide concentration was then determined using a calculated61 extinction coefficient at 205 nm of 91,880 M−1 cm−1, employing dH2O containing 1 mM TCEP as a blank reference. Starting from this solution, 15 two-fold serial dilutions were prepared into dH2O containing 1 mM TCEP. In addition, a 2X αvβ3 integrin/fluorescein-c(RGDyK)-IBB stock solution was prepared comprising 6 nM αvβ3 integrin, 10 nM fluorescein-c(RGDyK), 50 mM TrisHCl, 300 mM NaCl, 4 mM CaCl2, 2 mM MgCl2, 2 mM MnCl2, and 0.2% BSA at pH 8. For the assay, 40 μL of a peptide dilution was mixed with 40 μL of the 2X αvβ3 integrin/fluorescein-c(RGDyK)-IBB stock solution in a black 96-well plate to accomplish the aforementioned assay concentrations. After incubation for 1 h at RT, the samples were measured as described above.
Protease Stability Assays.
For testing the stability of linear and cyclized WT Pcn2.8 against different proteases, 5 μL of peptide sample (LysC pre-treated elution solution from the NiNTA column, see above) was mixed with 15 μL of respective protease buffer, 1 μL of a TCEP stock solution (20 mM in dH2O), and 2 μL of a stock solution of the respective protease (1 mg/mL in protease buffer). The assay mixtures were incubated overnight at 37 °C for elastase, GluC, and proteinase K, and at RT for chymotrypsin. For elastase, the buffer used contained 50 mM Tris at pH 9.0. For chymotrypsin and GluC, the buffer used contained 100 mM Tris and 10 mM CaCl2 at pH 8.0. For proteinase K, a buffer was used that contained 50 mM Tris at pH 7.5. Afterwards, the protease reactions were purified by using C18 ZipTips according to the manufacturer’s protocol and samples were eluted into 3 μL of 80% MeCN before analysis by MALDI-TOF-MS using sinapinic acid as matrix for sample preparation (Supporting Information Figure S77a).
For investigating the stability of Pcn2.8(5RGD) and Pcn2.8(15RGD) against trypsin, 50 μL of the elution solution from the NiNTA column was mixed with 1.1 μL of a CaCl2 stock solution (0.1 M) and 5 μL of a trypsin stock solution (3 mg/mL in 50 mM Tris at pH 7.5). After incubation overnight at RT, 20 μL of the protease reactions were prepared for and measured by MALDI-TOF MS as described above using C18 ZipTips (Supporting Information Figure S77b).
To assess the stability of cyclized and linear Pcn2.8(16RGD) against trypsin, defined amounts of purified peptides were employed. For the assay, 4 μL of a 5X trypsin buffer (250 mM Tris, 5 mM CaCl2 at pH 7.5) was mixed with 2.5 μL of a peptide stock solution (2 mg/mL in dH2O), 1 μL of a TCEP stock solution (20 mM in dH2O) and 12 μL of dH2O. After mixing, samples were incubated for 10 min at 50 °C to ensure full reduction of free thiol groups, cooled to RT and then 0.5 μL of trypsin stock solution (1 mg/mL in 50 mM Tris at pH 7.5) was added. After overnight incubation at RT, the samples were prepared for and measured by MALDI-TOF MS as described above using C18 ZipTips (Supporting Information Figure S77c).
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants from the National Institutes of Health (R37 GM 058822 to W.A.V.) and the Deutsche Forschungsgemeinschaft (DFG Research Fellowship 309199717 to J.D.H.). This study is dedicated to Prof. Ron Raines (MIT) on the occasion of his 60th birthday.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: XXX.
