

Abstract
Endothelial to mesenchymal transition (EndMT), where endothelial cells acquire mesenchymal characteristics has been implicated in several cardiopulmonary, vascular and fibrotic diseases. The most commonly studied molecular mechanisms involved in EndMT include TGFβ, Notch, interleukin, and interferon-γ signaling. As of today, the contributions of Akt1, an important mediator of TGFβ signaling and a key regulator of endothelial barrier function to EndMT remains unclear. By using the ShRNA based gene silencing approach and endothelial-specific inducible Akt1 knockdown (ECKOAkt1) mice, we studied the role of Akt1 in EndMT in vitro and pathological vascular remodeling in vivo. Stable, Akt1 silenced (ShAkt1) human microvascular endothelial cells (HMECs) indicated increased expression of mesenchymal markers such as N-cadherin and α-SMA, phosphorylation of Smad2/3, cellular stress via activation of p38 MAP Kinase and the loss of endothelial nitric oxide synthase (eNOS) accompanied by a change in the morphology of HMECs in vitro and co-localization of endothelial and mesenchymal markers promoting EndMT in vivo. EndMT as a result of Akt1 loss was associated with increased expression of TGFβ2, a potent inducer of EndMT and mesenchymal transcription factors Snail1, and FoxC2. We observed that hypoxia-induced lung vascular remodeling is exacerbated in ECKOAkt1 mice, which was reversed by pharmacological inhibition of β-catenin. Thus, we provide novel insights into the role of Akt1-mediated β-catenin signaling in EndMT and pathological vascular remodeling, and present β-catenin as a potential target for therapy for various cardiopulmonary diseases involving vascular remodeling.
Keywords: Akt1, EndMT, TGFβ2, β-catenin, vascular remodeling
Graphical Abstact
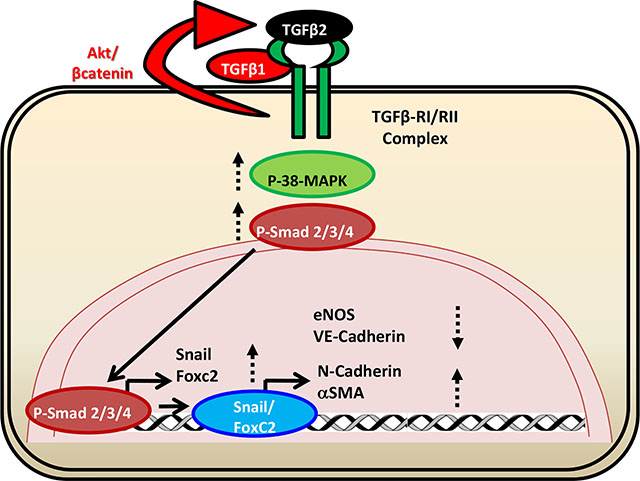
Endothelial Aktl loss promotes endothelial-to-mesenchymal transition (EndMT) via increased TGFβ2, FoxC2 and Snail expression and P38 MAP kinase activation in vitro and hypoxia- induced pathological vascular remodeling in the mouse lungs.
1. Introduction
Endothelial to mesenchymal transition (EndMT) is a phenomenon in which endothelial cells lose their endothelial markers and acquire mesenchymal properties [1, 2]. EndMT is characterized by loss of cell-cell adhesion and changes in cell polarity inducing a spindle-shaped morphology. EndMT is associated with loss of endothelial markers such as VE-cadherin and CD31, and increased expression of mesenchymal markers including fibroblast-specific protein-1 (FSP-1), alpha-smooth muscle actin (α-SMA), N-cadherin, and fibronectin [3] mediated by one or more of the transcription factors Snail, Slug, ZEB-1, SIP-1, Twist, and LEF-1 that suppress the transcription of genes encoding proteins involved in the formation of adherens and tight junctions which are critical in maintaining endothelial barrier integrity[4, 5]. EndMT is involved in embryonic development [4, 6, 7] and several cardiopulmonary diseases including but not limited to pulmonary arterial hypertension (PAH) [8–10], cardiac fibrosis [11, 12], idiopathic pulmonary fibrosis [13, 14], radiation-induced pulmonary fibrosis [15] transplant atherosclerosis and restenosis [16, 17] and chronic obstructive pulmonary disease [18].
A healthy pulmonary endothelial barrier is integral in maintaining vascular homeostasis. Endothelial barrier dysfunction occurs in response to inflammatory mediators such as IL-6 and tumor necrosis factor-α, as well as pathogens [19–21]. Loss of endothelial barrier integrity, disordered endothelial proliferation, and enhanced inflammatory cell infiltration are common features believed to contribute to the pathologic vascular remodeling [22, 23]. Pulmonary vascular remodeling, a hallmark of cardiopulmonary diseases is characterized by intimal thickening, medial hypertrophy, and plexiform lesions. [24]. However, the nature and origin of the cells contributing to the neointimal thickening and plexiform lesion formation remain controversial. Several groups have been trying to characterize the phenotype of these cells located in the pulmonary artery wall [25]. Increasing evidence suggests exposure of endothelial cells to chronic stress and inflammatory factors promotes endothelial to mesenchymal transition (EndMT) contributing to vascular smooth muscle cells (SMCs) and cardiac fibroblast populations during both embryogenesis and pathological conditions [11, 26, 27].
In endothelial cells, intracellular signaling pathways mediated by protein kinase Akt are greatly implicated in the regulation of cell survival, proliferation, migration, glucose metabolism, and gene expression [28, 29] and hence play an important role in the proliferation and migration of ECs, contributing to angiogenesis and transdifferentiation [29–31]. We have recently shown that endothelial loss of Akt1 enhances VEGF-induced barrier breakdown in vitro and promotes VEGF induced vascular leakage in mice ears [32, 33]. We have also shown that endothelial loss of Akt1 promotes lipopolysaccharide (LPS) induced acute lung injury [34] suggesting the involvement of Akt1 in vascular injury, a precursor for the pathological vascular remodeling [35, 36]. However, the role of Akt1, critical barrier integrity regulating kinase in EndMT and vascular remodeling remain largely unknown.
