

Abstract
While cancer is commonly described as “a disease of the genes”, it is also a disease of metabolism. Indeed, carcinogenesis and malignancy are highly associated with metabolic re-programming, and there is clinical evidence that interrupting a cancer’s metabolic program can improve patients’ outcomes. Notably, many of the metabolic adaptations observed in cancer are similar to the same perturbations observed in diabetic patients. For example, metformin is commonly used to reduce hyperglycemia in diabetic patients, and has been demonstrated to reduce cancer incidence. Treatment with PI3K inhibitors can induce hyperinsulinemia, which can blunt therapeutic efficacy if unchecked. While commonalities between metabolism in cancer and diabetes have been extensively reviewed, here we examine a less explored and emergent convergence between diabetic and cancer metabolism: the generation of lactic acid and subsequent acidification of the surrounding microenvironment. Extracellular lactic acidosis is integral in disease manifestation and is a negative prognostic in both disease states. In tumors, this results in important sequela for cancer progression including increased invasion and metastasis, as well as inhibition of immune surveillance. In diabetes, acidosis impacts the ability of insulin to bind to its receptor, leading to peripheral resistance and an exacerbation of symptoms. Thus, acidosis may be a relevant therapeutic target, and we describe three approaches for targeting: buffers, nanomedicine, and proton pump inhibitors.
Introduction
Acid-Base Balance
“The constancy of the internal environment (Milieu intérieur) is the condition for free and independent life” was noted by Claude Bernard in his Cahier Rouge in 1851 (translated to English in 19671). While he certainly envisioned that this included pH homeostasis, it was not until 1909 that Sørensen developed the pH scale and the pH meter, and Henderson contemporaneously developed equations (later elaborated by Hasselbach) to describe the relationships between acids and conjugate bases2. Since that time, the measurement of pH, especially applied to biological fluids, has been a fundamental component of modern medicine. Indeed the study of acid-base balance is currently an entire field of physiology, the review of which is beyond the scope and intent of the current chapter. However, there are a few fundamental observations (Table 1) that must be accommodated within any investigation of acid-base balance. In humans, arterial and venous bloods have a pH of 7.35–7.4 and 7.20–7.35, respectively. In the steady state, catabolism is oxidative, resulting in the conversion of fats (hydrocarbons), and carbohydrates (alcohols) into carbonic or keto acids. Although body pH is strictly regulated, several diseases besides cancer and diabetes involve significant excursions of intracellular (pHi) or extracellular (pHe) pH. For example, MELAS (mitochondrial myopathy, encephalopathy with lactic acidosis and stroke-like episodes) and epilepsy are both associated with a decrease in brain cell pHi3. In epilepsy, correction of pH balance has been shown to ameliorate episodic seizures in some patients4. Musculoskeletal and cardiac function are intimately coupled to pHi homeostasis5. Low pH is associated with inflammation and pain (nociception) as well6, and this is of paramount importance for both cancer and type II diabetes patients, because both inflammation and pain have a central role in the clinical manifestation of these diseases. This effect appears to have a pathogenetic mechanism for pain in the increased nociception due to the tissue acidosis7. As far as inflammation is concerned, a low pHe is related to the persistence of the inflammatory response and is interconnected with the sensitivity to pain9. Very often the symptoms may represent a central part of the disease, particularly when the disease become chronic. Primary nociceptors are acid stimulated ion channels, ASICs and hence, low pH has a key role in the transition from acute to chronic pain8. As we describe below, acidosis plays a major role in both cancer and diabetes, which share many pathways in common.
Table 1.
Fundamental observations regarding acid-base homeostasis
| 1. pH is the negative log of the H+ activity in solution |
| 2. Neutrality occurs when [H+] = [OH−] at a pH near 7.001. |
| 3. All energy catabolism is eventually oxidative: i.e. the conversion of hydrocarbons and alcohols to higher oxidations states, e.g. acids. |
| 4. Hence, the majority of regulatory systems in the body have evolved to deal with acidic pH excursions. |
| 5. Systemic acid-base balance is achieved through the lungs, which regulate CO2 tension, and kidneys, which regulate excretion of HCO3−. The aqueous CO2 -bicarbonate system is the most important biological buffer. |
| 6. Net diffusion of hydrogen ions in tissue occurs when bound to with mobile buffers: H+ must be accompanied by mobile anions for charge balance. |
| 7. Proteins provide significant non-mobile buffering power. |
| 8. Biological processes affected by pH are primarily mediated by histidine amino acid residues, which have pKa values in the physiological range2. |
| 9. Multiple pathways have evolved to export metabolically-derived acids from inside of cells to their environment. |
Measurement of intra- and extracellular pH, and its correlations with disease, is an extremely active area of research, because there is a compelling need for robust, accurate, and clinically-translatable methods of measuring pH in-vivo. Such technologies can be used as diagnostic, predictive, and/or response biomarkers, have been comprehensively reviewed elsewhere13. Of note, a recent study has shown that the MRI technique called chemical exchange saturation transfer, CEST, has recently been used to measure pH in a small set of human patients9.
Metabolic Commonalities of Diabetes and Cancer.
Epidemiological evidence indicate that patients with type 2 diabetes (T2D), especially those with metabolic syndrome of dyslipidemia, hyperinsulinemia, and hyperglycemia have increased risk of developing cancers of the GI tract (liver, pancreas, colon, and rectum), as well as breast, endometrium, and bladder10,11. Recent meta-analyses have indicated that the most convincing risk of T2D patients to develop a cancer is with colorectal cancer, and that, although there is a trend, the association was weaker for pancreatic, endometrial, hepatocellular and gallbladder carcinoma12,13. Epidemiological studies have shown that numerous risk factors are shared by diabetes and several cancer sites. Primary among these are obesity and smoking status, but also includes low physical activity and alcohol consumption. Pathophysiological mechanisms implicated in the association between T2D and cancer have been proposed for colorectal, pancreas and liver cancers. These include the T2D microenvironment, as represented by advanced glycation end-products, chronic local inflammation, hyperlipidemia, extracellular matrix disorders and altered microbiota that could predispose the development of colorectal cancer. However, despite the strong epidemiological evidence, the mechanisms of this association between diabetes and cancer are not understood.
The major sub-type of T2D is peripheral insulin resistance associated with obesity and central adiposity, leading to hyperinsulinemia and chronic inflammation, both of which have potential to exacerbate cancer risk. Hyperinsulinemia with hyperglycemia also contributes to accumulation of keto-acids, leading to chronic systemic metabolic acidosis, which is compensated by reducing HCO3- and reduced interstitial buffering capacity, making interstitial pH more fragile. Hyperinsulinemia is also associated with increased circulating levels of insulin-like growth factor-1 (IGF1), which is a potent mitogenic factor for neoplastic epithelial cells16. Binding of IGF1 to its receptor triggers activation of the PI3K → Akt → mTOR pathway, inducing metabolic activation and mitogenesis14.
The impact of activating these pathways on acid production is exacerbated by mitochondrial dysfunction, which is commonly observed in both T2D and cancers through activation of oncogenes or inactivation of tumor suppressors15,16. In cancer cells, mitochondria are one of the main signaling targets for oncogenic and tumor suppressors, resulting in reprogramming of cellular metabolism. This signaling process leads to dysfunctional shapes (fragmentation/fission) of mitochondria mediated by activation/upregulation of dynamin-related protein 1 (DRP1), matching the metabolic demands of the tumor cells17,18. Notably, T2D-associated hyperglycemia leads to the increased production of mitochondrial ROS, causing mitochondrial fragmentation/fission via activation/upregulation of a fission protein, dynamin-related protein 1 (DRP1), and downregulation of mitofusin 2 (Mfn2). Increased fragmentation/fission of mitochondria is linked to various types of symptoms observed in T2D including insulin resistance19. Although further information is required to clarify the role of mitochondrial fragmentation and fission in the symptoms observed inT2D and cancer, both T2D and cancer are associated with high proton production rates due to mitochondrial dysfunction.
