

Abstract
Patients with type 1 diabetes mellitus (T1DM) often suffer from osteopenia or osteoporosis. Although most agree that T1DM‐induced hyperglycemia is a risk factor for progressive bone loss, the mechanisms for the link between T1DM and bone loss still remain elusive. In this study, we found that bone marrow‐derived mesenchymal stem cells (BMSCs) isolated from T1DM donors were less inducible for osteogenesis than those from non‐T1DM donors and further identified a mechanism involving bone morphogenetic protein‐6 (BMP6) that was produced significantly less in BMSCs derived from T1DM donors than that in control cells. With addition of exogenous BMP6 in culture, osteogenesis of BMSCs from T1DM donors was restored whereas the treatment of BMP6 seemed not to affect non‐T1DM control cells. We also demonstrated that bone mineral density (BMD) was reduced in streptozotocin‐induced diabetic mice compared with that in control animals, and intraperitoneal injection of BMP6 mitigated bone loss and increased BMD in diabetic mice. Our results suggest that bone formation in T1DM patients is impaired by reduction of endogenous BMP6, and supplementation of BMP6 enhances osteogenesis of BMSCs to restore BMD in a mouse model of T1DM, which provides insight into the development of clinical treatments for T1DM‐assocaited bone loss. stem cells translational medicine 2019;8:522–534
Keywords: Type 1 diabetes mellitus, Hyperglycemia, Bone loss, Mesenchymal stem cell, Bone morphogenetic protein‐6, Osteogenesis
Significance Statement.
Patients with type 1 diabetes mellitus (T1DM) are likely to suffer from significant bone loss, which often compromises the structural integrity of bone to increase the risk of bone fracture. In this study, it has been shown that bone marrow‐derived mesenchymal stem cells isolated from T1DM donors exhibit a reduction in osteogenesis compared with those from non‐T1DM donors. This study further identified bone morphogenetic protein‐6 (BMP6) as a key molecule involved in the regulation of T1DM‐associated bone loss and successfully attenuated the bone loss in streptozotocin‐induced diabetic mice by administration of BMP6. The findings provide insight into the development of a viable treatment for T1DM‐associated bone loss.
Introduction
Type 1 diabetes mellitus (T1DM), or insulin dependent diabetes mellitus, is a disease characterized by autoimmune destruction of pancreatic β cells, resulting in insulin deficiency and hyperglycemia 1. The incidence of T1DM in the U.S. is roughly 20 in 100,000 annually with a prevalence of 800,000 patients 2, 3. Complications associated with poorly controlled T1DM include retinopathy, nephropathy, neuropathy, and cardiovascular diseases 1, 4. In addition, it has been shown that T1DM is linked with metabolic bone diseases, such as osteopenia and osteoporosis 5, 6. Studies have indicated that over half of T1DM patients also suffer from significantly reduced bone mineral density (BMD), which compromises bone integrity and increases risk of fracture 7, 8. Due to the high prevalence of T1DM, investigation into its relationship with bone loss is warranted.
Bone marrow‐derived mesenchymal stem cells (BMSCs) are multipotent cells capable of generating tissues with the mesodermal origin, including cartilage, bone, muscle, tendon, ligament, and fat 9. They play a vital role in bone formation during the process of bone remodeling in which the cell differentiates into osteoblasts to produce collagen type 1 and matrix proteins, such as osteocalcin and osteopontin, and hydroxyapatite is then deposited onto the protein matrix to form the mineralized bone structure 10, 11. Thus, medical conditions that impair BMSC activities or functions for bone formation often interrupt normal bone remodeling, causing bone loss 12, 13. In T1DM patients, bone loss can occur when osteoblastogenesis of BMSCs and osteoblast activity are hindered and/or osteoclast activity is promoted 14. Although several mechanisms have been suggested to explain T1DM‐associated bone loss 15, 16, 17, 18, 19, 20, 21, 22, recent evidence demonstrates that T1DM‐induced hyperglycemia can also attenuate the function of BMSCs in bone formation 23, 24, 25. For example, BMSCs isolated from streptozotocin (STZ)‐induced diabetic rodent exhibit decreased colony formation and cell proliferation and viability 23, 25, and the hyperglycemic condition in the BMSC niche increases levels of tissue necrosis factor‐α to induce cell apoptosis and limit fracture healing 24. These aforementioned studies have focused on effects of T1DM on bone physiology but few of them have investigated the underlying mechanism that governs osteogenesis of BMSCs affected by T1DM.
Members of the transforming growth factor‐β (TGFB) superfamily play an important role in bone and cartilage formation through regulating BMSC activities 26. We aimed to determine if the TGFB superfamily is involved in the pathogenesis of bone loss induced by T1DM by focusing on osteogenesis of BMSCs. We hypothesized that the capacity of osteogenesis and bone matrix formation of BMSCs isolated from T1DM donors is downregulated compared with that of the cell from non‐T1DM donors through regulation of the activity of TGFB superfamily members. Our experimental approach was to first compare the osteogenic capacity of BMSCs isolated from T1DM and non‐T1DM donors, identify key molecules involved in BMSC osteogenesis regulated by the disease, and evaluate bone formation in STZ‐induced diabetic mice treated with the identified molecules.
Materials and Methods
Isolation and Culture of BMSCs
Harvesting human bone marrow for BMSC isolation was approved by the Institutional Review Board at the University of Wisconsin‐Madison. BMSCs were isolated from the femoral head and neck of six patients (three T1DM and three non‐T1DM) undergoing total hip arthroplasty following our previously published protocol 27. Specifically, two female and one male donor with ages ranging from 45 to 65 were included for each group and none of them had an end‐stage renal disease. Donors of the diabetic cohort had received active treatment with diabetic medications to keep HbA1c values equal to or less than 7.5 at the time of surgery. The harvested cells were seeded in T75 tissue culture flasks containing medium composed of low‐glucose (1 g/ml) Dulbecco's modified Eagle's medium (DMEM), 10% fetal bovine serum (FBS; Atlanta Biologicals, Atlanta, GA), and 1% penicillin and streptomycin, and maintained at 37°C in a humidified 5% CO2 atmosphere. The culture medium was replaced every 3 days, and upon 70%–80% confluence, the cells were dissociated and passaged using 0.05% trypsin/EDTA (Life Technologies, Carlsbad, CA). The BMSCs were subsequently replated at a seeding density of 1,000 cells per cm2. Cells between passages 2 and 4 were used in this study.
Immunophenotypic Characterization of BMSCs
To characterize immunophenotypes, BMSCs isolated from T1DM and non‐T1DM donors were analyzed by the Attune NxT flow cytometer (Thermo Fisher Scientific, Waltham, MA). Briefly, cells were first trypsinized, washed three times with ice‐cold phosphate‐buffered saline (PBS) containing 1% bovine serum albumin and 5 mM EDTA, treated with conjugated phycoerythrin mouse anti‐human CD73, allophycocyanin mouse anti‐human CD90, fluorescein mouse anti‐human CD105, and PerCP‐CY5.5 mouse anti‐human CD45 (BD Biosciences, East Rutherford, NJ), and then incubated for 30 minutes at 4°C. After unbound antibodies were removed, cells were analyzed by flow cytometry. Data of flow cytometry were analyzed using FlowJo (TreeStar, OR). The Fluoresence Minus One method was used as a control to set up the gate.
Osteogenic Differentiation of BMSCs
BMSCs were induced for osteogenesis following our previously published protocol 28. Briefly, cells were trypsinized, seeded at a density of 5,000 cells per cm2, and induced for osteogenesis by medium composed of low‐glucose DMEM, 10% FBS, 1% penicillin and streptomycin, 10 mM β‐glycerophosphate, 50 μg/ml μM l‐ascorbic acid‐2‐phosphate, 0.1 μM dexamethasone, and 10 nM 1α,25‐dihydroxyvitamin D3 (Sigma–Aldrich, St. Louis, MO). The induction medium was changed every 3 days.
To determine if TGFB and/or bone morphogenetic protein (BMP) signaling involved in the regulation of osteogenesis of BMSCs are affected by diabetes, the cell from T1DM (T1DM‐BMSCs) and non‐T1DM donors (non‐T1DM‐BMSCs) were treated with 5 μM of dorsomorphin, an inhibitor of BMP type 1 receptor kinase, and/or 5 μM of SB431542, an inhibitor of TGFB receptor kinase (Abcam, Cambridge, MA) during 21‐day osteogenic differentiation.
