

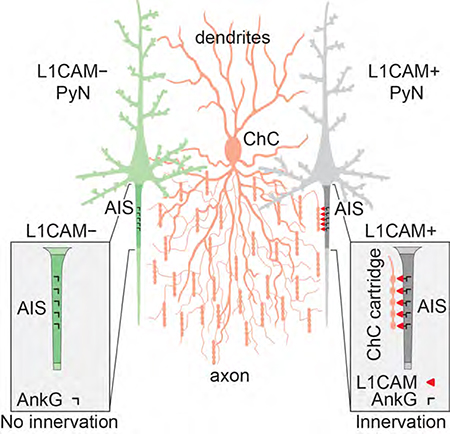
SUMMARY
Among the diverse interneuron subtypes in the neocortex, chandelier cells (ChCs) are the only population that selectively innervate pyramidal neurons (PyNs) at their axon initial segment (AIS), the site of action potential initiation, allowing them to exert powerful control over PyN output. Yet, mechanisms underlying their subcellular innervation of PyN AISs are unknown. To identify molecular determinants of ChC/PyN AIS innervation, we performed an in vivo RNAi screen of PyN-expressed axonal cell adhesion molecules (CAMs) and select Ephs/ephrins. Strikingly, we found the L1 family member L1CAM to be the only molecule required for ChC/PyN AIS innervation. We further show that L1CAM is required during both the establishment and maintenance of innervation and that selective innervation of PyN AISs by ChCs requires AIS anchoring of L1CAM by the cytoskeletal ankyrin-G/βIV-spectrin complex. Thus, our findings identify PyN-expressed L1CAM as a critical CAM required for innervation of neocortical PyN AISs by ChCs.
Graphical Abstract
eTOC Blurb
Chandelier cells (ChCs) are the only interneuron subtype that selectively innervate the axon initial segment (AIS) of pyramidal neurons (PyNs) in the neocortex; yet, the underlying mechanisms are unknown. Tai et al. reveal that neocortical ChC/PyN AIS innervation requires ankyrin-G-clustered L1CAM.
INTRODUCTION
Proper assembly and functioning of cortical circuits relies on the formation of specific synaptic connections between excitatory pyramidal neurons (PyNs) and different types of GABAergic interneurons (Bartolini et al., 2013; Huang et al., 2007; Kepecs and Fishell, 2014). At least ten GABAergic interneuron subtypes have been identified in the cerebral cortex, each with uniquely organized axonal arbors that selectively innervate distinct subcellular compartments to control the input, integration, and output of their target cells (DeFelipe et al., 2013; Tremblay et al., 2016). Among them, chandelier cells (ChCs), also referred to as axo-axonic cells, are arguably the most distinctive (Howard et al., 2005; Inan and Anderson, 2014; Jones, 1975; Somogyi, 1977; Szentagothai and Arbib, 1974; Woodruff et al., 2010). These cells, which predominantly derive from the ventral medial ganglionic eminence (vMGE) during late gestation (Inan et al., 2012; Taniguchi et al., 2013), exhibit a characteristic, highly-branched axon with multiple arrays of vertically oriented terminals, called cartridges, each harboring a string of synaptic boutons (Inda et al., 2007). Importantly, unlike other cortical interneurons that form somatodendritic synapses, ChC cartridges, typically 3–4 from 3–4 distinct ChCs, selectively innervate individual PyNs at their axon initial segment (AIS), the site of action potential initiation (DeFelipe et al., 1985; Somogyi, 1977). Furthermore, cartridges of single ChCs innervate hundreds of PyNs, which, combined with their exquisite subcellular specificity, makes them ideally suited to exert powerful control over PyN spiking and population output (DeFelipe et al., 1985; Howard et al., 2005; Inan et al., 2013; Woodruff et al., 2010). In line with this, recent studies have shown a critical role for ChCs in the synchronization of firing patterns of large populations of PyNs in different functional states (Glickfeld et al., 2009; Lu et al., 2017; Viney et al., 2013; Woodruff et al., 2011; Zhu et al., 2004). The importance of proper ChC function is further underscored by the association of ChC connectivity defects with brain disorders such as schizophrenia, epilepsy, and autism spectrum disorder (Ariza et al., 2018; Del Pino et al., 2013; Lewis, 2011; Ribak, 1985; Rocco et al., 2017). To date, however, the molecular mechanisms governing neocortical ChC/PyN AIS innervation remain entirely unknown. This has largely been due to the scarcity of ChCs and, most importantly, lack of unique ChC biochemical markers. Only recently have transgenic Nkx2.1-Cre mice become available which enable the reliable labeling of ChCs in the neocortex (Taniguchi et al., 2013; Xu et al., 2008).
Increasing evidence from other GABAergic interneuron subtypes indicates that the subcellular compartmentalization of synapses on principal neurons involves genetically determined mechanisms (Ango et al., 2004; Ashrafi et al., 2014; Di Cristo et al., 2004). In particular, cell adhesion molecules (CAMs) are emerging as key players in the axonal subcellular targeting of interneurons and the innervation of their postsynaptic cells (Ango et al., 2004; Ashrafi et al., 2014; Guan and Maness, 2010; Telley et al., 2016). For example, in the cerebellum, the L1 immunoglobulin (Ig) CAM family member neurofascin-186 (NF186), which is present at the soma and AIS of Purkinje cells (PCs), directs the navigation of basket interneuron axons from the PC soma to the AIS, where it then facilitates pinceau synapse formation (Ango et al., 2004). In addition, recent work in the spinal cord found that an Ig CAM complex of NB2 and Caspr4 on sensory neurons interacts with the L1 Ig family members NrCAM and CHL1 on GABApre interneurons to control synapse formation specifically at the axonal termini of sensory afferents (Ashrafi et al., 2014).
Given the above findings, we set out to perform in vivo screening of neocortical PyN-expressed axonal CAMs, including all those known to be enriched at the AIS, for their role(s) in ChC/PyN innervation (Ango et al., 2004; Hedstrom et al., 2007; Inda et al., 2006; Leterrier, 2016; Ogawa et al., 2008; Ogawa et al., 2010; Wimmer et al., 2015). In addition, as emerging evidence has implicated not only attractive but also repulsive mechanisms in the subcellular targeting/innervation of interneurons onto their target cells (Baohan et al., 2016; Brennaman et al., 2013; Kania and Klein, 2016), we also tested select members of the Eph and ephrin family of receptors/ligands expressed in the neocortex. To do so, we devised a strategy taking advantage of in utero electroporation (IUE) and RNA interference (RNAi) to individually knock down these molecules in neocortical PyNs while concurrently labeling ChCs using the recently generated Nkx2.1-CreER mouse line (Taniguchi et al., 2013). Strikingly, of all the candidates tested, we found the pan-axonally expressed CAM L1CAM to be the only molecule required for neocortical ChC/PyN AIS innervation, as knockdown of PyN L1CAM, but none of the other screened candidates, significantly reduced PyN AIS innervation by ChCs. Furthermore, we demonstrate a requirement for L1CAM during both the establishment and maintenance of neocortical ChC/PyN AIS innervation. Finally, we provide evidence that anchoring of L1CAM at the AIS by the ankyrin-G/βIV-spectrin AIS cytoskeletal complex is essential for ChC subcellular innervation of PyN AISs. Taken together, our findings identify L1CAM as the only PyN-expressed CAM known to date to regulate axo-axonic innervation of PyNs by ChCs in the neocortex.
