

Abstract
Pluripotent and tissue‐specific stem cells, such as blood‐forming stem cells, are maintained through a balance of quiescence, self‐renewal, and differentiation. Self‐renewal is a specialized cell division that generates daughter cells with the same features as the parental stem cell. Although many factors are involved in the regulation of self‐renewal, perhaps the most well‐known factors are members of the Krüppel‐like factor (KLF) family, especially KLF4, because of the landmark discovery that this protein is required to reprogram somatic cells into induced pluripotent stem cells. Because KLF4 regulates gene expression through transcriptional activation or repression via either DNA binding or protein‐to‐protein interactions, the outcome of KLF4‐mediated regulation largely depends on the cellular context, cell cycle regulation, chromatin structure, and the presence of oncogenic drivers. This study first summarizes the current understanding of the regulation of self‐renewal by KLF proteins in embryonic stem cells through a KLF circuitry and then delves into the potential function of KLF4 in normal hematopoietic stem cells and its emerging role in leukemia‐initiating cells from pediatric patients with T‐cell acute lymphoblastic leukemia via repression of the mitogen‐activated protein kinase 7 pathway. stem cells translational medicine 2019;8:568–574
Keywords: Self‐renewal, KLF4, Hematopoietic stem cells, Leukemia stem cells
Significance Statement.
Understanding the molecular regulation of self‐renewal in hematopoietic stem cells is essential not only for stem cell biology and regenerative medicine but also, most importantly, to address the resistance of hematological malignancies to drug therapy and develop novel therapies specific to leukemia stem cells, which represent an unmet medical need, because this population is believed to drive chemoresistance and relapse. The role of the reprogramming factor KLF4 in the regulation of self‐renewal is discussed in embryonic stem cells and normal and leukemic hematopoietic stem cells.
Introduction
Stem cells exist at the apex of tissue development and can orchestrate embryonic differentiation of various tissues in an adult organism and regulate tissue homeostasis and regeneration after injury. Embryonic stem cells (ESCs) are the most undifferentiated stem cells and are capable of generating all cell types within the organism, whereas somatic tissue‐specific stem cells (e.g., hematopoietic stem cells, HSCs) can only regenerate cells within the same tissue. In bone marrow, HSCs can produce blood cells on demand during homeostatic and regenerative hematopoiesis, and this capacity to produce the blood while maintaining the pool of HSCs is controlled by a delicate balance between self‐renewing and differentiating cell divisions. Self‐renewal is a specialized and highly regulated cell division producing one or two daughter cells with the same stem cell features as the parental stem cell. This multifaceted mechanism has been the subject of extensive research because of clear implications in tissue homeostasis, regenerative medicine, and cancer therapy. Leukemia is a cancer of the blood cells affecting either lymphoid or myeloid lineages that is caused by genetic and epigenetic alterations occurring in HSC or hematopoietic progenitor cell (HPC); the generated population of leukemic cells bear stem cell properties ensuring self‐preservation through their self‐renewal capacity while continuously feeding the neoplasm by differentiating into the bulk of leukemia cells. As a critical process regulating stem cell fate in normal and malignant hematopoiesis, self‐renewal is controlled by the specialized microenvironment, or niche, and intrinsic factors that guide the decision of a stem cell to either self‐renew or undergo differentiation, depending on the demand of the specific tissue. The Krüppel‐like factor (KLF) family of proteins encompasses 17 zinc‐finger transcription factors, of which at least 5 have been implicated in key stem cell functions, such as self‐renewal 1, 2, 3, 4, 5, pluripotency 1, 2, 3, 5, embryogenesis 6, and erythropoiesis 7. Like other members of the family, KLF4 contains activation and repression domains that mediate recruitment of coactivators or corepressors and three zinc fingers that bind to guanine‐cytosine‐rich sequences such as CACCC found in gene regulatory promoters and enhancers 8, 9. KLF4 has been increasingly studied since the landmark work describing its role in the reprogramming of somatic cells into induced pluripotent stem cells (iPSCs), and the contribution of KLF4 to this process suggests a potential function in the preservation of stemness in other tissue stem cells 10. KLF4 is expressed in a wide range of mammalian tissues and regulates diverse cellular processes during normal tissue homeostasis, including proliferation, survival, and differentiation. In fact, KLF4 regulates self‐renewal in stem cells from different tissues (e.g., embryonal, intestine, and skin) in both homeostasis and cancer 1, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23. Although KLF4 has been studied more extensively in ESCs and solid tumors, there is emerging evidence of KLF4 involvement in the process of blood formation by regulating normal hematopoiesis and leukemia stem cells (LSCs).