However the most important common feature between cancer and diabetes is the increased reliance on glucose fermentation. The continuous glucose fermentation can lead to lactate production and a significant local acidosis in both diabetic peripheral tissues and in tumors. Acidosis is exacerbated if combined with decreased perfusion, which can be a consequence of inflammation, peripheral vascular resistance, or dysangiogenesis, all common syndromes in cancer and diabetes. There is significant evidence, presented below, that this local acidosis in cancer, can promote tissue remodeling, local invasion, metastasis, and inhibition of immune surveillance. In diabetes, local and systemic acidosis reduces insulin’s affinity for its receptor, exacerbating the spiral of peripheral insulin resistance. Consequently, targeting acidosis would be an important therapeutic approach in both T2D and cancer, as discussed below.
A common pathway observed in both cancers and diabetes is the phosphatidyl inositol-4,5-bisphosphate 3-kinase, PI3K, cascade, resulting in production of inositoltris-phosphate, IP3, which is a potent activator of Ca2+ release from intracellular stores. Chronic activation of the inositol phosphate cascade can occur in either the PI3K catalytic subunit alpha, PIK3CA, or inactivation mutations of the phosphatase and tensin homolog, PTEN, gene, are among the most commonly observed metabolically-associated alterations in cancers20. Hence pharmacological inhibition of PI3K is an attractive pan-cancer therapeutic target. PI3K inhibitors have been in clinical trials, and clinical benefit has been reported in a phase I setting21,22. In these trials, however grade 3 or higher fasting hyperglycemia was a common (>30%) and sometimes limiting adverse event. As the PI3K enzyme is also responsible for insulin signal transduction, it was hypothesized that its inhibition would lead to blunting of the insulin signal (mimicking T2D), leading to hyperglycemia and hyperinsulinemia. This was recently investigated pre-clinically, where it was shown that the hyperinsulinemia could suppresses the effectiveness of PI3K inhibition in mouse models23. Further, suppressing the hyperglycemia/hyperinsulinemia response, most notably by a ketogenic diet, reduced levels of phosphorylated insulin receptor, pAkt and pS6 and could re-sensitize tumors to PI3K inhibition. This illustrates the close interplay in the metabolism of cancer and diabetes.
Tumor Acidity
Solid tumors, regardless of cancer type, are characterized as being highly heterogeneous, at the genomic, anatomic, physiologic and metabolic levels. The proximal cause of this heterogeneity is the “chaotic” and abnormal tumor vasculature, which leads to different microenvironments with different perfusion characteristics24. These perfusion deficits select for cells that express metabolic phenotypes that are most fit in these different environments, and these metabolic phenotypes are ultimately under control of the epigenetic and genetic landscape.
A common metabolic phenotype observed in solid tumors is elevated rates of fermentative glycolysis, i.e. the non-oxidative conversion of glucose to lactic acid. While this can be induced as an adaptive response to poor oxygenation (the “Pasteur Effect”), a remarkable century-old observation is that this glycolytic phenotype can be hardwired, and thus cancers ferment glucose, even in the presence of adequate oxygen (the “Warburg Effect”). This glycolytic switch likely occurs early in cancers, during the avascular phase of carcinoma in-situ, CIS25, wherein cancer cells that express this phenotype are more likely to survive than ones who do not26. Although the mechanism and drivers of aerobic glycolysis are still debated, it is an unequivocal fact that tumors produce copious amounts of non-oxidized acids as result of elevated glucose fermentation27. In combination with perfusion deficits, this results in the accumulation of acids in the extracellular environment and an acidic tumor pH, with values as low as pH 6.528-31.
An acidic microenvironment strongly influences cancer progression32,33. Although it is initiated early in carcinogenesis, this phenotype is retained as cancers become locally invasive, a process known as “niche engineering”. Acidosis promotes tumor progression by stimulating invasion and metastasis 26,32,34,35, can be toxic to normal cells and mediate degradation and remodeling of the extracellular matrix 36, can elevate angiogenesis through the release of VEGF37, and can inhibit immune surveillance by inducing T-cell stasis38.
An acidic environment has also been shown to induce genomic instability and is an evolutionary selection force for aggressive clones of cells that are acid-adapted, leading to genomic diversity 33. Further, the metabolic adaptations to acidity can result in large changes in the epigenome39. As acidity is evident in early cancers, it can be inferred that this contributes to intratumoral genetic heterogeneity40–43, which is a proximal cause of malignance and resistance44. In addition to genetic heterogeneity, it has been shown in imaging studies that tumors are also physiologically and anatomically heterogeneous and that this is related to poor prognoses 45–47. This can be quantified by converting medical images to mineable data (“radiomics”) of entire tumors46,48–50 or definable sub-regions in tumors with specific combinations of perfusion, cell density, and extracellular matrix (ECM)51,52. Notably, invasive edges of tumors have habitats that are physiologically distinct from the tumor cores, and the larger the differences, the worse the prognosis 53.
At the microscopic level, invasive edges are complex mixtures of multiple cell types, including cancer cells, fibroblasts and immune cells. The tumor cells at the invading edge have distinct protein expression patterns, compared to those in the tumor cores54,55. Specifically, cells at the edge are more proliferative, less apoptotic, and have increased expression of GLUT-1 and CA-IX relative to the tumor cells in the core. The membrane bound exofacial CA-IX is notable as it has a much lower pKa (<6.5) compared to other carbonic anhydrases CA-XII (7.1) and thus, CA-IX is more active at low pH 56,57, and acts to acidify the tumor-stroma interface58. Intravital microscopy shows that invading tumors secrete acid into their surrounding stroma36,59, which induces ECM remodeling and local invasion, driven by increased lysosomal turnover, the release of cathepsins, collagen re-organization and the release of inflammatory cytokines by stromal fibroblasts 60–64.
Another important sequelae of acid release into the stroma is immune evasion through inhibition of T-cell activation or induction of a macrophage phenotypic switch 38,65,66. Microenvironmental acidosis reduces the effector function of tumor infiltrating lymphocytes (TIL), with reduced secretion of IL-2, up-regulation of CD25, and activation of STA5/ERK signaling67–69. Recently it has been shown that acidic pH blocks the activation and anti-tumor functions of T-cells via inhibition of interferon-gamma translation and that this is associated with metabolic changes65.
Microenvironmental acidity is also involved in an increased rate of endosomal-lysosomal trafficking64,70, and increased release of extracellular vesicles (EVs) by tumor cells 71,72. This might be due to a need to eliminate waste, including excess acid 73. It is thus conceivable that cancer cells under the pressure of a very harsh microenvironment need to eliminate more toxic byproducts and one mechanism at their disposal is to use extracellular elimination through exosomes or vesicles. One example is the elimination of chemotherapeutics through exosomes74, thus participating to chemoresistance. However, the increased release of exosomes in in vitro acidic condition has been recently related to the in vivo condition comparing plasmatic exosomes from cancer patients to both healthy donors and patients with benign tumors. It is believed that exosomes may have a key role in tumor metastasis in both setting the metastatic niche and transforming stem cells contained in target organs75,76
Acidosis in Diabetes.
T2D is associated with mitochondrial dysfunction, altering the distribution of acid load15. Mitochondrial dysfunction observed in T2D leads patients to produce large amounts of lactic acid, due to loss of function of the TCA cycle, which contributes to a lowering of the interstitial fluid pH (Fig. 1). Additionally, systemic acid load can be exacerbated by intake of high amounts of protein, measured as potential renal acid load, PRAL, and net endogenous acid production, NEAP 77,78. In large meta-analyses, dietary acid load was strongly positively correlated with incidence of T2D, especially in women79,80. Further, in diabetes, due to this mitochondrial dysfunction and a lack of blood glucose, keto-acids such as beta-hydroxybutyric acid 81 are abundant and further contribute to acidosis 82. These ketone bodies are consumed in extra-hepatic muscle and brain for ATP synthesis 83. Thus, patients suffering from T2D with relatively normal function of hepatic mitochondria associated with no glucose availability show lowered pH in the interstitial fluid due to a large amount of ketone bodies produced from free fatty acids in addition to lactic acid.