Supporting figures and tables
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Arnison PG; Bibb MJ; Bierbaum G; Bowers AA; Bugni TS; Bulaj G; Camarero JA; Campopiano DJ; Challis GL; Clardy J; Cotter PD; Craik DJ; Dawson M; Dittmann E; Donadio S; Dorrestein PC; Entian KD; Fischbach MA; Garavelli JS; Göransson U; Gruber CW; Haft DH; Hemscheidt TK; Hertweck C; Hill C; Horswill AR; Jaspars M; Kelly WL; Klinman JP; Kuipers OP; Link AJ; Liu W; Marahiel MA; Mitchell DA; Moll GN; Moore BS; Müller R; Nair SK; Nes IF; Norris GE; Olivera BM; Onaka H; Patchett ML; Piel J; Reaney MJ; Rebuffat S; Ross RP; Sahl HG; Schmidt EW; Selsted ME; Severinov K; Shen B; Sivonen K; Smith L; Stein T; Süssmuth RD; Tagg JR; Tang GL; Truman AW; Vederas JC; Walsh CT; Walton JD; Wenzel SC; Willey JM; van der Donk WA Nat. Prod. Rep 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Evans RL 3rd; Latham JA; Xia Y; Klinman JP; Wilmot CM Biochemistry 2017, 56, 2735–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Klinman JP; Bonnot F Chem. Rev 2014, 114, 4343–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Haft DH; Pierce PG; Mayclin SJ; Sullivan A; Gardberg AS; Abendroth J; Begley DW; Phan IQ; Staker BL; Myler PJ; Marathias VM; Lorimer DD; Edwards TE Sci. Rep 2017, 7, 41074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ayikpoe R; Ngendahimana T; Langton M; Bonitatibus S; Walker LM; Eaton SS; Eaton GR; Pandelia ME; Elliott SJ; Latham JA Biochemistry 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Freeman MF; Gurgui C; Helf MJ; Morinaka BI; Uria AR; Oldham NJ; Sahl HG; Matsunaga S; Piel J Science 2012, 338, 387–390. [DOI] [PubMed] [Google Scholar]
- (7).Repka LM; Chekan JR; Nair SK; van der Donk WA Chem. Rev 2017, 117, 5457–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hegemann JD; Zimmermann M; Xie X; Marahiel MA Acc. Chem. Res 2015, 48, 1909–1919. [DOI] [PubMed] [Google Scholar]
- (9).Mohr KI; Volz C; Jansen R; Wray V; Hoffmann J; Bernecker S; Wink J; Gerth K; Stadler M; Müller R Angew. Chem. Int. Ed 2015, 54, 11254–11258. [DOI] [PubMed] [Google Scholar]
- (10).Hegemann JD; De Simone M; Zimmermann M; Knappe TA; Xie XL; Di Leva FS; Marinelli L; Novellino E; Zahler S; Kessler H; Marahiel MA J. Med. Chem 2014, 57, 5829–5834. [DOI] [PubMed] [Google Scholar]
- (11).Craik DJ; Du J Curr. Opin. Chem. Biol 2017, 38, 8–16. [DOI] [PubMed] [Google Scholar]
- (12).Camarero JA Bioorg. Med. Chem. Lett 2017, 27, 5089–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hudson GA; Mitchell DA Curr. Opin. Microbiol 2018, 45, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Himes PM; Allen SE; Hwang S; Bowers AA ACS Chem. Biol 2016, 11, 1737–1744. [DOI] [PubMed] [Google Scholar]
- (15).Biswas S; Garcia De Gonzalo CV; Repka LM; van der Donk WA ACS Chem. Biol 2017, 12, 2965–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Burkhart BJ; Kakkar N; Hudson GA; van der Donk WA; Mitchell DA ACS Cent. Sci 2017, 3, 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yang X; Lennard KR; He C; Walker MC; Ball AT; Doigneaux C; Tavassoli A; van der Donk WA Nat. Chem. Biol 2018, 14, 375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kakkar N; Perez JG; Liu WR; Jewett MC; van der Donk WA ACS Chem. Biol 2018, 13, 951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hetrick KJ; Walker MC; van der Donk WA ACS Cent. Sci 2018, 4, 458–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hegemann JD; Fage CD; Zhu S; Harms K; Di Leva FS; Novellino E; Marinelli L; Marahiel MA Mol. Biosyst 2016, 12, 1106–1109. [DOI] [PubMed] [Google Scholar]
- (21).Fage CD; Hegemann JD; Nebel AJ; Steinbach RM; Zhu S; Linne U; Harms K; Bange G; Marahiel MA Angew. Chem 2016, 55, 12717–12721. [DOI] [PubMed] [Google Scholar]
- (22).Hegemann JD; Zimmermann M; Zhu S; Steuber H; Harms K; Xie X; Marahiel MA Angew. Chem 2014, 53, 2230–2234. [DOI] [PubMed] [Google Scholar]
- (23).DiCaprio AJ; Firouzbakht A; Hudson GA; Mitchell DA J. Am. Chem. Soc 2019, 141, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Reyna-Gonzalez E; Schmid B; Petras D; Süssmuth RD; Dittmann E Angew. Chem. Int. Ed 2016, 55, 9398–9401. [DOI] [PubMed] [Google Scholar]
- (25).Weiz AR; Ishida K; Quitterer F; Meyer S; Kehr JC; Muller KM; Groll M; Hertweck C; Dittmann E Angew. Chem. Int. Ed 2014, 53, 3735–3738. [DOI] [PubMed] [Google Scholar]
- (26).Al Toma RS; Kuthning A; Exner MP; Denisiuk A; Ziegler J; Budisa N; Süssmuth RD Chembiochem 2015, 16, 503–509. [DOI] [PubMed] [Google Scholar]