Since Akt1 is essential for endothelial barrier integrity, we hypothesized that sustained endothelial loss of Akt1 will promote EndMT in vitro and pulmonary vascular remodeling in vivo. Our current study demonstrates that Akt1 suppression results in EndMT via increased gene expression of TGFβ2 and FoxC2 in vitro and exacerbates pulmonary vascular remodeling in vivo, which is reversed upon inhibition of β-catenin using ICG-001. Together, our study has identified a novel Akt1-mediated pathway regulating EndMT and pathological pulmonary vascular remodeling along with the therapeutic potential of β-catenin inhibitor ICG-001 for pulmonary vascular diseases.
2. Materials and Methods
2.1. Generation of ‘ECKOAkt1’ transgenic mouse model
All experiments were performed with approval by the Charlie Norwood VAMC Institutional Animal Care and Use Committee (Approval reference #13–09-062). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals. Carbon dioxide asphyxiation followed by cervical dislocation was performed for euthanasia. For our study, we generated an endothelial-specific, tamoxifen-inducible Akt1 knockout mouse model (ECKOAkt1) as previously described [32]. Age and sex-matched 8–12-week-old control wild-type (WT) and ECKOAkt1 mice on a C57BL/6 background were used in the study. 1 mg/10 g dose of Tamoxifen (Sigma, St. Louis, MO) was administered using a 27G needle via intraperitoneal (i.p.) injection every 24 hours for 5 consecutive days. Following this, the transgene was maintained with a custom-made Tamoxifen diet (Harlan, Madison, WI) for the duration of the experiments.
2.2. Cell culture and preparation of ShAkt1 stable cell lines
Human dermal (Telomerase-immortalized) microvascular endothelial cells (HMEC) (CRL-4025; ATCC, Manassas, VA) were maintained in Endothelial Cell Basal Medium-2 with a Growth Medium-2 Bullet Kit (Lonza; Walkersville, MD). All cultures were maintained in a humidified 5% CO2 incubator at 37 F and routinely passaged when 80– 90% confluent.
Stable ShControl, ShAkt1 (ACGCTTAACCTTTCCGCTG) HMEC cells were generated using SMART vector 2.0 lentivirus particles (109 pfu) (Thermo Scientific, Waltham, MA). Lentivirus particles were mixed in 1ml Hyclone SFM4Transfx-293 (Fisher, Hanover Park, IL) and added along with 1 μl Polybrene (10mg/ml, American Bioanalytical, Natick, MA). Three days later, transfection efficiency was tested through Turbo-GFP expression and subjected to 4 μg/ml puromycin (Life Technologies, Grand Island, NY) selection until all cells expressed GFP.
2.3. Human mesenchymal gene array analysis
Control and ShAkt1 HMEC lysates were used for the gene array analysis using human epithelial to mesenchymal transition gene arrays. Briefly, cells were lysed and RNA was isolated using RNeasy Mini plus Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions and the quality was confirmed by using Nanodrop 2000 spectrophotometer (Thermo Scientific). Next, cDNA was generated by RT2 First Strand Kit (SABiosciences, Frederick, MD), mixed with qPCR SyberGreen master mix and loaded into human EMT RT2 Profiler PCR Array plates. Reading was completed in Eppendorf Mastercycler realplex-2 equipment (Hauppauge, NY).
2.4. Cell Scattering Assay
Control and ShAkt1 HMECs were seeded at a low density in 6 well plates and were allowed to grow to form small colonies. After the formation of small scattered colonies, EBM-2 medium was replaced with fresh medium containing 5 % FBS and cells were subjected to either normoxia or hypoxia (0.1%) for 72 hours. Bright field images were captured to represent cell scattering using a phase contrast microscope (Zeiss, Oberkochen, Germany).
2.5. Western blot analysis
Cell lysates were prepared using complete lysis buffer (EMD Millipore, San Diego, CA) with protease and phosphatase inhibitor cocktails (Roche Diagnostics, Indianapolis, IN). Protein quantification was performed using the DC protein assay (Bio-Rad, Hercules, CA). Western blot analysis was performed as described previously [37, 38]. Antibodies used include Akt1, β-catenin (ser675), N-cadherin, VE-cadherin, Snail1, FoxC2, phospho- and total-Smad 2/3, phospho- and total-p-38 MAPK, GAPDH from Cell Signaling (Danvers, MA)., anti- β-actin, alpha-SMA, and TGFβ2 from Sigma Aldrich (St. Louis, MO) and eNOS from BD Pharmingen (SanDiego, CA). HRP-conjugated goat-anti-mouse and goat-anti-rabbit secondary antibodies were obtained from Bio-Rad (Hercules, CA). Densitometry was done using NIH Image J software.
2.6. Chronic Hypoxia and SUGEN-Hypoxia models
Chronic hypoxia exposure for 3 weeks has been widely used to induce pulmonary hypertension and vascular remodeling in rodents. Recently, the combination of chronic hypoxia and a vascular endothelial growth factor (VEGF) receptor antagonist, SUGEN5416 (SUGEN) has been proven to cause profound pulmonary hypertension in rats [39, 40]. In contrast to the chronic hypoxia model, the SUGEN-hypoxia combination causes angio-obliterative lesions in the pulmonary arterioles that are similar to the “plexiform” lesions found in human idiopathic pulmonary arterial hypertension [41]. In mice, it was reported that SUGEN-hypoxia developed severe pulmonary hypertension and vascular remodeling characterized by smooth muscle cell neointima and precapillary EC proliferation and obliteration, leading to increases in RV pressure, and RV hypertrophy [42]. Hence, we employed both the standard chronic hypoxia model and SUGEN-hypoxia model as a “second hit” model that exhibits hallmarks of severe human PAH and vascular remodeling. Briefly, both C57BL6 WT and ECKOAkt1 mice were injected with 10mg/kg Tamoxifen i.p for 5 consecutive days following which they were subjected to 21-day chronic hypoxia in a chamber with regulated nitrogen flow to maintain oxygen levels at 10%. In SUGEN-hypoxia model, the mice received an additional 10mg/kg SUGEN s.c. once a week along with 3 weeks of chronic hypoxia. Treatment groups also received 10mg/kg ICG-001, a β-catenin inhibitor for 21 days, i.p. Lungs were harvested and subjected to Masson’s trichrome staining. Images of lung sections were captured on a Zeiss confocal microscope (Thornwood, NY). Pulmonary blood vessel thickness has been measured using NIH ImageJ software in a blinded fashion. Briefly, medium vessels of size 200 micro meters (to maintain uniformity) have been randomly selected and the vessel wall thickness was measured at 4 regions (making a cross). An average of these four measurements was considered as the vessel thickness for one vessel.
