

Summary
Cancer is a complex disease that relies on both oncogenic mutations and non-mutated genes for survival, and therefore coined as oncogene and non-oncogene addictions. The need for more effective combination therapies to overcome drug resistance in oncology has been increasingly recognized, but the identification of potentially synergistic drugs at scale remains challenging. Here we propose a gene-expression-based approach, which uses the recurrent perturbation-transcript regulatory relationships inferred from a large compendium of chemical and genetic perturbation experiments across multiple cell lines, to engender a testable hypothesis for combination therapies. These transcript-level recurrences were distinct from known compound-protein target counterparts, were reproducible in external datasets, and correlated with small-molecule sensitivity. We applied these recurrent relationships to predict synergistic drug pairs for cancer and experimentally confirmed two unexpected drug combinations in vitro. Our results corroborate a gene-expression-based strategy for combinatorial drug screening as a way to target non-mutated genes in complex diseases.
Subject Areas: Bioinformatics, Cancer Systems Biology, Pharmacoinformatics
Graphical Abstract
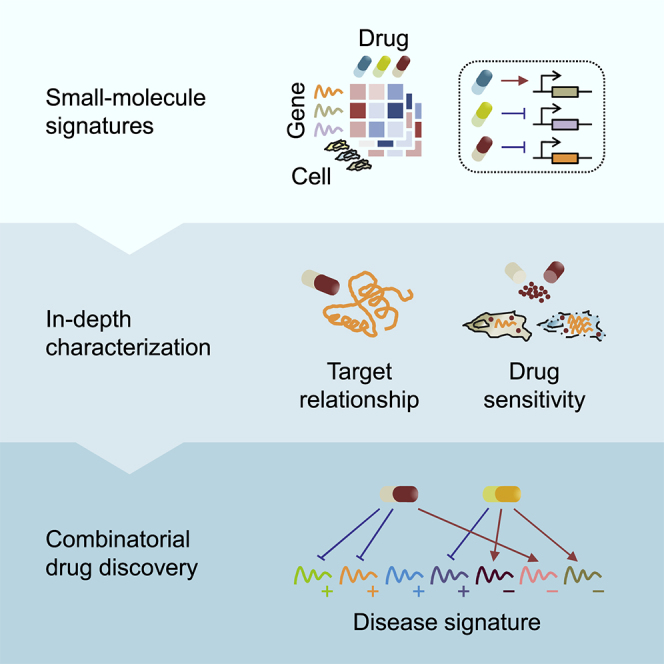
Highlights
-
•
Compound signatures targeting non-oncogene addiction for combinatorial drug discovery
-
•
These signatures are reproducible and linked to cancer hallmarks and drug sensitivity
-
•
Two synergistic drug combinations are experimentally confirmed in vitro
Bioinformatics; Cancer Systems Biology; Pharmacoinformatics
Introduction
Cancer is a genetic disease marked by extensive heterogeneities within the cells of a primary tumor (intratumoral heterogeneity) and also between the primary tumors of different patients (intertumoral heterogeneity) (Dagogo-Jack and Shaw, 2018). Our increasing comprehension of cancer genome has informed diverse mutational spectra that are responsible for the malignant transformation across various cancer types (Vogelstein et al., 2013). Together with the dependency on these oncogenic driver mutations for their growth and survival, cancer cells can develop an addiction to certain genes that are themselves not oncogenic but whose functions are required for maintenance of the tumorigenic state (Luo et al., 2009). These needs of both oncogenes and non-mutated genes for cell survival, coined as oncogene and non-oncogene addictions, respectively, add further complexity to the classical hallmarks and stress phenotypes of cancer (Hanahan and Weinberg, 2011, Luo et al., 2009). Strategies that interfere with this oncogenic circuitry have been proved to serve as attractive points for cancer therapies (Hanahan and Weinberg, 2011, Luo et al., 2009, Nagel et al., 2016).
Despite substantial efforts, anticancer drug resistance has become one of the biggest challenges in global human health (Holohan et al., 2013). Initial responses to conventional chemotherapeutics and molecularly targeted agents are greatly affected by intertumoral heterogeneity, whereas a gradual loss of drug sensitivity can often arise from intratumoral heterogeneity (Dagogo-Jack and Shaw, 2018, Saunders et al., 2012). Functional interplays between distinct hallmark capabilities are also enabling drug-tolerant cancer cells to evolve a resistance mechanism during the prolonged treatment (Hata et al., 2016, Ramirez et al., 2016). To thwart acquired resistance, combination drug therapies are now garnering much attention for their ability to cooperatively eliminate heterogeneous cancer cells within a single tumor (Al-Lazikani et al., 2012, Flemming, 2015, Jia et al., 2009, Webster, 2016). In view of high attrition rates in the de novo drug development (Waring et al., 2015), one tantalizing option for exploring combination opportunities in oncology is to consider already launched drugs that have well-documented safety profiles (Bertolini et al., 2015, Corsello et al., 2017). However, a critical step toward fully realizing the potential of combination therapy is to identify synergistic drug interactions before clinical testing (Lopez and Banerji, 2017). Given the tremendous number of all possible drug combinations, a systems-level method that can predict drug synergy to an unprecedented scale for every disease is urgently required (Kwong et al., 2013, Ryall and Tan, 2015, Sun et al., 2013).
Cellular molecular responses to small-molecule treatments have provided many possibilities for cancer drug discovery (Moffat et al., 2014). The complex polypharmacological interactions following small-molecule treatments would be reflected on variations in several molecular features (Feng et al., 2009), among which gene expression is popularly used for such efforts (Lamb et al., 2006). Although most molecular efficacy targets of approved drugs are rarely transcription factors (Santos et al., 2017), there is increasing evidence that changes in gene expression following small-molecule treatments are usually pervasive and may link to drug activity. For example, histone deacetylase (HDAC) inhibitors may provide neuroprotection by downregulating KEAP1 (kelch-like ECH-associated protein 1), a binding suppressor of the transcription factor NRF2 (nuclear factor, erythroid 2 like 2; also known as NFE2L2), thereby inducing nuclear translocation of NRF2 and its downstream transcriptional program (Wang et al., 2012). Inhibition of the mammalian target of rapamycin (mTOR) has been known to profoundly affect gene expression by regulating the activity of a wide range of transcription factors, such as peroxisome proliferator-activated receptor alpha in the blockade of hepatic ketogenesis (Laplante and Sabatini, 2013). Moreover, the transcription factor NRF1 (also known as NFE2L1) was found to be required for the transcriptional activation of proteasome subunit genes upon proteasome inhibition in mammalian cells (Radhakrishnan et al., 2010). These recent findings further strengthen the hypothesis that gene-expression changes can, to some extent, mirror drug activities and therefore be used to inform the selection of drug combinations.
Here we address this demand by introducing a strategy that combines the gene-expression signatures between chemical perturbations and disease statuses to provide a testable hypothesis for combination therapies (Figure 1). Using more than 1.3 million publicly accessible perturbational gene-expression profiles obtained from the Library of Integrated Network-Based Cellular Signatures (LINCS) (Subramanian et al., 2017), we first generated hundreds of thousands of recurrent perturbation-transcript regulatory associations inferred between 3,332 chemical and 3,934 genetic perturbagens and 12,494 transcripts across 10 cell types. These perturbation-transcript regulatory relationships are noticeably distinct from known annotated compound-target pairs, but can still be recapitulated in independent perturbation datasets. Our analysis also reveals shared and divergent transcriptional modules across small-molecule mechanisms of action (MoAs), of which some modules are common to many MoAs that target cancer-specific hallmarks. Together with the relationships that correlate chemical sensitivity across many cancer cell lines with basal (unperturbed) gene expression (Rees et al., 2016), the connections between chemical perturbations and recurrently regulated transcripts can further expand our understanding of the molecular mechanisms associated with chemical sensitivity. On the basis of the identified perturbational gene-expression signatures, we predicted synergistic drug combinations for cancer, using an algorithm to search for therapeutics that can reverse as many non-overlapping disease transcripts as possible. The synergistic inhibitory effects of two drug combinations—CD-437 (a retinoid) combined with sirolimus (an mTOR inhibitor) and narciclasine (a protein synthesis inhibitor) combined with purvalanol A (a cyclin-dependent kinase [CDK] inhibitor)—were confirmed in vitro, indicating their potential as the general combination therapies for cancer. This first large-scale, unbiased, in silico approach will offer significant improvements in our ability to rationalize combination therapies for clinical success.
Figure 1.