Regulation of Self‐Renewal by KLFs in Pluripotent Stem Cells
Because most of what is known on the regulation of self‐renewal by KLFs has been described in ESCs, we will briefly review the main contributions before discussing the role of KLF4 in normal and malignant HSCs. ESCs are derived from the inner cell mass of the blastocyst—an early stage of the preimplantation embryo—and characterized for their capacity for self‐renewal and pluripotent differentiation of ESCs into any tissue and cell type 24, 25. ESCs regulate self‐renewal and pluripotency properties through cell‐intrinsic (NANOG, OCT4, SOX2, and KLFs) and ‐extrinsic factors (leukemia inhibitory factor, LIF) 1, 26, 27, 28, 29, 30, 31, 32, 33, 34. OCT4, SOX2, and NANOG were originally identified as members of a core regulatory pathway involved in the preservation of stemness by promoting self‐renewal and preserving pluripotency. Several KLFs (KLF1, KLF2, KLF4, KLF5, and KLF17) regulate self‐renewal in the embryos, ESCs, hematopoietic cells, and bone marrow stromal cells in mice, humans, and zebrafish (Table 1). Among these KLFs, the most extensively studied and characterized are KLF2, KLF4, and KLF5, as they form part of a transcriptional circuitry that promotes self‐renewal in ESCs by activating pluripotency‐associated genes such as NANOG that inhibit their differentiation into primitive endoderm 1, 2. Abrogation of self‐renewal and terminal differentiation of ESCs by simultaneous knockdown of KLF2, KLF4, and KLF5 suggested a KLF regulatory circuitry, which was rescued by the introduction of RNAi‐resistant cDNA encoding these three factors 1. The KLF2/KLF4/KLF5 triad controls self‐renewal by regulating the expression of genes involved in self‐renewal (Oct4, Sox2, Myc, and Tcl1) and pluripotency (Nanog, Esrrb, and Oct4), facilitating the formation of autoregulatory loops among Oct4, Sox2, Nanog, and Sall4 in ESCs (Fig. 3) 1, 2. Although most KLF proteins (KLF1–KLF10) can bind to the regulatory regions of Nanog, only KLF2, KLF4, and KLF5 are able to maintain murine ESCs in an undifferentiated state in the absence of LIF 2. Based on chromatin immunoprecipitation and sequencing analysis, KLF4 and KLF5 inhibit differentiation of mesoderm and endoderm in ESCs by activating targets other than NANOG 18. These findings suggest that KLF2, KLF4, and KLF5 have overlapping functions but also exert distinct roles in self‐renewal of ESCs. Although KLF4 restores loss of stemness caused by deletion of Klf5 in murine ESCs 30, the fact that Klf4‐null embryos can develop to term suggests either that KLF4 is dispensable for embryogenesis or there is a functional compensation by other KLF proteins 1, 35. KLF4 and NANOG are among the first transcription factors to shut down their transcription when ESCs exit pluripotency, and nuclear export of KLF4 upon ERK activation is a critical first step to exit the naive pluripotent state and initiate ESC differentiation 16, 36. In addition, KLF4 is required for expression of the telomerase reverse transcriptase (TERT) in human ESCs and binds β‐catenin through protein‐to‐protein interaction, allowing the recruitment of this dimer to the Tert promoter in murine ESCs 17, 37. Finally, KLF4 acts as a fast mediator of LIF signaling through the activation of STAT3 to cooperate with OCT4 and SOX2 in activating the expression of NANOG while repressing the GATA6 and SOX17 genes, which are involved in endoderm differentiation 31. In summary, several KLF transcription factors regulate key processes of stem cell function in ESCs, among which KLF2, KLF4, and KLF5 play prominent roles. The formation of a KLF circuitry may be exclusive to ESCs, as this mechanism has not been described in other stem cells.
Table 1.
Roles of KLF in stem cell self‐renewal
| KLF proteins | Host organism | Tissue/cell analyzed | Key functionality | Genes regulated by KLFs |
|---|---|---|---|---|
| KLF1 | Mouse | Embryo 7 | Required for erythropoiesis 7 | Myc 7 |
| KLF2 | Mouse | Embryo and embryonic stem cells 1, 2, 7 | Promotes self‐renewal and pluripotency 1, 2
Required for erythropoiesis 7 |
Nanog and Esrrb
1, 2
Myc 1, 7 Oct4, Tcl1, Nr5a2, Tbx3, Rif1, Sox2, Tcf3, Mycn, and Foxd3 1 Stella 2 |
| Human | Bone marrow stromal cells 3 | Promotes self‐renewal and pluripotency 3 | Oct4, Nanog, and Rex1 3 | |
| Zebrafish | Embryo 6 | Required for embryogenesis 6 | Oct4 6 | |
| KLF4 | Mouse | Embryonic stem cells 1, 2 | Promotes self‐renewal and pluripotency 1, 2 |
Nanog and Esrrb
1, 2
Tcl1, Myc, Nr5a2, Tbx3, Nanog, Esrrb, Rif1, Oct4, Sox2, Tcf3, Mycn, and Foxd3 1 Stella 2 |
| Hematopoietic cells 4 | Promotes self‐renewal 4 | |||
| KLF5 | Mouse | Embryonic stem cells 1, 2, 5 | Promotes self‐renewal and pluripotency 1, 2, 5 |
Nanog
1, 2, 5
Esrrb 1, 2 Oct4 and Sox2 1, 5 Tcl1, Myc, Nr5a2, Tbx3, Esrrb, Rif1, Oct4, Tcf3, Mycn, and Foxd3 1 Stella 2 |
| KLF17 | Zebrafish | Embryo 6 | Required for embryogenesis 6 | Oct4 6 |
Abbreviation: KLF, Krüppel‐like factor.
Figure 3.
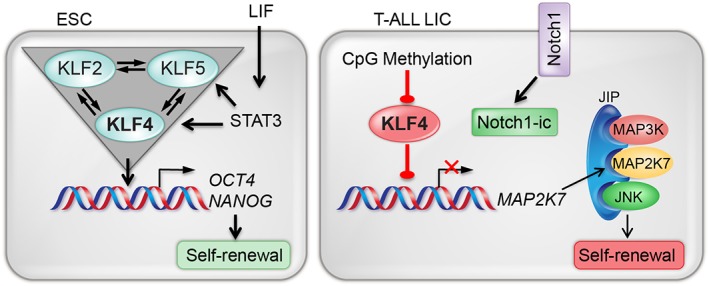
KLF4 regulates self‐renewal in ESCs and T‐ALL LICs. A KLF self‐regulated triad regulates self‐renewal in ESCs. In T‐ALL LICs, KLF4's repression of the kinase MAP2K7 is prevented by CpG methylation of the KLF4 promoter. Abbreviations: ESCs, embryonic stem cells; JIP, JNK‐interacting protein; KLF, Krüppel‐like factor; LIC, leukemia‐initiating cell; LIF, leukemia inhibitory factor; Notch1‐ic, Notch1 intracellular; MAP2K7, mitogen‐activated protein kinase kinase 7; MAP3K, mitogen‐activated protein kinase 3; T‐ALL, T‐cell acute lymphoblastic leukemia.