Fig. 1.
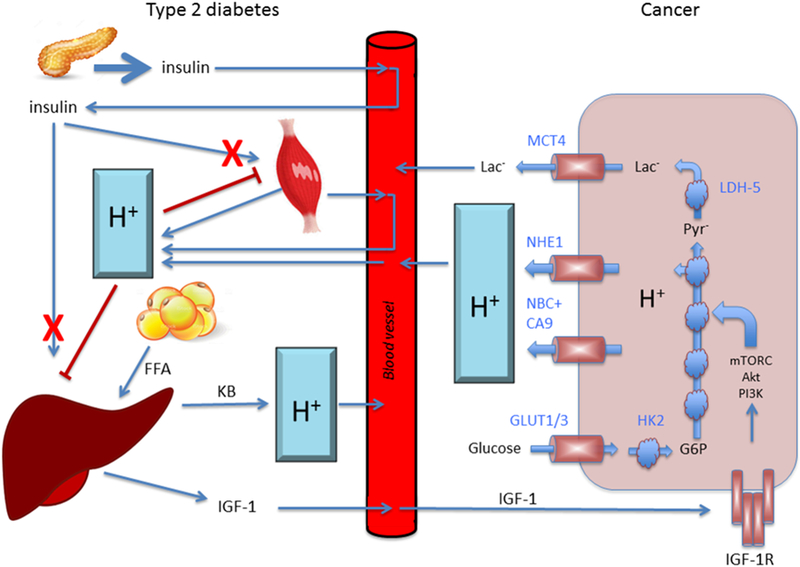
Regulation of pH and its roles in cancer and diabetic cells. The interstitial fluid pH is more highly variable compared with blood pH due to a lack of strong pH buffer. A large amount of proton and lactate- are produced via metabolism in cancer and diabetic cells. A large amount of proton (H+) produced systematically in diabetic cells of the whole body are circulated into other any organs in addition to cells themselves producing a large amount of proton (H+). The large amount of proton (H+) provides low pH environments surrounding cells in any organs including cancer and diabetic cells. These low pH environments produce insulin resistance by decreasing the affinity of insulin to its receptor, leading more proton (H+) production due to diminution of glucose availability caused by insulin resistance. In the liver, the insulin resistance produces ketone bodies (KB) from free fatty acids (FFT) leading to much lower pH environments, and also releases IGF-1, which accelerates the cancer cell growth producing more proton (H+).
Acidity is one of the most important therapeutic targets for diabetes mellitus 84. In diabetes, the chronic compensated metabolic acidosis is systemic rather than regional, as seen in cancer. Compensation results in loss of HCO3- buffering, which makes the interstitial pH more fragile. One consequence of the low extracellular low pH observed in diabetes mellitus is that it reduces the binding affinity of insulin to its receptor, exacerbating peripheral insulin resistance 85. Insulin resistance is one of the most essentially important symptoms observed in type 2 diabetes mellitus: patients of type 2 diabetes mellitus with insulin resistance develop hypertension, one of the most typical clinical symptoms frequently observed as cardiovascular disorders 86–91. Insulin resistance also leads to hyper-insulinemia, and develops vascular dysfunction, hyper-activation of sympathetic nerves, and renal failure; resulting in hypertension 91–97. Many researchers have been trying to develop various types of drugs for treatment of T2D such as sulfonylurea, biguanide, glucosidase inhibitors, thiazolidine, dipeptidyl-peptidase (PPD) IV inhibitors, and sodium/glucose co-transporter 2 (SGLT2) inhibitors based on the premise that reducing blood sugar levels can ameliorate many of the sequelae of T2D, with no significant results and needing new treatment approaches. The continuous hyperglycemia caused by the insulin-resistance-induced poor uptake of glucose into cells such as skeletal muscles, adipocytes, and hepatocytes stimulates insulin secretion from pancreatic β-cells98–104. Thus, continuous hyperglycemia exhausts pancreatic β-cells, resulting in development of dysfunction and damage. Indeed, reduction of blood sugar levels through, e.g. a ketogenic diet, prevents continuous hyper-secretion of insulin. As mentioned above, the fundamental prevention and treatment of T2D should be developed based on the idea of reducing insulin resistance. We hypothesize that insulin resistance is in part caused by lowered interstitial fluid pH observed in T2D105 and lowered interstitial (extracellular) fluid pH reduces the binding affinity of insulin to its receptor 85. Therefore, as a fundamental prevention and treatment for T2D, we propose to develop methods to maintain the interstitial fluid pH to normal levels, which is also a goal of cancer treatments.
Targeting Acidity
Numerous groups are working to develop therapeutic approaches to inhibit or target tumor acidity. These approaches can be divided into (1) direct approaches using oral buffers, diet, or targeted agents to raise tumor pH, (2) developing agents that sequester into cells that are in an acidic microenvironment; or (3) developing or repurposing acid-activated agents, such as proton pump inhibitors, PPIs.
Direct Targeting.
The most direct approach to neutralize tumor acidity is the administration of oral buffers, such as NaHCO3. In animal models oral buffers, such as sodium bicarbonate, specifically increase tumor pH without affecting systemic pH balance, and potently inhibits experimental or spontaneous metastases 106–109. Indeed, a commercially available mix of bicarbonate and carbonate salts (i.e. BasenPulver, Pascoe Germany) was able to control melanoma growth consistent with a buffering effect at both tumor and systemic level 110. These responses are due to buffer effects rather than to bicarbonate per se, as other buffers also work 111,112. Further, effects are also manifest in genetically engineered mouse cancer models (GEMMs). For example, in the TRAMP prostate model, the initiation of buffer therapy at 4 weeks of age prevents emergence of cancer113, but if administered after 10 weeks (after tumors are extracapsular), it has no effect on the primary tumors, but completely inhibits metastases114,115. As discussed below, despite the promising pre-clinical results, it is difficult to translate buffer therapy to the clinic. Recently, Another method to directly inhibit tissue acidity is through application of urease enzyme, which converts urea to 2 NH4+ and 1 HCO3-, producing a net local increase in pH. L-DOS47 is a Jack Bean urease targeted to CEACAM6 antigen116. An alternative agent to directly raise pH is TRC101, which is an oral non-digested nanoparticle that increases systemic buffering by absorbing HCl from the gut, leading to compensated metabolic alkalosis. This has been tested in successful clinical trials in patients with chronic kidney disease117.
Ion Trapping.
In normal tissues, the extracellular interstitial pH, pHe, is approx. 7.3 and the intracellular cytosolic pH, pHi, is ca. 7.2, or a pH gradient of −0.1. In acidic tissues, the pHe can be as low as 6.7, and the pHi will be maintained at 7.0–7.1, or a pH gradient of +0.3–0.4. This reverse pH gradient can be exploited to trap chemotherapeutics in the cytosol of cells that are in more acidic pHe environments. Ion trapping is well-characterized theoretically and empirically, and occurs if the non-ionized form of the drug is membrane permeant and the charged form is not. Hence, weak base chemotherapeutics are excluded from more alkaline compartments, and weak acids are sequestered118. Table 2 provides a summary of weakly acidic, weakly basic and complex chemotherapeutics that are commonly used to treat cancers. Notably, if therapy is designed to neutralize tumor acidity, the benefits of these agents would be reversed and hence, those with increased uptake due to tumor acidity would have reduced uptake and vice-versa. Thus, raising tumor pH with buffers or targeting would be expected to enhance efficacy of weakly basic drugs while reducing efficacy of weak acid agents 106,107.
Table 2.
Common chemotherapeutic agents that are affected by the acidic pH of tumors (decreased or increased by acidic pH).
| Weak bases (decreased uptake) |
| • Ifosfamide/Cyclophosphamide |
| • Erlotinib/Lapatinib/Gefitinib |
| • Tamoxifen |
| Weak Acids (increased uptake) |
| • Flurouricil |
| • Capcitabine |
| • Pemetrexed |
| Complex Ionization (decreased uptake) |
| • Melphelan |
| • Doxorubicin/Daunorubicin |
| • Imatinib |
| Complex ionization (increased uptake) |
| • Gemcitabine |
| • Irinotecan |
| • Sunitinib |
Acid-activated Agents.