- (27).Piscotta FJ; Tharp JM; Liu WR; Link AJ Chem. Commun 2015, 51, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Urban JH; Moosmeier MA; Aumüller T; Thein M; Bosma T; Rink R; Groth K; Zulley M; Siegers K; Tissot K; Moll GN; Prassler J Nat. Commun 2017, 8, 1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Schramma KR; Seyedsayamdost MR ACS Chem. Biol 2017, 12, 922–927. [DOI] [PubMed] [Google Scholar]
- (30).Allen CD; Link AJ J. Am. Chem. Soc 2016, 138, 14214–14217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).de Veer SJ; Weidmann J; Craik DJ Acc. Chem. Res 2017, 50, 1557–1565. [DOI] [PubMed] [Google Scholar]
- (32).Conibear AC; Chaousis S; Durek T; Rosengren KJ; Craik DJ; Schroeder CI Biopolymers 2016, 106, 89–100. [DOI] [PubMed] [Google Scholar]
- (33).Conibear AC; Bochen A; Rosengren KJ; Stupar P; Wang CN; Kessler H; Craik DJ ChemBioChem 2014, 15, 451–459. [DOI] [PubMed] [Google Scholar]
- (34).Pan SJ; Link AJ J. Am. Chem. Soc 2011, 133, 5016–5023. [DOI] [PubMed] [Google Scholar]
- (35).Meindl K; Schmiederer T; Schneider K; Reicke A; Butz D; Keller S; Guhring H; Vertesy L; Wink J; Hoffmann H; Bronstrup M; Sheldrick GM; Süssmuth RD Angew. Chem. Int. Ed 2010, 49, 1151–1154. [DOI] [PubMed] [Google Scholar]
- (36).Wang H; van der Donk WA ACS Chem. Biol 2012, 7, 1529–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wiebach V; Mainz A; Siegert MJ; Jungmann NA; Lesquame G; Tirat S; Dreux-Zigha A; Aszodi J; Le Beller D; Süssmuth RD Nat. Chem. Biol 2018, 14, 652–654. [DOI] [PubMed] [Google Scholar]
- (38).Ortega MA; Hao Y; Walker MC; Donadio S; Sosio M; Nair SK; van der Donk WA Cell Chem. Biol 2016, 23, 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ortega MA; Hao Y; Zhang Q; Walker MC; van der Donk WA; Nair SK Nature 2015, 517, 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Li B; Yu JP; Brunzelle JS; Moll GN; van der Donk WA; Nair SK Science 2006, 311, 1464–1467. [DOI] [PubMed] [Google Scholar]
- (41).Hegemann JD; van der Donk WA J. Am. Chem. Soc 2018, 140, 5743–5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Goto Y; Li B; Claesen J; Shi Y; Bibb MJ; van der Donk WA PLoS Biol 2010, 8, e1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Dong SH; Tang W; Lukk T; Yu Y; Nair SK; van der Donk WA eLife 2015, 4, e07607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Li B; Sher D; Kelly L; Shi Y; Huang K; Knerr PJ; Joewono I; Rusch D; Chisholm SW; van der Donk WA Proc. Natl. Acad. Sci. U.S.A 2010, 107, 10430–10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ma H; Gao Y; Zhao F; Wang J; Teng K; Zhang J; Zhong J Biochem. Biophys. Res. Commun 2014, 450, 1126–1132. [DOI] [PubMed] [Google Scholar]
- (46).Bobeica SC; van der Donk WA Methods Enzymol 2018, 604, 165–203. [DOI] [PubMed] [Google Scholar]
- (47).Tang W; van der Donk WA Biochemistry 2012, 51, 4271–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Mukherjee S; van der Donk WA J. Am. Chem. Soc 2014, 136, 10450–10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Liu W; Chan ASH; Liu H; Cochrane SA; Vederas JC J. Am. Chem. Soc 2011, 133, 14216–14219. [DOI] [PubMed] [Google Scholar]
- (50).Kido Y; Hamakado T; Yoshida T; Anno M; Motoki Y; Wakamiya T; Shiba TJ Antibiot 1983, 36, 1295–1299. [DOI] [PubMed] [Google Scholar]
- (51).Dechantsreiter MA; Planker E; Matha B; Lohof E; Holzemann G; Jonczyk A; Goodman SL; Kessler HJ Med. Chem 1999, 42, 3033–3040. [DOI] [PubMed] [Google Scholar]
- (52).Wang W; Wu Q; Pasuelo M; McMurray JS; Li C Bioconjugate Chem 2005, 16, 729–734. [DOI] [PubMed] [Google Scholar]
- (53).Silverman AP; Levin AM; Lahti JL; Cochran JR J. Mol. Biol 2009, 385, 1064–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Mas-Moruno C; Rechenmacher F; Kessler H Anticancer Agents Med. Chem 2010, 10, 753–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Kimura RH; Levin AM; Cochran FV; Cochran JR Proteins 2009, 77, 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Rink R; Arkema-Meter A; Baudoin I; Post E; Kuipers A; Nelemans SA; Akanbi MH; Moll GN J. Pharmacol. Toxicol. Methods 2010, 61, 210–218. [DOI] [PubMed] [Google Scholar]
- (57).Kluskens LD; Nelemans SA; Rink R; de Vries L; Meter-Arkema A; Wang Y; Walther T; Kuipers A; Moll GN; Haas MJ Pharmacol. Exp. Ther 2009, 328, 849–854. [DOI] [PubMed] [Google Scholar]
- (58).Chiu J; Tillett D; Dawes IW; March PE J. Microbiol. Methods 2008, 73, 195–198. [DOI] [PubMed] [Google Scholar]
- (59).Chiu J; March PE; Lee R; Tillett D Nucleic Acids Res 2004, 32, e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Bobeica SC; Dong S; Huo L; Mazo N; McLaughlin MIH; Jimenez-Oses G; Nair SK; van der Donk WA Elife 2019, 8, e42305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Anthis NJ; Clore GM Protein Sci 2013, 22, 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.