Cytochemistry Analysis
Cells induced for osteogenesis were fixed in 60% isopropanol and stained with alizarin red (Rowley Biochemical, Danvers, MA) for evaluation of mineral deposition. The matrix calcium content was extracted with 0.5 M hydrochloric acid and quantified using the LiquiColor kit (Stanbio, Boerne, TX) following the manufacturer's instructions. Alkaline phosphatase (ALP) was also stained using an ALP staining kit (Sigma–Aldrich). The activity level of ALP in osteogenic BMSCs was determined by digesting cells with a buffer solution containing 2% Triton X‐100, 1.5 mM Tris base, 1 mM ZnCl2 and 1 mM MgCl2·6H2O at pH 9.0 for 30 minutes at 37°C and measuring the digestion solution at the adsorption wavelength of 405 nm after reacting with Sigma‐104 Phosphatase Substrate (Sigma–Aldrich). The results were subsequently normalized to the cell number determined by the total DNA content quantified by the Quant‐iT PicoGreen dsDNA assay kit (Invitrogen, Carlsbad, CA), following the manufacturer's instructions.
Total RNA Extraction and Quantitative Reverse Transcriptase‐Polymerase Chain Reaction
Total RNA was extracted from cells using the Nucleo Spin RNA II kit (Clontech, Mountain View, CA) as per the manufacturer's instructions. The quantity and quality of RNA was determined using NanoDrop 1000 (Thermo Fisher Scientific). Complementary DNA prepared using the High Capacity cDNA Reverse Transcription kit (Applied Biosystem, Carlsbad, CA) was mixed with primers and the iQ SYBR Green Premix (Bio‐Rad, Hercules, CA) for quantitative reverse transcriptase‐polymerase chain reaction (qRT‐PCR) to detect the expression of mRNA transcripts. The primer sequences are listed in Supporting Information Table S1. Levels of relative mRNA expression were calculated using the 2−ΔCt method by referencing to those of the internal control ubiquitin.
Protein Isolation and Western Blotting
Cells were lysed using RIPA buffer composed of 50 mM Tris–HCl (pH 7.5), 0.25% Na‐deoxycholate, 1% Nonidet P‐40, 150 mM NaCl, 1 mM EDTA, and complete protease inhibitor cocktail (Roche, Indianapolis, IN). After centrifugation at 14,000 rpm for 10 minutes, total protein from the supernatant was collected and measured using the BCA protein assay kit (Pierce, Rockford, IL). The amount of 20 μg of protein sample was loaded on a 7.5% polyacrylamide gel (Bio‐Rad) for electrophoresis and then transferred onto a polyvinylidene fluoride membrane (Bio‐Rad). The membrane was incubated with blocking solution composed of 5% nonfat milk (Bio‐Rad) and 0.1% Tween 20 (Sigma–Aldrich) in Tris‐buffered saline overnight at 4°C with primary antibody against glyceraldehyde 3‐phosphate dehydrogenase (GAPDH), BMPR2, TGFBR2, SMAD3, pSMAD3, SMAD1, or pSMAD1/5 (Cell Signaling, Danvers, MA) before incubated with horseradish peroxidase‐linked secondary antibody (Cell Signaling) for 1 hour, detected by SuperSignal West Pico Chemiluminescent Substrate (Pierce), and imaged using the Kodak image station 4000R Pro (Kodak, Rochester, NY).
Quantification and Modulation of BMP6 Levels in Cell Culture Medium or Mouse Serum
The conditioned medium of BMSC culture was collected 10 and 21 days after induction of osteogenesis and then stored in a −20°C freezer before analysis. To collect serum from mice, blood was drawn from the submandibular vein using 4‐mm lancets (Fisher Scientific, Hampton, NH) and centrifuged in Vacutainer blood collection tubes (BD Biosciences) at 2,000g for 10 minutes. The supernatant was then stored in a −20°C freezer before analysis. Levels of BMP6 in BMSC culture or mouse serum were measured by ELISA kits containing human‐ (Raybiotech, Norcross, GA) or mouse‐ (MyBioSource, San Diego, CA) specific antibodies following the manufacturer's instructions.
To determine if BMP6 is a target molecule of interest involved in the regulation of osteogenesis of T1DM‐ or non‐T1DM‐BMSCs, the cell in osteogenic culture was supplemented with or without 10 ng/ml of recombinant BMP6 or neutralized with or without 2 μg/ml of BMP6 antibody (R&D, Minneapolis, MN).
Treatment of BMP6 in STZ‐Induced T1DM Animal Model
All mice handling and procedures were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin‐Madison. Six‐week‐old C57BL/6J male mice (Jackson Laboratories, Bar Harbor, ME) were randomly grouped into four experimental cohorts with six animals each: STZ−/BMP6− (vehicle control), STZ−/BMP6+, STZ+/BMP6−, and STZ+/BMP6+. During the first week, animals were treated with intraperitoneal injection of either sodium‐citrate buffer (vehicle) or 50 μg of STZ (Sigma) per gram of mouse weight for 5 consecutive days. All STZ‐treated mice became diabetic (>250 mg/dl blood glucose) after 9 days of drug administration, as confirmed by the Contour glucose monitoring system (Bayer, Whippany, NJ). Five weeks after STZ injections, the mice were then treated with either PBS (vehicle) or 10 ng of BMP6 (R&D) per gram of mouse weight triweekly for 9 weeks. The dose of BMP6 for injection was predetermined through a separate study in which 0, 1, 10, and 20 ng of the molecule per gram of mouse weight were evaluated to select a minimum dose required to cause a noticeable difference in BMD as determined by dual energy x‐ray absorptiometry (DEXA) imaging. The animals were euthanized by carbon dioxide‐induced asphyxiation and their femurs and tibias were carefully harvested and fixed in 10% neutral buffered formalin for 48 hours at 4°C. The tissue was then prepared for histological analysis and quantitative x‐ray imaging (qXRI).
Microcomputed Tomography Scanning
The hind limbs of mice were longitudinally imaged using microcomputed tomography (μCT) on weeks 1 and 6 (prior to the BMP6 treatment) and weeks 9, 12, and 15 (during the BMP6 treatment) to assess bone density in response to STZ and BMP6 treatment. The imaging provided the resolution and contrast needed to visualize and measure the changes in bone microstructure. Scanning was performed using a Siemens Inveon μCT scanner with the following parameters: 80 kVp, 10‐second of exposure time, 900 μA of current, 220 rotation steps with 441 projections, 15 minutes of scanning time, bin by 2, 50 μm focal spot size, and medium magnification yielding an overall reconstructed isotropic voxel size of approximately 40 μm, and analyzed by the Inveon Acquisition Workplace software and the Inveon Research Workplace General and three‐dimensional (3D) visualization analysis software (Siemens Medical Solutions USA, Inc., Knoxville, TN).
Raw data were processed with filtered back‐projection using the high‐speed COBRA reconstruction software (Exxim Computing Corporation, Pleasanton, CA). Hounsfield Units (HU), a scalar linear attenuation coefficient, was applied to each reconstruction to permit intersubject comparisons. 3D images were segmented using a minimum pixel intensity of 300 HU (limit of soft tissue) and a maximum intensity of 3,000 HU to represent bone density 29. After defining the region of interest, which was limited by 2.6 mm to distal metaphysis, the femur was electronically sectioned via the frontal plane, exposing the head, neck, and shaft trabecular tissue. The average pixel density was calculated for the given region, and averages for trabecular thickness (Tb.Th), number (Tb.N), separation (Tb.Sp), and bone volume density (BV/TV) were determined by the analysis software package.
Dual Energy X‐Ray Absorptiometry
The femur and whole body of mice were measured at weeks 1, 6, 9, 12, and 15 using the Lunar PIXImus Mouse Densitometer (General Electric, Boston, MA). Following isoflurane anesthesia, mice were placed on a scanner bed in the prone position with the limbs and tail stretched away from the body. Scans of PIXImus were acquired and analyzed with the PIXImus software (v. 1.45 for acquisition and v. 2.10 for analysis). A femoral region of interest was analyzed to determine the composition. Additionally, the entire body was analyzed to determine the percentage of total body fat. Prior to scanning, the manufacturer‐provided phantom was scanned and the instrument was calibrated for quality control tests.
Quantitative X‐Ray Imaging
The analysis of qXRI was performed following a published protocol 30. After femurs were harvested on week 15, the surrounding soft tissues were removed. The bones were then radiographed using the high‐resolution Faxitron (Model 43855A, Hewlett Packard, Tucson, AZ) imaging with the exposure parameters set to 30 kV at 3 mA for 1.5 minutes. An aluminum material standard was placed next to a tissue specimen for calibration. The Kodak Precision Line Film LPF7 (Eastman Kodak Company, Rochester, NY) was used for imaging, and the exposed film was then magnified by a stereomicroscope (Stemi SV11, Zeiss, Dublin, CA) and digitized (Axiocam, Zeiss). Data processing was performed using ImageJ (U.S. National Institutes of Health, Bethesda, MD). Absorbing material density was determined by referencing to the aluminum standard to define the gray scale distribution. Images were then converted to those with a pseudo‐color representation, and individual femurs were analyzed to produce a pixel density histogram. Average pixel density values were determined and used for comparison between groups.