Role of KLF4 in Normal HSCs
Regulation of HSC Self‐Renewal
The identification of mechanisms that promote ex vivo self‐renewing expansion is the Holy Grail in HSC research and is pursued by many groups aiming at bone marrow transplant and cell and gene therapy applications. The bone marrow milieu modulates stemness at different levels through secreted factors (stem cell factor, thrombopoietin, interleukin‐3 [IL3], IL‐6, IL‐11, and fms‐like tyrosine kinase 3 [FLT3]), inflammatory cytokines (e.g., tumor necrosis factor alpha and interferon gamma), hypoxia, the extracellular matrix, and topographic direction of the mitotic spindle with respect to cellular components of the niche during cell division, which could lead to losses of key cellular interactions and an asymmetric distribution of intracellular components. This specialized milieu delivers signals to HSCs through factors recognized by the corresponding receptors that translate information to nuclei, where transcription factors regulate the expression of genes involved in the control of self‐renewal. Some of the extrinsic mechanisms regulating HSCs are NOTCH1, hedgehog, WNT, EP receptor for prostaglandin E2, angiopoietin‐like protein 5, and pleiotrophin (review and references therein 38, 39, 40, 41) (Fig. 1). It is not clear whether KLF4 plays a role in the regulation of these extrinsic mechanisms (Fig. 1), although KLF4 can inhibit the WNT pathway in intestinal epithelium through interaction with β‐catenin and repress the expression of NOTCH1 in keratinocytes, whereas NOTCH1 inhibits the expression of KLF4 in intestinal epithelium 42, 43, 44.
Figure 1.
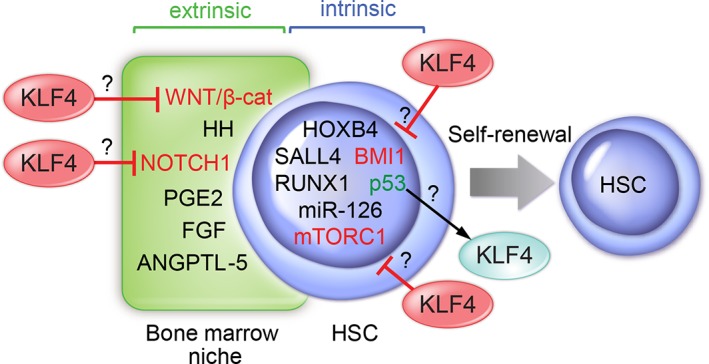
Self‐renewal is regulated by extrinsic (stem cell niche in bone marrow) and intrinsic factors in HSCs. A potential regulatory role of KLF4 is indicated based on reports in other cell types. Abbreviations: ANGPTL‐5, angiopoietin‐like protein 5; FGF, fibroblast growth factor; HH, hedgehog; HSC, hematopoietic stem cells; KLF, Krüppel‐like factor; PGE2, prostaglandin E2.
In addition to the regulation by stem cell niches, many intrinsic factors have been described as positive regulators of HSC self‐renewal (e.g., HOXB4, RUNX1, BMI1, p53, miR‐126, FLT3, STAT5A, HMGA2, and SALL4; Fig. 1) 45, 46, 47, 48, 49, 50, 51, 52. Collectively, intrinsic factors regulate cell fate during division at different levels, such as control of gene expression via transcriptional regulation (e.g., RUNX1 and STAT5A), metabolic sensing of nutrients and growth factors (e.g., mTORC1), response to hypoxia and metabolism (e.g., hypoxia‐inducible factor 1α), and development and aging (e.g., BMI1 and p16). Reflecting the complexity of self‐renewal regulation, many of these factors have interconnected functions; for example, TCF7 regulates the expression of RUNX1 independently of WNT signaling, and the histone H2A deubiquitinase MYSM1 drives the recruitment of RUNX1 into the GFI1 locus, another transcription factor involved in HSC self‐renewal 53, 54, 55. Although KLF4 has not been directly associated with the intrinsic regulation of HSC self‐renewal, a few reports suggest a potential role in steady‐state hematopoiesis, such as the inhibitory effect on BMI1 in intestinal cells, inhibition of mTORC1 during somatic cell reprogramming, and regulation of KLF4 by p53 in acute myeloid leukemia (AML) 56, 57, 58. In addition to transcriptional regulation, the expression of genes involved in self‐renewal can be mediated through epigenetic mechanisms such as CpG methylation and histone modifications. For example, mutations in DNMT3A lead to an increase in self‐renewal and upregulation of stemness genes in HSCs, and loss of DNMT3A promotes expansion of HSCs in the bone marrow 59, 60, 61. Interestingly, DNMT3A binds to the CpG island in the KLF4 promoter in endothelial cells, inducing DNA methylation and subsequent gene repression 62. In addition to DNMT3A, IDH1/2 and TET2 mutations, often found in hematologic malignancies, can deregulate the pattern of genomic DNA methylation and aberrantly increase self‐renewal 63. It was recently reported that TET2 binds to KLF4 through protein‐to‐protein interaction to drive locus‐specific demethylation during reprogramming of B cells into iPSCs 64. Lastly, maintenance of telomere length through telomerase activity also plays a critical role in self‐renewal, as loss of telomerase results in reduced self‐renewal capacity of HSCs, as evaluated by serial transplantation, in addition to promoting carcinogenesis via genomic instability 65. Interestingly, KLF4 activates TERT expression through interaction with β‐catenin in ESCs 37, 66. Finally, factors involved in the differentiation toward different lineages, not listed here, could be considered negative regulators of self‐renewal because the alternative fate during cell division is differentiation.
KLF4 Regulates Self‐Renewal in Adult HSCs
In the hematopoietic system, KLF4 promotes macrophage and monocyte differentiation, macrophage polarization, survival of natural killer cells, secondary antibody responses in memory B cells, and dendritic cell development, whereas KLF4 inhibits homeostatic proliferation of naïve T cells 4, 67, 68, 69, 70, 71, 72, 73, 74. The enrichment of KLF4 transcripts in human HSCs (CD34+ CD38lo Linlo) compared with HPCs (CD34+ CD38hi Linhi) led to the study of KLF4 in HSCs from fetal livers, because embryonic homozygous deletion results in postnatal lethality 35, 70, 75. Although clonogenic and competitive transplantation assays of fetal Klf4‐null HSCs showed normal colony formation in methylcellulose cultures and hematologic reconstitution of cytoablated recipient mice 70, the role of KLF4 in adult bone marrow HSCs has not been investigated. Further supporting a potential role of KLF4 in adult HSCs, loss of the cell fate determinant lethal giant larvae homolog 1 increases self‐renewal, resulting in elevated numbers of HSCs and a competitive advantage after transplantation that is associated with KLF4 repression 41. This finding suggests that inactivation of KLF4 might contribute to the regulation of self‐renewal in adult HSC and warrants the study of KLF4 using somatic gene deletion.