Study Workflow
To identify recurrent relationships between perturbations and regulated transcripts, we used the gene-expression profiles of more than 7,000 different chemical and genetic perturbagens across 10 selected cell lines from the LINCS L1000 resource. The inferred transcript-level recurrences were first compared with known small molecule-target annotations, benchmarked against independent perturbation datasets, characterized at the small-molecule-class regulatory level, and then connected with small-molecule sensitivity. We hypothesized that drug synergy can be informed by correlating a given disease signature with the combined patterns of gene reversal achieved by small-molecule-regulated recurring transcripts. To investigate this possibility, we applied this method to predict synergistic drug combinations across cancer types and validated the predictions in vitro.
Results
Recurrent Perturbation-Transcript Regulatory Relationships
We used publicly available large-scale chemical and genetic perturbation datasets for 10 cell types in LINCS, for which three perturbation types—exposure to small molecules or drugs for 6 h (denoted as d6), exposure to small molecules or drugs for 24 h (d24), and exposure to short hairpin RNAs (shRNAs) for 96 h (sh96)—comprised the majority of perturbation experiments (Table S1) (Huang et al., 2018). To investigate recurrent relationships between perturbations and regulated transcripts, we adapted a method and rank-based statistical score, the REC score (Jacobsen et al., 2013), to evaluate the recurrence of a given perturbation-transcript association across selected cell types (Transparent Methods). Among more than 100 million perturbation-transcript pairs (>10,000 chemical and genetic perturbations times 12,949 measured transcripts), we identified 77,691, 30,151, and 162,628 significantly recurring interactions for d6, d24, and sh96 perturbation types, respectively (Benjamini-Hochberg [BH]-corrected false discovery rate [FDR] < 0.001) (Figure 2 and Table S2). Several of these significantly recurring pairs were recovered in the analysis with resampling of 60% cell types, indicating their general robustness to variations in cell identity in our screen (Figure S1). We observed that whereas most perturbations tended to regulate a balanced number of genes in each direction (up- or downregulation), most genes exhibited a propensity to be either up- or downregulated by different effective perturbations in an exclusive manner (Figure 2A). For chemical perturbations, drugs or small molecules appeared to consistently regulate significant transcripts at both 6 and 24 h (Figure 2B). Of 3,190 genes targeted by the LINCS shRNA library and also measured for their expression, only 440 were recurrently downregulated by their corresponding “on-target” shRNAs, whose associated recurrent pairs accounted for 20.1% of all identified sh96 relationships (Figure 2C), probably in part due to the “off-target” activity (Jackson and Linsley, 2010) or other indirect responses 96 h after specific perturbations. Despite this observation, we found that the strongest “on-target” negative associations were captured in the most significantly recurring pairs (Figure 2D). Moreover, we note that some known small-molecule-regulated transcripts were captured by our analysis—for example, KEAP1 downregulated by HDAC inhibitors (such as vorinostat and dacinostat) (Wang et al., 2012) and ATF3 (activating transcription factor 3) upregulated by proteasome inhibitors (such as bortezomib and MLN-2238) (Zimmermann et al., 2000) (Table S2). These data substantiate our approach for identifying biologically meaningful relationships.
Figure 2.

Recurrent Perturbation-Transcript Regulatory Relationships
(A) Summary of the recurrent relationships between the chemical and genetic perturbations and the regulated transcripts. The upper panels show the number of significant relationships per perturbation, whereas the lower panels show the same per gene. d6, small-molecule or drug treatment for 6 h; d24, small-molecule or drug treatment for 24 h; sh96, short hairpin RNA treatment for 96 h. REC, recurrence score.
(B) Comparison between d6 and d24 recurrent relationships. FDR, false discovery rate.
(C) On-target sh96 recurrent relationships. The REC scores for significant on-target pairs are shown in a violin plot; line, median.
(D) Top 10 significant d6, d24, and sh96 recurrent pairs.
Comparison to Annotated Compound-Target Pairs
To assess the degree of correlation between small-molecule-regulated transcripts and their assigned protein targets, we compared the transcript-level recurrences for chemical perturbations with those compound-target annotations made by DrugBank (Figure S2 and Table S3) (Law et al., 2014). Of 11,022 annotated compound-target pairs, nearly 71% had no available REC scores, owing to the lack of either the target transcript or the matched compound for a given pair in LINCS (Figure 3A, as “not available”). The REC scores for an additional <10% pairs (<1,000) were not defined due to the insufficient number of cross-cell-type perturbation experiments per compound that is required for robust statistical inference (for a given compound-transcript pair, the REC score is defined only if the compound has been treated in at least half of the 10 cell lines analyzed in LINCS; Transparent Methods; Figure 3A, as “not defined”). Of the remaining compound-target pairs with correspondent REC scores (∼2,500; Figure 3A, the first two categories), only no more than 20 displayed statistically significant transcript-level recurrences (BH-corrected FDR < 0.001) (Figure 3B) and there was no apparent bias in the distribution of REC scores for each compound action type (for example, many outlier relationships for the “inhibitor” or “antagonist” and the “agonist” categories were readily found in both directions of regulation) (Figure 3C). For those compounds with at least one annotated target that exhibited a significant transcript-level recurrence (BH-corrected FDR < 0.001), not all other target transcripts for each compound were necessarily significantly regulated. For example, the pan-CDK inhibitor alvocidib seemed not to recurrently regulate all CDK transcripts at 6 h (only for CDK1 and CDK7; Figure 3D), ditto for the promiscuous kinase inhibitor staurosporine (only for CSK and GSK3B; Figure 3E) and the proteasome inhibitor bortezomib (only for PSMA1 and PSMD1; Figure 3F). However, we highlighted some intriguing observations from annotated targets shared by compounds that harbored at least one significantly recurring relationship toward the target transcript—topoisomerase inhibitors (teniposide, amsacrine, and idarubicin and, less significantly, mitoxantrone and etoposide) were most likely to downregulate TOP2A (DNA topoisomerase II alpha) (Figure 3G), heat shock protein 90 (HSP90) inhibitors (radicicol and geldanamycin) inclined to upregulate HSP90AB1 (heat shock protein 90 alpha family class B member 1) (Figure 3H), and 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase (HMGCR) inhibitors (simvastatin, atorvastatin, and lovastatin and, less significantly, mevastatin, fluvastatin, and pravastatin) tended to upregulate HMGCR (Figure 3I). Overall, the transcript-level recurrences identified by our analysis represent completely distinct relationships from those known compound-target annotations.
Figure 3.

Comparison with Annotated Compound-Target Relationships
(A) Distribution of REC scores in DrugBank.
(B) The significant d6 or d24 pairs (FDR < 0.001) overlapping with DrugBank.
(C) Distribution of available REC scores for each compound action type. Box plots depict the interquartile range (IQR), and whiskers represent 1.5 × IQR. Blue vertical lines indicate the REC scores for which FDR = 0.001.
(D–I) Distribution of REC scores in LINCS for each of selected compounds (D–F) or genes (G–I) with at least one significant REC score overlapping with compound-target annotations. For each plot, all available DrugBank annotations are shown, and horizontal dashed lines indicate the REC scores for which FDR = 0.001.
Validating Transcript-Level Recurrences in Independent Datasets
We next sought to determine whether the recurrent perturbation-transcript interactions inferred with the LINCS resource can be reflected in independent datasets while generalizable to other cell lines. To this end, we performed differential gene-expression analysis in compound perturbed and unperturbed samples and looked at the overlap between these externally derived, differentially expressed (DE) genes and the corresponding transcript-level recurrences. A significant overlap of concordantly regulated DE (two-sided unpaired Student's t test, BH-corrected p < 0.05) and REC (BH-corrected FDR < 0.001 or 10−6) genes for each direction was observed for many compound treatments in MCF7 cells, a human breast adenocarcinoma cell line included for evaluation of transcript-level recurrence (hypergeometric p < 0.05) (Figures 4A–4D and S3). In other two cell lines, the Ishikawa (human endometrial adenocarcinoma) and HepaRG (human hepatoma) cells, both of which were not used in this study, the patterns of perturbation-induced changes in gene expression also overlapped substantially with the inferred transcript-level recurrences (Figures 4E–4H and S3). This suggests that the recurring perturbation-transcript associations that we inferred from a few core cell lines are sufficiently characteristic of the unique compound actions that depend less on cell identity.
Figure 4.