RESULTS
AIS-enriched CAM NF186 is Dispensable for Neocortical PyN AIS Innervation by ChCs
Given the unique subcellular innervation of PyN AISs by ChCs, we reasoned that a CAM localized at the AIS of PyNs likely mediates this selective form of synaptic targeting in the neocortex. To explore this, we initially focused our attention on the AIS-enriched CAM NF186 given its previously demonstrated roles in governing GABAergic innervation of cerebellar PC AISs (Ango et al., 2004) and in clustering inhibitory postsynaptic proteins at the AIS of hippocampal granule cells (GCs) (Kriebel et al., 2011).
To determine whether NF186 is required for neocortical ChC/PyN AIS innervation, we combined IUE with RNAi in Nkx2.1-CreER;Rosa26-loxpSTOPloxp-tdTomato (Ai9) mice (Taniguchi et al., 2013) to knock down PyN-expressed NF186 and label ChCs in the same neocortical layer (Fig. 1A). In brief, vectors coexpressing EGFP and miR30-based shRNAs capable of efficiently depleting NF186 protein levels (miR.NF#1 & #2) were generated (Fig. S1A and B) and independently transfected into nascent layer II/III PyNs in Nkx2.1-CreER;Ai9 animals via IUE at embryonic day 15.5 (E15.5). Tamoxifen (TMX) was then administrated to the embryos at E18.5 via oral gavage of the pregnant mother to trigger expression of tdTomato red fluorescent protein (RFP) in a sparse population of layer II ChCs (Fig. 1A; Taniguchi et al., 2013). Brains from electroporated Nkx2.1-CreER;Ai9 animals were collected at postnatal day 28 (P28) when ChCs are fully matured, sectioned coronally, and immunostained with an anti-ankyrin-G (AnkG) antibody to label the AIS. Confocal images were then acquired and the average percentage of GFP+ PyNs innervated at their AIS by single RFP+ ChCs in 200 μm × 200 μm fields of view (FOVs) (50 μm depth) in the somatosensory cortex was calculated (see Methods and Fig. S5 for details).
Figure 1. PyN AIS-enriched NF186 is Dispensable for Neocortical ChC/PyN AIS Innervation.

(A1-A3) Experimental strategy to target/manipulate PyN gene expression and label ChCs in the same neocortical layer. (A1) Schematic drawing of E15.5 in utero electroporation (IUE) targeting nascent layer II/III (LII/III) PyNs in embryos from Swiss Webster (SW) females that were bred with Nkx2.1-CreER+/−;Rosa26-loxpSTOPloxp-tdTomato (Ai9)+/+ males. The position of the positive (+) and negative (−) electrodes used to target neocortical progenitors in the ventricular zone (VZ) is depicted. (A2) Tamoxifen (TMX) administration at E18.5 induces Cre activity and excision of a STOP cassette resulting in tdTomato red fluorescent protein (RFP) expression in ChC progenitors. (A3) Representative 200 μm × 200 μm confocal image of a single RFP+ ChC and neighboring electroporated GFP+ PyNs in LII of somatosensory cortex. Scale bar, 20 μm. Enlarged view of the boxed area showing a GFP+ PyN innervated at its AIS by an RFP+ ChC cartridge (arrow) is depicted on the right. Scale bar, 5 μm. AISs are visualized by immunostaining for ankyrin-G (AnkG) (blue).
(B) Representative images of PyNs innervated by ChC cartridges in LII of somatosensory cortex from Nkx2.1-CreER;Ai9 mice electroporated at E15.5 with plasmids expressing EGFP and miR30-based shRNAs against NF186 (miR.NF#1 or miR.NF#2) or Renilla luciferase (miR.Ctrl) and sacrificed at P28. Scale bar, 10 μm.
(C) Quantification of the percentage of GFP+ PyNs innervated by single RFP+ ChCs at P28. Innervation percentages are indicated for each condition (in Fig. 1C and E). 6–8 ChCs and 37–131 GFP+ PyNs per ChC from 3 animals were analyzed for each condition; one-way ANOVA, post hoc Tukey-Kramer test.
(D) Representative images of PyNs innervated by ChC cartridges in LII of somatosensory cortex from Nkx2.1-CreER;Ai9 mice coelectroporated at E15.5 with plasmids expressing EGFP and Cas9 together with an sgRNA against NF186 (crNF186) or LacZ (crLacZ), as a control, and sacrificed at P28. Endogenous NF186 is visualized by immunostaining for NF186. Scale bars, 10 μm.
(E) Quantification of the percentage of GFP+ PyNs innervated by single RFP+ ChCs at P28. Of note, crNF186-electroporated PyNs still expressing NF186 (crNF186 NF186+) were also analyzed and included as an additional control. 8–9 ChCs and 17–73 GFP+ PyNs per ChC from 3 animals were analyzed for each condition; one-way ANOVA, post hoc Tukey-Kramer test.
For all images of ChC/PyN AIS innervation, stars and arrows indicate GFP+ PyNs innervated by RFP+ ChC cartridges and the site of GFP+ PyN AIS innervation, respectively.
n.s. (not significant) indicates p ≥ 0.05. Data are mean ± SEM. See also Fig. S1.
Surprisingly, we found that knockdown of PyN NF186 using either miR.NF#1 or miR.NF#2 had no effect on ChC/PyN AIS innervation (Fig. 1B and C). In both groups, the percentage of GFP+ PyN AISs innervated by single RFP+ ChCs was similar to that observed in the control (miR.Ctrl) group (Fig. 1B and C), implying that NF186 is not involved in neocortical ChC/PyN AIS innervation. To further corroborate these findings given NF186’s demonstrated roles at the AIS of PCs and GCs in the cerebellum and hippocampus, respectively (Ango et al., 2004; Kriebel et al., 2011), we combined the CRISPR/Cas9 system (Cong et al., 2013) and IUE to selectively knockout NF186 in layer II/III neocortical PyNs. Specifically, we constructed a pX330 CRISPR plasmid expressing Cas9 and a single-guide RNA (sgRNA) against the NF186 gene (crNF186) and electroporated this plasmid together with an EGFP-expressing vector into Nkx2.1-CreER;Ai9 embryos at E15.5. Loss of NF186 protein in layer II/III GFP+ PyNs was verified by immunohistochemistry (Fig. 1D) and the percentage of GFP+, NF186− PyN AISs innervated by single RFP+ ChCs was subsequently calculated. Consistent with the results obtained for PyN NF186 knockdown, CRISPR/Cas9-mediated knockout of NF186 in PyNs did not affect ChC/PyN AIS innervation (Fig. 1D and E). Together, these data unequivocally demonstrate that NF186 is dispensable for proper neocortical PyN AIS innervation by ChCs, indicating that another/other molecule(s) must govern this unique form of axo-axonic innervation in the neocortex.
RNAi Screen of PyN-expressed Cell Surface Molecules Identifies L1CAM as a Regulator of Neocortical ChC/PyN AIS Innervation
To identify the molecular factors required for neocortical ChC/PyN AIS innervation, we initiated an in vivo RNAi screen of PyN-expressed cell surface molecules present in the neocortex using shRNAs. We began by screening additional members of the L1 family of Ig CAMs (NrCAM, L1CAM, and CHL1) (Maness and Schachner, 2007) as well as other previously reported AIS-enriched adhesion molecules (TAG-1, SCN1B, ADAM22, and CASPR2) present in the neocortex (Fig. 2A and Table S1) (Inda et al., 2006; Ogawa et al., 2008; Ogawa et al., 2010; Wimmer et al., 2015). Furthermore, we also tested select Eph receptors and ephrins (Efns) (Fig. 2A and Table S1) due to their well-documented roles in mediating attraction, repulsion, and/or adhesion through interactions with their respective ligand(s)/receptor(s) (EphA3, EphA4, EphB2, EfnB1, EfnB2, and EfnB3) (Kania and Klein, 2016).
Figure 2. RNAi Screen of PyN-expressed Cell Surface Molecules Identifies L1CAM as a Regulator of Neocortical ChC/PyN AIS Innervation.