Role of KLF4 in Leukemic Stem Cells
KLF4 in Cancer
KLF4 can behave as a tumor suppressor and an oncogene in a cell context‐dependent manner because of its dual properties as activator and repressor of gene expression. For example, KLF4 has a tumor suppressor function in the gastrointestinal tract, whereas in breast cancer KLF4 has oncogenic properties 76, 77. The potential mechanisms of KLF4 duality in solid‐tumor carcinogenesis have been reviewed and depend on multiple factors, such as cell cycle status (expression of p21 and p53), presence of coactivators or corepressors, chromatin accessibility, the epigenome, regulation of oncogenic pathways (e.g., WNT, RAS, TGFβ, and NOTCH1), and interplay with oncogenic drivers 76, 77, 78, 79. In contrast to solid tumors, the role of KLF4 in hematological malignancies has not been well‐studied, in large part because in the genomic era, no widespread genetic alterations (mutations, chromosomal translocations) have been found in patients, although epigenetic regulation of tumor suppressors and oncogenes also contributes to the leukemogenic process. At least two inactivating mutations of KLF4 have been identified in childhood acute lymphoblastic leukemia (ALL), both in the 3 prime untranslated region, abolishing a miR‐2909 regulatory domain and a zinc‐finger motif; the latter inactivates its DNA‐binding capacity 80. However, most of the role of KLF4 in cancer appears to be mediated through epigenetic or post‐transcriptional gene inactivation. Low levels of KLF4 transcript have been detected in AML, B‐cell non‐Hodgkin and Hodgkin lymphomas, multiple myeloma, and T‐cell ALL (T‐ALL), suggesting potential tumor‐suppressive properties 81, 82, 83, 84. Our group and others found that KLF4 gene expression is silenced by promoter hypermethylation in B‐cell lymphomas and T‐ALL 81, 82. In addition to CpG methylation, downregulation of KLF4 is associated with deregulation of microRNAs such as miR‐10a and miR‐10b, and the transcriptional repression of KLF4 by CDX2 in AML is associated with inhibition of PPARγ signaling 84, 85, 86. Conversely, when human CD34+ cells are transduced with ZMYM2‐FGFR, the fusion protein product of the t(8;13)(p11;q12) chromosomal translocation found in myeloproliferative neoplasm, and transplanted into NSG mice, the mice display elevated KLF4, suggesting a potential oncogenic function 87. In contrast, AML patients with a low level of HDAC1, which is negatively correlated with KLF4 level, exhibit better prognosis 88. More recently, KLF4 expression has been identified in resistant clones in chronic lymphocytic leukemia evaluated by performing single‐cell RNA sequencing during diagnosis, treatment, and relapse 89. Therefore, KLF4 may be involved in the emergence of aggressive leukemic clones during clonal evolution. Collectively, these findings indicate that KLF4 likely regulates the maintenance of LSCs in leukemia.
KLF4 in Leukemia Stem/Initiating Cells
LSCs and leukemia‐initiating cells (LICs) are rare populations considered leukemia reservoirs, because they can survive chemotherapy and induce relapses and therefore are considered important targets for the development of curative therapies. In stem cell leukemias, LSCs are generated by transformation of a single HSC with a genetic driver mutation that retains the self‐renewal property (e.g., chronic myeloid leukemia), whereas LICs are originated by transformation of HPCs that are devoid of self‐renewal capacity, and therefore their transformation must be accompanied by acquisition of self‐renewal (Fig. 2). The study of the molecular regulation that maintains LSCs/LICs has the main goal of uncovering potential targets for therapy and involves testing the ability of enriched populations to initiate disease after transplantation into secondary wild‐type mice, which is the gold‐standard assay to evaluate self‐renewal capacity.
Figure 2.
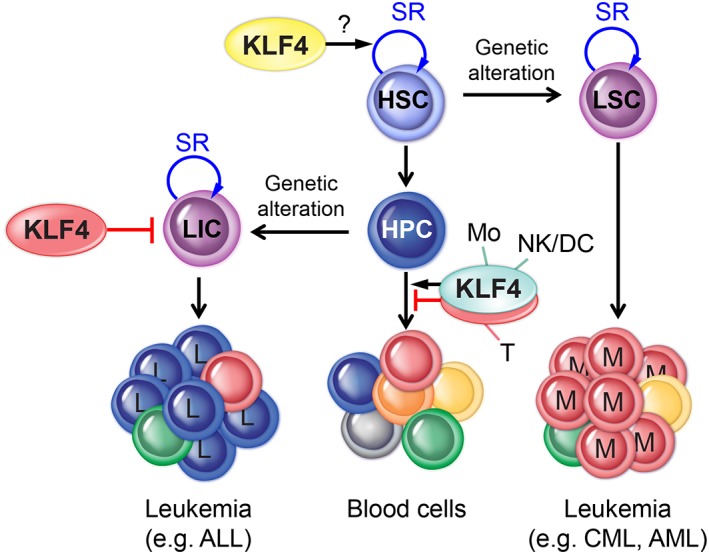
Transformation of HSC/HPC and generation of LSC/LIC. Genetic alterations (e.G., gene mutation, deletion, and translocation) transform normal HSCs into LSCs, preserving the capacity of SR that drives expansion of M in stem cell leukemia. In contrast, LICs feeding L leukemias are generated by transformation of HPCs with acquisition of SR. the roles of KLF4 in HSCs, LICs, and blood cells (Mo, NK, DC, and T) are indicated. Abbreviations: ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CML, chronic myeloid leukemia; DC, dendritic cell; HPC, hematopoietic progenitor cell; HSC, hematopoietic stem cell; KLF, Krüppel‐like factor; L, lymphoid; LIC, leukemia‐initiating cell; LSC, leukemia stem cell; M, myeloid cell; Mo, monocytes; NK, natural killer; SR, self‐renewal; T, T cell.