External Validation of Transcript-Level Recurrences
For each dataset corresponding to a given small-molecule perturbation in a given cell line for indicated time duration, the differentially expressed (DE) genes (two-sided unpaired Student's t test, Benjamini-Hochberg (BH)-corrected p < 0.05) were compared with corresponding transcript-level recurrences (FDR < 0.001 or 10−6) for upregulated and downregulated genes separately using the hypergeometric test. (A–D) Validation of transcript-level recurrences for vorinostat (A), vinblastine (B), triptolide (C), or clobetasol (D) treated in MCF7 cells. (E) Validation of transcript-level recurrences for vorinostat treated in Ishikawa cells. (F) Validation of transcript-level recurrences for vinblastine treated in Ishikawa cells. (G) Validation of transcript-level recurrences for vorinostat treated in HepaRG cells. (H) Validation of transcript-level recurrences for clobetasol treated in Ishikawa cells. For detailed information about the comparison with different criteria, see Figure S3.
Regulatory Modules across Small-Molecule Classes
We next aimed at exploring the transcriptional regulatory landscape for drugs and small molecules. For each compound, we first manually annotated its primary MoA and searched for, if available, the associated World Health Organization Anatomical Therapeutic Chemical Classification codes (ATC codes) as the standard therapeutic indicator (Figure S4 and Table S1). For each class label (a primary MoA or an ATC code at any level), we calculated the proportion of hits of recurring regulation for each gene targeted by the tested compounds belonging to the class. This process has highlighted many transcriptional aspects of small-molecule-class actions that might be of therapeutic importance. For example, we found that many glucocorticoids (>10) can recurrently upregulate NFKBIA (nuclear factor-κB inhibitor alpha), consistent with the previously reported promoter activation by the glucocorticoid dexamethasone (Deroo and Archer, 2001), and DDIT4 (DNA damage-inducible transcript 4), whose upregulation by dexamethasone has been shown to mediate the drug-induced autophagy in lymphocytes (Molitoris et al., 2011) (Figures 5A and S5). Several genes involved in cholesterol homeostasis, including INSIG1 (insulin induced gene 1), HMGCS1 (HMG-CoA synthase 1), FDFT1 (farnesyl-diphosphate farnesyltransferase 1), SQLE (squalene epoxidase), HMGCR, and LDLR (low-density lipoprotein receptor), were found to be recurrently regulated by many antipsychotics (Figure S6), supporting the lipid disturbance associated with antipsychotic medications (Perez-Iglesias et al., 2009, Wiklund et al., 2010). In line with our observation (Figure 3H), HSP inhibitors are prone to transcriptionally upregulating several HSP family members, including HSPH1, HSPA6, HSP90AB1, HSPD1, HSPE1, HSPB1, and HSP90AA1 (Figure S7).
Figure 5.

Regulatory Gene Modules across Diverse Drug Classes
(A) Recurrently regulated genes by glucocorticoids.
(B) Transcriptional modules across small-molecule primary MoAs. For detailed descriptions of primary MoAs, see Table S1.
(C) Three ubiquitous gene clusters identified in (B). For each small-molecule-class hit, the number of within-class small molecules accounting for the hit is shown.
(D) Enrichments of gene regulatory modules for cancer hallmarks.
To gain a global picture of small-molecule transcriptional responses, we focused on genes frequently targeted at the level of the primary MoA or ATC code separately (for a given class, proportion of hits ≥20% and consistent hits by ≥ 3 compounds). Hierarchical clustering of these frequently targeted transcripts revealed 226 gene modules across 50 primary MoAs (Figure 5B), covering 140 gene singletons and three gene clusters harboring ubiquitously regulated genes among primary MoAs (Figure 5C and Table S4). Of these gene modules, some displayed specific enrichments for cancer hallmarks. For example, one ubiquitous cluster of 63 frequently downregulated genes by several cytotoxic drugs and kinase inhibitors (ucl1) was enriched for mitotic cell cycle regulation (E2F targets, BH-corrected hypergeometric p = 4.15 × 10−15; G2/M checkpoint, p = 1.95 × 10−13; and mitotic spindle, p = 0.0167), whereas another cluster of 15 ubiquitously upregulated genes (ucl2) was enriched for stress phenotypes (mTOR complex 1 [mTORC1] signaling, p = 1.76 × 10−6; p53 pathway, p = 4.21 × 10−8; hypoxia, p = 0.00174; apoptosis, p = 6.72 × 10−4; and unfolded protein response, p = 0.00505) (Figure 5D). Using the same criteria for analysis on ATC categories, we, nevertheless, found the limited sharing of gene modules across therapeutic areas (Figure S8 and Table S5), owing to the relatively less emphasis on small-molecule action in the ATC classification system. This allowed us to distinguish extensively targeted transcripts across different ATC classes, for example, NFKBIA and DDIT4, which were prone to being downregulated by drugs approved in the dermatological (category D), alimentary (category A), respiratory (category R), and sensory (category S) systems (Figure S8C). Overall, through our systematic and unbiased analysis, these small-molecule transcriptional modules can advance further studies to identify novel compound action.
Correlation with Small-Molecule Sensitivity
We next studied the relationships between compound-induced transcriptional responses and chemical sensitivity patterns generated by correlating an area under the dose-response curve metric for each of 481 tested compounds with 18,543 basal transcripts across 823 cancer cell lines (Rees et al., 2016). By overlapping significant perturbation-transcript regulatory interactions (BH-corrected FDR <0.001) and sensitivity-expression associations (two-sided Bonferroni-corrected p < 0.05), we identified 9,413 compound-transcript connections (6,431 for d6 plus 2,982 for d24) that could help enhance the molecular interpretation of small-molecule action (Figure 6A and Table S6). For example, we found that high expression of TIMELESS, a circadian regulator whose overexpression has been implicated in various cancer types (Mao et al., 2013), was strongly correlated with sensitivity to the HDAC inhibitor vorinostat, which, in turn, can recurrently downregulate TIMELESS after the treatment (Figures S9A and S9B). In contrast, low expression of BAG3 (BCL2-associated athanogene 3) (Rosati et al., 2011) was correlated with sensitivity to the proteasome inhibitor bortezomib that can upregulate BAG3 after the treatment (Figures S9C and S9D), consistent with those observed in leukemia cells where BAG3 knockdown restored sensitivity to bortezomib (Liu et al., 2009). To gain a broader insight into the relationships between expression-sensitivity correlation and reciprocal gene regulation, we extended this analysis to the level of compound MoA (Figure 6B and Table S7). Interestingly, SYNCRIP (synaptotagmin-binding cytoplasmic RNA-interacting protein), an RNA-binding protein recently shown to be required for the development of leukemia, was strongly correlated with sensitivity to topoisomerase and HDAC inhibitors, both of which can recurrently downregulate SYNCRIP (Figure 6B) (Vu et al., 2017). Overall, these data have enabled us to explore molecular mechanisms associated with small-molecule sensitivity for the treatment of disease.
Figure 6.

Comparison with Small-Molecule Sensitivity Data
(A) Overlapping transcript-perturbagen (REC scores with FDR <0.001, identified by our analysis; y axis) and expression-sensitivity (Z-scored area under the dose-response-curve correlation strength with Bonferroni-corrected two-sided p < 0.05, identified by Rees et al (Rees et al., 2016); x axis) relationships. See Table S6.
(B) Overlapping relationships at the level of primary MoA. Shown are significant MoA-transcript overlapping pairs with occurrences ≥3 and the proportion ≥50%, or simply with occurrences ≥8, as defined in Table S7.