2.7. Statistical Analysis
All the data are presented as Mean ± SD and were calculated from multiple independent experiments performed in quadruplicates. For normalized data analysis, data was confirmed that normality assumption was satisfied and analyzed using paired sample t-test (dependent t-test) and/or further confirmed with non-parametric test Wilcoxon signed rank test. For all other analysis, Student’s two-tailed t-test or ANOVA test were used to determine significant differences between treatment and control values using the GraphPad Prism 4.03 software and SPSS 17.0 software.
3. Results
3.1. Endothelial Loss of Akt1 promotes upregulation of mesenchymal markers and downregulation of endothelial markers in vitro
Western blot analysis of the HMEC lysates transfected with ShRNA targeting Akt1 showed that endothelial loss of Akt1 promotes the expression of mesenchymal markers N-Cadherin and α-SMA (Figure 1A–C), and loss of endothelial marker eNOS (Figure 1A, D). Endothelial loss of Akt1 also upregulates the phosphorylation of stress mediator and fibrotic transcription factors of the TGFβ pathway, p-38 MAP Kinase, and Smad2/3, respectively (Figure 1A, E, F). Together, these results indicate that loss of Akt1 induces EndMT in vitro.
Figure 1. Endothelial loss of Akt1 induces EndMT in vitro.

(A) Representative Western blot images and the corresponding bar graph of band densitometry showing increased expression of N-cadherin (B), and αSMA (C) and a decrease in eNOS (D) accompanied by changes in the expression of pP38 MAP kinase (E) and Smad2/3 (F) compared to their total proteins in HMECs transfected with ShRNA targeting Akt1. Unpaired t-test was performed and data are represented as mean ± SD. *p<0.05; $p<0.0001.
3.2. Endothelial Loss of Akt1 promotes the mRNA and protein expression of mesenchymal genes and transcription factors
Gene array and Western blot analysis of the HMECs transfected with ShRNA targeting Akt1 showed that endothelial loss of Akt1 upregulates the expression of mesenchymal genes fibronectin 1 (FN1), keratin 7 (KRT7), and mesenchymal transcription factor FoxC2. Interestingly, we observed a 9-fold increase in the gene expression of a potent EndMT inducing TGFβ isoform, TGFβ2 (Figure 2A). This was further confirmed by Western blot analysis of the cell lysates, which showed an increase in the expression of TGFβ2 (Figure 2B, D) and the mesenchymal transcription factor Snail1 (Figure 2C, E). These results suggest that endothelial loss of Akt1 induces the expression of TGFβ2 which further activates both canonical and non-canonical signaling pathways promoting EndMT via p-38 MAPK and Smad 2/3. To examine the effect of endothelial loss of Akt1 on morphology, HMECs transfected with ShRNA targeting Akt1 were cultured in 6 well plates and subjected to cell scattering assay. Endothelial loss of Akt1 induced a change in morphology of HMECs where the normoxic cobble shaped cells acquired a spindle-shaped and elongated morphology which is very similar to that of untransfected HMECs exposed to 0.1% hypoxia for 72 hours (Figure 2F). These results indicate that endothelial loss of Akt1 promotes the expression of mesenchymal transcription factors, cell differentiation cytokines and hence a change in the morphology of HMECs contributing to EndMT.
Figure 2. Endothelial loss of Akt1 upregulates mesenchymal genes and transcription factors in vitro.

(A) Gene array analysis of the ShCtrl and ShAkt1 HMEC cell lysates demonstrating changes in the expression of mesenchymal genes FN1and KRT7, mesenchymal transcription factors Foxc2, and a 9-fold increase in the expression of TGFβ2. (B-C) Representative Western blot images and the corresponding bar graph of band densitometry showing increased expression of blot analysis of TGFβ2 (D), Snail (E) with endothelial loss of Akt1. (F) Representative images of normoxic ShControl and ShAkt1 HMECs and normal HMECs exposed to 72-hour hypoxia demonstrating changes in the morphology of HMECs to an elongated spindle shape. One-way ANOVA was performed for figures A and G; Unpaired t-test was performed for figures D and E. Data are represented as mean ± SD. *p<0.05; #p<0.01; $p<0.0001.
3.3. Endothelial Loss of Akt1 and Hypoxia induce EndMT in vivo
Immunofluorescent staining of lung sections of WT and ECKOAkt1 mice subjected to either normoxia or hypoxia revealed that hypoxia promotes co-staining of endothelial marker CD31 and mesenchymal marker α-SMA in WT mice compared to that of normoxia (Figure 3A and C, respectively). Interestingly, in ECKOAkt1 mice, we noticed that endothelial loss of Akt1 resulted in co-expression of CD31 and α-SMA even under normoxic conditions, and the same was observed when they were subjected to 21-day chronic hypoxia (Figure 3B and D, respectively). Together, these results demonstrate that endothelial loss of Akt1 induces EndMT in vivo similar to that of hypoxia.
Figure 3. ECKOAkt1 mice exhibit EndMT in vivo.

(A-B) Representative immuno-fluorescent confocal images of WT and ECKOAkt1 mouse lung vasculature under normoxic condition co-stained with endothelial marker CD31 (in Red) and mesenchymal marker αSMA (in Green). (C-D) Representative immunofluorescent images of WT and ECKOAkt1 mouse lung vasculature under 10% hypoxia co-stained with endothelial marker CD31 and mesenchymal marker αSMA.
3.4. Endothelial Loss of Akt1 exacerbates Hypoxia-induced vascular remodeling which was reversed by β-catenin suppression in vivo
Masson’s trichrome staining of lung sections of ECKOAkt1 mice injected with Tamoxifen and subjected to either normoxia or chronic hypoxia showed that endothelial-specific loss of Akt1 alone induces pulmonary vascular remodeling by increasing blood vessel thickness of the pulmonary microvasculature which was further exacerbated in hypoxia (Figure 4A, C). Masson’s trichrome staining of lung sections of WT and ECKOAkt1 mice subjected to SUGEN-hypoxia and treated with either vehicle or ICG-001, a β-catenin inhibitor showed that inhibition of β-catenin reduced the SUGEN-hypoxia-induced vessel thickening significantly in WT and moderately in ECKOAkt1 mice (Figure 4B, D), indicating that Akt1 mediated β-catenin signaling is involved in inducing EndMT thus contributing to pathological pulmonary vascular remodeling.