(A) Schematic depicting the subcellular localization (i.e. axonal or AIS-enriched) of screened cell surface molecules in neocortical PyNs.
(B) In vivo RNAi screen of PyN-expressed axonal and AIS-enriched cell adhesion molecules and select members of the Eph and ephrin family of receptors/ligands present in the neocortex. Quantification of the percentage of GFP+ PyNs innervated by single RFP+ ChCs at P28 in LII of somatosensory cortex from Nkx2.1-CreER;Ai9 mice electroporated at E15.5 with plasmids expressing EGFP and shRNAs targeting indicated cell surface molecules. Innervation percentages are indicated for each condition. 6–16 ChCs and 12–99 GFP+ PyNs per ChC from 3 animals were analyzed for each condition; one-way ANOVA, post hoc Tukey-Kramer test.
(C) Representative images of PyNs innervated by ChC cartridges in LII of somatosensory cortex from Nkx2.1-CreER;Ai9 mice electroporated at E15.5 with plasmids expressing EGFP and shCtrl, shNrCAM, shLICAM, or shTAG-1 and sacrificed at P28. Scale bar, 10 μm.
***p < 0.001. Data are mean ± SEM. See also Fig. S1 and Table S1.
For the screening of the above molecules, we used a strategy similar to the one described for NF186. Namely, vectors expressing EGFP and validated shRNAs targeting individual mouse cell surface molecules were constructed (Fig. S1C-H and L-N; Anderson et al., 2012; Cisse et al., 2011; Khodosevich et al., 2011; McClelland et al., 2009; Nishikimi et al., 2011) and then individually transfected into nascent layer TT/TTT PyNs of E15.5 Nkx2.1-CreER;Ai9 embryos by IUE prior to TMX administration at E18.5. Electroporated brains were collected at P28, immunostained for AnkG and imaged using a confocal microscope, and the average percentage of GFP+ PyNs innervated at their AIS by single RFP+ ChCs was calculated for each condition. Strikingly, of all the molecules tested, we found that only PyN knockdown of pan-axonally-expressed L1CAM caused a marked reduction in PyN AIS innervation by ChCs (Fig. 2B and C). Compared to the control shRNA group where approximately 33% of GFP+ PyNs were innervated by single RFP+ ChCs, only about 7% of GFP+ PyNs were innervated in the L1CAM shRNA group (Fig. 2B). In all other groups, the percentage of GFP+ PyN AISs innervated by single RFP+ ChCs was not significantly different from that of the control group (Fig. 2B and C). Together, these findings identify L1CAM as a strong candidate molecule critical for ChC/PyN AIS innervation in the neocortex.
Postsynaptic PyN-expressed L1CAM is Essential for Proper Neocortical PyN AIS Innervation by ChCs
Following our initial screening results, we set out to ensure that the observed impairment in ChC/PyN AIS innervation was due to specific knockdown of PyN L1CAM. To this end, we tested a second independent L1CAM shRNA (shL1CAM#2) as well as a miR30-based shRNA driven by the CAG promoter (CAG-miR.L1CAM). Both shRNAs substantially reduced endogenous L1CAM protein levels in cultured cortical neurons (Fig. S1C and I). Consistent with the results obtained for the L1CAM shRNA used in our RNAi screen (shL1CAM#1), we found that expression of either shL1CAM#2 or CAG-miR.L1CAM in neocortical PyNs significantly reduced the percentage of GFP+ PyN AISs innervated by single RFP+ ChCs (Fig. 3A and B). In addition, we performed rescue experiments using a human L1CAM cDNA (hL1CAM) (Hlavin and Lemmon, 1991), which harbors three base pair mismatches with the mouse targeted shL1CAM#1 sequence making it resistant to shL1CAM#1-mediated L1CAM knockdown (Fig. S1J). Coexpression of hL1CAM with shL1CAM#1 in neocortical PyNs completely rescued the RNAi-induced innervation phenotype (Fig. 3A and B). Of note, expression of hL1CAM alone (hL1CAM overexpression (OE)) did not have any effect on ChC/PyN AIS synaptic innervation (Fig. S2A-D). Combined, these data demonstrate that the effect of L1CAM RNAi on ChC/PyN AIS innervation is specific.
Figure 3. PyN L1CAM is Required for Neocortical PyN AIS Synaptic Innervation by ChC Cartridges.

(A) Representative images of PyNs innervated by ChC cartridges in LII of somatosensory cortex from Nkx2.1-CreER;Ai9 mice electroporated at E15.5 with plasmids expressing EGFP and shCtrl, shL1CAM#1, shL1CAM#2, or shL1CAM#1 + human L1CAM (hL1CAM) and sacrificed at P28. Scale bar, 10 μm.
(B) Quantification of the percentage of GFP+ PyNs innervated by single RFP+ ChCs at P28. Innervation percentages are indicated for each condition (in Fig. 3B and D). 9–16 ChCs and 17–126 GFP+ PyNs per ChC from 3 animals were analyzed for each condition; one-way ANOVA, post hoc Tukey-Kramer test (left) and Student’s t test (right).
(C) Representative images of PyNs innervated by ChC cartridges in LII of somatosensory cortex from Nkx2.1-CreER;Ai9 mice electroporated at E15.5 with plasmids expressing EGFP and CAMKII-miR.Ctrl or CAMKII-miR.L1CAM and sacrificed at P28. Scale bar, 10 μm.
(D) Quantification of the percentage of GFP+ PyNs innervated by single RFP+ ChCs at P28. 10–14 ChCs and 32–147 GFP+ PyNs per ChC from 3 animals were analyzed for each condition; Student’s t test.
(E, G) Representative images of GFP+ PyN AISs in LII of somatosensory cortex from CD1 mice electroporated at E15.5 with plasmids expressing EGFP and shCtrl or shL1CAM#1 and sacrificed at P28. Inhibitory synapses are visualized by immunostaining for the GABAergic presynaptic marker VGAT (red; E) or the GABAergic postsynaptic marker gephyrin (Gphn; red; G). Scale bars, 5 μm.
(F, H) Quantification of the average number of VGAT (F) or gephyrin (H) puncta per GFP+ PyN AIS at P28. 33–43 AISs from 3 animals were analyzed for each condition; Student’s t tests.
(I) Quantification of the average number of VGAT puncta per μm on GFP+ PyN cell bodies at P28. 30 cell bodies from 3 animals were analyzed for each condition; Student’s t test.
(J, L) Quantification of the average number of VGAT (J) or gephyrin (L) puncta per μm on the dendrites of GFP+ PyNs at P28. 24–51 dendrites from 3 animals were analyzed for each condition; Student’s t tests.
(K) Quantification of the average fluorescence intensity (AU) of gephyrin on the cell body of GFP+ PyNs at P28. 34 cell bodies from 3 animals were analyzed for each condition; Student’s t test.
(M) Representative traces of mIPSCs recorded from GFP+ PyNs in LII/III of somatosensory cortex from CD1 mice electroporated at E15.5 with plasmids expressing EGFP and shCtrl or shL1CAM#1 and sacrificed at P27-P30.
(N) Quantification of mIPSC amplitude (left) and frequency (right). 16 GFP+ PyNs from 4 animals were analyzed for each condition; Student’s t tests.
n.s. indicates p ≥ 0.05, *p < 0.05, ***p < 0.001. Data are mean ± SEM. See also Fig. S1, S2, and S3.