Combinatorial Drug Discovery for Cancer and Experimental Validation
We hypothesized that the perturbation-transcript regulatory recurrences could be exploited to predict synergistic drug interactions by correlating the combined patterns of perturbation-regulated recurring transcripts with a given disease gene signature. Specifically, we developed an algorithm that examines the extent of gene reversal achieved by a combination of small molecules with individual transcript-level recurrences in reference to the disease signature, in which the combination is called synergistic if it reverses a significant proportion of disease transcripts in a nearly non-overlapping manner (Figure S10, Transparent Methods). The therapeutic score by this approach provides a rough interpretation of gene reversal such that suppose there are 1,000 disease genes and a given perturbation can reverse exactly 100 genes with REC scores for which all FDRs are 0.001, the therapeutic score is 0.1. To address this possibility, we used gene-expression signatures between primary tumors and tumor-adjacent normal tissues among eight cancer types from The Cancer Genome Atlas (TCGA) to inform pan-cancer synergistic drug combinations (RNA sequencing reads with Bonferroni-corrected p < 0.05, |log2(fold change)| > 1, and log2(count per million) > 3) (Table S8) (Aran et al., 2017). Using our expression-based approach, we first identified ∼300 single agents that could significantly reverse disease gene signatures across many cancer types, including several chemotherapeutics and investigational drugs that target cancer-related pathways (BH-corrected p < 0.05, by permutation analysis; 377 of 2,628 tested single agents had at least one significant therapeutic score) (Figure 7A and Table S9). Based on the patterns of gene reversal for these individual therapeutics (n = 218 and 162 for d6 and d24 perturbations, respectively, corresponding to a union of 288 compounds with at least one therapeutic score ≥0.01 with BH-corrected p < 0.05), we then predicted synergistic drug pairs for cancer in general, most of which were combinations of a topoisomerase inhibitor and an mTOR or phosphoinositide 3-kinase (PI3K) inhibitor (BH-corrected p < 0.05, by permutation analysis; 20,050 of 32,508 tested combinations had at least one significant therapeutic score) (Figures 7B and 7C and Table S10), consistent with the reported synergistic toxicity in some cancer cells (Babichev et al., 2016, Itamochi et al., 2011).
Figure 7.

Predicted Synergistic Drug Combinations across Cancer Types
(A) Expression-based single therapeutics for cancers with corresponding median cell line sensitivity available from the Cancer Therapeutics Response Portal (CTRP). Shown are small molecules with at least one significant therapeutic score ≥0.05 (BH-corrected p < 0.05) across cancers types. For full results, see Table S9.
(B) Top 15 synergistic drug combinations across cancer types. For full results, see Table S10.
(C) Occurrences of the combinations of small-molecule classes in top 100.
(D) Validation of two predicted synergistic drug pairs in breast and lung cancer cells using clonogenic assays. Drug synergy was evaluated using the delta score (Yadav et al., 2015). For full results, see Figures S11 and S12.
Among the top 15 predicted synergistic drug pairs, we selected two unexpected interactions—a retinoid CD-437 in combination with an mTOR inhibitor sirolimus and a protein synthesis inhibitor narciclasine in combination with a CDK inhibitor purvalanol A—for testing in tumor-type-matched breast (BRCA; n = 2) and lung (LUAD; n = 2) cancer cell lines (Figure 7B). To evaluate drug synergy, we used the delta score, which has been shown to exhibit superior performance over other commonly used drug interaction scores, for example, based on the Loewe additivity or Bliss independence model (Yadav et al., 2015). The delta score is symmetric, ranging from −1 (antagonism) through 0 (additive effect) to +1 (synergy), and also biologically meaningful, which can inform the additional effect produced over an expected drug interaction. For example, a delta score of 0.1 indicates 10% of additional effects of growth inhibition in a cytotoxic assay for a given drug combination, whereas a delta score of −0.1 indicates antagonism with 10% effects below the expected interaction. For known synergistic drug combinations, high delta scores of 0.03 or more over the dose-response matrices were confirmed by Yadav et al (Yadav et al., 2015). Of eight tested conditions in this study (two drug combinations for four cell lines), five revealed synergistic antiproliferative effects (average delta score ≥0.04; Figures 7D and S11). The observed interaction patterns of synergy were also reproducible across biological replicates (Figure S12). For the most synergistic case (CD-437 combined with sirolimus in H1299 cells), the average and maximum delta scores (0.14 and 0.54, respectively) were comparable with those of the top synergistic interaction revealed by Yadav et al. (0.176 and 0.40, respectively). We also note that the degree of synergy varied across sensitive cell lines for each drug combination, consistent with cell-type-specific responses to most anticancer drugs (Niepel et al., 2017).
The concept of targeting non-oncogene dependencies based on gene expression is being actively explored for combinatorial drug discovery. A recent analysis of breast cancer cell lines and molecularly targeted agents reveals that drug synergy may arise from compounds not only between different gene-expression clusters but also within the same cluster (Niepel et al., 2017). With compound screen data, it seems feasible that predictive drug-response gene biomarkers may translate to synergistic drug combinations. Jiang et al. have developed a computational method to predict gene interactions of drug response and show that several genes are frequently associated with resistance or sensitivity to multiple targeted drugs (Jiang et al., 2018). For example, their approach identified AXL to be a resistance gene for the ALK inhibitor NVP-TAE684, consistent with our observation that low expression of AXL correlated with sensitivity to NVP-TAE684, which can in turn downregulate AXL after treatment (Table S6). However, these predictive methodologies are limited in their ability to provide a general solution to identifying efficacious drug combinations at a sufficiently large scale.
Recent studies are revealing the potential to inform new drugs through their ability to invert the non-oncogene dependency program. It has been demonstrated that reversal of cancer gene expression positively correlates, although not outstandingly, with drug efficacy (Chen et al., 2017). This has led to the identification of the top four compounds that have not been previously reported for liver cancer—a cardiac glycoside strophanthidin, a mitochondrial uncoupler FCCP, an ATM/ATR kinase inhibitor CGK-733, and an anthelmintic drug pyrvinium pamoate—all of which were effectively recapitulated by our approach in liver cancer (Table S9). Indeed, the compounds predicted for liver cancer in this study were highly enriched in the top compound list generated by Chen et al. (p ≈ 0, by enrichment analysis, Figure S13) (Chen et al., 2017), further verifying our approach for identifying effective drug therapies. More recently, a precision oncology method has been proposed to identify compounds that could achieve the global inversion of master regulator protein activities in gastroenteropancreatic neuroendocrine tumors (GEP-NETs), a rare malignancy originating in the pancreas, small bowel, and rectum (Alvarez et al., 2018). Of 107 compounds that were validated in vitro to be differentially active in GEP-NET-related cells, 68 were included in our inference analysis of transcript-level recurrences. Of these, 63% (43 of 68) achieved significant reversal of at least one cancer signature in this study, including entinostat (an HDAC inhibitor, which was selected for validation in vivo by Alvarez et al.), CD-437, alvocidib, bortezomib, and GDC-0941 (also known as pictilisib, a PI3K inhibitor) (Figures 7A and 7B and Table S11). This validates that our gene-expression-based approach is able to identify small-molecule compounds that could effectively target non-oncogene dependencies. However, these current methodologies still cannot be extended to combinatorial drug discovery, owing to the shared limitation of all gene-expression-based similarity approaches to connect more than one perturbation (query) to a given disease of interest (reference) at a time. Inspired by the fact that a given perturbation should only affect a restricted panel of genes (Felix and Barkoulas, 2015), our approach leverages small-molecule-regulated recurring transcripts that more closely represent the unique compound mechanisms independent of cell identity, which further enables us to predict effective drug pairs by comparing the compound mechanisms mirrored in their gene-expression signatures. Together, these data demonstrate the feasibility of using transcript-level recurrences for unbiased data-driven prediction of synergistic drug interactions.
Discussion
Our study presents a systematic computational approach for combinatorial drug discovery by correlating the patterns of gene reversal in a given disease signature achieved by small-molecule-regulated recurring transcripts. These transcript-level recurrences inferred within the LINCS resource are reproducible enough in external datasets to represent the unique small-molecule action and are clearly distinct from annotated compound-target relationships. In-depth analysis revealed that some small-molecule-class transcriptional modules can regulate common hallmarks and stress phenotypes of cancer and that some recurrently regulated transcripts are associated with chemical and small-molecule-class sensitivity that warrants further exploration.
Although gene expression provides a basis for predicting drug synergy, the extent of gene reversal by single agents does not necessarily correlate with chemical sensitivity, suggesting that cellular processes to which cancer cells are addicted may shape the response to cancer therapies. In addition, although we predicted synergistic drug combinations using “bulk” patient gene-expression profiles, cell-line-intrinsic characteristics (for example, coding mutations that alter actionable sites for drug binding or other mechanisms related to drug inactivation) may also influence the evaluation of drug synergy in our experimental setting. However, the presented approach can only capture some synergistic drug pairs that exhibited nearly non-overlapping patterns of gene reversal and it is possible for two drugs without significant reversal effects to display synergistic killing. Interestingly, this approach may also be combined with a drug similarity network approach to expand the repertoire of potential synergistic drug pairs using co-clustering relationships (Huang et al., 2018), as suggested by a recent study showing that synergy can be observed with pairs of drugs that elicit both similar and distinct transcriptional responses (Niepel et al., 2017).