Histological Analysis of Bone
Tissue samples were collected at week 15 after euthanasia. Femurs were first fixed in 10% formalin for 48 hours and were decalcified in 10% formic acid for approximately 7 days. Decalcified samples were washed with ethanol repeatedly and then embedded in paraffin. The specimens were sliced into 8‐μm thick sections using a microtome and stained with hematoxylin and eosin (H&E) solutions (Sigma) and the Masson's trichome staining kit (Abcam).
Statistical Analysis
This study used BMSCs from six donors (three T1DM and three non‐T1DM patients) to perform in vitro quantitative assays with three biological replicates per group and three sample replicates per donor. The data were presented as mean ± SE. For in vivo assays, six animals per experimental cohort were used and the quantitative data were represented as mean ± SE. A Student's paired t test or two‐way analysis of variance with post hoc Turkey's test was performed for statistical comparison. A p‐value less than .05 was considered statistically significant.
Results
BMSCs from T1DM Donors Are Less Inducible for Osteogenesis
Immunophenotypic characterization by flow cytometry showed that 99.9% and 98.9% of the cell population isolated from non‐T1DM and T1DM donors, respectively, were CD45−/CD90+/CD73+/CD105+ BMSCs (Supporting Information Fig. S1). We then induced BMSCs isolated from non‐T1DM and T1DM donors for osteogenesis to determine their osteogenic potential. After 21 days of induction, we performed histological evaluation for ALP and matrix mineralization. The level of ALP activity detected by a biochemical assay was significantly lower in T1DM‐BMSCs than that in non‐T1DM‐BMSCs (Fig. 1A). The same trend was also shown in the result of ALP staining. Similarly, matrix mineralization was reduced in T1DM‐BMSCs, as shown by significantly lower calcium content and lighter alizarin red staining, compared with that in non‐T1DM‐BMSCs (Fig. 1B). These results suggest that the osteogenic potential of BMSCs is impaired by T1DM.
Figure 1.
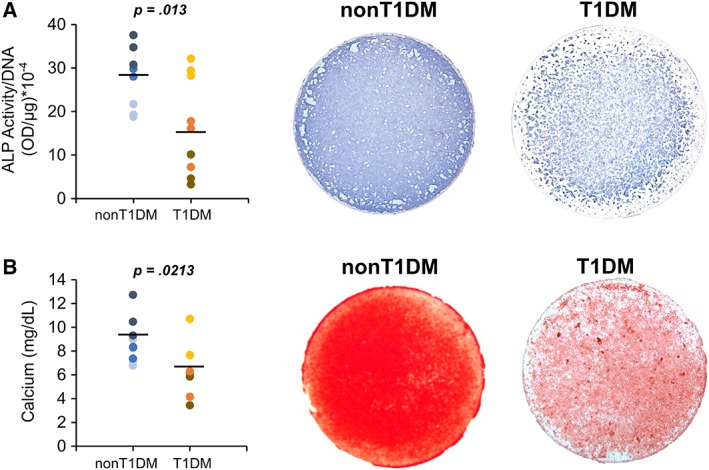
Osteogenic differentiation of bone marrow‐derived mesenchymal stem cells (BMSCs) harvested from type 1 diabetes mellitus (T1DM) and non‐T1DM donors. (A): Quantification and cytological staining of alkaline phosphatase activity of the cell after osteogenic induction. (B): Quantification of calcium content and cytological staining of alizarin red to detect matrix mineralization in BMSC culture induced for osteogenesis. n = 3 biological replicates. Each color of dots represents one donor.
Production of TGFBs and BMPs Are Differentially Regulated Between T1DM‐BMSCs and Non‐T1DM‐BMSCs
The TGFB superfamily is essential to osteogenesis and plays a critical role in the regulation of bone metabolism 31. We examined the expression of endogenous TGFB1, 2, and 3 and BMP2, 4, 6, and 7 in T1DM‐ and non‐T1DM‐BMSCs 7 days after induction of osteogenesis. The results of RT‐PCR showed that there was no significant difference in the levels of BMP2, 4, 7 and TGFB1 and 2 over the induction period between both cells whereas the levels of BMP6 and TGFB3 were consistently lower during the period of osteogenesis in T1DM‐BMSCs than those in non‐T1DM controls (Fig. 2A). To exclude the possibility that the glucose concentration of culture medium plays a role in dictating BMSC response in this assay, T1DM‐ and non‐T1DM‐BMSCs were also cultured in high‐ and low‐glucose medium, respectively, in a separate experiment and then collected for the same analysis. The results showed similar trends in the mRNA expression levels of all TGFBs and BMPs except BMP2 (Supporting Information Fig. S2), which along with the results of Figure 2A demonstrated that the response of T1DM‐ and non‐T1DM‐BMSCs was not affected by the glucose concentration of medium in culture. Taken together, our results suggest that a reduction in TGF‐β3 and/or BMP6 in T1DM‐BMSCs may be associated with downregulation of osteogenesis in the cell.
Figure 2.
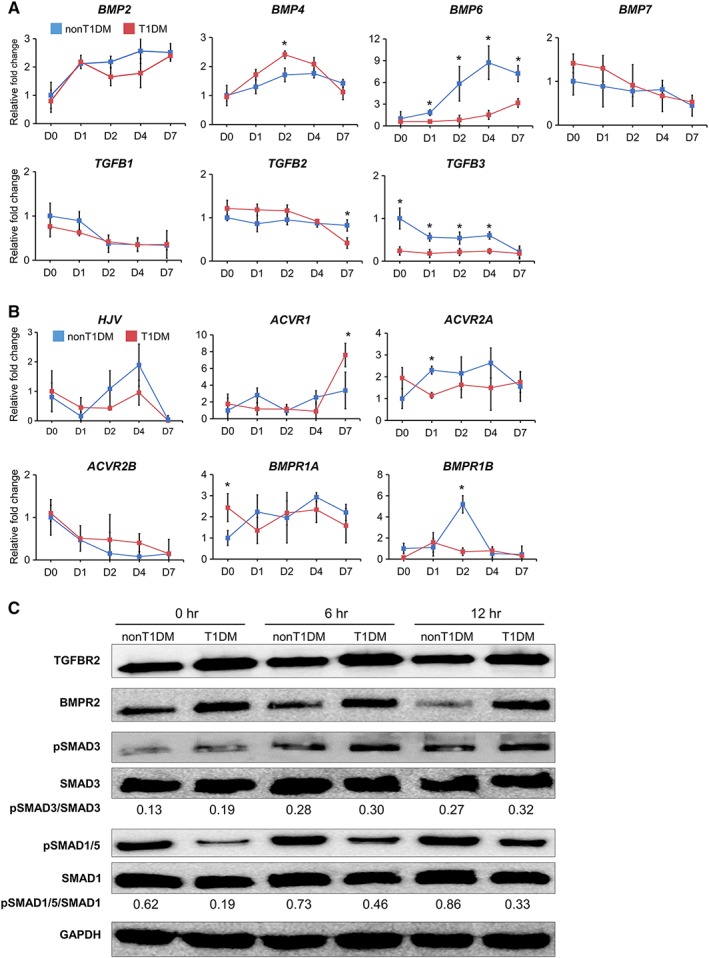
Transcript or protein expression of endogenous growth factors and their receptors and downstream signaling molecules of type 1 diabetes mellitus (T1DM)‐ and non‐T1DM‐bone marrow‐derived mesenchymal stem cells (BMSCs). (A): Expression levels of BMPs and TGFBs in T1DM‐BMSCs during osteogenic induction were compared with those in non‐T1DM‐BMSCs. (B): Expression levels of transcripts of BMP6 receptors HJV, ACVR1, ACVR2A, ACVR2B, BMPR1A, and BMPR1B in T1DM‐BMSCs during osteogenic induction were compared with those in non‐T1DM‐BMSCs. (C): Expression levels of BMPR2 and TGFBR2 and their downstream signaling SMADs and phospho‐SMADs in T1DM‐BMSCs during osteogenic induction were compared with those in non‐T1DM‐BMSCs. *p < .05; n = 3 technical replicates.