Figure 4. Endothelial loss of Akt1 exacerbates hypoxia-induced vascular remodeling in vivo through β-catenin.

(A) Representative images of Masson’s trichrome staining and (C) Bar graph indicating the blood vessel thickness of lung sections of WT and ECKOAkt1 mice subjected to either normoxia or 10% hypoxia for 3 weeks. (B) Representative images of Masson’s trichrome staining and (D) Bar graph indicating the blood vessel thickness of lung sections of WT and ECKOAkt1 mice injected with SUGEN and subjected to hypoxia for 3 weeks. The treatment group received ICG001, a β-catenin inhibitor. Vessel thickness was measured using NIH Image J software. One-way ANOVA was performed and data are represented as mean ± SD. *p<0.05; #p<0.01.
3.5. SU5416 causes irreversible pulmonary vascular remodeling via Akt suppression
Masson’s trichrome staining of lung sections of WT and ECKOAkt1 mice subjected to hypoxia alone or in combination with SUGEN, a VEGFR2 inhibitor revealed that SUGEN induces irreversible pulmonary vascular remodeling in mice even as evident from the increased blood vessel thickening in WT mice compared to that of ECKOAkt1 mice. Treatment with SUGEN exacerbated the hypoxia-induced vascular remodeling in ECKOAkt1 mice (Figure 5A, B).
Figure 5. SUGEN causes irreversible pulmonary vascular remodeling via Akt suppression.

(A) Representative images of Masson’s trichrome staining and (B) Bar graph of vessel thickness of lung sections from ECKOAkt1 mice subjected to hypoxia alone or SUGEN-hypoxia and WT mice subjected to SUGEN-hypoxia. (C-D) Representative Western blot images and (E-F) bar graphs of the band densitometry of HMECs treated with 10 μM SUGEN for 24 and 72 hours, respectively showing that SUGEN treatment directly inhibits Akt. One-way ANOVA was performed for figure B and Unpaired t-test was performed for figures C and D. Data are represented as mean ± SD. (n=3–5), *p<0.05; **p<0.01.
Surprisingly, there was no significant difference between WT mice and ECKOAkt1 mice in vascular remodeling when subjected to SUGEN-Hypoxia given that even normoxic ECKOAkt1 mice exhibited exacerbated vascular thickening compared to that of Hypoxic WT mice. We found that SUGEN inhibits Akt in both short (Figure 5C, E) and long term (Figure 5D, F) in vitro hence contributing to vascular remodeling in vivo.
3.6. Inhibition of β-catenin reverses EndMT induced by the endothelial loss of Akt1 in vitro.
Western blot analysis of HMECs transfected with ShRNA targeting Akt1 and treated with ICG-001 showed that inhibition of β-catenin suppressed the upregulation of mesenchymal markers NCadherin (Figure 6A, C) and ɑ-SMA (Figure 6B, D) induced by the endothelial loss of Akt1. These results reveal the integral role of β-catenin in the promotion of EndMT as a result of Akt1 suppression.
Figure 6. EndMT in ShAkt1 HMECs is blunted by treatment with β-catenin inhibitor ICG-001.

(A-B) Representative Western blot images of untreated ShControl and ShAkt1 HMECs, as well as ShAkt1 HMECs, treated with ICG-001 indicating the changes in the expression of mesenchymal markers N-cadherin and αSMA. (C-D) Bar graph showing the changes in the expression of mesenchymal markers N-cadherin and αSMA in untreated ShControl and ShAkt1 HMECs, as well as ShAkt1 HMECs, treated with ICG-001. (E) Schematic diagram showing the working hypothesis on the molecular mechanisms by which endothelial loss of Akt1 contributes to EndMT. One-way ANOVA was performed and data are represented as mean ± SD. *p<0.05; **p<0.01.
4. Discussion
Endothelial acquisition of mesenchymal characteristics [43, 44], a process essential in the embryonic process [4, 6, 7] has recently been implicated in several cardiopulmonary diseases [8–18]. Although TGFβ, Notch, interleukin, and interferon-γ have been identified as common inducers of EndMT [45–49], the downstream signaling pathways of EndMT are not fully characterized. Although TGFβ remains to be the widely studied EndMT inducing cytokine, the contributions of Akt pathway, an important mediator of TGFβ signaling and a key regulator of endothelial barrier function [29, 50] in EndMT is unclear. In the current study, we examined the role of Akt1, the predominant endothelial isoform [50] in EndMT, investigated its downstream signaling mediators and determined how EndMT can be pharmacologically targeted. We first observed that silencing Akt1 gene in endothelial cells results in increased expression of mesenchymal markers and reduced expression of eNOS accompanied by activation of p38 MAP Kinase as well as increased expression of TGFβ-regulated transcription factors Smad2/3 and mesenchymal transcription factors Snail1 and FoxC2. Next, our results showed that ShAkt1 endothelial cells express increased TGFβ2 mRNA and protein. The analysis of mouse lung vasculature indicated co-localization of endothelial marker CD31 and mesenchymal marker αSMA in WT-hypoxia, ECKOAkt1-normoxia, and ECKOAkt1-hypoxia, compared to WT-normoxia controls, accompanied by significant increase in the vascular thickness, which was blunted by co-treatment with β-catenin inhibitor ICG-001. Lastly, our data revealed that pharmacological inhibition of β-catenin reverses EndMT induced by endothelial loss of Akt1 in vitro. Thus, our study has demonstrated that endothelial Akt1 suppression promotes EndMT via TGFβ2 and β-catenin signaling in vitro and vascular remodeling in vivo, which can be reversed upon pharmacological inhibition of βcatenin.