To predict effective drug combinations for cancer in this study, we assumed that all disease transcripts are equally important except their differential expression values. However, the predicting algorithm can be improved by adjusting the weights of disease transcripts (for example, by integrative analysis of the cancer transcriptome to narrow down the genes essential for maintaining the tumorigenic state) or by imposing additional constraints that require some specific transcripts to be targeted. The need for more refined cancer signatures was reflected on the generally low therapeutic scores (of around 0.1) in this study, indicating that only a small proportion of genes in a given signature (roughly 10% for a therapeutic score of 0.1) can be reversed by small-molecule treatments. Alternatively, our gene-expression-based approach provides a rational framework on which to combine standard cancer therapies (chemotherapeutic agents, molecularly targeted drugs, or immunotherapies) with a drug that would be able to reverse the signature of an emerging resistance mechanism, which may relate to the acquired addiction of cancer cells to non-mutated genes that are hard to decipher by genetic analysis (Flemming, 2015).
In addition to being placed in the context of a global reversal of a disease signature for predicting synergistic drug interactions, these transcript-level recurrences can also be integrated with small-molecule sensitivity to develop a combinatorial strategy for treating diseases. For example, in cases where low expression of a given transcript correlates with sensitivity to a small molecule or a small-molecule class that can in turn upregulate the transcript after treatment (i.e., the relationships in the first quadrant in each panel of Figure 6A and those connected with blue edges in the network of Figure 6B), a strategy that combines the small-molecule treatment with the inhibition of the correlated transcript could augment the small-molecule sensitivity in cancer cells harboring high level of the transcript (for example, the bortezomib:BAG3 pair for which BAG3 knockdown sensitized leukemia cells to bortezomib-induced apoptosis; Liu et al., 2009). Conversely, for pairs for which a given small-molecule treatment is able to downregulate a target transcript whose expression positively correlates with the small-molecule sensitivity (i.e., the relationships in the third quadrant in each panel of Figure 6A and those connected with red edges in the network of Figure 6B), enforced overexpression of the target transcript may enhance the cytotoxic effects in small-molecule-sensitive cell lines.
To benchmark our inferred transcript-level regulatory recurrences, we used independent perturbation profiles to gauge the reproducibility while, at the same time, using annotated compound-target relationships to unveil a disparate regulatory nature of the direct target proteins and the corresponding transcripts. We observed examples in which a protein inhibitor may either upregulate (HSP inhibitors versus HSP family members) or downregulate (topoisomerase inhibitors versus TOP2A) the expression of the corresponding transcript and also found evidence of no regulatory bias in target transcripts for each small-molecule-binding type. In contrast to the ability of expression-sensitivity correlations made between unperturbed gene-expression data and small-molecule sensitivity profiles across cell lines to identify some direct target connections (Rees et al., 2016), our transcript-level regulatory recurrences, in a way, represent small-molecule mechanisms at the level of treatment-induced transcriptional changes other than direct target associations. Moreover, these transcript-level recurrences could be integrated with hallmark network models to map the small-molecule mechanisms onto cancer cell vulnerabilities and predict effective drug treatments (Wang et al., 2015, Zaman et al., 2013). The transcriptional modules identified in our analysis further bring to light several unexplored relationships that might be of potential therapeutic relevance (Molitoris et al., 2011, Perez-Iglesias et al., 2009, Wiklund et al., 2010).
We also note that the cell line identities have a great influence on the composition of the inferred transcript-level regulatory recurrences. For example, recurrent findings might be favored in situations in which the cell lines used for inference can respond similarly to a given perturbation, but less likely in situations when most of the cell lines lack the expression of a transcript relevant to the mechanism of a given perturbation. Consistent with this notion, the compounds with the strongest transcript-level regulatory relationships inferred in this study were less enriched for some molecularly targeted drugs that are thought to be effective in only a subset of cancer cells harboring the relevant gene mutations or pathway activities, such as the receptor tyrosine kinase epidermal growth factor receptor or HER2 inhibitors lapatinib, neratinib, and erlotinib (Figure 1A and Table S2). This is in contrast to those studies where targeted therapies are the main focus and tested in responsive cells (Jiang et al., 2018, Niepel et al., 2017). Despite this concern, the recurring regulatory relationships identified in this study can adequately reproduce some small-molecule transcriptional mechanisms in a separate panel of cell lines. Nonetheless, there are many circumstances in which this inference method can be applied—for example, in a desired subset of immune cell types to identify therapeutic opportunities for manipulating the immune function or in selected cell lines to develop effective combination therapies against a specific cancer type or subtype for which some molecularly targeted drugs are deemed more likely to induce similar transcriptional responses.
Importantly, our approach has potential clinical applicability to guide future combination therapy in individual patients with cancer. To this end, a tumor sample and its matched normal tissue for each patient are required to generate a cancer gene-expression signature that represents the transition from normal to malignant state, thus enabling the prediction of drug combinations that can effectively reverse this transition in a patient-specific manner. Given the observed variation of gene expression within human tissues (GTEx Consortium, 2015), it is often necessary for our approach to use patient-specific tumor-adjacent normal tissues rather than a pooled reference from patients with the same tumor type. In this study, we chose to predict pan-cancer drugs by comparison between pooled tumors and normal tissues for each cancer type, owing to the general paucity of the matched normal samples in TCGA. However, in vitro testing of the top predictions in randomly chosen tumor-type-matched lines verified our proof-of-concept analysis, indicating that these top prioritized drug combinations may be effective in a large fraction of patients with cancer.
In summary, we establish a systems-level computational strategy through gene signature analysis to inform testable predictions for effective combination therapies. The small-molecule-regulated recurring transcripts identified here might not only be particularly useful for targeting non-oncogene addiction from which a resistance mechanism arises but also enable further investigation of the transcriptional mechanisms that underlie small-molecule sensitivity and side effects. Our findings provide a resource for future research on small-molecule mechanisms and will pave the way for more rational combination therapies in clinical trials.
Limitations of the Study
For precision oncology, this approach requires a tumor sample (or a cancer cell line) and its matched normal tissue of each patient (or a lineage-matched untransformed cell line with similar genetic background) to generate a cancer gene-expression signature that represents the transition from the normal to malignant state, which enables the prediction of drug combinations that can effectively reverse this transition. Therefore it might not be appropriate to use a pooled reference from those tumor-adjacent normal tissues of patients with the same tumor type (or a pooled reference from all other cancer cell lines in pan-cancer analysis). In addition, the quality of a disease gene-expression signature will affect the ability of this approach to identify effective therapies.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by the Ministry of Science and Technology (MOST 105-2320-B-002-057-MY3 and MOST 106-2320-B-002-053-MY3) and the National Health Research Institutes (NHRI-EX107-10530PI and NHRI-EX107-10709BI) in Taiwan.
Author Contributions
H.-C.H. and H.-F.J. defined the research theme and supervised the work. C.-T.H. analyzed all data. C.-H.H. and Y.-H.C. performed the drug combination experiments. Y.-J.O. helped with data analysis. C.-T.H., H.-C.H., and H.-F.J. conceived the research, interpreted the results, and wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: May 31, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.04.039.
Contributor Information
Hsuan-Cheng Huang, Email: hsuancheng@ym.edu.tw.
Hsueh-Fen Juan, Email: yukijuan@ntu.edu.tw.