Development of agents that are only active under relatively acidic conditions is an area of active investigation. There is a tremendous interest in developing nanoparticles that are induced to release therapeutic agents in areas at low pH of tumors, and this has recently been reviewed119,120. Multiple chemistries can be used in these particles, such as lipid and polymeric shells made of ionizable groups that are designed to disassemble at low pH, releasing chemotherapeutic agents 121–123 .
A well-developed class of agents that are activated by low pH are the Proton Pump Inhibitors, PPIs. As discussed previously, maintenance of intracellular pH is accomplished by a series of proton exchangers, including vacuolar ATPases (V-ATPase), Na+/H+ exchanger (NHE), monocarboxylate transporters (MCTs) and carbonic anhydrase 9 124. These proton-extruding mechanisms remove metabolic acid into the extracellular matrix and are thus key to survival, especially in an acidic milieu. Thus, depriving cancer cells of the functions exploited by these exchangers should inevitably lead to cell death due to internal acidification. V-ATPases actively participate in this process in tumor cells by pumping H+ both from the cytosol to internal vacuoles that are rapidly turned over in the face of acidity 125. While direct inhibition of V-ATPases with specific agents, such as bafilomycin, are effectively cytotoxic in vitro, they have too much toxicity in vivo to be effectively used therapeutically, with an LD50 (mice) of ~0.45 mg/kg. An alternative approach would be to use proton pump inhibitors (PPI) that are used worldwide as very potent antacids (i.e. omeprazole, esomeprazole, lansoprazole, pantoprazole and rabeprazole). Importantly, these are well tolerated, even in prolonged treatments and at very high dosages, as in patients with Zollinger and Hellison syndrome and other conditions 124–127. .
PPIs are cysteine targeting Tetracyclic Sulfonamide prodrugs that require protonation in an acidic milieu to be activated to covalently bind to free sulfhydryls. Thus, they are effective in the acid pH of the stomach, and hence the major intended target is the gastric H+/K+ATPase, which is abundant and has a vulnerable cysteine. In cells of extragastric tissues, acid pH is limited to lysosomes and hence, V-ATPases are also targeted, as they have vulnerable cysteines as well128,129. Tumor targeting may well come from the well-characterized effect of acidic extracellular environments to stimulate lysosomogenesis with increased lysosomal turnover 64,70,130,131. . A series of preclinical investigations have shown that PPI sensitize tumor cells and tumors to the action of chemotherapeutics, even at low doses. Further, PPIs as monotherapy can exert a potent antitumor activity, and can be associated with in vivo modulation of tumor pH. As with buffers, PPIs have been shown to reverse tumor immune escape, through a clear increase of the immune infiltrate within the tumor mass 66. These preclinical data represented the background for a series of clinical studies aimed at supporting the use of PPI as chemosensitizers.
In addition, pre-clinical studies have clearly shown that Carbonic Anhydrase-9, CA-IX, is critically important in maintaining an acidic pHe, and that inhibitors of this proton exchanger may be successfully used as anti-cancer agents124. Multiple inhibitors of CA-IX have been developed but have in common a sulfonamide to target the active site, as well as a large size to prevent it from crossing the plasma membrane, as the active site of CA-IX (as well as CA-IV and CA-XII) is exofacial 132,133. Moreover, recent evidence suggests that proton pump inhibitors and CA-IX inhibitors may be successfully combined in the treatment of human cancers, particularly under low pH conditions133, thus leading us to re-think about the use of combinations between proton exchangers inhibitors134. As far as the involvement of CAs in diabetes there is clear evidence that CA inhibition may represent an effective prevention and treatment of obesity that we know as a common pathway of cancer and diabetes135. Thus, what we are learning on cancer treatment may be highly useful in the future treatment of diabetes.
Clinical Studies
Direct Targeting of Acidity.
To translate the provocative responses of animal tumors to buffers into the clinic, phase I/II clinical trials of sodium bicarbonate monotherapy were initiated, and are described in 136. Because of widespread complaints of GI discomfort and potential effects on edema, it has been determined that NaHCO3 alone is insufficient as a buffer therapy. As an alternative to buffer therapy, Helix Biopharma has recently developed a urease targeted to CEACAM-6 (L-DOS47) to raise pH by converting endogenous urea to two NH4+ and one HCO3-. This was investigated in a clinical trial (NCT02309892) in non-small cell lung cancer patients and was shown to be well-tolerated 137. TRC101117 has just completed a phase III pivotal trail in patients with chronic kidney disease (NCT03317444), but not yet been investigated in relation to diabetes or cancer.
PPIs in the clinic.
In 2014, Papagerakis et al. published a large retrospective metanalysis of outcomes in 596 previously untreated head and neck squamous cell carcinoma (HNSCC) patients138. The major findings of this study were strong univariate associations between both histamine receptor-2 antagonists (H2RAs) and proton pump inhibitors (PPIs) with treatment outcomes showing that both PPIs and H2RAs were significantly positive prognostic factors for overall survival.
Prospective clinical studies in osteosarcomas 139 and metastatic breast cancer have been published 140. In osteosarcomas, the clinical goal was to improve the effect of neoadjuvant chemotherapy, NAC, on the tumor lesion in the resected bone with the addition of esomeprazole the two days before the combined chemotherapy. The results showed that pre-treatment with PPI increased the effectiveness of NAC in osteosarcoma patients, particularly in the chondroblastic variant. In breast cancer, patients were divided into 3 arms: one receiving a standard cisplatin and paclitaxel (C+P) chemotherapy; and 2 arms with second with C+P plus either 120 mg or 160 mg esomeprazole the days before chemotherapy. After the course of C+P was completed, patients were further divided into two arms: one continuing esomeprazole for the following year and the other discontinuing. Results showed that the 45% of the patients that continued esomeprazole after of chemotherapy were alive at the end of the study, particularly the triple negative patients, with a significant increase of both the time to progression (TTP) and the overall survival (OS). More recently, a case series study in refractory gastro-intestinal cancer have shown that the addition of PPI to chemotherapy increased the TTP in these patients140. Further, PPIs were shown to increase the efficacy of standard chemotherapy and significantly improved the quality of life of treated companion animals with in either standard treatment142 or metronomic regimens143.
Despite this promise, the mechanism by which PPIs exert their effects remain unknown. An intriguing hypothesis might be that PPI, as the buffer therapies as well, may induce their effect through a buffering effect on the stomach 144. This hypothesis is based on the high level of anti-acidic effect of PPI at the gastric level, but this of course will be a matter for future studies. Additionally PPIs have been shown to have off target effects, specifically to dopamine and serotonin receptors 145. However, it has also been shown that P-type H+/K+ ATPases may be expressed by cancer cells of non-gastric origin146,147, but also in extragastric non-cancer conditions148. In any event, PPIs are well-tolerated and have shown clinical benefit.
CONCLUSIONS
Diabetes and Cancer and, indeed other pathological conditions, share a phenotype of disrupted acid-base homeostasis. In both cases, the ensuing extracellular acidity has been shown to be relevant to the diseases’ etiopathology. In both bases, targeting the extracellular acidity directly has been shown to ameliorate some symptoms, providing pre-clinical or clinical benefit. Despite this promise, targeting acidity and defining the mechanisms driving acidity are still nascent areas of investigation.