Two recent studies from our group have demonstrated that 1-day TGFβ1 treatment inhibits the activity of Akt1 in endothelial cells [33] and that 3-day TGFβ1 treatment results in EndMT in vitro [51]. Three other recent studies from our group have also demonstrated that treatment with LPS results in Akt1 inhibition in endothelial cells, in turn, leading to endothelial-barrier breakdown in vitro and pulmonary vascular injury in vivo via reduced expression of tight-junction claudins and increased expression of stromelysin1 akin to the phenotypic changes in ShAkt1 endothelial cells and ECKOAkt1 mouse lung vasculature [32, 34, 52]. Intriguingly, two recent studies by Suzuki et al have indicated that LPS-induced lung injury in mice is accompanied by EndMT [53, 54]. In the current study, we observed that silencing Akt1 gene in human microvascular endothelial cells results in the upregulation of mesenchymal markers, and downregulation of eNOS, indicating the negative regulation of EndMT by Akt1. The reduced expression of eNOS upon loss of Akt1 supports the previous observations that Akt1 plays an important role in the regulation of blood pressure via phosphorylation of eNOS at Ser1179 [55, 56], which indeed implies that that loss of Akt1 could be associated with an increase in blood pressure which itself might contribute to microvascular remodeling. Activation of p38MAP Kinase and Smad2/3 transcription factors observed in ShAkt1 endothelial cells have also been implicated in the TGFβ-induced EndMT [51, 57]. Together, these results indicate that Akt1, which is essential for endothelial homeostasis and barrier function negatively regulates EndMT and that long-term suppression of Akt1 may lead to vascular abnormalities.
Although two different processes, EndMT shares several similarities with epithelial to mesenchymal transition (EMT) in molecular signaling as well as the morphological [58] and pathophysiological processes [59]. A human EMT gene array analysis of ShAkt1 compared to ShControl HMECs revealed an increase in the mRNA expression of several mesenchymal genes fibronectin 1 (FN1), keratin 7 (KRT7), FoxC2 and a dramatic 9-fold increase in the expression of TGFβ2 isoform. We have previously demonstrated that although all three TGFβ isoforms induce EndMT in vitro, TGFβ2 is the most potent among them [51]. In addition, we noticed a significant decrease in the expression of bone morphogenic protein 2 (BMP2), one of the TGFβ superfamily ligands that bind to BMP Receptor-2 (BMPR2) and prevents vascular pathologies [60]. BMPR2 is highly mutated in familial and idiopathic PAH patients [61, 62] as well as in several other vascular diseases [63] indicating that the molecular signatures of EndMT as a result of Akt1 suppression is well aligned with those identified by others in the vascular disease pathologies. Apart from the mesenchymal gene expression, EndMT is also characterized by a change in the endothelial cell morphology [64]. Akt1 deficient endothelial cells were also observed to be more elongated and their distribution scattered, very similar to that of the morphology induced by hypoxia, a well-known inducer of EndMT [65] once again highlighting the importance of basal Akt1 activity in preventing EndMT and associated vascular pathologies.
Co-localization of endothelial and mesenchymal markers is a hallmark feature that defines EndMT in experimental animal models. Reduced expression of endothelial markers, increased expression of mesenchymal markers and their co-expression indicate a transitional phase of endothelial cells [10, 66]. Our results showed that chronic hypoxia promotes co-staining of endothelial marker CD31 and mesenchymal marker αSMA in WT mice compared to that of normoxia indicating that hypoxia induces EndMT in vivo. Interestingly, we noticed that endothelial loss of Akt1 in ECKOAkt1 exhibited co-staining of CD31 and α-SMA even in normoxic conditions and the same was also observed when these mice were subjected to chronic hypoxia, which was accompanied by vascular remodeling similar to what has been reported in PAH [67, 68]. Normoxic ECKOAkt1 mice displayed increased thickness of the pulmonary microvasculature compared to normoxic WT mice, which is further exacerbated by hypoxia. These changes, however, were difficult to capture in the mouse lung vasculature as compared to the magnitude of changes observed with EndMT in vitro. This could be because of the differences between the mouse and human lung vasculature where the mouse pulmonary vasculature displays minimal medial thickening with no apparent complex plexiform lesions, a hallmark in human PAH [41, 69–71]. Together, these results demonstrate that endothelial Akt1 suppression induces EndMT and exacerbates hypoxia-induced vascular remodeling in vivo.
In order to focus on molecular characterization of vascular EndMT in vivo specifically to endothelial cells, we utilized a more suitable SUGEN-Hypoxia model of PAH, where SUGEN, a VEGFR2 inhibitor exacerbates endothelial injury to promote vascular remodeling in vivo [41]. Both WT and ECKOAkt1 mice subjected to SUGEN-Hypoxia exhibited pulmonary vascular thickening, which was reversed by the pharmacological inhibition of β-catenin, a transcription factor that we have previously demonstrated to be activated with Akt1 suppression [32, 52]. Hypoxia has also been shown to promote β-catenin transcriptional activity [72], and inhibition of β-catenin transcriptional activity by ICG-001 treatment in ShAkt1 endothelial cells in vitro resulted in the inhibition of N-Cadherin and ɑ-SMA expression. Surprisingly, we observed no significant difference between the WT and ECKOAkt1 pulmonary vascular thickening SUGEN-hypoxia given that normoxic ECKOAkt1 mice exhibited exacerbated vascular thickening compared to that of Hypoxic WT mice. Upon further investigation, we found the reason for this observation as the ability of SUGEN to directly inhibit Akt, thus masking the effect of endothelial Akt1 loss in ECKOAkt1 mice.
In conclusion, we have identified a novel role of Akt1-β-catenin pathway in EndMT. To our knowledge, this is the first study demonstrating that Akt1 loss in endothelial cells will result in EndMT in vitro and vascular remodeling in vivo through changes in the expression of several mesenchymal genes including TGFβ2. Our study also demonstrates that EndMT in vitro and vascular remodeling in vivo can be pharmacologically targeted using β-catenin inhibitor ICG-001 indicating its therapeutic benefits for various vascular pathologies.
ACKNOWLEDGMENTS
Funds were provided by the NHLBI grant R01HL103952, NCATS grant UL1TR002378, Wilson Pharmacy Foundation (intramural) and Translational Research Initiative grant (intramural). This work has been accomplished using the resources and facilities at the VA Medical Center in Augusta, GA. The funders had no role in the study design, data collection, analysis, and decision to publish the data. The contents of the manuscript do not represent the views of the Department of Veteran Affairs or the United States Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
Authors declare that there are no financial or any other conflicts of interests exist.