Supplemental Information
References
- Al-Lazikani B., Banerji U., Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012;30:679–692. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]; Al-Lazikani, B., Banerji, U., and Workman, P. (2012). Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 30, 679-692. [DOI] [PubMed]
- Alvarez M.J., Subramaniam P.S., Tang L.H., Grunn A., Aburi M., Rieckhof G., Komissarova E.V., Hagan E.A., Bodei L., Clemons P.A. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 2018;50:979–989. doi: 10.1038/s41588-018-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; Alvarez, M.J., Subramaniam, P.S., Tang, L.H., Grunn, A., Aburi, M., Rieckhof, G., Komissarova, E.V., Hagan, E.A., Bodei, L., Clemons, P.A., et al. (2018). A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 50, 979-989. [DOI] [PMC free article] [PubMed]
- Aran D., Camarda R., Odegaard J., Paik H., Oskotsky B., Krings G., Goga A., Sirota M., Butte A.J. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 2017;8:1077. doi: 10.1038/s41467-017-01027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; Aran, D., Camarda, R., Odegaard, J., Paik, H., Oskotsky, B., Krings, G., Goga, A., Sirota, M., and Butte, A.J. (2017). Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 8, 1077. [DOI] [PMC free article] [PubMed]
- Babichev Y., Kabaroff L., Datti A., Uehling D., Isaac M., Al-Awar R., Prakesch M., Sun R.X., Boutros P.C., Venier R. PI3K/AKT/mTOR inhibition in combination with doxorubicin is an effective therapy for leiomyosarcoma. J. Transl. Med. 2016;14:67. doi: 10.1186/s12967-016-0814-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; Babichev, Y., Kabaroff, L., Datti, A., Uehling, D., Isaac, M., Al-Awar, R., Prakesch, M., Sun, R.X., Boutros, P.C., Venier, R., et al. (2016). PI3K/AKT/mTOR inhibition in combination with doxorubicin is an effective therapy for leiomyosarcoma. J. Transl. Med. 14, 67. [DOI] [PMC free article] [PubMed]
- Bertolini F., Sukhatme V.P., Bouche G. Drug repurposing in oncology–patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015;12:732–742. doi: 10.1038/nrclinonc.2015.169. [DOI] [PubMed] [Google Scholar]; Bertolini, F., Sukhatme, V.P., and Bouche, G. (2015). Drug repurposing in oncology-patient and health systems opportunities. Nat. Rev. Clin. Oncol. 12, 732-742. [DOI] [PubMed]
- Chen B., Ma L., Paik H., Sirota M., Wei W., Chua M.S., So S., Butte A.J. Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nat. Commun. 2017;8:16022. doi: 10.1038/ncomms16022. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chen, B., Ma, L., Paik, H., Sirota, M., Wei, W., Chua, M.S., So, S., and Butte, A.J. (2017). Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nat. Commun. 8, 16022. [DOI] [PMC free article] [PubMed]
- Corsello S.M., Bittker J.A., Liu Z., Gould J., McCarren P., Hirschman J.E., Johnston S.E., Vrcic A., Wong B., Khan M. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat. Med. 2017;23:405–408. doi: 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]; Corsello, S.M., Bittker, J.A., Liu, Z., Gould, J., McCarren, P., Hirschman, J.E., Johnston, S.E., Vrcic, A., Wong, B., Khan, M., et al. (2017). The Drug Repurposing Hub: a next-generation drug library and information resource. Nat. Med. 23, 405-408. [DOI] [PMC free article] [PubMed]
- Dagogo-Jack I., Shaw A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018;15:81–94. doi: 10.1038/nrclinonc.2017.166. [DOI] [PubMed] [Google Scholar]; Dagogo-Jack, I., and Shaw, A.T. (2018). Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81-94. [DOI] [PubMed]
- Deroo B.J., Archer T.K. Glucocorticoid receptor activation of the I kappa B alpha promoter within chromatin. Mol. Biol. Cell. 2001;12:3365–3374. doi: 10.1091/mbc.12.11.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]; Deroo, B.J., and Archer, T.K. (2001). Glucocorticoid receptor activation of the I kappa B alpha promoter within chromatin. Mol. Biol. Cell 12, 3365-3374. [DOI] [PMC free article] [PubMed]
- Felix M.A., Barkoulas M. Pervasive robustness in biological systems. Nat. Rev. Genet. 2015;16:483–496. doi: 10.1038/nrg3949. [DOI] [PubMed] [Google Scholar]; Felix, M.A., and Barkoulas, M. (2015). Pervasive robustness in biological systems. Nat Rev Genet. 16, 483-496. [DOI] [PubMed]
- Feng Y., Mitchison T.J., Bender A., Young D.W., Tallarico J.A. Multi-parameter phenotypic profiling: using cellular effects to characterize small-molecule compounds. Nat. Rev. Drug Discov. 2009;8:567–578. doi: 10.1038/nrd2876. [DOI] [PubMed] [Google Scholar]; Feng, Y., Mitchison, T.J., Bender, A., Young, D.W., and Tallarico, J.A. (2009). Multi-parameter phenotypic profiling: using cellular effects to characterize small-molecule compounds. Nat. Rev. Drug Discov. 8, 567-578. [DOI] [PubMed]
- Flemming A. Anticancer drugs: finding the perfect combination. Nat. Rev. Drug Discov. 2015;14:13. doi: 10.1038/nrd4524. [DOI] [PubMed] [Google Scholar]; Flemming, A. (2015). Anticancer drugs: Finding the perfect combination. Nat. Rev. Drug Discov. 14, 13. [DOI] [PubMed]
- GTEx Consortium The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]; GTEx Consortium (2015). The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648-660. [DOI] [PMC free article] [PubMed]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]; Hanahan, D., and Weinberg, R.A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646-674. [DOI] [PubMed]
- Hata A.N., Niederst M.J., Archibald H.L., Gomez-Caraballo M., Siddiqui F.M., Mulvey H.E., Maruvka Y.E., Ji F., Bhang H.E., Krishnamurthy Radhakrishna V. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016;22:262–269. doi: 10.1038/nm.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hata, A.N., Niederst, M.J., Archibald, H.L., Gomez-Caraballo, M., Siddiqui, F.M., Mulvey, H.E., Maruvka, Y.E., Ji, F., Bhang, H.E., Krishnamurthy Radhakrishna, V., et al. (2016). Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 22, 262-269. [DOI] [PMC free article] [PubMed]
- Holohan C., Van Schaeybroeck S., Longley D.B., Johnston P.G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]; Holohan, C., Van Schaeybroeck, S., Longley, D.B., and Johnston, P.G. (2013). Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714-726. [DOI] [PubMed]
- Huang C.T., Hsieh C.H., Oyang Y.J., Huang H.C., Juan H.F. A large-scale gene expression intensity-based similarity metric for drug repositioning. iScience. 2018;7:40–52. doi: 10.1016/j.isci.2018.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; Huang, C.T., Hsieh, C.H, Oyang, Y.J., Huang, H.C., and Juan, H.F. (2018). A large-scale gene expression intensity-based similarity metric for drug repositioning. iScience 7, 40-52. [DOI] [PMC free article] [PubMed]
- Itamochi H., Oishi T., Shimada M., Sato S., Uegaki K., Naniwa J., Sato S., Nonaka M., Terakawa N., Kigawa J. Inhibiting the mTOR pathway synergistically enhances cytotoxicity in ovarian cancer cells induced by etoposide through upregulation of c-Jun. Clin. Cancer Res. 2011;17:4742–4750. doi: 10.1158/1078-0432.CCR-11-0190. [DOI] [PubMed] [Google Scholar]; Itamochi, H., Oishi, T., Shimada, M., Sato, S., Uegaki, K., Naniwa, J., Sato, S., Nonaka, M., Terakawa, N., Kigawa, J., et al. (2011). Inhibiting the mTOR pathway synergistically enhances cytotoxicity in ovarian cancer cells induced by etoposide through upregulation of c-Jun. Clin. Cancer Res. 17, 4742-4750. [DOI] [PubMed]
- Jackson A.L., Linsley P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]; Jackson, A.L., and Linsley, P.S. (2010). Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 9, 57-67. [DOI] [PubMed]
- Jacobsen A., Silber J., Harinath G., Huse J.T., Schultz N., Sander C. Analysis of microRNA-target interactions across diverse cancer types. Nat. Struct. Mol. Biol. 2013;20:1325–1332. doi: 10.1038/nsmb.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jacobsen, A., Silber, J., Harinath, G., Huse, J.T., Schultz, N., and Sander, C. (2013). Analysis of microRNA-target interactions across diverse cancer types. Nat. Struct. Mol. Biol. 20, 1325-1332. [DOI] [PMC free article] [PubMed]