Acknowledgments
This work was supported by The AntiCancer Fund (RJG), US PHS NIH grants R01 CA077575 (RJG) and U54 CA193489 (RJG); JSPS KAKENHI Grants JP15K15034 (YM) and JP18H03182 (YM), Salt Science 1235 (YM), KIT-KPUM-KPU-KPhU Collaborative Research Grant (YM), Kyoto-Funding for Innovation in Health-related R&D Fields (YM), Fuji Foundation for Protein Research (YM), and Cell Research Conference (YM) and The Italian Ministry of Health
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCESS
- 1.Bernard C, H.H., H., Guillemin L & Guillemin R The cahier rouge of Claude Bernard. /Translated by Hoff Hebbel H., Guillemin Lucienne and Guillemin Roger, (Schenkman Pub. Co., 1967). [Google Scholar]
- 2.Corey HE Stewart and beyond: new models of acid-base balance. Kidney international 64, 777–787, doi: 10.1046/j.1523-1755.2003.00177.x (2003). [DOI] [PubMed] [Google Scholar]
- 3.Andrade CS et al. Widespread pH abnormalities in patients with malformations of cortical development and epilepsy: a phosphorus-31 brain MR spectroscopy study. Brain & development 36, 899–906, doi: 10.1016/j.braindev.2013.12.010 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Yuen AWC, Walcutt IA & Sander JW An acidosis-sparing ketogenic (ASK) diet to improve efficacy and reduce adverse effects in the treatment of refractory epilepsy. Epilepsy Behav 74, 15–21, doi: 10.1016/j.yebeh.2017.05.032 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Kemp GJ, Ahmad RE, Nicolay K & Prompers JJ Quantification of skeletal muscle mitochondrial function by 31P magnetic resonance spectroscopy techniques: a quantitative review. Acta physiologica 213, 107–144, doi: 10.1111/apha.12307 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Reeh PW & Steen KH Tissue acidosis in nociception and pain. Prog Brain Res 113, 143–151 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Steen KH, Issberner U & Reeh PW Pain due to experimental acidosis in human skin: evidence for non-adapting nociceptor excitation. Neurosci Lett 199, 29–32 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Sun WH & Chen CC Roles of Proton-Sensing Receptors in the Transition from Acute to Chronic Pain. J Dent Res 95, 135–142, doi: 10.1177/0022034515618382 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Jones KM et al. Clinical Translation of Tumor Acidosis Measurements with AcidoCEST MRI. Mol Imaging Biol 19, 617–625, doi: 10.1007/s11307-016-1029-7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giovannucci E et al. Diabetes and cancer: a consensus report. Diabetes Care 33, 1674–1685, doi: 10.2337/dc10-0666 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esposito K, Chiodini P, Colao A, Lenzi A & Giugliano D Metabolic syndrome and risk of cancer: a systematic review and meta-analysis. Diabetes Care 35, 2402–2411, doi: 10.2337/dc12-0336 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez N et al. 2017 update on the relationship between diabetes and colorectal cancer: epidemiology, potential molecular mechanisms and therapeutic implications. Oncotarget 8, 18456–18485, doi: 10.18632/oncotarget.14472 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sacerdote C & Ricceri F Epidemiological dimensions of the association between type 2 diabetes and cancer: A review of observational studies. Diabetes Res Clin Pract, doi: 10.1016/j.diabres.2018.03.002 (2018). [DOI] [PubMed]
- 14.Ter Braak B et al. Insulin-like growth factor 1 receptor activation promotes mammary gland tumor development by increasing glycolysis and promoting biomass production. Breast Cancer Res 19, 14, doi: 10.1186/s13058-017-0802-0 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hesselink MK, Schrauwen-Hinderling V & Schrauwen P Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nature reviews. Endocrinology 12, 633–645, doi: 10.1038/nrendo.2016.104 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Feng Z & Levine AJ The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol 20, 427–434, doi: 10.1016/j.tcb.2010.03.004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674, doi: 10.1016/j.cell.2011.02.013 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Trotta AP & Chipuk JE Mitochondrial dynamics as regulators of cancer biology. Cell Mol Life Sci 74, 1999–2017, doi: 10.1007/s00018-016-2451-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams M & Caino MC Mitochondrial Dynamics in Type 2 Diabetes and Cancer. Front Endocrinol (Lausanne) 9, 211, doi: 10.3389/fendo.2018.00211 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Millis SZ, Ikeda S, Reddy S, Gatalica Z & Kurzrock R Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19784 Diverse Solid Tumors. JAMA Oncol 2, 1565–1573, doi: 10.1001/jamaoncol.2016.0891 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Mayer IA et al. A Phase Ib Study of Alpelisib (BYL719), a PI3Kalpha-Specific Inhibitor, with Letrozole in ER+/HER2-Metastatic Breast Cancer. Clin Cancer Res 23, 26–34, doi: 10.1158/1078-0432.CCR-16-0134 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Juric D et al. Phase I Dose-Escalation Study of Taselisib, an Oral PI3K Inhibitor, in Patients with Advanced Solid Tumors. Cancer Discov 7, 704–715, doi: 10.1158/2159-8290.CD-16-1080 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hopkins BD & al., e. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature (in press) (2018). [DOI] [PMC free article] [PubMed]
- 24.Gillies RJ, Brown JS, Anderson ARA & Gatenby RA Eco-evolutionary causes and consequences of temporal changes in intratumoural blood flow. Nature reviews. Cancer 18, 576–585, doi: 10.1038/s41568-018-0030-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barathova M et al. Alternative splicing variant of the hypoxia marker carbonic anhydrase IX expressed independently of hypoxia and tumour phenotype. British journal of cancer 98, 129–136, doi: 10.1038/sj.bjc.6604111 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gatenby RA & Gillies RJ Why do cancers have high aerobic glycolysis? Nature reviews. Cancer 4, 891–899, doi: 10.1038/nrc1478 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Vander Heiden MG & DeBerardinis RJ Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669, doi: 10.1016/j.cell.2016.12.039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hashim AI, Zhang X, Wojtkowiak JW, Martinez GV & Gillies RJ Imaging pH and metastasis. NMR in biomedicine 24, 582–591, doi: 10.1002/nbm.1644 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swietach P, Vaughan-Jones RD, Harris AL & Hulikova A The chemistry, physiology and pathology of pH in cancer. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 369, 20130099, doi: 10.1098/rstb.2013.0099 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gatenby RA & Gawlinski ET A reaction-diffusion model of cancer invasion. Cancer research 56, 5745–5753 (1996). [PubMed] [Google Scholar]
- 31.Stubbs M, McSheehy PM, Griffiths JR & Bashford CL Causes and consequences of tumour acidity and implications for treatment. Molecular medicine today 6, 15–19 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Gatenby RA & Gillies RJ A microenvironmental model of carcinogenesis. Nature reviews. Cancer 8, 56–61, doi: 10.1038/nrc2255 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Gillies RJ, Verduzco D & Gatenby RA Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nature reviews. Cancer 12, 487–493, doi: 10.1038/nrc3298 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Damaghi M, Wojtkowiak JW & Gillies RJ pH sensing and regulation in cancer. Frontiers in physiology 4, 370, doi: 10.3389/fphys.2013.00370 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moellering RE et al. Acid treatment of melanoma cells selects for invasive phenotypes. Clinical & experimental metastasis 25, 411–425, doi: 10.1007/s10585-008-9145-7 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Estrella V et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer research 73, 1524–1535, doi: 10.1158/0008-5472.CAN-12-2796 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu L, Fukumura D & Jain RK Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: mechanism of low pH-induced VEGF. The Journal of biological chemistry 277, 11368–11374, doi: 10.1074/jbc.M108347200 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Lardner A The effects of extracellular pH on immune function. Journal of leukocyte biology 69, 522–530 (2001). [PubMed] [Google Scholar]