References
- [1].Boyer AS, Ayerinskas II, Vincent EB, McKinney LA, Weeks DL, Runyan RB, TGFbeta2 and TGFbeta3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart, Developmental biology 208(2) (1999) 530–45. [DOI] [PubMed] [Google Scholar]
- [2].Azhar M, Runyan RB, Gard C, Sanford LP, Miller ML, Andringa A, Pawlowski S, Rajan S, Doetschman T, Ligand-specific function of transforming growth factor beta in epithelialmesenchymal transition in heart development, Developmental dynamics : an official publication of the American Association of Anatomists 238(2) (2009) 431–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Potenta S, Zeisberg E, Kalluri R, The role of endothelial-to-mesenchymal transition in cancer progression, British journal of cancer 99(9) (2008) 1375–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, Dejana E, Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse, The Journal of cell biology 166(3) (2004) 359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Medici D, Hay ED, Olsen BR, Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3, Molecular biology of the cell 19(11) (2008) 4875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Arciniegas E, Neves CY, Carrillo LM, Zambrano EA, Ramirez R, Endothelial-mesenchymal transition occurs during embryonic pulmonary artery development, Endothelium 12(4) (2005) 193–200. [DOI] [PubMed] [Google Scholar]
- [7].Hall SM, Hislop AA, Pierce CM, Haworth SG, Prenatal origins of human intrapulmonary arteries: formation and smooth muscle maturation, Am J Respir Cell Mol Biol 23(2) (2000) 194–203. [DOI] [PubMed] [Google Scholar]
- [8].Arciniegas E, Frid MG, Douglas IS, Stenmark KR, Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension, Am J Physiol Lung Cell Mol Physiol 293(1) (2007) L1–8. [DOI] [PubMed] [Google Scholar]
- [9].Sakao S, Tatsumi K, Crosstalk between endothelial cell and thrombus in chronic thromboembolic pulmonary hypertension: perspective, Histol Histopathol 28(2) (2013) 185–93. [DOI] [PubMed] [Google Scholar]
- [10].Stenmark KR, Frid M, Perros F, Endothelial-to-Mesenchymal Transition: An Evolving Paradigm and a Promising Therapeutic Target in PAH, Circulation 133(18) (2016) 1734–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R, Endothelial-tomesenchymal transition contributes to cardiac fibrosis, Nat Med 13(8) (2007) 952–61. [DOI] [PubMed] [Google Scholar]
- [12].Zhang Y, Wu X, Li Y, Zhang H, Li Z, Zhang Y, Zhang L, Ju J, Liu X, Chen X, Glybochko PV, Nikolenko V, Kopylov P, Xu C, Yang B, Endothelial to mesenchymal transition contributes to arsenic-trioxide-induced cardiac fibrosis, Sci Rep 6 (2016) 33787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hashimoto N, Phan SH, Imaizumi K, Matsuo M, Nakashima H, Kawabe T, Shimokata K, Hasegawa Y, Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis, Am J Respir Cell Mol Biol 43(2) (2010) 161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nataraj D, Ernst A, Kalluri R, Idiopathic pulmonary fibrosis is associated with endothelial to mesenchymal transition, Am J Respir Cell Mol Biol 43(2) (2010) 129–30. [DOI] [PubMed] [Google Scholar]
- [15].Choi SH, Hong ZY, Nam JK, Lee HJ, Jang J, Yoo RJ, Lee YJ, Lee CY, Kim KH, Park S, Ji YH, Lee YS, Cho J, Lee YJ, A Hypoxia-Induced Vascular Endothelial-to-Mesenchymal Transition in Development of Radiation-Induced Pulmonary Fibrosis, Clin Cancer Res 21(16) (2015) 3716–26. [DOI] [PubMed] [Google Scholar]
- [16].Beranek JT, Vascular endothelium-derived cells containing smooth muscle actin are present in restenosis, Lab Invest 72(6) (1995) 771. [PubMed] [Google Scholar]
- [17].Beranek JT, Cavarocchi NC, Undifferentiated vascular endothelial cells in coronary allograft atherosclerosis, Int J Cardiol 28(1) (1990) 127–8. [DOI] [PubMed] [Google Scholar]
- [18].Sohal SS, Endothelial to mesenchymal transition (EndMT): an active process in Chronic Obstructive Pulmonary Disease (COPD)?, Respir Res 17 (2016) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lerman A, Zeiher AM, Endothelial function: cardiac events, Circulation 111(3) (2005) 363–8. [DOI] [PubMed] [Google Scholar]
- [20].Rajashekhar G, Shivanna M, Kompella UB, Wang Y, Srinivas SP, Role of MMP-9 in the breakdown of barrier integrity of the corneal endothelium in response to TNF-alpha, Exp Eye Res 122 (2014) 77–85. [DOI] [PubMed] [Google Scholar]
- [21].Fairaq A, Goc A, Artham S, Sabbineni H, Somanath PR, TNFalpha induces inflammatory stress response in microvascular endothelial cells via Akt- and P38 MAP kinase-mediated thrombospondin-1 expression, Mol Cell Biochem 406(1–2) (2015) 227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Budhiraja R, Tuder RM, Hassoun PM, Endothelial dysfunction in pulmonary hypertension, Circulation 109(2) (2004) 159–65. [DOI] [PubMed] [Google Scholar]
- [23].Burton VJ, Ciuclan LI, Holmes AM, Rodman DM, Walker C, Budd DC, Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function, Blood 117(1) (2011) 333–41. [DOI] [PubMed] [Google Scholar]
- [24].Derrett-Smith EC, Dooley A, Gilbane AJ, Trinder SL, Khan K, Baliga R, Holmes AM, Hobbs AJ, Abraham D, Denton CP, Endothelial injury in a transforming growth factor beta-dependent mouse model of scleroderma induces pulmonary arterial hypertension, Arthritis Rheum 65(11) (2013) 2928–39. [DOI] [PubMed] [Google Scholar]
- [25].Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F, Mechanisms of disease: pulmonary arterial hypertension, Nat Rev Cardiol 8(8) (2011) 443–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Frid MG, Kale VA, Stenmark KR, Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis, Circ Res 90(11) (2002) 1189–96. [DOI] [PubMed] [Google Scholar]