Next, we verified that the innervation phenotype caused by PyN L1CAM depletion was not an indirect consequence of alterations in PyN AIS structure and/or protein composition. To this end, we analyzed the length and area of the AIS as well as the distribution and levels of AIS-enriched proteins, including AnkG, βlV-spectrin, NF186, NrCAM and Nav1.6, under PyN L1CAM knockdown conditions (Fig. S3A-E). We found that none of these parameters were significantly different between the control and the shL1CAM#1 groups, indicating that the LICAM-dependent innervation phenotype did not result from altered PyN AIS structure/composition. Next, to rule out that the observed phenotype was secondary to defects in L1CAM-associated neurodevelopmental processes, such as neuronal migration and axon formation/guidance (Appel et al., 1993; Cohen et al., 1998; Lemmon et al., 1989; Lindner et al., 1983), we examined whether silencing of PyN L1CAM at a later developmental stage affects ChC/PyN AIS innervation. To accomplish this, we generated a vector that coexpresses EGFP and the miR30-based L1CAM shRNA from the CaMKII promoter (CAMKII-miR.L1CAM) (Fig. S1K). This promoter becomes active in the second postnatal week at which time the above neurodevelopmental processes are largely completed (data not shown). Analysis of electroporated brain slices at P28 revealed that CaMKII-driven L1CAM knockdown caused a significant reduction in ChC/PyN AIS percent innervation (Fig. 3C and D), indicating that the L1CAM RNAi-induced innervation phenotype was not secondary to PyN L1CAM-dependent defects in earlier neurodevelopmental processes. Finally, to ensure that the observed innervation phenotype was not due to a reduction in ChC cartridge number, we counted the number of cartridges of single RFP+ ChCs in 200 μm × 200 μm FOVs (50 μm depth) in the somatosensory cortex of P28 Nkx2.1-CreER;Ai9 animals electroporated at E15.5 with shCtrl- or shL1CAM#1-expressing constructs. No difference in ChC cartridge number was found between the shCtrl and shL1CAM#1 groups (Fig. S3F). Together, these data unveil that PyN-expressed L1CAM is indispensable for neocortical PyN AIS innervation by ChCs.
PyN-expressed L1CAM Selectively Regulates ChC/PyN GABAergic Synapses at the AIS
To corroborate and extend our above findings, we performed immunohistochemistry on brain slices with antibodies against AnkG and vesicular GABA transporter (VGAT), a presynaptic marker for GABAergic synaptic vesicles, or gephyrin, a postsynaptic marker for GABAergic synapses, to visualize axo-axonic synapses made by ChCs onto the AIS of PyNs. In particular, brain sections from P28 animals electroporated at E15.5 with vectors expressing EGFP and shCtrl or shL1CAM#1 were coimmunostained for AnkG and VGAT or gephyrin and the number of VGAT- or gephyrin-positive puncta per GFP+ AIS was calculated. In line with our findings for ChC/PyN AIS innervation, we found that the number of VGAT and gephyrin puncta at the AIS of GFP+ PyNs was markedly reduced in the shL1CAM#1 group as compared to the shCtrl group (Fig. 3E-H). On the other hand, the number of VGAT and gephyrin puncta along the somatodendritic compartment of GFP+ PyNs was not altered in the shL1CAM#1 group relative to the shCtrl group (Fig. 3I-L), implying that PyN L1CAM selectively regulates ChC/PyN AIS synapses and not the synaptic innervation of other PyN subcellular domains by other interneuron subtypes. In this regard, it is also worth noting that knockdown of L1CAM in PyNs did not induce off-target ChC/PyN innervation at the somatodendritic compartment or distal axon of such shL1CAM#1-electroporated GFP+ PyNs (Fig. S3G). Instead, under these knockdown conditions, neighboring non-electroporated GFP-PyNs with wild-type levels of L1CAM were preferentially innervated selectively at their AIS by ChCs (Fig. S3H). In the same vein, we found that ectopic expression of hL1CAM in neocortical PyNs, which failed to induce any changes in ChC/PyN AIS percent innervation, also did not drive ChC/PyN innervation outside the AIS (Figure S2B).
Given our findings demonstrating a marked reduction in GABAergic synapse number at the AIS of L1CAM-depleted PyNs in addition to recent reports demonstrating ChC-mediated inhibition of PyN firing (Lu et al., 2017; Woodruff et al., 2011), we next investigated whether inhibitory synaptic transmission onto L1CAM-depleted PyNs is altered. To this end, whole-cell patch-clamp recordings were performed to measure miniature inhibitory postsynaptic currents (mIPSCs) in layer II/III GFP+ PyNs in the somatosensory cortex of P27-P30 mice electroporated at E15.5 with plasmids expressing EGFP and shCtrl or shL1CAM#1. Interestingly, we found that both the frequency and amplitude of mIPSCs were decreased in the shL1CAM#1 group relative to the shCtrl group (Fig. 3M and N). Combined with our findings that the number of GABAergic synapses on PyN AISs, but not along their somatodendritic compartment, is decreased under PyN L1CAM knockdown conditions, these results suggest that silencing of PyN L1CAM reduces the inhibitory input from ChCs onto L1CAM-depleted PyNs.
Altogether, these data establish PyN-expressed L1CAM as a critical factor required for proper neocortical ChC/PyN AIS GABAergic synaptic innervation. To the best of our knowledge, L1CAM is so far the only PyN-expressed adhesion molecule identified to date to govern PyN AIS innervation by ChCs in the neocortex.
L1CAM is Required for Both the Establishment and Maintenance of Neocortical ChC/PyN AIS Innervation
To further define the role of L1CAM in neocortical ChC/PyN AIS innervation, we asked when L1CAM is required during the innervation process. Namely, is it required early on for the establishment and/or later on for the maintenance of this unique form of subcellular innervation? To address this, we first needed to determine the temporal profile of neocortical PyN AIS innervation by ChCs. To this end, brains from E18.5 TMX-induced Nkx2.1-CreER;Ai9 animals were collected at time points spanning P8 to P28, sectioned and coimmunostained for AnkG and gephyrin, and analyzed for the percentage of PyN AISs innervated by single RFP+ ChCs in 200 μm × 200 μm FOVs (Fig 4A and B). In addition, the number of gephyrin puncta per PyN AIS and the percentage of RFP+ ChC boutons overlapping with PyN AIS gephyrin puncta were quantified (Fig. 4C and D).
Figure 4. Developmental Profiling of ChC Axonal Morphology and Connectivity.

(A) Temporal profile of PyN AIS innervation by ChCs in LII of somatosensory cortex from Nkx2.1-CreER;Ai9 mice. Representative images of RFP+ ChC cartridges, AISs of neighboring PyNs, and gephyrin puncta at time points spanning P8 to P28. Scale bar, 5 μm.
(B-D) Quantification of the percentage of PyN AISs innervated by single RFP+ ChCs (B), the average number of gephyrin puncta per PyN AIS (C), and the percentage of RFP+ ChC boutons overlapping with AIS gephyrin puncta (D) from P8 to P28. 5–9 ChCs and 177–321 GFP+ PyNs (B) or 8–25 PyN AISs (C, D) per ChC from 3 animals were analyzed for each time point.
(E) Representative maximum projection renderings from 3D reconstructions of individual neocortical RFP+ ChC somas and axons at time points spanning P8 to P14. Scale bar, 20 μm.
(F) Scholl analysis comparing axonal arbors of ChCs at time points spanning P8 to P14. 4–7 ChCs from 3 animals were analyzed for each time point.
(G-I) Morphometric analyses of ChC axonal complexity index (G), branch points (H), and length (I) at time points spanning P8 to P14. 4–7 ChCs from 3 animals were analyzed for each time point.
Data are mean ± SEM.