- 39.Kinnaird A, Zhao S, Wellen KE & Michelakis ED Metabolic control of epigenetics in cancer. Nature reviews. Cancer 16, 694–707, doi: 10.1038/nrc.2016.82 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Gerlinger M et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366, 883–892, doi: 10.1056/NEJMoa1113205 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sottoriva A et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A 110, 4009–4014, doi: 10.1073/pnas.1219747110, 1219747110 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baird RD & Caldas C Genetic heterogeneity in breast cancer: the road to personalized medicine? BMC Med 11, 151, doi: 10.1186/1741-7015-11-151, 1741–7015-11–151 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yachida S & Iacobuzio-Donahue CA Evolution and dynamics of pancreatic cancer progression. Oncogene 32, 5253–5260, doi: 10.1038/onc.2013.29, onc201329 [pii] (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ercan D et al. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene 29, 2346–2356, doi: 10.1038/onc.2009.526onc2009526[pii] (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y et al. Radiological Image traits Predictive of Cancer Status in Pulmonary Nodules. Clin Cancer Res, doi: 10.1158/1078-0432.CCR-15-3102 (2016). [DOI] [PMC free article] [PubMed]
- 46.Hawkins S et al. Predicting Malignant Nodules from Screening CT Scans. J Thorac Oncol, doi: 10.1016/j.jtho.2016.07.002 (2016). [DOI] [PMC free article] [PubMed]
- 47.Balagurunathan Y et al. Reproducibility and Prognosis of Quantitative Features Extracted from CT Images. Translational oncology 7, 72–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aerts HJ et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nature communications 5, 4006, doi: 10.1038/ncomms5006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chaudhury B et al. Heterogeneity in intratumoral regions with rapid gadolinium washout correlates with estrogen receptor status and nodal metastasis. Journal of magnetic resonance imaging : JMRI, doi: 10.1002/jmri.24921 (2015). [DOI] [PMC free article] [PubMed]
- 50.Liu Y et al. CT Features Associated with Epidermal Growth Factor Receptor Mutation Status in Patients with Lung Adenocarcinoma. Radiology 280, 271–280, doi: 10.1148/radiol.2016151455 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gatenby RA, Grove O & Gillies RJ Quantitative imaging in cancer evolution and ecology. Radiology 269, 8–15, doi: 10.1148/radiol.13122697 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gillies RJ, Kinahan PE & Hricak H Radiomics: Images Are More than Pictures, They Are Data. Radiology 278, 563–577, doi: 10.1148/radiol.2015151169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grove O et al. Quantitative computed tomographic descriptors associate tumor shape complexity and intratumor heterogeneity with prognosis in lung adenocarcinoma. PloS one 10, e0118261, doi: 10.1371/journal.pone.0118261 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lloyd MC et al. Vascular measurements correlate with estrogen receptor status. BMC cancer 14, 279, doi: 10.1186/1471-2407-14-279 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lloyd MC et al. Darwinian Dynamics of Intratumoral Heterogeneity: Not Solely Random Mutations but Also Variable Environmental Selection Forces. Cancer research 76, 3136–3144, doi: 10.1158/0008-5472.CAN-15-2962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tafreshi NK, Lloyd MC, Bui MM, Gillies RJ & Morse DL Carbonic anhydrase IX as an imaging and therapeutic target for tumors and metastases. Sub-cellular biochemistry 75, 221–254, doi: 10.1007/978-94-007-7359-2_12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mahon BP et al. The Structure of Carbonic Anhydrase IX Is Adapted for Low-pH Catalysis. Biochemistry 55, 4642–4653, doi: 10.1021/acs.biochem.6b00243 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee SH et al. Carbonic anhydrase IX is a pH-stat that sets an acidic tumour extracellular pH in vivo. British journal of cancer 119, 622–630, doi: 10.1038/s41416-018-0216-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B & Gillies RJ Acid-mediated tumor invasion: a multidisciplinary study. Cancer research 66, 5216–5223, doi: 10.1158/0008-5472.CAN-05-4193 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Rothberg JM et al. Acid-mediated tumor proteolysis: contribution of cysteine cathepsins. Neoplasia 15, 1125–1137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Avnet S et al. Cancer-associated mesenchymal stroma fosters the stemness of osteosarcoma cells in response to intratumoral acidosis via NF-κB activation. Intl. J. Cancer (in press) (2016). [DOI] [PMC free article] [PubMed]
- 62.Wojtkowiak JW et al. Chronic Autophagy Is a Cellular Adaptation to Tumor Acidic pH Microenvironments. Cancer Res (cover) 72, 3938–3947 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saftig P & Klumperman J Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nature reviews. Molecular cell biology 10, 623–635, doi: 10.1038/nrm2745 (2009). [DOI] [PubMed] [Google Scholar]
- 64.Damaghi M et al. Chronic acidosis in the tumour microenvironment selects for overexpression of LAMP2 in the plasma membrane. Nature communications 6, 8752, doi: 10.1038/ncomms9752 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pilon-Thomas S et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer research 76, 1381–1390, doi: 10.1158/0008-5472.CAN-15-1743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Calcinotto A et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer research 72, 2746–2756, doi: 10.1158/0008-5472.CAN-11-1272 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Grundstrom S, Dohlsten M & Sundstedt A IL-2 unresponsiveness in anergic CD4+ T cells is due to defective signaling through the common gamma-chain of the IL-2 receptor. J Immunol 164, 1175–1184, doi:ji_v164n3p1175 [pii] (2000). [DOI] [PubMed] [Google Scholar]
- 68.Wells AD, Walsh MC, Sankaran D & Turka LA T cell effector function and anergy avoidance are quantitatively linked to cell division. J Immunol 165, 2432–2443, doi:ji_v165n5p2432 [pii] (2000). [DOI] [PubMed] [Google Scholar]
- 69.Demotte N et al. Restoring the association of the T cell receptor with CD8 reverses anergy in human tumor-infiltrating lymphocytes. Immunity 28, 414–424, doi:S1074–7613(08)00070–8 [pii], 10.1016/j.immuni.2008.01.011 (2008). [DOI] [PubMed] [Google Scholar]
- 70.Glunde K et al. Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia 5, 533–545 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Logozzi M et al. Increased PSA expression on prostate cancer exosomes in in vitro condition and in cancer patients. Cancer Lett 403, 318–329, doi: 10.1016/j.canlet.2017.06.036 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Parolini I et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. The Journal of biological chemistry 284, 34211–34222, doi: 10.1074/jbc.M109.041152 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yanez-Mo M et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 4, 27066, doi: 10.3402/jev.v4.27066 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Federici C et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PloS one 9, e88193, doi: 10.1371/journal.pone.0088193 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lugini L et al. Exosomes from human colorectal cancer induce a tumor-like behavior in colonic mesenchymal stromal cells. Oncotarget 7, 50086–50098, doi: 10.18632/oncotarget.10574 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao H et al. The key role of extracellular vesicles in the metastatic process. Biochim Biophys Acta 1869, 64–77, doi: 10.1016/j.bbcan.2017.11.005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Remer T & Manz F Potential renal acid load of foods and its influence on urine pH. J Am Diet Assoc 95, 791–797, doi: 10.1016/S0002-8223(95)00219-7 (1995). [DOI] [PubMed] [Google Scholar]