In addition to the analysis of expression of the TGFB superfamily ligands, we also assayed the expression of BMP, TGFB, and activin receptors. The results of RT‐PCR indicated that there was either no significant or transient difference in expression levels and trends of HJV, ACVR1, ACVR2A, ACVR2B, BMPR1A, and BMPR1B between non‐T1DM and T1DM‐BMSCs over a period of 7‐day culture (Fig. 2B). We then focused our analysis on the expression levels of TGFB receptor type 2 (TGFBR2) and BMP receptor type 2 (BMPR2), membrane receptors for TGFB3 and BMP6, respectively, on BMSCs during osteogenesis 32. Interestingly, Western blot results showed reduced levels of TGFBR2 and BMPR2 in non‐T1DM‐BMSCs than those in T1DM‐BMSCs (Fig. 2C). To further determine activation levels of their downstream signaling pathways, we measured the expression of SMAD3, phosphorylated SMAD3, SMAD1, and phosphorylated SMAD1/5 33, 34. Although the total protein levels of SMAD3 and SMAD1/5 were comparable between T1DM and non‐T1DM‐BMSCs up to 12 hours after osteogenic induction, there were greater amounts of phosphorylated SMAD1/5 in non‐T1DM‐BMSCs than those in T1DM‐BMSCs, and no noticeable difference in the levels of phosphorylated SMAD3 between the two groups was observed. The results suggest that during osteogenesis T1DM‐BMSCs undergo decreased activation of BMP6/SMAD1/5 signaling, despite an increased level of the membrane receptor, compared with non‐T1DM‐BMSCs.
BMP6 Plays a Critical Role in Regulating Osteogenesis of T1DM‐BMSCs
Since the expression levels of TGFB3 and BMP6 during osteogenesis were significantly different between T1DM‐ and non‐T1DM‐BMSCs, we would then like to confirm their roles in the regulation by treating the cell with SB431542 to inhibit TGFB receptors, dorsomorphin to inhibit BMP receptors 35, 36, or both during 21‐day osteogenic induction and evaluating their ALP activity and matrix mineralization. Consistent with the results of Figure 1A, 1B, reduced ALP activity and matrix mineralization, as shown by significantly lower expression levels and less intensity of cytological staining of bone‐associated markers, were found in the culture with T1DM‐BMSCs compared with those with control cells (Fig. 3A, 3B). The ALP activity and calcium content of T1DM‐ or non‐T1DM‐BMSCs treated with dorsomorphin or dorsomorphin/SB431542 were significantly decreased compared with those of cells of the control group whereas the ALP activity and calcium content were significantly reduced only in non‐T1DM‐BMSCs but not in T1DM‐BMSCs treated with SB431542 compared with those in cells of the control group. Notably, the reduction through blocking TGFB receptors was much less than that through blocking BMP receptors in both T1DM‐ and non‐T1DM‐BMSCs. The treatment of SB431542 and dorsomorphin combination did not result in a further reduction in the ALP activity and calcium content of T1DM‐ or non‐T1DM‐BMSCs compared with the treatment of dorsomorphin alone, suggesting that BMP signaling is more dominant than TGFB signaling in regulating osteogenesis of both T1DM‐ and non‐T1DM‐BMSCs.
Figure 3.
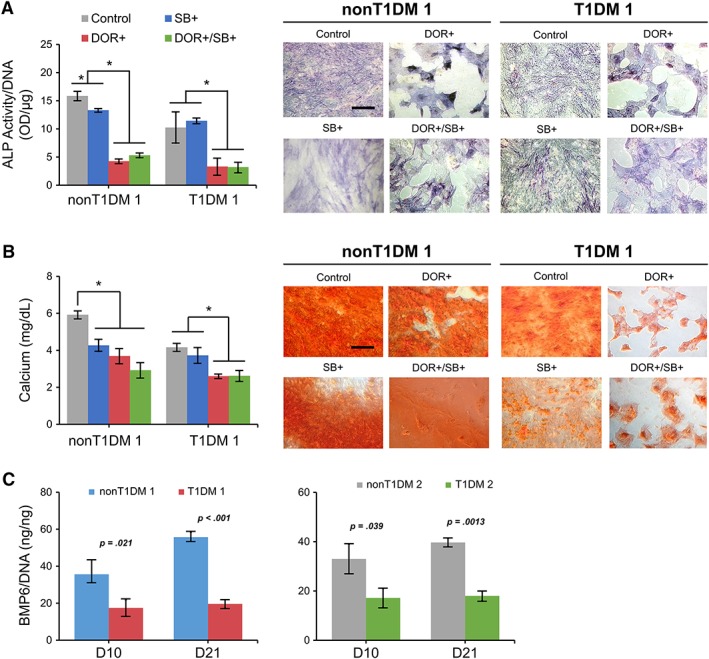
Osteogenesis of type 1 diabetes mellitus (T1DM)‐ and non‐T1DM‐bone marrow‐derived mesenchymal stem cells (BMSCs) modulated by receptor antagonists. (A): Quantification and cytological staining of alkaline phosphatase activity of the cell treated with dorsomorphin (DOR), a bone morphogenetic protein receptor antagonist, and/or SB431542 (SB), a transforming growth factor‐β receptor antagonist during osteogenesis. (B): Quantification of calcium content and cytological staining of alizarin red to detect matrix mineralization in BMSC culture treated with or without DOR and/or SB during osteogenic induction. (C): Amounts of soluble BMP6 in T1DM‐ and non‐T1DM‐BMSC culture. Concentration levels of BMP6 released from the cell in the cultured medium detected by ELISA during osteogenesis induction. *p < .05; n = 3 biological replicates, values represented as mean ± SEM. Scale bar: 100 μm.
Therefore, we focused on investigating whether BMP6 is involved in the mechanism through which osteogenesis of T1DM‐BMSCs is downregulated. The results of ELISA analysis showed that in osteogenic culture the amount of BMP6 produced by T1DM‐BMSCs was significantly less than that produced by non‐T1DM‐BMSCs at days 10 and 21 (Fig. 3C), which are consistent with the results of RT‐PCR shown in Figure 2A. To further determine the role of BMP6 in the regulation of osteogenesis of T1DM‐BMSCs, cells were treated with or without recombinant BMP6 during osteogenic induction. Without BMP6 treatment, the ALP staining intensity and activity level in T1DM‐BMSCs were less than those in non‐T1DM‐BMSCs but with the induction of BMP6 the trend was reversed: the ALP activity level in T1DM‐BMSCs treated with BMP6 was significantly higher than that in the cell without BMP6 (Fig. 4A). The same trends were also observed in results of matrix mineralization staining and total calcium content. Additionally, the analysis of mRNA expression of osteopontin (OPN), a bone‐associated marker, also showed consistent results while that of core‐binding factor α 1 (CBFA1), a bone transcription factor, revealed no difference between T1DM‐BMSCs treated with and without BMP6 (Fig. 4B). Notably, there was no significant difference in levels of ALP activity, calcium content, and CBFA1 and OPN expression between non‐T1DM‐BMSCs treated with and without BMP6. These results suggest that unlike non‐T1DM‐BMSCs, T1DM‐BMSCs are unable to produce an adequate amount of endogenous BMP6 so the addition of exogenous BMP6 replenishes the BMP6 shortage in culture for osteogenic induction; on the other hand, the addition of exogenous BMP6 makes no significant difference in non‐T1DM‐BMSC culture with already‐adequate levels of endogenous BMP6.
Figure 4.
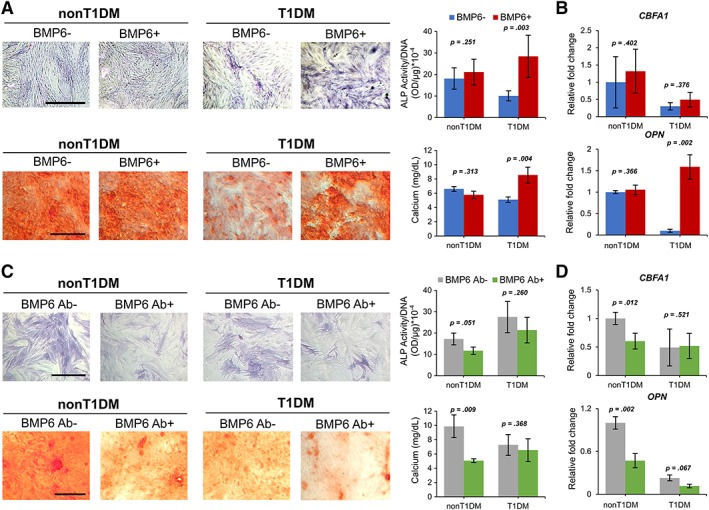
Osteogenesis of type 1 diabetes mellitus (T1DM)‐ and non‐T1DM‐bone marrow‐derived mesenchymal stem cells (BMSCs) treated with or without bone morphogenetic protein‐6 (BMP6) or BMP6‐neutralizing antibody. (A): Cytological staining and quantification of corresponding alkaline phosphatase (ALP) activity and calcium content. (B): Expression levels of CBFA1 and OPN transcripts. (C): Cytological staining and quantification of corresponding ALP activity and calcium content. (D): Expression levels of CBFA1 and OPN transcripts. n = 3 biological replicates, values represented as mean ± SEM. Scale bar: 100 μm.