- Jia J., Zhu F., Ma X., Cao Z., Cao Z.W., Li Y., Li Y.X., Chen Y.Z. Mechanisms of drug combinations: interaction and network perspectives. Nat. Rev. Drug Discov. 2009;8:111–128. doi: 10.1038/nrd2683. [DOI] [PubMed] [Google Scholar]; Jia, J., Zhu, F., Ma, X., Cao, Z., Cao, Z.W., Li, Y., Li, Y.X., and Chen, Y.Z. (2009). Mechanisms of drug combinations: interaction and network perspectives. Nat. Rev. Drug Discov. 8, 111-128. [DOI] [PubMed]
- Jiang P., Lee W., Li X., Johnson C., Liu J.S., Brown M., Aster J.C., Liu X.S. Genome-scale signatures of gene interaction from compound screens predict clinical efficacy of targeted cancer therapies. Cell Syst. 2018;6:343–354.e5. doi: 10.1016/j.cels.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jiang, P., Lee, W., Li, X., Johnson, C., Liu, J.S., Brown, M., Aster, J.C., and Liu, X.S. (2018). Genome-Scale Signatures of Gene Interaction from Compound Screens Predict Clinical Efficacy of Targeted Cancer Therapies. Cell Syst. 6, 343-354.e5. [DOI] [PMC free article] [PubMed]
- Kwong L.N., Heffernan T.P., Chin L. A systems biology approach to personalizing therapeutic combinations. Cancer Discov. 2013;3:1339–1344. doi: 10.1158/2159-8290.CD-13-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kwong, L.N., Heffernan, T.P., and Chin, L. (2013). A systems biology approach to personalizing therapeutic combinations. Cancer Discov. 3, 1339-1344. [DOI] [PMC free article] [PubMed]
- Lamb J., Crawford E.D., Peck D., Modell J.W., Blat I.C., Wrobel M.J., Lerner J., Brunet J.P., Subramanian A., Ross K.N. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]; Lamb, J., Crawford, E.D., Peck, D., Modell, J.W., Blat, I.C., Wrobel, M.J., Lerner, J., Brunet, J.P., Subramanian, A., Ross, K.N., et al. (2006). The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929-1935. [DOI] [PubMed]
- Laplante M., Sabatini D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013;126:1713–1719. doi: 10.1242/jcs.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]; Laplante, M., and Sabatini, D.M. (2013). Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell. Sci. 126, 1713-1719. [DOI] [PMC free article] [PubMed]
- Law V., Knox C., Djoumbou Y., Jewison T., Guo A.C., Liu Y., Maciejewski A., Arndt D., Wilson M., Neveu V. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]; Law, V., Knox, C., Djoumbou, Y., Jewison, T., Guo, A.C., Liu, Y., Maciejewski, A., Arndt, D., Wilson, M., Neveu, V., et al. (2014). DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 42, D1091-1097. [DOI] [PMC free article] [PubMed]
- Liu P., Xu B., Li J., Lu H. BAG3 gene silencing sensitizes leukemic cells to Bortezomib-induced apoptosis. FEBS Lett. 2009;583:401–406. doi: 10.1016/j.febslet.2008.12.032. [DOI] [PubMed] [Google Scholar]; Liu, P., Xu, B., Li, J., and Lu, H. (2009). BAG3 gene silencing sensitizes leukemic cells to Bortezomib-induced apoptosis. FEBS Lett. 583, 401-406. [DOI] [PubMed]
- Lopez J.S., Banerji U. Combine and conquer: challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017;14:57–66. doi: 10.1038/nrclinonc.2016.96. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lopez, J.S., and Banerji, U. (2017). Combine and conquer: challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 14, 57-66. [DOI] [PMC free article] [PubMed]
- Luo J., Solimini N.L., Elledge S.J. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; Luo, J., Solimini, N.L., and Elledge, S.J. (2009). Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823-837. [DOI] [PMC free article] [PubMed]
- Mao Y., Fu A., Leaderer D., Zheng T., Chen K., Zhu Y. Potential cancer-related role of circadian gene TIMELESS suggested by expression profiling and in vitro analyses. BMC Cancer. 2013;13:498. doi: 10.1186/1471-2407-13-498. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mao, Y., Fu, A., Leaderer, D., Zheng, T., Chen, K., and Zhu, Y. (2013). Potential cancer-related role of circadian gene TIMELESS suggested by expression profiling and in vitro analyses. BMC Cancer 13, 498. [DOI] [PMC free article] [PubMed]
- Moffat J.G., Rudolph J., Bailey D. Phenotypic screening in cancer drug discovery - past, present and future. Nat. Rev. Drug Discov. 2014;13:588–602. doi: 10.1038/nrd4366. [DOI] [PubMed] [Google Scholar]; Moffat, J.G., Rudolph, J., and Bailey, D. (2014). Phenotypic screening in cancer drug discovery - past, present and future. Nat. Rev. Drug Discov. 13, 588-602. [DOI] [PubMed]
- Molitoris J.K., McColl K.S., Swerdlow S., Matsuyama M., Lam M., Finkel T.H., Matsuyama S., Distelhorst C.W. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J. Biol. Chem. 2011;286:30181–30189. doi: 10.1074/jbc.M111.245423. [DOI] [PMC free article] [PubMed] [Google Scholar]; Molitoris, J.K., McColl, K.S., Swerdlow, S., Matsuyama, M., Lam, M., Finkel, T.H., Matsuyama, S., and Distelhorst, C.W. (2011). Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J. Biol. Chem. 286, 30181-30189. [DOI] [PMC free article] [PubMed]
- Nagel R., Semenova E.A., Berns A. Drugging the addict: non-oncogene addiction as a target for cancer therapy. EMBO Rep. 2016;17:1516–1531. doi: 10.15252/embr.201643030. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nagel, R., Semenova, E.A., and Berns, A. (2016). Drugging the addict: non-oncogene addiction as a target for cancer therapy. EMBO Rep. 17, 1516-1531. [DOI] [PMC free article] [PubMed]
- Niepel M., Hafner M., Duan Q., Wang Z., Paull E.O., Chung M., Lu X., Stuart J.M., Golub T.R., Subramanian A. Common and cell-type specific responses to anti-cancer drugs revealed by high throughput transcript profiling. Nat. Commun. 2017;8:1186. doi: 10.1038/s41467-017-01383-w. [DOI] [PMC free article] [PubMed] [Google Scholar]; Niepel, M., Hafner, M., Duan, Q., Wang, Z., Paull, E.O., Chung, M., Lu, X., Stuart, J.M., Golub, T.R., Subramanian, A., et al. (2017). Common and cell-type specific responses to anti-cancer drugs revealed by high throughput transcript profiling. Nat. Commun. 8, 1186. [DOI] [PMC free article] [PubMed]
- Perez-Iglesias R., Mata I., Pelayo-Teran J.M., Amado J.A., Garcia-Unzueta M.T., Berja A., Martinez-Garcia O., Vazquez-Barquero J.L., Crespo-Facorro B. Glucose and lipid disturbances after 1 year of antipsychotic treatment in a drug-naive population. Schizophr. Res. 2009;107:115–121. doi: 10.1016/j.schres.2008.09.028. [DOI] [PubMed] [Google Scholar]; Perez-Iglesias, R., Mata, I., Pelayo-Teran, J.M., Amado, J.A., Garcia-Unzueta, M.T., Berja, A., Martinez-Garcia, O., Vazquez-Barquero, J.L., and Crespo-Facorro, B. (2009). Glucose and lipid disturbances after 1 year of antipsychotic treatment in a drug-naive population. Schizophr. Res. 107, 115-121. [DOI] [PubMed]
- Radhakrishnan S.K., Lee C.S., Young P., Beskow A., Chan J.Y., Deshaies R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell. 2010;38:17–28. doi: 10.1016/j.molcel.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; Radhakrishnan, S.K., Lee, C.S., Young, P., Beskow, A., Chan, J.Y., and Deshaies, R.J. (2010). Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 38, 17-28. [DOI] [PMC free article] [PubMed]
- Ramirez M., Rajaram S., Steininger R.J., Osipchuk D., Roth M.A., Morinishi L.S., Evans L., Ji W., Hsu C.H., Thurley K. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016;7:10690. doi: 10.1038/ncomms10690. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ramirez, M., Rajaram, S., Steininger, R.J., Osipchuk, D., Roth, M.A., Morinishi, L.S., Evans, L., Ji, W., Hsu, C.H., Thurley, K., et al. (2016). Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 7, 10690. [DOI] [PMC free article] [PubMed]
- Rees M.G., Seashore-Ludlow B., Cheah J.H., Adams D.J., Price E.V., Gill S., Javaid S., Coletti M.E., Jones V.L., Bodycombe N.E. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016;12:109–116. doi: 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rees, M.G., Seashore-Ludlow, B., Cheah, J.H., Adams, D.J., Price, E.V., Gill, S., Javaid, S., Coletti, M.E., Jones, V.L., Bodycombe, N.E., et al. (2016). Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 12, 109-116. [DOI] [PMC free article] [PubMed]