- 78.Remer T Influence of diet on acid-base balance. Semin Dial 13, 221–226 (2000). [DOI] [PubMed] [Google Scholar]
- 79.Kiefte-de Jong JC et al. Diet-dependent acid load and type 2 diabetes: pooled results from three prospective cohort studies. Diabetologia 60, 270–279, doi: 10.1007/s00125-016-4153-7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jayedi A & Shab-Bidar S Dietary acid load and risk of type 2 diabetes: A systematic review and dose-response meta-analysis of prospective observational studies. Clin Nutr ESPEN 23, 10–18, doi: 10.1016/j.clnesp.2017.12.005 (2018). [DOI] [PubMed] [Google Scholar]
- 81.Persson B Determination of plasma acetoacetate and D-beta-hydroxybutyrate in new-born infants by an enzymatic fluorometric micro-method. Scandinavian Journal of Clinical and Laboratory Investigation 25, 9–18 (1970). [DOI] [PubMed] [Google Scholar]
- 82.Gosmanov AR, Gosmanova EO & Dillard-Cannon E Management of adult diabetic ketoacidosis. Diabetes, metabolic syndrome and obesity : targets and therapy 7, 255–264, doi: 10.2147/dmso.s50516 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dedkova EN & Blatter LA Role of beta-hydroxybutyrate, its polymer poly-beta-hydroxybutyrate and inorganic polyphosphate in mammalian health and disease. Frontiers in physiology 5, 260, doi: 10.3389/fphys.2014.00260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aoi W et al. Improvement of insulin resistance, blood pressure and interstitial pH in early developmental stage of insulin resistance in OLETF rats by intake of propolis extracts. Biochemical and biophysical research communications 432, 650–653, doi: 10.1016/j.bbrc.2013.02.029 (2013). [DOI] [PubMed] [Google Scholar]
- 85.Hayata H, Miyazaki H, Niisato N, Yokoyama N & Marunaka Y Lowered extracellular pH is involved in the pathogenesis of skeletal muscle insulin resistance. Biochemical and biophysical research communications 445, 170–174, doi: 10.1016/j.bbrc.2014.01.162 (2014). [DOI] [PubMed] [Google Scholar]
- 86.Cederholm J & Wibell L Glucose intolerance in middle-aged subjects--a cause of hypertension? Acta medica Scandinavica 217, 363–371 (1985). [DOI] [PubMed] [Google Scholar]
- 87.Eriksson KF & Lindgarde F Contribution of estimated insulin resistance and glucose intolerance to essential hypertension. Journal of internal medicine. Supplement 735, 75–83 (1991). [PubMed] [Google Scholar]
- 88.Bao W, Srinivasan SR & Berenson GS Persistent elevation of plasma insulin levels is associated with increased cardiovascular risk in children and young adults. The Bogalusa Heart Study. Circulation 93, 54–59 (1996). [DOI] [PubMed] [Google Scholar]
- 89.Haffner SM, Ferrannini E, Hazuda HP & Stern MP Clustering of cardiovascular risk factors in confirmed prehypertensive individuals. Hypertension 20, 38–45 (1992). [DOI] [PubMed] [Google Scholar]
- 90.Lahera V, de Las Heras N, Lopez-Farre A, Manucha W & Ferder L Role of Mitochondrial Dysfunction in Hypertension and Obesity. Current hypertension reports 19, 11, doi: 10.1007/s11906-017-0710-9 (2017). [DOI] [PubMed] [Google Scholar]
- 91.Horita S et al. The role of renal proximal tubule transport in the regulation of blood pressure. Kidney research and clinical practice 36, 12–21, doi: 10.23876/j.krcp.2017.36.1.12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Marunaka Y, Hagiwara N & Tohda H Insulin activates single amiloride-blockable Na channels in a distal nephron cell line (A6). Am. J. Physiol. Renal Physiol 263, F392–400 (1992). [DOI] [PubMed] [Google Scholar]
- 93.Mills E, Kuhn CM, Feinglos MN & Surwit R Hypertension in CB57BL/6J mouse model of non-insulin-dependent diabetes mellitus. The American journal of physiology 264, R73–78 (1993). [DOI] [PubMed] [Google Scholar]
- 94.Edwards JG & Tipton CM Influences of exogenous insulin on arterial blood pressure measurements of the rat. Journal of applied physiology (Bethesda, Md. : 1985) 67, 2335–2342 (1989). [DOI] [PubMed] [Google Scholar]
- 95.Meehan WP, Buchanan TA & Hsueh W Chronic insulin administration elevates blood pressure in rats. Hypertension 23, 1012–1017 (1994). [DOI] [PubMed] [Google Scholar]
- 96.Marunaka Y et al. Regulation of epithelial sodium transport via epithelial Na+ channel. J Biomed Biotechnol 2011, 978196 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marunaka Y Characteristics and pharmacological regulation of epithelial Na+ channel (ENaC) and epithelial Na+ transport. J. Pharmacol. Sci 126, 21–36, doi: 10.1254/jphs.14R01SR (2014). [DOI] [PubMed] [Google Scholar]
- 98.Utriainen T et al. Insulin resistance characterizes glucose uptake in skeletal muscle but not in the heart in NIDDM. Diabetologia 41, 555–559, doi: 10.1007/s001250050946 (1998). [DOI] [PubMed] [Google Scholar]
- 99.Kraus LM, Traxinger R & Kraus AP Uremia and insulin resistance: N-carbamoyl-asparagine decreases insulin-sensitive glucose uptake in rat adipocytes. Kidney international 65, 881–887, doi: 10.1111/j.1523-1755.2004.00456.x (2004). [DOI] [PubMed] [Google Scholar]
- 100.Abdul-Ghani MA & DeFronzo RA Pathogenesis of insulin resistance in skeletal muscle. J Biomed Biotechnol 2010, 476279, doi: 10.1155/2010/476279 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ciccarelli M et al. G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation 123, 1953–1962, doi: 10.1161/circulationaha.110.988642 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hanssen MJ et al. Glucose uptake in human brown adipose tissue is impaired upon fasting-induced insulin resistance. Diabetologia 58, 586–595, doi: 10.1007/s00125-014-3465-8 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Malkki H Alzheimer disease: Insulin resistance could be linked to risk of AD via reduced glucose uptake. Nature reviews. Neurology 11, 485, doi: 10.1038/nrneurol.2015.147 (2015). [DOI] [PubMed] [Google Scholar]
- 104.Willette AA et al. Association of Insulin Resistance With Cerebral Glucose Uptake in Late Middle-Aged Adults at Risk for Alzheimer Disease. JAMA neurology 72, 1013–1020, doi: 10.1001/jamaneurol.2015.0613 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marunaka Y The proposal of molecular mechanisms of weak organic acids intake-induced improvement of insulin resistance in diabetes mellitus via elevation of interstitial fluid pH. Int J Mol Sci 19(10):3244, 2018, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mahoney BP, Raghunand N, Baggett B & Gillies RJ Tumor acidity, ion trapping and chemotherapeutics. I. Acid pH affects the distribution of chemotherapeutic agents in vitro. Biochemical pharmacology 66, 1207–1218 (2003). [DOI] [PubMed] [Google Scholar]
- 107.Raghunand N, Mahoney BP & Gillies RJ Tumor acidity, ion trapping and chemotherapeutics. II. pH-dependent partition coefficients predict importance of ion trapping on pharmacokinetics of weakly basic chemotherapeutic agents. Biochemical pharmacology 66, 1219–1229 (2003). [DOI] [PubMed] [Google Scholar]
- 108.Robey IF et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer research 69, 2260–2268, doi: 10.1158/0008-5472.CAN-07-5575 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Silva AS, Yunes JA, Gillies RJ & Gatenby RA The potential role of systemic buffers in reducing intratumoral extracellular pH and acid-mediated invasion. Cancer research 69, 2677–2684, doi: 10.1158/0008-5472.CAN-08-2394 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Azzarito T et al. Effect of Modified Alkaline Supplementation on Syngenic Melanoma Growth in CB57/BL Mice. PloS one 11, e0159763, doi: 10.1371/journal.pone.0159763 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ibrahim Hashim A. et al. Reduction of metastasis using a non-volatile buffer. Clinical & experimental metastasis 28, 841–849, doi: 10.1007/s10585-011-9415-7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ribeiro M et al. Buffer Therapy for Cancer. J. Nutr. Food. Sci S2, 1–7 (2012). [PMC free article] [PubMed] [Google Scholar]