Interestingly, we found that there was minimal innervation of PyN AISs by ChCs in layer II/III of the somatosensory cortex from P8 to P11 but that a sharp increase in innervation occurred at P12 (Fig. 4A and B). Notably, this sharp increase was accompanied by a simultaneous rise in the average number of gephyrin puncta per PyN AIS and the percentage of AIS-associated RFP+ ChC boutons overlapping with gephyrin puncta (Fig. 4A, C, and D). In line with this, we found that such a spike in ChC/PyN AIS innervation paralleled the development of neocortical ChC axons, which undergo a marked increase in branching/complexity starting at P11/P12 (Fig. 4E-I). The increase in ChC/PyN AIS innervation and gephyrin puncta per PyN AIS continued into the third/fourth postnatal week before stabilizing at approximately P28 (Fig. 4B and C). Importantly, the increase in the average number of gephyrin puncta per PyN AIS starting at P12 likely reflects the establishment of true ChC/PyN synapses as the percentage of AIS-associated RFP+ ChC boutons overlapping with gephyrin puncta approached 100% by P13/P14 (Fig. 4D). Together, these data reveal that neocortical ChC/PyN AIS innervation starts at approximately P12 and reaches final maturity by P28.
Next, we examined whether L1CAM expression in PyNs is correlated with and required early on during the onset of ChC/PyN AIS innervation. To this end, we began by performing coimmunostainings of L1CAM and AnkG on mouse brain cryosections at time points spanning P10 to P13. Significantly, we found L1CAM expression to increase at the AIS of layer II/III neocortical PyNs from P10 to P13 (Fig. 5A and B), coinciding with the time window when ChCs first start to innervate the AIS of PyNs. Moreover, as a complementary approach, we took advantage of the novel genome-editing technique SLENDR (single-cell labeling of endogenous proteins by CRISPR/Cas9-mediated homology-directed repair) (Mikuni et al., 2016) to insert an HA-encoding epitope tag into the genomic locus of L1CAM. Single-cell genome editing in neocortical PyNs is achieved by delivering the editing machinery to dividing PyN progenitors via IUE. By coimmunostaining for HA and AnkG in electroporated brain slices collected from P10 and P13 mice, we found a similar increase in L1CAM-HA expression at the AIS of transfected PyNs during this developmental time window (Fig. 5C and D), further corroborating our in vivo L1CAM immunohistochemistry findings. We then investigated whether knockdown of PyN L1CAM affects AIS innervation at P14 when synaptic connections between ChCs and PyN AISs are just starting to form. Brains of either Nkx2.1-CreER;Ai9 or CD1 mice electroporated at E15.5 with shL1CAM#1- or shCtrl-expressing vectors were collected at P14, sectioned and coimmunostained for AnkG with or without gephyrin, and analyzed as described above. Similar to our findings at P28, we observed a significant decrease in ChC/PyN AIS percent innervation and correspondingly in the average number of gephyrin puncta per GFP+ PyN AIS in the shL1CAM#1 group relative to the control group at P14 (Fig. 5E-H), suggesting that PyN L1CAM is needed during early postnatal development for the establishment of neocortical ChC/PyN AIS innervation.
Figure 5. PyN L1CAM Is Required for the Establishment and Maintenance of Neocortical ChC/PyN AIS Innervation.

(A) Temporal expression profile of endogenous L1CAM at the AIS of PyNs in LII of somatosensory cortex from CD1 mice. Representative images of PyN AISs at time points spanning P10-P13. L1CAM is visualized by immunostaining for L1CAM (L1; red). Scale bar, 5 μm.
(B) Quantification of the relative fluorescence intensity of endogenous L1CAM at the AIS of PyNs at time points spanning P10-P13. 90–155 AISs from 3 animals were analyzed for each time point.
(C) Temporal expression profile of C-terminal HA-tagged endogenous L1CAM (L1CAM-HA) at the AIS of transfected PyNs in LII of somatosensory cortex from CD1 mice coelectroporated at E13.5 with pCAG-YFP, SLENDR construct pX330-crL1CAM, and ssODN-LlCAM. Representative images of electroporated PyN AISs at P10 and P13. L1CAM-HA is visualized by immunostaining for HA (red). Scale bar, 5 μm.
(D) Quantification of the relative fluorescence intensity of L1CAM-HA at the AIS of electroporated PyNs at P10 and P13. 38–42 AISs from 3 animals were analyzed for each time point; Student’s t test.
(E) Representative images of PyNs innervated by ChC cartridges in LII of somatosensory cortex from Nkx2.1-CreER;Ai9 mice electroporated at E15.5 with plasmids expressing EGFP and shCtrl or shL1CAM#1 and sacrificed at P14. Scale bar, 10 μm.
(F) Quantification of the percentage of GFP+ PyNs innervated by single RFP+ ChCs at P14. Innervation percentages are indicated for each condition (in Fig. 5F and M). 12–13 ChCs and 12–86 GFP+ PyNs per ChC from 3 animals were analyzed for each condition; Student’s t test.
(G) Representative images of GFP+ PyN AISs in LII of somatosensory cortex from CD1 mice electroporated at E15.5 with plasmids expressing EGFP and shCtrl or shL1CAM#1 and sacrificed at P14. Inhibitory synapses are visualized by immunostaining for gephyrin (red). Scale bar, 5 μm.
(H) Quantification of the average number of gephyrin puncta per GFP+ PyN AIS at P14. 33–49 AISs from 3 animals were analyzed for each condition; Student’s t test.
(I) Cre-dependent RNAi expression vector used for the temporal regulation of L1CAM knockdown, based on previously described “FLIP vectors” (Stern et al., 2008). Cre expression inverts the EGFP-miR.L1CAM sequence, allowing for its regulated expression, and at the same time deletes the Puro-2A-Thy1.1 encoding sequences.
(J) Knockdown of L1CAM using inducible L1CAM RNAi. Western blot of total lysates from HEK293T cells co-transfected with plasmids expressing mouse L1CAM (mLlCAM) and CAG-driven Cre, FLIP-miR.Ctrl, and/or FLIP-miR.LlCAM, probed with antibodies to L1CAM and γ-tubulin. Data shown are representative of three independent experiments.
(K) Scheme of experimental workflow to test whether L1CAM is required for the maintenance of neocortical ChC/PyN AIS innervation. First, ChC progenitors in CD1 embryos were labeled via vMGE-directed IUE at E13.5 with a tdTomato-expressing plasmid. The same embryos were then subjected to VZ-directed IUE at E15.5 to introduce FLIP-miR.L1CAM- or FLIP-miR.Ctrl-expressing vectors together with a CAG-ER CreER-expressing plasmid into PyN progenitors. Positions of the positive (+) and negative (−) electrodes used during vMGE- and VZ-directed IUEs are depicted. Animals were administered TMX via intraperitoneal (i.p.) injection at P28 to induce L1CAM RNAi expression and sacrificed at P40.
(L) Representative images of PyNs innervated by ChC cartridges in LII of somatosensory cortex from CD1 mice subjected to dual IUEs, induced at P28 with TMX, and sacrificed at P40 (as described in Fig. 5K). Scale bar, 10 μm.
(M) Quantification of the percentage of GFP+ PyNs innervated by single RFP+ ChCs at P40. 15–18 ChCs and 4–40 GFP+ PyNs per ChC from 3 animals were analyzed for each condition; Student’s t test.
(N) Representative images of GFP+ PyN AISs in LII of somatosensory cortex from CD1 mice coelectroporated with pCAG-ER CreER and plasmids expressing FLIP-miR.L1CAM or FLIP-miR.Ctrl, TMX induced at P28, and sacrificed at P40. Inhibitory synapses are visualized by immunostaining for gephyrin (red). Scale bar, 5 μm.
(O) Quantification of the average number of gephyrin puncta per GFP+ PyN AIS at P40. 27–30 AISs from 3 animals were analyzed for each condition; Student’s t test.