We then further confirmed the role of BMP6 in regulating osteogenesis of T1DM‐BMSCs by blocking the activity of endogenous BMP6 released from BMSCs in culture during osteogenesis. The treatment of BMP6‐neutralizing antibody decreased ALP activity and calcium deposition in non‐T1DM‐BMSCs but did not affect those in T1DM‐BMSCs (Fig. 4C). Consistent results were also found in the analysis of mRNA expression of CBFA1 and OPN in non‐T1DM‐ and T1DM‐BMSCs treated with or without BMP6‐neutralizing antibody (Fig. 4D). These results again imply that in non‐T1DM‐BMSC culture, an adequate amount of endogenous BMP6 is produced for osteogenic induction so knocking down BMP6 levels in culture decreases osteogenesis of the cell whereas in T1DM‐BMSC culture BMP6‐neutralizing antibody cannot further reduce an already‐low level of BMP6 to downregulate osteogenesis.
Supplementation of BMP6 Attenuates Reduction of Bone Density in STZ‐Induced Diabetic Mice
To explore the therapeutic potential of BMP6 for T1DM‐associated reduction in bone density, an in vivo study with a T1DM animal model established by treating C57BL/6J mice with STZ was performed (Fig. 5A). Hyperglycemia induced by the compound STZ that selectively damages insulin‐secreting pancreatic β cells in mice was confirmed with the level of blood glucose exceeding 250 mg/dl during the entire course of the in vivo study, including the periods with and without BMP6 treatment (Supporting Information Fig. S3A). In this study, there were four groups, STZ−/BMP6−, STZ+/BMP6−, STZ−/BMP6+, and STZ+/BMP6+, evaluated with six mice per group. Levels of BMP6 in the serum of STZ+/BMP6− mice detected by ELISA were significantly lower than those in the serum of STZ−/BMP6− mice 8 weeks after the final STZ injection (Fig. 5B), consistent with the in vitro results of Figure 3C showing that T1DM‐BMSCs secreted less BMP6 than non‐T1DM‐BMSCs. These results suggest that diabetic mice produced less BMP6 than nondiabetic mice. Additionally, 5 weeks after STZ treatment, the mice began to show a significant decrease in BMD in the femur as determined by DEXA compared with control mice without STZ treatment (Fig. 5C). During the 9‐week treatment course with or without BMP6 injection, STZ−/BMP6− and STZ−/BMP6+ mice showed no significant difference in their BMD whereas BMD of STZ+/BMP6+ mice was significantly greater than that of STZ+/BMP6− mice, suggesting that BMP6 treatment is capable of attenuating the reduction of BMD induced by hyperglycemia. Notably, the results demonstrating that BMP6 improved BMD of diabetic mice but did not affect that of nondiabetic mice are in agreement with the results of osteogenesis of BMSCs in response to BMP6 induction in Figure 4A, 4B. In addition to lower BMD, it was also found that STZ+/BMP6− mice weighed significantly less and exhibits reduced lean and fat mass (Supporting Information Fig. S3B). Interestingly, treatment with BMP6 reduced the magnitudes of weight loss associated with STZ‐induced diabetes.
Figure 5.
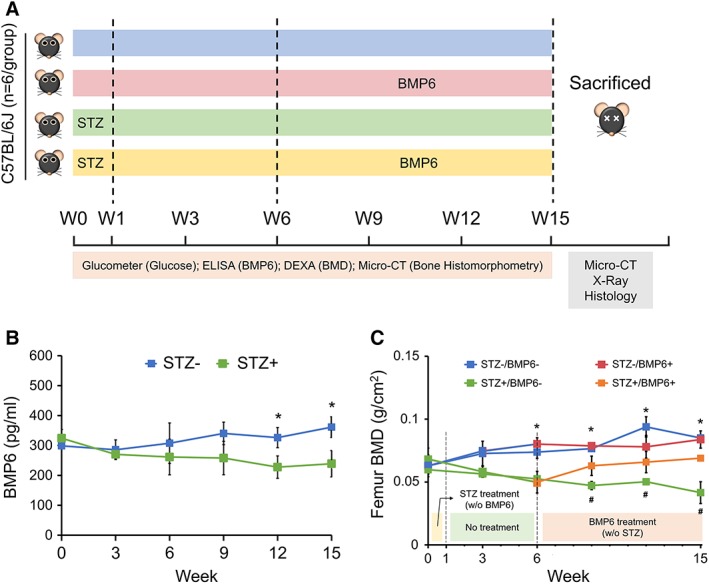
Treatment of bone loss in streptozotocin (STZ)‐induced diabetic mice by bone morphogenetic protein‐6 (BMP6). (A): Experimental design illustrating time points for inducing diabetes by STZ, establishing bone loss, and treating bone loss by BMP6 administration in the animal model as well as for performing a number of analyses. (B): Serum levels of BMP6 in animals with or without STZ induction determined by ELISA. (C): Bone mineral density in femur of mice induced with or without STZ and then treated with or without BMP6. *p < .05; n = 6 biological replicates, values represented as mean ± SEM.
After 9 weeks of BMP6 treatment, femurs of the mice were collected for structural and histochemical analyses. Cross‐sectional and regional images of μCT scanning are shown in Figure 6A. The trabecular bone density of STZ+/BMP6− mice was obviously reduced compared with that of control STZ−/BMP6− mice. However, with the administration of BMP6, the reduction of bone density in STZ‐induced diabetic mice was attenuated; STZ+/BMP6+ mice showed comparable levels of bone density to control STZ−/BMP6− mice. Quantitative analysis of these images to determine the structural property of bone revealed that values of BV/TV and Tb.Th were lesser but values of Tb.Sp were greater in STZ+/BMP6− mice than those in control STZ−/BMP6− mice (Fig. 6A); no difference were observed in trabecular number (Tb.N). With the injection of BMP6, values of BV/TV and Tb.Th of STZ‐induced diabetic mice were significantly increased whereas those of the Tb.Sp were significantly decreased to the level similar to those of control STZ−/BMP6− and STZ−/BMP6+ mice (Fig. 6A). Quantitative x‐ray imaging also showed consistent findings that the femoral bone density of STZ+/BMP6− mice was significantly lower than that of STZ−/BMP6− or STZ+/BMP6+ mice (Fig. 6B). Histological staining of H&E revealed that a reduction in trabecular bone thickness was evident in femurs of STZ+/BMP6− mice compared with those of control STZ−/BMP6− mice but the reduction was attenuated by BMP6 treatment (Fig. 7). Furthermore, the amount and distribution of collagen fibers in bone of STZ+/BMP6− mice detected by Masson's trichrome were much less and limited than that in bone of STZ−/BMP6− and STZ+/BMP6+ mice.
Figure 6.
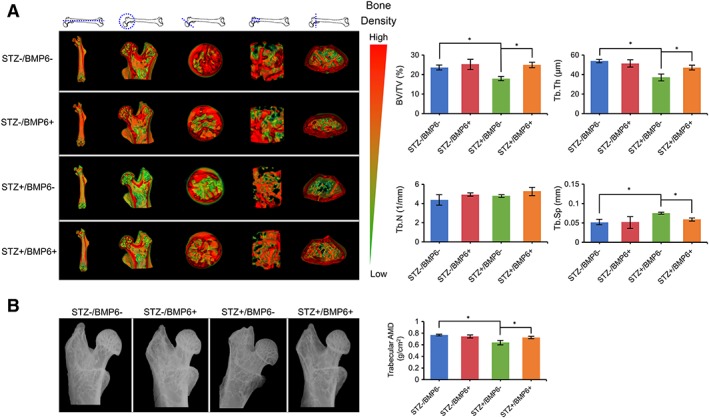
Structural and bone density analyses of mouse femurs. (A): Microcomputed tomography‐generated three‐dimensional images with pseudo color representing bone density taken from cross‐sections of different regions of the bone. (B): Bone histomorphometric analysis with quantification of bone volume density, trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular separation (Tb.Sp) of femurs between different experimental groups. (C): Radiographic imaging and quantification of absorptive material density of the femoral head and shaft. *p < .05; n = 6 biological replicates, values represented as mean ± SEM.
Figure 7.