- 113.Ibrahim-Hashim A et al. Systemic buffers inhibit carcinogenesis in TRAMP mice. The Journal of urology 188, 624–631, doi: 10.1016/j.juro.2012.03.113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ibrahim-Hashim A et al. Defining cancer subpopulations by adaptive strategies rather than molecular properties provides novel insights into intratumoral evolution. Cancer research, doi: 10.1158/0008-5472.CAN-16-2844 (2017). [DOI] [PMC free article] [PubMed]
- 115.Astigiano S, Puglisi A, Mastracci L, Fais S & Barbieri O Systemic alkalinisation delays prostate cancer cell progression in TRAMP mice. J Enzyme Inhib Med Chem 32, 363–368, doi: 10.1080/14756366.2016.1252760 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tian B et al. Production and characterization of a camelid single domain antibody-urease enzyme conjugate for the treatment of cancer. Bioconjug Chem 26, 1144–1155, doi: 10.1021/acs.bioconjchem.5b00237 (2015). [DOI] [PubMed] [Google Scholar]
- 117.Bushinsky DA et al. Randomized, Controlled Trial of TRC101 to Increase Serum Bicarbonate in Patients with CKD. Clin J Am Soc Nephrol 13, 26–35, doi: 10.2215/CJN.07300717 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wojtkowiak JW, Verduzco D, Schramm KJ & Gillies RJ Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Molecular pharmaceutics 8, 2032–2038, doi: 10.1021/mp200292c (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shi J, Kantoff PW, Wooster R & Farokhzad OC Cancer nanomedicine: progress, challenges and opportunities. Nature reviews. Cancer 17, 20–37, doi: 10.1038/nrc.2016.108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Upreti M, Jyoti A & Sethi P Tumor microenvironment and nanotherapeutics. Transl Cancer Res 2, 309–319, doi: 10.3978/j.issn.2218-676X.2013.08.11 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Meng F, Zhong Y, Cheng R, Deng C & Zhong Z pH-sensitive polymeric nanoparticles for tumor-targeting doxorubicin delivery: concept and recent advances. Nanomedicine 9, 487–499, doi: 10.2217/nnm.13.212 (2014). [DOI] [PubMed] [Google Scholar]
- 122.Du JZ, Mao CQ, Yuan YY, Yang XZ & Wang J Tumor extracellular acidity-activated nanoparticles as drug delivery systems for enhanced cancer therapy. Biotechnol Adv 32, 789–803, doi: 10.1016/j.biotechadv.2013.08.002 (2014). [DOI] [PubMed] [Google Scholar]
- 123.Yang XZ et al. Sheddable ternary nanoparticles for tumor acidity-targeted siRNA delivery. ACS Nano 6, 771–781, doi: 10.1021/nn204240b (2012). [DOI] [PubMed] [Google Scholar]
- 124.Spugnini EP et al. Proton channels and exchangers in cancer. Biochim Biophys Acta 1848, 2715–2726, doi: 10.1016/j.bbamem.2014.10.015 (2015). [DOI] [PubMed] [Google Scholar]
- 125.Fais S, De Milito A, You H & Qin W Targeting vacuolar H+-ATPases as a new strategy against cancer. Cancer research 67, 10627–10630, doi: 10.1158/0008-5472.CAN-07-1805 (2007). [DOI] [PubMed] [Google Scholar]
- 126.Izumi H et al. Cellular pH regulators: potentially promising molecular targets for cancer chemotherapy. Cancer Treat Rev 29, 541–549 (2003). [DOI] [PubMed] [Google Scholar]
- 127.Taylor S et al. Microenvironment acidity as a major determinant of tumor chemoresistance: Proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist Updat 23, 69–78, doi: 10.1016/j.drup.2015.08.004 (2015). [DOI] [PubMed] [Google Scholar]
- 128.De Milito A et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int J Cancer 127, 207–219, doi: 10.1002/ijc.25009 (2010). [DOI] [PubMed] [Google Scholar]
- 129.De Milito A, Marino ML & Fais S A rationale for the use of proton pump inhibitors as antineoplastic agents. Curr Pharm Des 18, 1395–1406 (2012). [DOI] [PubMed] [Google Scholar]
- 130.Steffan JJ, Snider JL, Skalli O, Welbourne T & Cardelli JA Na+/H+ exchangers and RhoA regulate acidic extracellular pH-induced lysosome trafficking in prostate cancer cells. Traffic 10, 737–753, doi: 10.1111/j.1600-0854.2009.00904.x (2009). [DOI] [PubMed] [Google Scholar]
- 131.Rozhin J, Sameni M, Ziegler G & Sloane BF Pericellular pH affects distribution and secretion of cathepsin B in malignant cells. Cancer research 54, 6517–6525 (1994). [PubMed] [Google Scholar]
- 132.Nocentini A & Supuran CT Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008–2018). Expert Opin Ther Pat 28, 729–740, doi: 10.1080/13543776.2018.1508453 (2018). [DOI] [PubMed] [Google Scholar]
- 133.Supuran CT Carbonic Anhydrase Inhibition and the Management of Hypoxic Tumors. Metabolites 7, doi: 10.3390/metabo7030048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Iessi E et al. Rethinking the Combination of Proton Exchanger Inhibitors in Cancer Therapy. Metabolites 8, doi: 10.3390/metabo8010002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Scozzafava A, Supuran CT & Carta F Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 23, 725–735, doi: 10.1517/13543776.2013.790957 (2013). [DOI] [PubMed] [Google Scholar]
- 136.Pilot C, Mahipal A & Gillies RJ Buffer Theray-->Buffer Diet. K. Nutr. Food Sci 8, 684–688 (2018). [Google Scholar]
- 137.Ramlau R et al. Phase I/II Dose Escalation Study of L-DOS47 as a Monotherapy in Non-Squamous Non-Small Cell Lung Cancer Patients. J. Thorac. Oncol 12, S1017–S1072 (2017). [Google Scholar]
- 138.Papagerakis S et al. Proton pump inhibitors and histamine 2 blockers are associated with improved overall survival in patients with head and neck squamous carcinoma. Cancer Prev Res (Phila) 7, 1258–1269, doi: 10.1158/1940-6207.CAPR-14-0002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ferrari S et al. Proton pump inhibitor chemosensitization in human osteosarcoma: from the bench to the patients’ bed. J Transl Med 11, 268, doi: 10.1186/1479-5876-11-268 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang BY et al. Intermittent high dose proton pump inhibitor enhances the antitumor effects of chemotherapy in metastatic breast cancer. J Exp Clin Cancer Res 34, 85, doi: 10.1186/s13046-015-0194-x (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Falcone R et al. High-doses of proton pump inhibitors in refractory gastro-intestinal cancer: A case series and the state of art. Dig Liver Dis 48, 1503–1505, doi: 10.1016/j.dld.2016.08.126 (2016). [DOI] [PubMed] [Google Scholar]
- 142.Spugnini EP et al. Lansoprazole as a rescue agent in chemoresistant tumors: a phase I/II study in companion animals with spontaneously occurring tumors. J Transl Med 9, 221, doi: 10.1186/1479-5876-9-221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Spugnini EP et al. High dose lansoprazole combined with metronomic chemotherapy: a phase I/II study in companion animals with spontaneously occurring tumors. J Transl Med 12, 225, doi: 10.1186/s12967-014-0225-y (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Spugnini E & Fais S Proton pump inhibition and cancer therapeutics: A specific tumor targeting or it is a phenomenon secondary to a systemic buffering? Semin Cancer Biol 43, 111–118, doi: 10.1016/j.semcancer.2017.01.003 (2017). [DOI] [PubMed] [Google Scholar]
- 145.Campillos M, Kuhn M, Gavin AC, Jensen LJ & Bork P Drug target identification using side-effect similarity. Science 321, 263–266, doi: 10.1126/science.1158140 (2008). [DOI] [PubMed] [Google Scholar]
- 146.Yang T et al. Benzimidazole derivative, BMT-1, induces apoptosis in multiple myeloma cells via a mitochondrial-mediated pathway involving H+/K+-ATPase inhibition. Oncol Rep 31, 2743–2750, doi: 10.3892/or.2014.3122 (2014). [DOI] [PubMed] [Google Scholar]
- 147.Streif D et al. Expression of the non-gastric H+/K+ ATPase ATP12A in normal and pathological human prostate tissue. Cell Physiol Biochem 28, 1287–1294, doi: 10.1159/000335860 (2011). [DOI] [PubMed] [Google Scholar]
- 148.Matsui MS et al. Omeprazole, a gastric proton pump inhibitor, inhibits melanogenesis by blocking ATP7A trafficking. J Invest Dermatol 135, 834–841, doi: 10.1038/jid.2014.461 (2015) [DOI] [PubMed] [Google Scholar]