**p < 0.01, ***p < 0.001. Data are mean ± SEM.
Finally, we sought to determine whether L1CAM is also required for the maintenance of ChC/PyN AIS innervation at later postnatal stages. To do so, we engineered a Cre-dependent inducible RNAi expression vector targeted against L1CAM (FLIP-miR.LlCAM) that allows for the temporal control of L1CAM knockdown postnatally via TMX administration (Fig. 5I-J) (Tai et al., 2014). Of note, due to the inducible nature of the FLIP-miR.LlCAM construct, this experiment could not be performed in Nkx2.1-CreER;Ai9 mice since TMX induction at E18.5 to label ChCs would turn on expression of miR.L1CAM embryonically. Hence, we instead employed a double IUE approach separately targeting neocortical ChCs and PyNs in wild-type CD1 animals. Namely, in the same embryos, we performed vMGE-directed IUEs at E13.5 to label nascent layer II ChCs with a tdTomato fluorescent marker (Tai et al., 2014) and E15.5 VZ-directed IUEs to co-transfect nascent layer II/III PyNs with FLIP-miR.L1CAM, or FLIP-miR.Ctrl, and a vector expressing a TMX-inducible form of Cre recombinase (ER CreER; Fig. 5K). Such doubly electroporated animals were then given TMX at P28 to turn on expression of miR.L1CAM and their brains were collected at P40 for analysis. Interestingly, we found that both the percentage of GFP+ PyN AISs innervated by single RFP+ ChCs and the average number of gephyrin puncta per GFP+ PyN AIS were significantly reduced in the FLIP-miR.L1CAM group relative to the FLIP-miR.Ctrl group (Fig. 5L-O). Together, these data indicate that L1CAM is not only required during the establishment of neocortical ChC/PyN AIS innervation but also for the maintenance of such innervation into adulthood.
L1CAM Anchoring at the AIS via the Ankyrin-G/βIV-spectrin Cytoskeletal Complex is Necessary for PyN AIS Innervation by ChCs
While our above findings demonstrate a critical role for PyN L1CAM in governing neocortical ChC/PyN AIS innervation, an outstanding question is how L1CAM mediates this unique form of subcellular innervation given that it is expressed throughout the axon (Boiko et al., 2007; Scotland et al., 1998; Winckler et al., 1999). Due to the stark difference in ChC/PyN innervation between PyN axonal subdomains (AIS and distal axon), we postulated that AIS-localized L1CAM differs from the pool of L1CAM in the distal axon via its binding to an AIS-specific protein(s). Of particular relevance in this regard is L1CAM’s interaction with the AIS-localized scaffolding protein AnkG (Boiko et al., 2007; Davis and Bennett, 1994; Hortsch et al., 2009; Needham et al., 2001), the master organizer of the AIS, which plays a critical role in linking AIS transmembrane components to the actin cytoskeleton via its interaction with βIV-spectrin (Leterrier, 2016; Yang et al., 2007). Moreover, previous work found AnkG to promote L1CAM oligomerization and limit its diffusibility selectively at the AIS in vitro (Boiko et al., 2007; Davis and Bennett, 1994; Winckler et al., 1999). Based on these findings, and our data showing an enrichment of surface L1CAM at the AIS relative to the distal axon of primary cultured cortical PyNs (Fig. S4A and E), we postulated that L1CAM-AnkG-βIV-spectrin interactions facilitate the clustering and enrichment of surface L1CAM at the AIS of PyNs, thereby enabling proper subcellular innervation by ChCs.
To test this possibility, we investigated whether perturbing L1CAM-AnkG-βIV-spectrin interactions affects the subcellular distribution of surface L1CAM and most importantly ChC/PyN AIS innervation. To this end, we utilized a previously described human L1CAM mutant containing a disease-linked mutation in its cytoplasmic FIGQY motif (Y1229H) that disrupts its interaction with AnkG (Needham et al., 2001). In addition, we used miR30-based shRNAs (miR.βIV#1 & #2) capable of efficiently reducing βIV-spectrin protein expression (Fig. S4D) as another means to disrupt L1CAM-AnkG-βIV-spectrin interactions at the AIS. To assess the effects of the Y1229H mutation and βIV-spectrin knockdown on the subcellular distribution of surface L1CAM, we performed live cell surface labeling of L1CAM on primary cultured cortical PyNs. In line with a critical role for AnkG and βIV-spectrin in regulating the anchoring/clustering of L1CAM at the AIS, we found that the levels of surface L1CAM at the AIS were significantly reduced in neurons expressing L1CAM-Y1229H or βIV-spectrin shRNA as compared to control neurons expressing L1CAM-WT or miR.Ctrl shRNA, respectively (Fig. S4A-C and E-G).
Next, we tested the ability of the Y1229H mutant to rescue the L1CAM RNAi-induced defect in ChC/PyN AIS innervation. To this end, a vector expressing RNAi-resistant human (h) L1CAM-Y1229H was co-transfected with shL1CAM#1 into nascent layer TT/TTT PyNs of Nkx2.1-CreER;Ai9 animals via E15.5 IUE and the percentage of GFP+ PyN AISs innervated by single RFP+ ChCs was calculated at P14 and P28. We found that coexpression of hL1CAM-Y1229H with shL1CAM#1 failed to rescue the L1CAM RNAi-induced innervation defect at both P14 and P28, in contrast to control hL1CAM-WT (Fig. 6A-C). Finally, we examined the effects of PyN βIV-spectrin knockdown on ChC/PyN AIS innervation using the aforementioned miR.βIV#1 & #2 shRNAs. Expression of either miR.βIV#1 or miR.βIV#2 in layer TT/TTT PyNs significantly reduced the percentage of GFP+ PyNs innervated by single RFP+ ChCs at P14 and P28 (Fig. 6D-F). Together, these data demonstrate that LICAM’s interaction with the AnkG-βIV-spectrin AIS cytoskeletal complex is critical for its role in mediating selective innervation of neocortical PyN AISs by ChCs.
Figure 6. L1CAM’s Interaction with the AnkG-βIV-Spectrin AIS Cytoskeletal Complex is Necessary for PyN AIS Innervation by ChCs.

(A) Representative images of PyNs innervated by ChC cartridges in LII of somatosensory cortex from Nkx2.1-CreER;Ai9 mice electroporated at E15.5 with plasmids expressing EGFP and shCtrl, shL1CAM#1, shL 1 CAM# 1 +hL 1CAM-WT, or shL1CAM#1+hL1CAM-Y1229H and sacrificed at P28. Scale bar, 10 μm.
(B, C) Quantification of the percentage of GFP+ PyNs innervated by single RFP+ ChCs at P14 (B) and P28 (C). Innervation percentages are indicated for each condition at each time point (in Fig. 6B, C, E, and F). 10–16 ChCs and 12–110 GFP+ PyNs per ChC from 3 animals were analyzed for each condition; one-way ANOVAs, post hoc Tukey-Kramer tests.
(D) Representative images of PyNs innervated by ChC cartridges in LII of somatosensory cortex from Nkx2.1-CreER;Ai9 mice electroporated at E15.5 with plasmids expressing EGFP and miR.Ctrl, miR.βIV#1, or miR.βIV#2 and sacrificed at P28. Scale bar, 10 μm.
(E, F) Quantification of the percentage of GFP+ PyNs innervated by single RFP+ ChCs at P14 (E) and P28 (F). 6–10 ChCs and 24–132 GFP+ PyNs per ChC from 3 animals were analyzed for each condition; one-way ANOVAs, post hoc Tukey-Kramer tests.