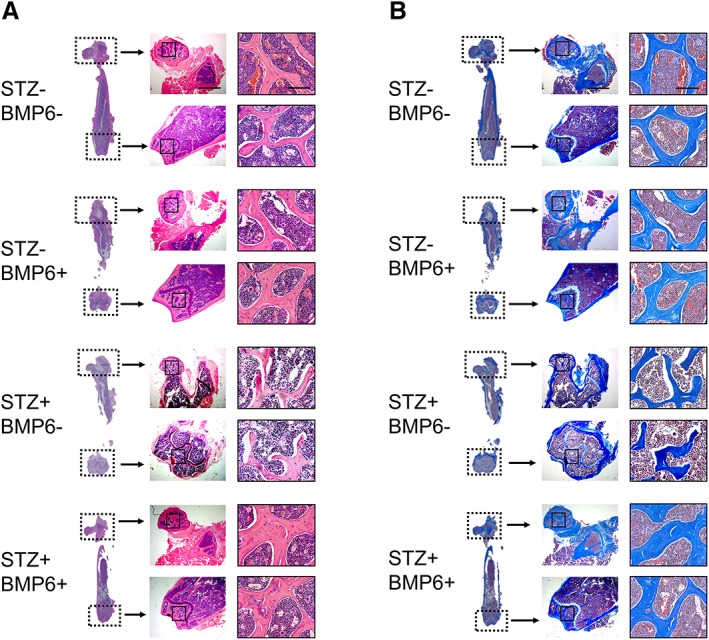
Histological analysis of mouse femurs. Trabecular bone in the metaphyseal and epiphyseal regions of the proximal or distal femurs detected by hematoxylin and eosin staining (A) and Masson's trichrome staining (B). Images in the right column are those of respective boxed areas in the central column at a higher magnification; images in the central column are those of respective boxed areas in the left column at a higher magnification. Scale bar: 1 mm or 100 μm.
Discussion
Alteration of bone structure and quality leading to bone fracture is a complication often seen in diabetic patients 37, 38. Although both T1DM and T2DM have been shown to affect bone quality in different ways with T1DM decreasing and T2DM increasing BMD, both types of diabetes result in an increased risk of bone fracture 7, 39. Studies have shown that T2DM patients tend to have greater BMD but are approximately 1.7‐fold more likely to suffer from bone fracture than their age‐ and sex‐matched controls 40. On the other hand, T1DM patients prone to fracturing their bone tend to have lower BMD than non‐T1DM ones 8, 17. In this study, our finding showing a significant reduction in BMD of STZ‐induced T1DM animals is consistent with those previously published 41, 42.
Several studies have suggested that hyperglycemia affects the biology and function of BMSCs. Specifically, hyperglycemia reduces proliferation and increases cellular senescence and apoptosis of BMSCs in vitro 43, 44. Studies have also shown that diabetes‐induced hyperglycemia inhibits osteoblast activities but promotes adipocyte activities 45, 46. Moreover, it has been reported that T1DM affects lineage‐specific differentiation of BMSCs to modulate bone quality 47, 48. Specifically, the disease decreases osteoblastogenesis but increases adipogenesis of the cell, resulting in formation of less bone and more fat, suggesting that T1DM may affect fate commitment of BMSCs. In this study, we have demonstrated that bone loss in STZ‐induced T1DM animals is accompanied by reduced total weight and body fat mass (Supporting Information Fig. S3), consistent with previously published findings 49, 50. This may be due to increased catabolism of non‐sugar molecules, such as proteins in muscle and fats, for energy consumption, induced by STZ‐induced hyperglycemia in the model. Taken together, these findings suggest that in STZ‐induced T1DM mice breakdown of fats outpaces increased production of the molecules facilitated by biased adipo‐lineage commitment, resulting in a net loss of body fat mass. It is also possible that the deposition of increased fats produced by BMSCs is limited to bone marrow. To confirm these hypotheses, further investigations are warranted.
It is known that BMP6 is capable of inducing bone formation. A study has shown that BMP6 knockout mice exhibit growth retardation with reduced trabecular bone volume 51. Another study has demonstrated that adipose‐derived MSCs overexpressing BMP6 are capable of repairing critical‐size bone defects of a porcine model by improving cell survival and increasing bone thickness and mineral density 52. It has also been shown that BMP6, when injected systemically to ovariectomized rats, increases bone volume as well as enhances mechanical properties of trabecular and cortical bone 53. In addition to a positive role in the regulation of MSC osteoblastogenesis, it is likely that BMP6 mitigates T1DM‐associated bone loss by directing BMSC differentiation away from the adipogenic lineage to lean toward osteogenic lineage commitment. This is supported by previous findings that BMPs are involved in downregulation of adipogenesis and lipid formation in embryo or in brown adipose tissue and inhibition of BMPs by gremlin‐1, a BMP antagonist, leads to an increase in production of lipids 54, 55.
Our results have demonstrated that BMP6 supplementation can restore BMD in STZ‐induced diabetic mice. The finding brings up an interesting question whether the restoration of BMD is directly resulted from BMP6 induction or indirectly through modulation of glucose concentration originated from BMP6 induction. A previous study by Vukicevic and Grgurevic has shown that knocking out BMP6 causes a reduction in the number of pancreatic β cells, which in turn increases the glucose level in mice 56, suggesting that manipulation of BMP6 production affects pancreatic β cells and glucose concentration in the body. Other than BMP6, Chen et al. have reported that BMP9 is also capable of modulating glucose production to improve glucose tolerance in diabetic mice 57. Nonetheless, in this study we did not find the similar result. Our data have shown that the glucose level of STZ+ or STZ− mice is not affected by BMP6 treatment, indicating that the restoration of BMD in STZ‐induced T1DM mice treated with BMP6 is unlikely attributed to modulation of glucose concentration.
Considering the potential of our findings for developing effective treatments for bone loss in T1DM patients, it is important to ensure that injection of BMP6 would not cause unintended complications such as heterotopic ossification (HO) in the body. This may be achieved by optimizing the dose and frequency of injection. It is known that treatments of high‐dose recombinant BMPs, particularly BMP2, are associated with increased risk of HO, abnormal bone formation in soft tissues 58, 59. In our study, we did not observe gross appearance of HO at the peritoneal injection site with the dose we selected. Moreover, given that previous studies investigating systemic treatment with doses of BMP6 ranging from 1 to 50 ng per gram of mouse weight reported no HO was found 53, 60, the dose of 10 ng per gram of animal weight seems to be safe and we think there is still room for increasing the dose of injection before safety is compromised. The frequency of injection is another critical variable that should be considered for safety and effectiveness of BMP6 injection. Since many growth factors have a short in vivo half‐life, like that BMP2 has a half‐life of 7–16 minutes in the circulatory system of a rat or nonhuman primate 61, increasing the frequency of injection may help prolong therapeutic effects of BMP6 in vivo, which may also allow the use of a lower dose to reduce the risk of HO.
Studies have shown that BMP6 binds to receptors, such as BMPR2, activin A receptor type 2 (ACVR2), BMPR1A, and BMPR1B, to regulate osteoblast differentiation 34, 35. BMPR2 or ACVR2 complexes with ACVR1 upon ligand binding to activate SMAD1/5 32 that in turn interacts with RUNX2 to regulate osteogenesis 62. Based on our RT‐PCR results showing comparable expression levels of BMP6 receptors, including HJV, ACVR2A, ACVR2B, ACVR1, BMPR1A, and BMPR1B, between non‐T1DM‐ and T1DM‐BMSCs, it is suggested that the difference in regulation of BMSC osteogenesis between the two groups may not be resulted from differential expression of these receptors. Although downregulation of the expression level of BMPR2 in response to hyperglycemia has been reported by Yeh et al. in their study using STZ‐induced T1DM rats 63, interestingly we did not find the similar result with T1DM‐BMSCs in this study. We have shown that BMSCs isolated from T1DM donors produce less endogenous BMP6 whereas expressing more BMPR2 than those from non‐T1DM donors. It is possible that in response to a decrease in the production of endogenous BMP6, T1DM‐BMSCs compensate the reduction by increasing the amount of BMPR2 available on cell surface for activation. Furthermore, we have found that the increased expression of BMPR2 on T1DM‐BMSCs does not lead to increased activation of downstream SMAD1/5 signaling. Our findings suggest that activation of the BMP6/SMAD1/5 signaling pathway during osteogenesis of BMSCs is impaired by attenuated production of BMP6 associated with T1DM‐induced hyperglycemia.
An important point to consider is that while many T1DM patients are able to adequately control their blood glucose levels with diet and insulin, they are still subject to bone loss. This suggests that high glucose may not be the main determinant of bone loss in T1DM patients. Given that insulin and insulin‐like growth factor 1 (IGF1) are insufficient in T1DM patients and both of them play a crucial role in regulation of bone growth 64, 65, future studies to examine whether insulin or IGF1 is a downstream molecule regulated by BMP6 or the other way around to modulate osteogenesis of T1DM‐BMSCs and bone formation in T1DM animal models.