Research on the role of KLF4 in the maintenance of malignant HSCs has not been actively pursued despite mounting evidence supporting a potential function in the regulation of self‐renewal in LSCs or LICs. We recently reported that loss of KLF4 promotes activation of a kinase pathway that drives expansion of LICs in pediatric patients suffering from T‐ALL, a lethal blood cancer. Despite significant improvements in the management of children with T‐ALL through risk‐adaptive therapy, central nervous system‐directed chemotherapy, and supportive care, relapse patients have a poor prognosis, and the rate of complete remission drops significantly in each marrow relapse 90. The chemoresistance capacity of LICs in addition to their ability to self‐renew and induce relapses has made this rare population a target of multiple studies aimed at the development of targeted therapy that is not available yet for T‐ALL or any other leukemia 91, 92, 93, 94, 95. Inactivation of KLF4 by somatic gene deletion in mouse models accelerates the development of NOTCH1‐induced leukemia by enhancing the G1‐to‐S transition in leukemic cells and promoting the expansion of LICs (Fig. 3) 96. An analysis of global gene expression and genome‐wide binding of KLF4 in murine T‐ALL cells revealed that KLF4 represses the gene encoding the dual‐specificity mitogen‐activated protein kinase kinase 7 (MAP2K7) (Fig. 3). In contrast to ESCs, a network of KLF proteins was not identified in T‐ALL LICs. MAP2K7 is part of a three‐tiered signaling unit consisting of upstream MAP3K and downstream MAPK, held together by a scaffolding protein known as c‐Jun N‐terminal kinases (JNK)‐interacting protein, and although the kinase upstream of MAP2K7 has not been identified, particularly in leukemic T cells, the only known downstream substrate of MAP2K7 is JNK 97. JNK activation in turn phosphorylates the final effectors of this pathway, ATF2 and c‐Jun, which are involved in the control of cellular proliferation and believed to fuel expansion of LICs. Strikingly, patients exhibit low levels of KLF4 that was associated with hypermethylation of the KLF4 promoter and aberrant activation of the MAP2K7 pathway, similar to T‐ALL mice deficient in KLF4, because normally KLF4 represses the MAP2K7 gene 96. The inactivation of KLF4 across pediatric T‐ALL patients and the frequency of NOTCH1 mutations (approximately 50%) suggest that MAP2K7 activation is likely independent of the driver mutation used to induce T‐ALL in mice. The fact that deletion of KLF4 results in upregulation of both total and phosphorylated MAP2K7 led to the hypothesis that either leukemic cells exhibit basal activation of this pathway or KLF4 somehow additionally represses MAP2K7 activation. Because KLF4‐deficient T‐ALL mice show an increased frequency of LICs, defined as CD4− CD8− CD25+ IL7Rα+ and CD25+ c‐Myc+ leukemic cells, which was confirmed by limiting‐dose transplantation of leukemic bone marrow cells, it is possible that inhibition of the MAP2K7 pathway can target the LIC population in high‐risk T‐ALL patients. As a proof of concept, pharmacological inhibition of JNK in T‐ALL cells with the CC401 and AS602801 compounds showed significant reduction in the leukemia burden in cell line‐based xenograft and patient‐derived xenograft models 96. However, JNK inhibitors show low potency, with antileukemic properties in the micromolar range, and thus one current focus is on the identification of more potent and specific MAP2K7 inhibitors.
Concluding Remarks
Despite a vast literature describing the role of KLF4 in the self‐renewal of ESCs, the function of KLF4 in normal and malignant HSCs is less well‐known. This review provides evidence that KLF4 regulates hematopoiesis and self‐renewal of T‐ALL LICs through repression of the MAP2K7 pathway and does not involve a KLF circuitry as in ESCs or direct regulation of the cell cycle by KLF4 as seen in solid tumors. A broader knowledge of KLF4 function in blood stem/progenitor cells is necessary to understand the leukemogenic process and how leukemia is driven during treatment and relapses.
Author Contributions
C.S.P., A.L., T.C.: manuscript writing, table preparation; D.L.: conception/design, figure design, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Acknowledgments
We apologize to those authors whose work we have not cited due to space constrains. We thank Karen Prince for the preparation of figures. This work was supported by grants from the National Cancer Institute to D.L. (RO1 CA207086‐01), the NIGMS T32 to T.C. (GM008231), and the Baylor College of Medicine Comprehensive Cancer Training Program from the Cancer Prevention and Research Institute of Texas to A.L. (RP160283).