***p < 0.001. Data are mean ± SEM. See also Fig. S4.
DISCUSSION
The striking selectivity of ChC innervation at the AIS of PyNs has intrigued neuroscientists ever since the initial discovery of ChCs in the 1970s (Jones, 1975; Somogyi, 1977; Szentagothai and Arbib, 1974). This interest has heightened with accruing evidence showing that this unique form of subcellular innervation allows ChCs to exert powerful yet precise control over cortical PyN spiking and population output (Howard et al., 2005; Inan and Anderson, 2014; Lu et al., 2017), and, in particular, given increasing reports linking ChC connectivity defects to neurological conditions, such as schizophrenia and epilepsy (Ariza et al., 2018; Del Pino et al., 2013; Lewis, 2011; Ribak, 1985; Rocco et al., 2017). Despite these findings, to date little to nothing is known about the mechanisms underlying the subcellular innervation of neocortical PyN AISs by ChCs. While the atypical Rac activator DOCK7 and the receptor tyrosine kinase ErbB4 have been implicated in the regulation of ChC cartridge bouton size/density (Fazzari et al., 2010; Tai et al., 2014), depletion of these proteins in ChCs does not affect the percentage of PyN AISs innervated by ChCs (Fig. S6A-D). To identify the molecular factors required for ChC/PyN AIS innervation in the neocortex, we devised a strategy in this study to target/manipulate PyN gene expression and label ChCs in the same neocortical layer. This strategy enabled us to screen PyN-expressed axonal CAMs in addition to select Ephs and ephrins present in the neocortex for their involvement in ChC/PyN AIS innervation in vivo. Strikingly, our results revealed that silencing of only L1CAM, and none of the other molecules tested, caused a marked reduction in neocortical PyN AIS innervation by ChCs. In line with this, we found the number of VGAT and gephyrin puncta at the AIS, but not along the somatodendritic compartment, to be concomitantly reduced in L1CAM-depleted PyNs, indicating that PyN L1CAM selectively regulates ChC/PyN AIS synaptic innervation and not the subcellular targeting of other PyN subcellular domains by other interneuron subtypes. Our data further reveal that PyN L1CAM plays a dual role in this process as it is required for both the establishment and maintenance of neocortical ChC/PyN AIS innervation postnatally. Regarding establishment, we found that 1) L1CAM expression at the AIS of PyNs markedly increases during the time window of initial ChC/PyN AIS innervation and 2) depleting PyN L1CAM perturbs ChC/PyN AIS innervation during the same time frame. With regards to maintenance, silencing of PyN L1CAM after the majority of ChC/PyN axoaxonic synapses have already been established still causes a significant decrease in ChC/PyN innervation and gephyrin puncta number per AIS. Thus, to the best of our knowledge, our results identify PyN L1CAM as the first known adhesion molecule required for the establishment and maintenance of neocortical ChC/PyN AIS innervation.
L1CAM is the founding member of the L1 family of CAMs which also includes NrCAM, CHL1, and Neurofascin in vertebrates (Maness and Schachner, 2007). The L1CAM protein was initially described as a neuronal cell adhesion molecule involved in neurite/axon outgrowth (Appel et al., 1993; Lemmon et al., 1989; Rathjen and Schachner, 1984). Subsequent studies have also implicated L1CAM in processes such as neuronal migration, axon guidance, synapse development/growth, and the arborization of presynaptic terminals (Castellani et al., 2000; Cohen et al., 1998; Enneking et al., 2013; Guan and Maness, 2010; Lindner et al., 1983). While these previous studies primarily focused on aspects of early development and/or the presynaptic function of L1CAM, our findings uncover a novel, previously unrecognized postsynaptic role for PyN L1CAM in regulating ChC/PyN AIS synaptic innervation/function in the neocortex. Importantly, this postsynaptic function of L1CAM is not compensated for by other L1 family members, as knockdown of L1CAM on its own is sufficient to cause the innervation phenotype. Moreover, depletion of neither NF186, NrCAM, nor CHL1 affected neocortical ChC/PyN AIS innervation. Our findings that L1CAM, but not NF186 or NrCAM, is required for innervation is intriguing, especially given that L1CAM is localized pan-axonally and not restricted to the AIS like NF186 and NrCAM. A likely explanation for this is that the presynaptic partner(s) of L1CAM on ChC axon terminals or in the surrounding environment is/are not recognized by the other CAMs. At present, the identity of L1CAM’s presynaptic binding partner(s) on ChC cartridges remains unknown. In this regard, a number of molecules have been reported to interact in trans with L1CAM but not with NF186 or NrCAM, including L1CAM itself, other Ig-domain containing proteins (e.g. ALCAM), extracellular matrix proteins (e.g. neurocan), and integrins (e.g. αVβ3, αIIbβ3) (Blaess et al., 1998; DeBernardo and Chang, 1996; Friedlander et al., 1994; Oleszewski et al., 1999). We can preclude the involvement of a trans L1CAM homophilic interaction, as silencing of L1CAM in ChCs did not affect ChC/PyN AIS innervation (data not shown). Exploring whether any other known L1CAM trans-interacting molecules are expressed on ChC cartridges or in the surrounding milieu and mediate the innervation of PyN AISs would be a worthwhile endeavor. Alternatively, we cannot exclude the possibility that a novel L1CAM ligand present on ChC cartridges or in the surrounding extracellular milieu mediates ChC/PyN AIS innervation. Future studies will be required to identify the presynaptic partner(s) of L1CAM necessary for proper neocortical ChC/PyN AIS innervation.
How does L1CAM govern selective PyN AIS innervation despite its pan-axonal localization? Our findings support a model in which the AIS AnkG-βIV-spectrin cytoskeletal complex anchors and clusters L1CAM at the AIS to promote high-affinity cell adhesion between ChC cartridges and PyN AISs, thereby facilitating axo-axonic synapse formation/stabilization. Indeed, we found that disruption of L1CAM-AnkG-βIV-spectrin interactions perturbs the subcellular distribution of surface L1CAM, reducing its enrichment at the AIS, and most importantly impairs PyN AIS innervation by ChCs. In contrast, the AnkG free pool of L1CAM in the distal portion of the axon is likely more diffusible and not clustered (Boiko et al., 2007), rendering it incapable of mediating ChC/PyN synaptic connections. In line with our findings, previous work showed that cerebellar PC-expressed NF186 also requires association with AnkG to mediate AIS pinceau synapse formation (Ango et al., 2004), suggesting a conserved role for AnkG in the anchoring/clustering of L1 family members at the AIS. It is noteworthy though, that while an AnkG-free form of NF186 at PC soma helps basket axon terminals navigate from the PC soma to the AIS, there is no indication that the AnkG-free form of L1CAM in the distal portion of the axon directs ChC cartridges from the distal axon of PyNs to their AIS. Possibly, direct innervation of neocortical PyN AISs by ChC cartridges occurs as a result of the unique, axonal arborization of ChCs. These cells undergo exuberant axonal overgrowth and branching followed by pruning (Inan and Anderson, 2014; Steinecke et al., 2017; Taniguchi et al., 2013); this is not the case for basket cells, which directionally orient their axon collaterals towards PC soma (Ango et al., 2004). In this regard, our data show that neocortical ChC axonal branching and complexity vastly increase at P11/P12, which interestingly coincides with an increase in PyN L1CAM expression at the AIS and the onset of ChC/PyN AIS innervation. It is therefore plausible that such morphometric changes bring ChC cartridges in close proximity to their target cells, allowing them to contact the AIS, bind AnkG-anchored L1CAM, and stabilize their connections. In contrast, the lesser-branched basket cell axons may require a molecular guide to help navigate to the AIS of cerebellar PCs (Ango et al., 2004; Telley et al., 2016). We do not exclude the possibility, however, that another/other molecule(s) contribute(s) to ChC axon targeting to PyN AISs either by engaging in repulsive mechanisms at subcellular compartments other than the AIS or by functioning at the AIS itself where it may or may not act in cis with L1CAM. So far, though, we have found that besides L1CAM, silencing of any of the other transmembrane adhesion molecules known to be localized at the AIS of PyNs did not disrupt neocortical ChC/PyN AIS innervation. Future work will be needed to fully elucidate the targeting mechanisms underlying neocortical PyN AIS innervation by ChCs.