Conclusion
We have demonstrated here both in vitro and in vivo evidence that BMP6 is a key molecule significantly reduced in BMSCs isolated from T1DM patients and a reduction of BMP6 in T1DM‐BMSCs leads to decreased osteogenesis. By supplementing BMP6, bone loss observed in STZ‐induced T1DM mice can be attenuated. Our findings suggest that BMP6 plays an important role in etiology of T1DM‐associated bone loss and therapies based on the molecule can be developed to treat the T1DM complication.
Author Contributions
J.F.W.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; M.‐S.L., D.J.T.: collection and assembly of data, data analysis and interpretation, final approval of manuscript; T.‐L.T.: conception and design, collection and assembly of data, data analysis and interpretation, final approval of manuscript; E.M.L.: provision of study material, collection and assembly of data, data analysis and interpretation, final approval of manuscript; M.W.S.: provision of study material, final approval of manuscript; W.‐J.L.: conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
M.W.S. declared Intellectual property rights with Zimmer‐Biomet. W.‐J.L. declared financial relationship as editor of an academic book for a publisher group. The other authors indicated no potential conflicts of interest.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supporting information
Supplemental Figure 1: Density plots of flow cytometry analyzing expression of surface markers on cells isolated from nonT1DM and T1DM donors. Live cells and single cells (two left columns) were selected by side scatter and forward scatter gating. Cells of both groups did not express CD45 (central column) but abundant CD73, CD90, and CD105 (two right columns).
Supplemental Figure 2: Expression levels of transcripts of BMPs and TGFBs in nonT1DM‐ and T1DM‐BMSCs during osteogenic induction. Prior to differentiation induction, nonT1DM‐BMSCs were maintained in low‐glucose culture whereas T1DM‐BMSCs were maintained in high‐glucose culture. *p < .05; n = 3 technical replicates.
Supplemental Figure 3: Measurements of physiological parameters of C57BL/6J mice treated with or without STZ and/or BMP6. Blood glucose (A) and weight and total body fat (B) at the end of the experimental period of 15 weeks were recorded. *p < .05; n = 6 biological replicates.
Supplemental Table: Primer sequences for analysis of quantitative PCR
Acknowledgments
This work was partially supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R01 AR064803. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Dr. Ricki Coleman and Justin Jeffery for their assistance in acquiring DEXA and μCT data.
References
- 1. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet 2014;383:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Group SfDiYS , Liese AD, D'Agostino RB Jr et al. The burden of diabetes mellitus among US youth: Prevalence estimates from the SEARCH for Diabetes in Youth Study. Pediatrics 2006;118:1510–1518. [DOI] [PubMed] [Google Scholar]
- 3. Maahs DM, West NA, Lawrence JM et al. Epidemiology of type 1 diabetes. Endocrinol Metab Clin North Am 2010;39:481–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lightfoot YL, Chen J, Mathews CE. Immune‐mediated β‐cell death in type 1 diabetes: Lessons from human β‐cell lines. Eur J Clin Invest 2012;42:1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leidig‐Bruckner G, Grobholz S, Bruckner T et al. Prevalence and determinants of osteoporosis in patients with type 1 and type 2 diabetes mellitus. BMC Endocr Disord 2014;14:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weber DR, Schwartz G. Epidemiology of skeletal health in type 1 diabetes. Curr Osteoporos Rep 2016;14:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nicodemus KK, Folsom AR. Type 1 and type 2 diabetes and incident hip fractures in postmenopausal women. Diabetes Care 2001;24:1192–1197. [DOI] [PubMed] [Google Scholar]
- 8. Ronnemaa T, Impizaara O, Puukka P et al. Bone mineral density and diabetes. Diabetes Care 2000;23:564. [DOI] [PubMed] [Google Scholar]
- 9. Oreffo RO, Cooper C, Mason C et al. Mesenchymal stem cells. Stem Cell Rev 2005;1:169–178. [DOI] [PubMed] [Google Scholar]
- 10. Siddiqui JA, Partridge NCJP. Physiological bone remodeling: Systemic regulation and growth factor involvement. Physiology 2016;31:233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blair HC, Larrouture QC, Li Y et al. Osteoblast differentiation and bone matrix formation in vivo and in vitro. Tissue Eng Part B Rev 2017;23:268–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu G, Xu R, Zhang P et al. Estrogen regulates stemness and senescence of bone marrow stromal cells to prevent osteoporosis via ERβ‐SATB2 pathway. J Cell Physiol 2018;233:4194–4204. [DOI] [PubMed] [Google Scholar]
- 13. Fan J‐Z, Yang L, Meng G‐L et al. Estrogen improves the proliferation and differentiation of hBMSCs derived from postmenopausal osteoporosis through notch signaling pathway. Mol Cell Biochem 2014;392:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Williams JP, Blair HC, McDonald JM et al. Regulation of osteoclastic bone resorption by glucose. Biochem Biophys Res Commun 1997;235:646–651. [DOI] [PubMed] [Google Scholar]
- 15. Hofbauer LC, Brueck CC, Singh SK et al. Osteoporosis in patients with diabetes mellitus. J Bone Miner Res 2007;22:1317–1328. [DOI] [PubMed] [Google Scholar]
- 16. McNair P, Christensen M, Madsbad S et al. Hypoparathyroidism in diabetes mellitus. Acta Endocrinol 1981;96:81–86. [DOI] [PubMed] [Google Scholar]
- 17. Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—A meta‐analysis. Osteoporos Int 2007;18:427–444. [DOI] [PubMed] [Google Scholar]
- 18. Hein G, Weiss C, Lehmann G et al. Advanced glycation end product modification of bone proteins and bone remodelling: Hypothesis and preliminary immunohistochemical findings. Ann Rheum Dis 2006;65:101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyata T, Kawai R, Taketomi S et al. Possible involvement of advanced glycation end‐products in bone resorption. Nephrol Dial Transplant 1996;11:54–57. [DOI] [PubMed] [Google Scholar]
- 20. Miyata T, Notoya K, Yoshida K et al. Advanced glycation end products enhance osteoclast‐induced bone resorption in cultured mouse unfractionated bone cells and in rats implanted subcutaneously with devitalized bone particles. J Am Soc Nephrol 1997;8:260–270. [DOI] [PubMed] [Google Scholar]
- 21. Kayal RA, Tsatsas D, Bauer MA et al. Diminished bone formation during diabetic fracture healing is related to the premature resorption of cartilage associated with increased osteoclast activity. J Bone Miner Res 2007;22:560–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsentidis C, Gourgiotis D, Kossiva L et al. Higher levels of s‐RANKL and osteoprotegerin in children and adolescents with type 1 diabetes mellitus may indicate increased osteoclast signaling and predisposition to lower bone mass: A multivariate cross‐sectional analysis. Osteoporos Int 2016;27:1631–1643. [DOI] [PubMed] [Google Scholar]
- 23. Jin P, Zhang X, Wu Y et al. Streptozotocin‐induced diabetic rat‐derived bone marrow mesenchymal stem cells have impaired abilities in proliferation, paracrine, antiapoptosis, and myogenic differentiation. Transplant Proc 2010;42:2745–2752. [DOI] [PubMed] [Google Scholar]
- 24. Ko KI, Coimbra LS, Tian C et al. Diabetes reduces mesenchymal stem cells in fracture healing through a TNFα‐mediated mechanism. Diabetologia 2015;58:633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stolzing A, Sellers D, Llewelyn O et al. Diabetes induced changes in rat mesenchymal stem cells. Cells Tissues Organs 2010;191:453–465. [DOI] [PubMed] [Google Scholar]
- 26. Reddi AH. Bone and cartilage differentiation. Curr Opin Genet Dev 1994;4:737–744. [DOI] [PubMed] [Google Scholar]
- 27. Tsai T‐L, Manner P, Li W‐J. Regulation of mesenchymal stem cell chondrogenesis by glucose through protein kinase C/transforming growth factor signaling. Osteoarthr Cartil 2013;21:368–376. [DOI] [PubMed] [Google Scholar]
- 28. Tsai T‐L, Wang B, Squire MW et al. Endothelial cells direct human mesenchymal stem cells for osteo‐and chondro‐lineage differentiation through endothelin‐1 and AKT signaling. Stem Cell Res Ther 2015;6:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shapurian T, Damoulis PD, Reiser GM et al. Quantitative evaluation of bone density using the Hounsfield index. Int J Oral Maxillofac Implants 2006;21:290–297. [PubMed] [Google Scholar]