References
- 1. Jiang J, Chan YS, Loh YH et al. A core Klf circuitry regulates self‐renewal of embryonic stem cells. Nat Cell Biol 2008;10:353–360. [DOI] [PubMed] [Google Scholar]
- 2. Jeon H, Waku T, Azami T et al. Comprehensive identification of Kruppel‐like factor family members contributing to the self‐renewal of mouse embryonic stem cells and cellular reprogramming. PLoS One 2016;11:e0150715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang H, Zhou Y, Yu D et al. Klf2 contributes to the stemness and self‐renewal of human bone marrow stromal cells. Cytotechnology 2016;68:839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schuetz A, Nana D, Rose C et al. The structure of the Klf4 DNA‐binding domain links to self‐renewal and macrophage differentiation. Cell Mol Life Sci 2011;68:3121–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhao T, Liu C, Chen L. Roles of Klf5 acetylation in the self‐renewal and the differentiation of mouse embryonic stem cells. PLoS One 2015;10:e0138168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kotkamp K, Mossner R, Allen A et al. A Pou5f1/Oct4 dependent Klf2a, Klf2b, and Klf17 regulatory sub‐network contributes to EVL and ectoderm development during zebrafish embryogenesis. Dev Biol 2014;385:433–447. [DOI] [PubMed] [Google Scholar]
- 7. Pang CJ, Lemsaddek W, Alhashem YN et al. Kruppel‐like factor 1 (KLF1), KLF2, and Myc control a regulatory network essential for embryonic erythropoiesis. Mol Cell Biol 2012;32:2628–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghaleb AM, Yang VW. Kruppel‐like factor 4 (KLF4): What we currently know. Gene 2017;611:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Swamynathan SK. Kruppel‐like factors: Three fingers in control. Hum Genomics 2010;4:263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663–676. [DOI] [PubMed] [Google Scholar]
- 11. Yamane M, Ohtsuka S, Matsuura K et al. Overlapping functions of Kruppel‐like factor family members: Targeting multiple transcription factors to maintain the naive pluripotency of mouse embryonic stem cells. Development 2018;145:dev162404. [DOI] [PubMed] [Google Scholar]
- 12. Wei Z, Gao F, Kim S et al. Klf4 organizes long‐range chromosomal interactions with the oct4 locus in reprogramming and pluripotency. Cell Stem Cell 2013;13:36–47. [DOI] [PubMed] [Google Scholar]
- 13. Kim MO, Kim SH, Cho YY et al. ERK1 and ERK2 regulate embryonic stem cell self‐renewal through phosphorylation of Klf4. Nat Struct Mol Biol 2012;19:283–290. [DOI] [PubMed] [Google Scholar]
- 14. Li Y, McClintick J, Zhong L et al. Murine embryonic stem cell differentiation is promoted by SOCS‐3 and inhibited by the zinc finger transcription factor Klf4. Blood 2005;105:635–637. [DOI] [PubMed] [Google Scholar]
- 15. Chan KK, Zhang J, Chia NY et al. KLF4 and PBX1 directly regulate NANOG expression in human embryonic stem cells. Stem Cells 2009;27:2114–2125. [DOI] [PubMed] [Google Scholar]
- 16. Zhang P, Andrianakos R, Yang Y et al. Kruppel‐like factor 4 (Klf4) prevents embryonic stem (ES) cell differentiation by regulating Nanog gene expression. J Biol Chem 2010;285:9180–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wong CW, Hou PS, Tseng SF et al. Kruppel‐like transcription factor 4 contributes to maintenance of telomerase activity in stem cells. Stem Cells 2010;28:1510–1517. [DOI] [PubMed] [Google Scholar]
- 18. Aksoy I, Giudice V, Delahaye E et al. Klf4 and Klf5 differentially inhibit mesoderm and endoderm differentiation in embryonic stem cells. Nat Commun 2014;5:3719. [DOI] [PubMed] [Google Scholar]
- 19. Yu F, Li J, Chen H et al. Kruppel‐like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene 2011;30:2161–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan Y, Li Z, Kong X et al. KLF4‐mediated suppression of CD44 signaling negatively impacts pancreatic cancer stemness and metastasis. Cancer Res 2016;76:2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu T, Chen X, Zhang W et al. KLF4 regulates adult lung tumor‐initiating cells and represses K‐Ras‐mediated lung cancer. Cell Death Differ 2016;23:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li X, Zhao Z, Zhang X et al. Klf4 reduces stemness phenotype, triggers mesenchymal‐epithelial transition (MET)‐like molecular changes, and prevents tumor progression in nasopharygeal carcinoma. Oncotarget 2017;8:93924–93941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu XY, Wang L, Luan SH et al. The PGI‐KLF4 pathway regulates self‐renewal of glioma stem cells residing in the mesenchymal niches in human gliomas. Neoplasma 2014;61:401–410. [DOI] [PubMed] [Google Scholar]
- 24. Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981;292:154–156. [DOI] [PubMed] [Google Scholar]
- 25. Kleinman HK, Klebe RJ, Martin GR. Role of collagenous matrices in the adhesion and growth of cells. J Cell Biol 1981;88:473–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith AG, Heath JK, Donaldson DD et al. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature 1988;336:688–690. [DOI] [PubMed] [Google Scholar]
- 27. Williams RL, Hilton DJ, Pease S et al. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 1988;336:684–687. [DOI] [PubMed] [Google Scholar]
- 28. Cartwright P, McLean C, Sheppard A et al. LIF/STAT3 controls ES cell self‐renewal and pluripotency by a Myc‐dependent mechanism. Development 2005;132:885–896. [DOI] [PubMed] [Google Scholar]
- 29. Chambers I, Colby D, Robertson M et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 2003;113:643–655. [DOI] [PubMed] [Google Scholar]
- 30. Ema M, Mori D, Niwa H et al. Kruppel‐like factor 5 is essential for blastocyst development and the normal self‐renewal of mouse ESCs. Cell Stem Cell 2008;3:555–567. [DOI] [PubMed] [Google Scholar]
- 31. Masui S, Nakatake Y, Toyooka Y et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol 2007;9:625–635. [DOI] [PubMed] [Google Scholar]
- 32. Matsuda T, Nakamura T, Nakao K et al. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J 1999;18:4261–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mitsui K, Tokuzawa Y, Itoh H et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 2003;113:631–642. [DOI] [PubMed] [Google Scholar]