- Rosati A., Graziano V., De Laurenzi V., Pascale M., Turco M.C. BAG3: a multifaceted protein that regulates major cell pathways. Cell Death Dis. 2011;2:e141. doi: 10.1038/cddis.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rosati, A., Graziano, V., De Laurenzi, V., Pascale, M., and Turco, M.C. (2011). BAG3: a multifaceted protein that regulates major cell pathways. Cell Death Dis. 2, e141. [DOI] [PMC free article] [PubMed]
- Ryall K.A., Tan A.C. Systems biology approaches for advancing the discovery of effective drug combinations. J. Cheminform. 2015;7:7. doi: 10.1186/s13321-015-0055-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ryall, K.A., and Tan, A.C. (2015). Systems biology approaches for advancing the discovery of effective drug combinations. J. Cheminform. 7, 7. [DOI] [PMC free article] [PubMed]
- Santos R., Ursu O., Gaulton A., Bento A.P., Donadi R.S., Bologa C.G., Karlsson A., Al-Lazikani B., Hersey A., Oprea T.I. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017;16:19–34. doi: 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]; Santos, R., Ursu, O., Gaulton, A., Bento, A.P., Donadi, R.S., Bologa, C.G., Karlsson, A., Al-Lazikani, B., Hersey, A., Oprea, T.I., et al. (2017). A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19-34. [DOI] [PMC free article] [PubMed]
- Saunders N.A., Simpson F., Thompson E.W., Hill M.M., Endo-Munoz L., Leggatt G., Minchin R.F., Guminski A. Role of intratumoural heterogeneity in cancer drug resistance: molecular and clinical perspectives. EMBO Mol. Med. 2012;4:675–684. doi: 10.1002/emmm.201101131. [DOI] [PMC free article] [PubMed] [Google Scholar]; Saunders, N.A., Simpson, F., Thompson, E.W., Hill, M.M., Endo-Munoz, L., Leggatt, G., Minchin, R.F., and Guminski, A. (2012). Role of intratumoural heterogeneity in cancer drug resistance: molecular and clinical perspectives. EMBO Mol. Med. 4, 675-684. [DOI] [PMC free article] [PubMed]
- Subramanian A., Narayan R., Corsello S.M., Peck D.D., Natoli T.E., Lu X., Gould J., Davis J.F., Tubelli A.A., Asiedu J.K. A next generation Connectivity Map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171:1437–1452.e17. doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]; Subramanian, A., Narayan, R., Corsello, S.M., Peck, D.D., Natoli, T.E., Lu, X., Gould, J., Davis, J.F., Tubelli, A.A., Asiedu, J.K., et al. (2017). A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 171, 1437-1452.e17. [DOI] [PMC free article] [PubMed]
- Sun X., Vilar S., Tatonetti N.P. High-throughput methods for combinatorial drug discovery. Sci. Transl. Med. 2013;5:205rv201. doi: 10.1126/scitranslmed.3006667. [DOI] [PubMed] [Google Scholar]; Sun, X., Vilar, S., and Tatonetti, N.P. (2013). High-throughput methods for combinatorial drug discovery. Sci. Transl. Med. 5, 205rv201. [DOI] [PubMed]
- Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A., Jr., Kinzler K.W. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]; Vogelstein, B., Papadopoulos, N., Velculescu, V.E., Zhou, S., Diaz, L.A., Jr., and Kinzler, K.W. (2013). Cancer genome landscapes. Science 339, 1546-1558. [DOI] [PMC free article] [PubMed]
- Vu L.P., Prieto C., Amin E.M., Chhangawala S., Krivtsov A., Calvo-Vidal M.N., Chou T., Chow A., Minuesa G., Park S.M. Functional screen of MSI2 interactors identifies an essential role for SYNCRIP in myeloid leukemia stem cells. Nat. Genet. 2017;49:866–875. doi: 10.1038/ng.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]; Vu, L.P., Prieto, C., Amin, E.M., Chhangawala, S., Krivtsov, A., Calvo-Vidal, M.N., Chou, T., Chow, A., Minuesa, G., Park, S.M., et al. (2017). Functional screen of MSI2 interactors identifies an essential role for SYNCRIP in myeloid leukemia stem cells. Nat. Genet. 49, 866-875. [DOI] [PMC free article] [PubMed]
- Wang B., Zhu X., Kim Y., Li J., Huang S., Saleem S., Li R.C., Xu Y., Dore S., Cao W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic. Biol. Med. 2012;52:928–936. doi: 10.1016/j.freeradbiomed.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang, B., Zhu, X., Kim, Y., Li, J., Huang, S., Saleem, S., Li, R.C., Xu, Y., Dore, S., and Cao, W. (2012). Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic. Biol. Med. 52, 928-936. [DOI] [PMC free article] [PubMed]
- Wang E., Zaman N., McGee S., Milanese J.S., Masoudi-Nejad A., O'Connor-McCourt M. Predictive genomics: a cancer hallmark network framework for predicting tumor clinical phenotypes using genome sequencing data. Semin. Cancer Biol. 2015;30:4–12. doi: 10.1016/j.semcancer.2014.04.002. [DOI] [PubMed] [Google Scholar]; Wang, E., Zaman, N., McGee, S., Milanese, J.S., Masoudi-Nejad, A., and O'Connor-McCourt, M. (2015). Predictive genomics: a cancer hallmark network framework for predicting tumor clinical phenotypes using genome sequencing data. Semin. Cancer Biol. 30, 4-12. [DOI] [PubMed]
- Waring M.J., Arrowsmith J., Leach A.R., Leeson P.D., Mandrell S., Owen R.M., Pairaudeau G., Pennie W.D., Pickett S.D., Wang J. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015;14:475–486. doi: 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]; Waring, M.J., Arrowsmith, J., Leach, A.R., Leeson, P.D., Mandrell, S., Owen, R.M., Pairaudeau, G., Pennie, W.D., Pickett, S.D., Wang, J., et al. (2015). An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 14, 475-486. [DOI] [PubMed]
- Webster R.M. Combination therapies in oncology. Nat. Rev. Drug Discov. 2016;15:81–82. doi: 10.1038/nrd.2016.3. [DOI] [PubMed] [Google Scholar]; Webster, R.M. (2016). Combination therapies in oncology. Nat. Rev. Drug Discov. 15, 81-82. [DOI] [PubMed]
- Wiklund E.D., Catts V.S., Catts S.V., Ng T.F., Whitaker N.J., Brown A.J., Lutze-Mann L.H. Cytotoxic effects of antipsychotic drugs implicate cholesterol homeostasis as a novel chemotherapeutic target. Int. J. Cancer. 2010;126:28–40. doi: 10.1002/ijc.24813. [DOI] [PubMed] [Google Scholar]; Wiklund, E.D., Catts, V.S., Catts, S.V., Ng, T.F., Whitaker, N.J., Brown, A.J., and Lutze-Mann, L.H. (2010). Cytotoxic effects of antipsychotic drugs implicate cholesterol homeostasis as a novel chemotherapeutic target. Int. J. Cancer 126, 28-40. [DOI] [PubMed]
- Yadav B., Wennerberg K., Aittokallio T., Tang J. Searching for drug synergy in complex dose-response landscapes using an interaction potency model. Comput. Struct. Biotechnol. J. 2015;13:504–513. doi: 10.1016/j.csbj.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yadav, B., Wennerberg, K., Aittokallio, T., and Tang, J. (2015). Searching for Drug Synergy in Complex Dose-Response Landscapes Using an Interaction Potency Model. Comput. Struct. Biotechnol. J. 13, 504-513. [DOI] [PMC free article] [PubMed]
- Zaman N., Li L., Jaramillo M.L., Sun Z., Tibiche C., Banville M., Collins C., Trifiro M., Paliouras M., Nantel A. Signaling network assessment of mutations and copy number variations predict breast cancer subtype-specific drug targets. Cell Rep. 2013;5:216–223. doi: 10.1016/j.celrep.2013.08.028. [DOI] [PubMed] [Google Scholar]; Zaman, N., Li, L., Jaramillo, M.L., Sun, Z., Tibiche, C., Banville, M., Collins, C., Trifiro, M., Paliouras, M., Nantel, A., et al. (2013). Signaling network assessment of mutations and copy number variations predict breast cancer subtype-specific drug targets. Cell Rep. 5, 216-223. [DOI] [PubMed]
- Zimmermann J., Erdmann D., Lalande I., Grossenbacher R., Noorani M., Furst P. Proteasome inhibitor induced gene expression profiles reveal overexpression of transcriptional regulators ATF3, GADD153 and MAD1. Oncogene. 2000;19:2913–2920. doi: 10.1038/sj.onc.1203606. [DOI] [PubMed] [Google Scholar]; Zimmermann, J., Erdmann, D., Lalande, I., Grossenbacher, R., Noorani, M., and Furst, P. (2000). Proteasome inhibitor induced gene expression profiles reveal overexpression of transcriptional regulators ATF3, GADD153 and MAD1. Oncogene 19, 2913-2920. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.