- 30. Bassett JD, van der Spek A, Gogakos A et al. Quantitative X‐ray imaging of rodent bone by Faxitron. Methods Mol Biol 2012;816:499–506. [DOI] [PubMed] [Google Scholar]
- 31. Gautschi OP, Frey SP, Zellweger R. Bone morphogenetic proteins in clinical applications. ANZ J Surg 2007;77:626–631. [DOI] [PubMed] [Google Scholar]
- 32. Lavery K, Swain P, Falb D et al. BMP‐2/4 and BMP‐6/7 differentially utilize cell surface receptors to induce osteoblastic differentiation of human bone marrow‐derived mesenchymal stem cells. J Biol Chem 2008;283:20948–20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Attisano L, Wrana JL. Signal transduction by the TGF‐β superfamily. Science 2002;296:1646–1647. [DOI] [PubMed] [Google Scholar]
- 34. Ebisawa T, Tada K, Kitajima I et al. Characterization of bone morphogenetic protein‐6 signaling pathways in osteoblast differentiation. J Cell Sci 1999;112:3519–3527. [DOI] [PubMed] [Google Scholar]
- 35. Inman GJ, Nicolás FJ, Callahan JF et al. SB‐431542 is a potent and specific inhibitor of transforming growth factor‐β superfamily type I activin receptor‐like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol 2002;62:65–74. [DOI] [PubMed] [Google Scholar]
- 36. Paul BY, Hong CC, Sachidanandan C et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol 2008;4:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miao J, Brismar K, Nyrén O et al. Elevated hip fracture risk in type 1 diabetic patients. Diabetes Care 2005;28:2850–2855. [DOI] [PubMed] [Google Scholar]
- 38. Notarnicola A, Maccagnano G, Tafuri S et al. Epidemiology of diabetes mellitus in the fragility fracture population of a region of Southern Italy. J Biol Regul Homeost Agents 2016;30:297–302. [PubMed] [Google Scholar]
- 39. Hamann C, Kirschner S, Günther K‐P et al. Bone, sweet bone—Osteoporotic fractures in diabetes mellitus. Nat Rev Endocrinol 2012;8:297–305. [DOI] [PubMed] [Google Scholar]
- 40. Strotmeyer ES, Cauley JA, Schwartz AV et al. Nontraumatic fracture risk with diabetes mellitus and impaired fasting glucose in older white and black adults: The health, aging, and body composition study. Arch Intern Med 2005;165:1612–1617. [DOI] [PubMed] [Google Scholar]
- 41. Martin LM, McCabe LR. Type I diabetic bone phenotype is location but not gender dependent. Histochem Cell Biol 2007;128:125–133. [DOI] [PubMed] [Google Scholar]
- 42. Silva MJ, Brodt MD, Lynch MA et al. Type 1 diabetes in young rats leads to progressive trabecular bone loss, cessation of cortical bone growth, and diminished whole bone strength and fatigue life. J Bone Miner Res 2009;24:1618–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang T‐C, Hsu M‐F, Wu KK. High glucose induces bone marrow‐derived mesenchymal stem cell senescence by upregulating autophagy. PLoS One 2015;10:e0126537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li Y‐M, Schilling T, Benisch P et al. Effects of high glucose on mesenchymal stem cell proliferation and differentiation. Biochem Biophys Res Commun 2007;363:209–215. [DOI] [PubMed] [Google Scholar]
- 45. Cao B, Liu N, Wang W. High glucose prevents osteogenic differentiation of mesenchymal stem cells via lncRNA AK028326/CXCL13 pathway. Biomed Pharmacother 2016;84:544–551. [DOI] [PubMed] [Google Scholar]
- 46. Wang A, Midura RJ, Vasanji A et al. Hyperglycemia diverts dividing osteoblastic precursor cells to an adipogenic pathway and induces synthesis of a hyaluronan matrix that is adhesive for monocytes. J Biol Chem 2014;289:11410–11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fowlkes JL, Bunn RC, Liu L et al. Runt‐related transcription factor 2 (RUNX2) and RUNX2‐related osteogenic genes are down‐regulated throughout osteogenesis in type 1 diabetes mellitus. Endocrinology 2007;149:1697–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thrailkill KM, Liu L, Wahl EC et al. Bone formation is impaired in a model of type 1 diabetes. Diabetes 2005;54:2875–2881. [DOI] [PubMed] [Google Scholar]
- 49. Hebert SL, Nair KS. Protein and energy metabolism in type 1 diabetes. Clin Nutr 2010;29:13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hibbert‐Jones E. Fat and protein counting in type 1 diabetes. Pract Diabetes 2016;33:243–247. [Google Scholar]
- 51. Kugimiya F, Kawaguchi H, Kamekura S et al. Involvement of endogenous bone morphogenetic protein (BMP) 2 and BMP6 in bone formation. J Biol Chem 2005;280:35704–35712. [DOI] [PubMed] [Google Scholar]
- 52. Sheyn D, Tawackoli W, Gazit Z et al. Allogeneic, BMP6 gene‐modified, bone marrow MSCs induce vertebral fracture repair in a porcine model: A pilot study. Spine J 2013;13:S71–S72. [Google Scholar]
- 53. Simic P, Culej JB, Orlic I et al. Systemically administered bone morphogenetic protein‐6 restores bone in aged ovariectomized rats by increasing bone formation and suppressing bone resorption. J Biol Chem 2006;281:25509–25521. [DOI] [PubMed] [Google Scholar]
- 54. Gustafson B, Hammarstedt A, Hedjazifar S et al. BMP4 and BMP antagonists regulate human white and beige adipogenesis. Diabetes 2015;64:1670–1680. [DOI] [PubMed] [Google Scholar]
- 55. Hino J, Miyazawa T, Miyazato M et al. Bone morphogenetic protein‐3b (BMP‐3b) is expressed in adipocytes and inhibits adipogenesis as a unique complex. Int J Obes 2012;36:725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vukicevic S, Grgurevic L. BMP‐6 and mesenchymal stem cell differentiation. Cytokine Growth Factor Rev 2009;20:441–448. [DOI] [PubMed] [Google Scholar]
- 57. Chen C, Grzegorzewski KJ, Barash S et al. An integrated functional genomics screening program reveals a role for BMP‐9 in glucose homeostasis. Nat Biotechnol 2003;21:294–301. [DOI] [PubMed] [Google Scholar]
- 58. Shi L, Sun W, Gao F et al. Heterotopic ossification related to the use of recombinant human BMP‐2 in osteonecrosis of femoral head. Medicine 2017;96:27(e7413). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Arzeno A, Wang T, Huddleston JI III. Abundant heterotopic bone formation following use of rhBMP‐2 in the treatment of acetabular bone defects during revision hip arthroplasty. Arthroplast Today 2018;4:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pauk M, Bordukalo‐Niksic T, Brkljacic J et al. A novel role of bone morphogenetic protein 6 (BMP6) in glucose homeostasis. Acta Diabetol 2018;55:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Poynton AR, Lane JM. Safety profile for the clinical use of bone morphogenetic proteins in the spine. Spine 2002;27:S40–S48. [DOI] [PubMed] [Google Scholar]
- 62. Huang W, Yang S, Shao J et al. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front Biosci 2007;12:3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yeh C‐H, Chang C‐K, Cheng M‐F et al. Decrease of bone morphogenetic protein‐7 (BMP‐7) and its type II receptor (BMP‐RII) in kidney of type 1‐like diabetic rats. Horm Metab Res 2009;41:605–611. [DOI] [PubMed] [Google Scholar]
- 64. Hou C‐J, Liu J‐L, Li X et al. Insulin promotes bone formation in augmented maxillary sinus in diabetic rabbits. Int J Oral Maxillofac Surg 2012;41:400–407. [DOI] [PubMed] [Google Scholar]
- 65. Niu T, Rosen CJ. The insulin‐like growth factor‐I gene and osteoporosis: A critical appraisal. Gene 2005;361:38–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Density plots of flow cytometry analyzing expression of surface markers on cells isolated from nonT1DM and T1DM donors. Live cells and single cells (two left columns) were selected by side scatter and forward scatter gating. Cells of both groups did not express CD45 (central column) but abundant CD73, CD90, and CD105 (two right columns).
Supplemental Figure 2: Expression levels of transcripts of BMPs and TGFBs in nonT1DM‐ and T1DM‐BMSCs during osteogenic induction. Prior to differentiation induction, nonT1DM‐BMSCs were maintained in low‐glucose culture whereas T1DM‐BMSCs were maintained in high‐glucose culture. *p < .05; n = 3 technical replicates.
Supplemental Figure 3: Measurements of physiological parameters of C57BL/6J mice treated with or without STZ and/or BMP6. Blood glucose (A) and weight and total body fat (B) at the end of the experimental period of 15 weeks were recorded. *p < .05; n = 6 biological replicates.
Supplemental Table: Primer sequences for analysis of quantitative PCR
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.