- 34. Nichols J, Zevnik B, Anastassiadis K et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 1998;95:379–391. [DOI] [PubMed] [Google Scholar]
- 35. Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet 1999;22:356–360. [DOI] [PubMed] [Google Scholar]
- 36. Dhaliwal NK, Miri K, Davidson S et al. KLF4 Nuclear Export Requires ERK Activation and Initiates Exit from Naive Pluripotency. Stem Cell Rep 2018;10:1308–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoffmeyer K, Raggioli A, Rudloff S et al. Wnt/beta‐catenin signaling regulates telomerase in stem cells and cancer cells. Science 2012;336:1549–1554. [DOI] [PubMed] [Google Scholar]
- 38. Sands WA, Copland M, Wheadon H. Targeting self‐renewal pathways in myeloid malignancies. Cell Commun Signal 2013;11:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Orford KW, Scadden DT. Deconstructing stem cell self‐renewal: Genetic insights into cell‐cycle regulation. Nat Rev Genet 2008;9:115–128. [DOI] [PubMed] [Google Scholar]
- 40. Goyama S, Wunderlich M, Mulloy JC. Xenograft models for normal and malignant stem cells. Blood 2015;125:2630–2640. [DOI] [PubMed] [Google Scholar]
- 41. Molofsky AV, Pardal R, Morrison SJ. Diverse mechanisms regulate stem cell self‐renewal. Curr Opin Cell Biol 2004;16:700–707. [DOI] [PubMed] [Google Scholar]
- 42. Evans PM, Chen X, Zhang W et al. KLF4 interacts with beta‐catenin/TCF4 and blocks p300/CBP recruitment by beta‐catenin. Mol Cell Biol 2010;30:372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ghaleb AM, Aggarwal G, Bialkowska AB et al. Notch inhibits expression of the Kruppel‐like factor 4 tumor suppressor in the intestinal epithelium. Mol Cancer Res 2008;6:1920–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lambertini C, Pantano S, Dotto GP. Differential control of Notch1 gene transcription by Klf4 and Sp3 transcription factors in normal versus cancer‐derived keratinocytes. PLoS One 2010;5:e10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kumano K, Kurokawa M. The role of Runx1/AML1 and Evi‐1 in the regulation of hematopoietic stem cells. J Cell Physiol 2010;222:282–285. [DOI] [PubMed] [Google Scholar]
- 46. Miyake N, Brun AC, Magnusson M et al. HOXB4‐induced self‐renewal of hematopoietic stem cells is significantly enhanced by p21 deficiency. Stem Cells 2006;24:653–661. [DOI] [PubMed] [Google Scholar]
- 47. Wilson A, Laurenti E, Trumpp A. Balancing dormant and self‐renewing hematopoietic stem cells. Curr Opin Genet Dev 2009;19:461–468. [DOI] [PubMed] [Google Scholar]
- 48. Milyavsky M, Gan OI, Trottier M et al. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis‐independent role for p53 in self‐renewal. Cell Stem Cell 2010;7:186–197. [DOI] [PubMed] [Google Scholar]
- 49. Lechman ER, Gentner B, Ng SWK et al. miR‐126 regulates distinct self‐renewal outcomes in normal and malignant hematopoietic stem cells. Cancer Cell 2016;29:602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moore MA, Dorn DC, Schuringa JJ et al. Constitutive activation of Flt3 and STAT5A enhances self‐renewal and alters differentiation of hematopoietic stem cells. Exp Hematol 2007;35:105–116. [DOI] [PubMed] [Google Scholar]
- 51. Copley MR, Babovic S, Benz C et al. The Lin28b‐let‐7‐Hmga2 axis determines the higher self‐renewal potential of fetal haematopoietic stem cells. Nat Cell Biol 2013;15:916–925. [DOI] [PubMed] [Google Scholar]
- 52. Yang J, Aguila JR, Alipio Z et al. Enhanced self‐renewal of hematopoietic stem/progenitor cells mediated by the stem cell gene Sall4. J Hematol Oncol 2011;4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu JQ, Seay M, Schulz VP et al. Tcf7 is an important regulator of the switch of self‐renewal and differentiation in a multipotential hematopoietic cell line. PLoS Genet 2012;8:e1002565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang T, Nandakumar V, Jiang XX et al. The control of hematopoietic stem cell maintenance, self‐renewal, and differentiation by Mysm1‐mediated epigenetic regulation. Blood 2013;122:2812–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zeng H, Yucel R, Kosan C et al. Transcription factor Gfi1 regulates self‐renewal and engraftment of hematopoietic stem cells. EMBO J 2004;23:4116–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wu Y, Li Y, Zhang H et al. Autophagy and mTORC1 regulate the stochastic phase of somatic cell reprogramming. Nat Cell Biol 2015;17:715–725. [DOI] [PubMed] [Google Scholar]
- 57. Yu T, Chen X, Zhang W et al. Regulation of the potential marker for intestinal cells, Bmi1, by beta‐catenin and the zinc finger protein KLF4: Implications for colon cancer. J Biol Chem 2012;287:3760–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Seipel K, Marques MT, Bozzini MA et al. Inactivation of the p53‐KLF4‐CEBPA axis in acute myeloid leukemia. Clin Cancer Res 2016;22:746–756. [DOI] [PubMed] [Google Scholar]
- 59. Ley TJ, Ding L, Walter MJ et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010;363:2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shah MY, Licht JD. DNMT3A mutations in acute myeloid leukemia. Nat Genet 2011;43:289–290. [DOI] [PubMed] [Google Scholar]
- 61. Challen GA, Sun D, Jeong M et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 2011;44:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhu X, Du J, Yu J et al. LncRNA NKILA regulates endothelium inflammation by controlling a NF‐kappaB/KLF4 positive feedback loop. J Mol Cell Cardiol 2019;126:60–69. [DOI] [PubMed] [Google Scholar]
- 63. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell 2014;14:275–291. [DOI] [PubMed] [Google Scholar]
- 64. Sardina JL, Collombet S, Tian TV et al. Transcription factors drive Tet2‐mediated enhancer demethylation to reprogram cell fate. Cell Stem Cell 2018;23:727e9–741e9. [DOI] [PubMed] [Google Scholar]
- 65. Allsopp RC, Morin GB, DePinho R et al. Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood 2003;102:517–520. [DOI] [PubMed] [Google Scholar]
- 66. Hsieh MH, Chen YT, Chen YT et al. PARP1 controls KLF4‐mediated telomerase expression in stem cells and cancer cells. Nucleic Acids Res 2017;45:10492–10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Park CS, Shen Y, Lewis A et al. Role of the reprogramming factor KLF4 in blood formation. J Leukoc Biol 2016;99:673–685. [DOI] [PubMed] [Google Scholar]