- [27].Rieder F, Kessler SP, West GA, Bhilocha S, de la Motte C, Sadler TM, Gopalan B, Stylianou E, Fiocchi C, Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis, Am J Pathol 179(5) (2011) 2660–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Coffer PJ, Jin J, Woodgett JR, Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation, The Biochemical journal 335 ( Pt 1) (1998) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shiojima I, Walsh K, Role of Akt signaling in vascular homeostasis and angiogenesis, Circ Res 90(12) (2002) 1243–50. [DOI] [PubMed] [Google Scholar]
- [30].Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K, The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals, Nat Med 6(9) (2000) 1004–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kawasaki K, Smith RS Jr., Hsieh CM, Sun J, Chao J, Liao JK, Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis, Molecular and cellular biology 23(16) (2003) 5726–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gao F, Artham S, Sabbineni H, Al-Azayzih A, Peng XD, Hay N, Adams RH, Byzova TV, Somanath PR, Akt1 promotes stimuli-induced endothelial-barrier protection through FoxO-mediated tight-junction protein turnover, Cell Mol Life Sci 73(20) (2016) 3917–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gao F, Sabbineni H, Artham S, Somanath PR, Modulation of long-term endothelial-barrier integrity is conditional to the cross-talk between Akt and Src signaling, J Cell Physiol (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Artham S, Gao F, Verma A, Alwhaibi A, Sabbineni H, Hafez S, Ergul A, Somanath PR, Endothelial stromelysin1 regulation by the forkhead box-O transcription factors is crucial in the exudative phase of acute lung injury, Pharmacol Res 141 (2019) 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kanemura N, Shibata R, Ohashi K, Ogawa H, Hiramatsu-Ito M, Enomoto T, Yuasa D, Ito M, Hayakawa S, Otaka N, Murohara T, Ouchi N, C1q/TNF-related protein 1 prevents neointimal formation after arterial injury, Atherosclerosis 257 (2017) 138–145. [DOI] [PubMed] [Google Scholar]
- [36].Tanaka LY, Laurindo FR, Vascular remodeling: A redox-modulated mechanism of vessel caliber regulation, Free Radic Biol Med (2017). [DOI] [PubMed] [Google Scholar]
- [37].Abdalla M, Goc A, Segar L, Somanath PR, Akt1 mediates alpha-smooth muscle actin expression and myofibroblast differentiation via myocardin and serum response factor, J Biol Chem 288(46) (2013) 33483–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Goc A, Sabbineni H, Abdalla M, Somanath PR, p70 S6-kinase mediates the cooperation between Akt1 and Mek1 pathways in fibroblast-mediated extracellular matrix remodeling, Biochim Biophys Acta 1853(7) (2015) 1626–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM, Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension, FASEB J 15(2) (2001) 427–38. [DOI] [PubMed] [Google Scholar]
- [40].Nicolls MR, Mizuno S, Taraseviciene-Stewart L, Farkas L, Drake JI, Al Husseini A, Gomez-Arroyo JG, Voelkel NF, Bogaard HJ, New models of pulmonary hypertension based on VEGF receptor blockade-induced endothelial cell apoptosis, Pulm Circ 2(4) (2012) 434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vitali SH, Hansmann G, Rose C, Fernandez-Gonzalez A, Scheid A, Mitsialis SA, Kourembanas S, The Sugen 5416/hypoxia mouse model of pulmonary hypertension revisited: long-term follow-up, Pulm Circ 4(4) (2014) 619–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW, Jarai G, Walker C, Westwick J, Thomas M, A novel murine model of severe pulmonary arterial hypertension, Am J Respir Crit Care Med 184(10) (2011) 1171–82. [DOI] [PubMed] [Google Scholar]
- [43].Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G, Baker AH, Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review, J Am Coll Cardiol 73(2) (2019) 190–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jackson AO, Zhang J, Jiang Z, Yin K, Endothelial-to-mesenchymal transition: A novel therapeutic target for cardiovascular diseases, Trends Cardiovasc Med 27(6) (2017) 383–393. [DOI] [PubMed] [Google Scholar]
- [45].Bianchini F, Peppicelli S, Fabbrizzi P, Biagioni A, Mazzanti B, Menchi G, Calorini L, Pupi A, Trabocchi A, Triazole RGD antagonist reverts TGFbeta1-induced endothelial-to-mesenchymal transition in endothelial precursor cells, Mol Cell Biochem 424(1–2) (2017) 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Perez L, Munoz-Durango N, Riedel CA, Echeverria C, Kalergis AM, Cabello-Verrugio C, Simon F, Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions, Cytokine Growth Factor Rev 33 (2017) 41–54. [DOI] [PubMed] [Google Scholar]
- [47].Shafiee A, Patel J, Wong HY, Donovan P, Hutmacher DW, Fisk NM, Khosrotehrani K, Priming of endothelial colony-forming cells in a mesenchymal niche improves engraftment and vasculogenic potential by initiating mesenchymal transition orchestrated by NOTCH signaling, FASEB J 31(2) (2017) 610–624. [DOI] [PubMed] [Google Scholar]
- [48].Song S, Zhang M, Yi Z, Zhang H, Shen T, Yu X, Zhang C, Zheng X, Yu L, Ma C, Liu Y, Zhu D, The role of PDGF-B/TGF-beta1/neprilysin network in regulating endothelial-to-mesenchymal transition in pulmonary artery remodeling, Cell Signal 28(10) (2016) 1489–501. [DOI] [PubMed] [Google Scholar]
- [49].Wang Z, Han Z, Tao J, Wang J, Liu X, Zhou W, Xu Z, Zhao C, Ju X, Wang Z, Tan R, Gu M, Transforming Growth Factor-beta1 Induces Endothelial-to-Mesenchymal Transition via Akt Signaling Pathway in Renal Transplant Recipients with Chronic Allograft Dysfunction, Ann Transplant 21 (2016) 775–783. [DOI] [PubMed] [Google Scholar]