- 68. Park CS, Lee PH, Yamada T et al. Kruppel‐like factor 4 (KLF4) promotes the survival of natural killer cells and maintains the number of conventional dendritic cells in the spleen. J Leukoc Biol 2012;91:739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yamada T, Park CS, Mamonkin M et al. Transcription factor ELF4 controls the proliferation and homing of CD8+ T cells via the Kruppel‐like factors KLF4 and KLF2. Nat Immunol 2009;10:618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Alder JK, Georgantas RW 3rd, Hildreth RL et al. Kruppel‐like factor 4 is essential for inflammatory monocyte differentiation in vivo. J Immunol 2008;180:5645–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Feinberg MW, Wara AK, Cao Z et al. The Kruppel‐like factor KLF4 is a critical regulator of monocyte differentiation. EMBO J 2007;26:4138–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kurotaki D, Osato N, Nishiyama A et al. Essential role of the IRF8‐KLF4 transcription factor cascade in murine monocyte differentiation. Blood 2013;121:1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liao X, Sharma N, Kapadia F et al. Kruppel‐like factor 4 regulates macrophage polarization. J Clin Invest 2011;121:2736–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Good KL, Tangye SG. Decreased expression of Kruppel‐like factors in memory B cells induces the rapid response typical of secondary antibody responses. Proc Natl Acad Sci USA 2007;104:13420–13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Georgantas RW 3rd, Tanadve V, Malehorn M et al. Microarray and serial analysis of gene expression analyses identify known and novel transcripts overexpressed in hematopoietic stem cells. Cancer Res 2004;64:4434–4441. [DOI] [PubMed] [Google Scholar]
- 76. Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context‐dependent oncogene. Nat Cell Biol 2005;7:1074–1082. [DOI] [PubMed] [Google Scholar]
- 77. Rowland BD, Peeper DS. KLF4, p21 and context‐dependent opposing forces in cancer. Nat Rev Cancer 2006;6:11–23. [DOI] [PubMed] [Google Scholar]
- 78. Tetreault MP, Yang Y, Katz JP. Kruppel‐like factors in cancer. Nat Rev Cancer 2013;13:701–713. [DOI] [PubMed] [Google Scholar]
- 79. Zhao W, Hisamuddin IM, Nandan MO et al. Identification of Kruppel‐like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene 2004;23:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Malik D, Kaul D, Chauhan N et al. miR‐2909‐mediated regulation of KLF4: A novel molecular mechanism for differentiating between B‐cell and T‐cell pediatric acute lymphoblastic leukemias. Mol Cancer 2014;13:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yasunaga J, Taniguchi Y, Nosaka K et al. Identification of aberrantly methylated genes in association with adult T‐cell leukemia. Cancer Res 2004;64:6002–6009. [DOI] [PubMed] [Google Scholar]
- 82. Guan H, Xie L, Leithauser F et al. KLF4 is a tumor suppressor in B‐cell non‐Hodgkin lymphoma and in classic Hodgkin lymphoma. Blood 2010;116:1469–1478. [DOI] [PubMed] [Google Scholar]
- 83. Schoenhals M, Kassambara A, Veyrune JL et al. Kruppel‐like factor 4 blocks tumor cell proliferation and promotes drug resistance in multiple myeloma. Haematologica 2013;98:1442–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Faber K, Bullinger L, Ragu C et al. CDX2‐driven leukemogenesis involves KLF4 repression and deregulated PPARgamma signaling. J Clin Invest 2013;123:299–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zou Q, Tan S, Yang Z et al. The human nucleophosmin 1 mutation A inhibits myeloid differentiation of leukemia cells by modulating miR‐10b. Oncotarget 2016;7:71477–71490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bryant A, Palma CA, Jayaswal V et al. miR‐10a is aberrantly overexpressed in Nucleophosmin1 mutated acute myeloid leukaemia and its suppression induces cell death. Mol Cancer 2012;11:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ren M, Qin H, Wu Q et al. Development of ZMYM2‐FGFR1 driven AML in human CD34+ cells in immunocompromised mice. Int J Cancer 2016;139:836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Huang Y, Chen J, Lu C et al. HDAC1 and Klf4 interplay critically regulates human myeloid leukemia cell proliferation. Cell Death Dis 2014;5:e1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhao Z, Goldin L, Liu S et al. Evolution of multiple cell clones over a 29‐year period of a CLL patient. Nat Commun 2016;7:13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Schrappe M, Hunger SP, Pui CH et al. Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med 2012;366:1371–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cox CV, Martin HM, Kearns PR et al. Characterization of a progenitor cell population in childhood T‐cell acute lymphoblastic leukemia. Blood 2007;109:674–682. [DOI] [PubMed] [Google Scholar]
- 92. Chiu PP, Jiang H, Dick JE. Leukemia‐initiating cells in human T‐lymphoblastic leukemia exhibit glucocorticoid resistance. Blood 2010;116:5268–5279. [DOI] [PubMed] [Google Scholar]
- 93. Gerby B, Clappier E, Armstrong F et al. Expression of CD34 and CD7 on human T‐cell acute lymphoblastic leukemia discriminates functionally heterogeneous cell populations. Leukemia 2011;25:1249–1258. [DOI] [PubMed] [Google Scholar]
- 94. Diamanti P, Cox CV, Blair A. Comparison of childhood leukemia initiating cell populations in NOD/SCID and NSG mice. Leukemia 2012;26:376–380. [DOI] [PubMed] [Google Scholar]
- 95. Diamanti P, Cox CV, Moppett JP et al. Parthenolide eliminates leukemia‐initiating cell populations and improves survival in xenografts of childhood acute lymphoblastic leukemia. Blood 2013;121:1384–1393. [DOI] [PubMed] [Google Scholar]
- 96. Shen Y, Park CS, Suppipat K et al. Inactivation of KLF4 promotes T‐cell acute lymphoblastic leukemia and activates the MAP2K7 pathway. Leukemia 2017;31:1314–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Shen Y, Chen TJ, Lacorazza HD. Novel tumor‐suppressor function of KLF4 in pediatric T‐cell acute lymphoblastic leukemia. Exp Hematol 2017;53:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]