- [50].Somanath PR, Razorenova OV, Chen J, Byzova TV, Akt1 in endothelial cell and angiogenesis, Cell Cycle 5(5) (2006) 512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sabbineni H, Verma A, Somanath PR, Isoform-specific effects of transforming growth factor beta on endothelial-to-mesenchymal transition, J Cell Physiol 233(11) (2018) 8418–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gao F, Alwhaibi A, Artham S, Verma A, Somanath PR, Endothelial Akt1 loss promotes prostate cancer metastasis via beta-catenin-regulated tight-junction protein turnover, British journal of cancer 118(11) (2018) 1464–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Suzuki T, Tada Y, Nishimura R, Kawasaki T, Sekine A, Urushibara T, Kato F, Kinoshita T, Ikari J, West J, Tatsumi K, Endothelial-to-mesenchymal transition in lipopolysaccharide-induced acute lung injury drives a progenitor cell-like phenotype, Am J Physiol Lung Cell Mol Physiol 310(11) (2016) L1185–98. [DOI] [PubMed] [Google Scholar]
- [54].Suzuki T, Tada Y, Gladson S, Nishimura R, Shimomura I, Karasawa S, Tatsumi K, West J, Vildagliptin ameliorates pulmonary fibrosis in lipopolysaccharide-induced lung injury by inhibiting endothelial-to-mesenchymal transition, Respir Res 18(1) (2017) 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC, Regulation of endothelium-derived nitric oxide production by the protein kinase Akt, Nature 399(6736) (1999) 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Michell BJ, Griffiths JE, Mitchelhill KI, Rodriguez-Crespo I, Tiganis T, Bozinovski S, de Montellano PR, Kemp BE, Pearson RB, The Akt kinase signals directly to endothelial nitric oxide synthase, Curr Biol 9(15) (1999) 845–8. [DOI] [PubMed] [Google Scholar]
- [57].Goumans MJ, Lebrin F, Valdimarsdottir G, Controlling the angiogenic switch: a balance between two distinct TGF-b receptor signaling pathways, Trends Cardiovasc Med 13(7) (2003) 301–7. [DOI] [PubMed] [Google Scholar]
- [58].Wesseling M, Sakkers TR, de Jager SCA, Pasterkamp G, Goumans MJ, The morphological and molecular mechanisms of epithelial/endothelial-to-mesenchymal transition and its involvement in atherosclerosis, Vascul Pharmacol 106 (2018) 1–8. [DOI] [PubMed] [Google Scholar]
- [59].Lamouille S, Xu J, Derynck R, Molecular mechanisms of epithelial-mesenchymal transition, Nat Rev Mol Cell Biol 15(3) (2014) 178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yadin D, Knaus P, Mueller TD, Structural insights into BMP receptors: Specificity, activation and inhibition, Cytokine Growth Factor Rev 27 (2016) 13–34. [DOI] [PubMed] [Google Scholar]
- [61].Machado RD, James V, Southwood M, Harrison RE, Atkinson C, Stewart S, Morrell NW, Trembath RC, Aldred MA, Investigation of second genetic hits at the BMPR2 locus as a modulator of disease progression in familial pulmonary arterial hypertension, Circulation 111(5) (2005) 607–13. [DOI] [PubMed] [Google Scholar]
- [62].Aldred MA, Vijayakrishnan J, James V, Soubrier F, Gomez-Sanchez MA, Martensson G, Galie N, Manes A, Corris P, Simonneau G, Humbert M, Morrell NW, Trembath RC, BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension, Hum Mutat 27(2) (2006) 212–3. [DOI] [PubMed] [Google Scholar]
- [63].Liu D, Liu QQ, Guan LH, Jiang X, Zhou DX, Beghetti M, Qu JM, Jing ZC, BMPR2 mutation is a potential predisposing genetic risk factor for congenital heart disease associated pulmonary vascular disease, Int J Cardiol 211 (2016) 132–6. [DOI] [PubMed] [Google Scholar]
- [64].Krenning G, Barauna VG, Krieger JE, Harmsen MC, Moonen JR, Endothelial Plasticity: Shifting Phenotypes through Force Feedback, Stem Cells Int 2016 (2016) 9762959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhang B, Niu W, Dong HY, Liu ML, Luo Y, Li ZC, Hypoxia induces endothelialmesenchymal transition in pulmonary vascular remodeling, Int J Mol Med 42(1) (2018) 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hong L, Du X, Li W, Mao Y, Sun L, Li X, EndMT: A promising and controversial field, Eur J Cell Biol 97(7) (2018) 493–500. [DOI] [PubMed] [Google Scholar]
- [67].Reid LM, The Third Grover Conference on the pulmonary Circulation. The control of cellular proliferation in the pulmonary circulation, Am Rev Respir Dis 140(5) (1989) 1490–3. [DOI] [PubMed] [Google Scholar]
- [68].Mecham RP, Whitehouse LA, Wrenn DS, Parks WC, Griffin GL, Senior RM, Crouch EC, Stenmark KR, Voelkel NF, Smooth muscle-mediated connective tissue remodeling in pulmonary hypertension, Science 237(4813) (1987) 423–6. [DOI] [PubMed] [Google Scholar]
- [69].Frank DB, Lowery J, Anderson L, Brink M, Reese J, de Caestecker M, Increased susceptibility to hypoxic pulmonary hypertension in Bmpr2 mutant mice is associated with endothelial dysfunction in the pulmonary vasculature, Am J Physiol Lung Cell Mol Physiol 294(1) (2008) L98–109. [DOI] [PubMed] [Google Scholar]
- [70].Stenmark KR, Fagan KA, Frid MG, Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms, Circ Res 99(7) (2006) 675–91. [DOI] [PubMed] [Google Scholar]
- [71].Dempsey EC, Wick MJ, Karoor V, Barr EJ, Tallman DW, Wehling CA, Walchak SJ, Laudi S, Le M, Oka M, Majka S, Cool CD, Fagan KA, Klemm DJ, Hersh LB, Gerard NP, Gerard C, Miller YE, Neprilysin null mice develop exaggerated pulmonary vascular remodeling in response to chronic hypoxia, Am J Pathol 174(3) (2009) 782–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Luo D, Wang J, Li J, Post M, Mouse snail is a target gene for HIF, Mol Cancer Res 9(2) (2011) 234–45. [DOI] [PubMed] [Google Scholar]