Taken together, our findings unveil a critical role for PyN L1CAM, assembled by the AnkG-βIV-spectrin cytoskeletal complex at the AIS, in the axo-axonic innervation of neocortical PyNs by ChCs, thereby providing novel mechanistic insight into this unique form of subcellular innervation in the neocortex. Importantly, given our findings that L1CAM is required for proper neocortical ChC/PyN AIS innervation, this study also sheds light on the molecular mechanisms of connectivity defects underlying brain disorders. Interestingly, in this regard, mutations in L1CAM have been reported in patients with a broad spectrum of neurological abnormalities and mental retardation, which together are termed L1 syndrome (Jouet et al., 1994; Kenwrick et al., 1996; Weller and Gartner, 2001). While major corpus callosum agenesis, retardation, adducted thumbs, shuffling gait, and hydrocephalus (CRASH) defects associated with various L1CAM mutations are likely due to perturbations in early developmental processes, such as neuronal migration and neurite outgrowth, several genetic alterations in the L1CAM gene are also associated with schizophrenia and epilepsy (Kurumaji et al., 2001; Poltorak et al., 1997; Weller and Gartner, 2001), which commonly involve defects in neuronal connectivity. Thus, our findings that loss of L1CAM function causes postnatal defects in neocortical ChC/PyN AIS innervation and ultimately connectivity may provide a mechanistic explanation for some of the neurological aspects of L1 syndrome.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Linda Van Aelst (vanaelst@cshl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All experiments were carried out in accordance with the guidelines of the Animal Care and Use Committee of Cold Spring Harbor Laboratory (CSHL). Nkx2.1-CreER (JAX Stock 014552) and Rosa26-loxpSTOPloxp-TdTomato (Ai9) (JAX Stock 007905) were provided by Z.J. Huang (CSHL). To genetically label a sparse population of neocortical chandelier cells (ChCs), we crossed Nkx2.1-CreER+/− mice with Rosa26-loxpSTOPloxp-tdTomato (Ai9+/+) reporter mice. To properly identify embryonic days 15.5 (E15.5) and E18.5 for in utero electroporations (IUEs) and tamoxifen (TMX) inductions, respectively, Swiss Webster females (Charles River) were housed with Nkx2.1-CreER+/−;Ai9+/+ males overnight and checked for a vaginal plug between 8:00–9:00 AM the next morning. Positive plug identification was designated E0.5. Timed-pregnant female CD1 mice (Charles River) were used for E13.5 and E15.5 double IUE experiments targeting ChCs and pyramidal neurons (PyNs), respectively. Mice were housed in groups of five per cage or singly if pregnant under standard vivarium conditions with a 12 h light/dark cycle. Animals were fed ad libitum and their health status were routinely monitored. Both male and female animals, except for SLENDR studies (see below), were used in all experiments.
HEK293T and Neuro-2a cell cultures
HEK293T (ATCC, RRID: CVCL_0063) and Neuro-2a cells (ATCC, RRID: CVCL_0470) were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS) (HyClone), 100 IU/ml penicillin (Thermo Fisher Scientific), and 100 μg/ml streptomycin (Thermo Fisher Scientific) in a humidified incubator at 37°C with 5% CO2. The cell lines used in this study were not further authenticated and not found to be on the list of commonly misidentified cell lines (International Cell Line Authentication Committee).
Mouse cortical neuron culture
For the preparation of primary mouse cortical neuron cultures, cortices were dissected from E15.5 CD1 embryos of both sexes and digested in Hank’s Balanced Salt Solution (HBSS) (Thermo Fisher Scientific) containing 0.25% trypsin (Sigma-Aldrich) and 0.1 mg/ml DNase (New England Biolabs) at 37°C for 15 min. Digested cortices were then resuspended in Minimal Essential Medium (MEM) (Thermo Fisher Scientific) containing 10% FBS, triturated with a glass Pasteur pipette, and filtered using a 70 μm cell strainer to obtain a single-cell suspension. The cell suspension was centrifuged at 1000 × g for 5 min at room temperature (RT) and the pellet was resuspended in MEM containing 10% FBS. Neurons were plated at a density of 1×10 /cm on 12 mm glass coverslips (coated with 0.1 mg/ml poly-D-lysine (Sigma-Aldrich)) for immunofluorescence experiments or at a density of 2×106 per well of a 6-well plate (coated with 0.1 mg/ml poly-D-lysine) for Western blot experiments. 2–4 h after plating, cell culture medium was replaced with Neurobasal medium (Thermo Fisher Scientific) supplemented with 2% B27 (Thermo Fisher Scientific), 100 IU/ml penicillin, 100 μg/ml streptomycin, and 4 mM L-glutamine (Thermo Fisher Scientific). Neurons were maintained in a humidified incubator at 37°C with 5% CO2. Half of the medium was replaced with fresh medium every 7 days.
METHOD DETAILS
Experimental Design
Animals were randomly allocated to experimental groups prior to experimental manipulation(s), and the experimenter was blinded to the condition until the experiment and quantifications/analyses were complete. For all IUE experimental conditions, analyses were performed on brains from a minimum of 3 electroporated animals. For all immunostaining experiments (in vivo and in vitro), quantifications/analyses were performed on cells from at least 3 individual animals. All western blotting experiments were performed in triplicate and depicted images are representative. No statistical methods were used to predetermine sample size in experiments. No data or animals were excluded from analysis.
Experimental constructs
The pCAG-MCS vector is a derivative of pCAGGS and was made by first blunting and religating the Xhol site of pCAGGS, thus abolishing the Xhol site, and then inserting a multiple cloning site (MCS) into the EcoRI site, abolishing the EcoRI site in the process. Human L1CAM and AnkG-binding mutant human L1CAM-Y1229H expression vectors (pCAG-hL1CAM and pCAG-hL1CAM-Y1229H, respectively) were generated by subcloning full-length L1CAM cDNA fragments from pcDNA3.1-hL1CAM (AddGene#12307) and pcDNA3.1-hL1CAM-Y1229H (gift from P. Maness), respectively, into the EcoRI and Pmel sites of pCAG-MCS-4, which is a derivative of pCAG-MCS with an EcoRI- and PmeI-containing MCS inserted into the PacI and NheI sites of pCAG-MCS. A full-length SCN1B cDNA fragment was obtained via RT-PCR of rat cortical mRNA (isolated from P14 rat cortices) and the cDNA was subcloned into the XhoI and EcoRI sites of pcDNA3.1/myc-His-B (Thermo Fisher Scientific). For RNA interference (RNAi) experiments, DNA fragments encoding control short hairpin RNA (shRNA) (Ctrl: 5’-GCTATACGGGATCGAAAGA-3’) or shRNAs directed against mouse L1CAM (L1CAM#1: 5 ‘-GCATCCACTTCAAACCCAA-3 ‘; L1CAM#2 5’-AGCCTTACCAGAAGGGAAA-3’), mouse NrCAM (5’-GCAATGCCTCTAACAAATA-3’), mouse TAG-1 (5’-CCTGCTTTGCTGAGAACTT-3’), mouse CASPR2 (5’-GATTAGAGCCAGAGGGAAT-3’), mouse EphA3 (5’-GGACCTATGTTGATCCACATA-3’), mouse EphA4 (5’-GCAGCACCATCATCCATTG-3’), mouse EphB2 (5’-ACGAGAACATGAACACTAT-3’), mouse ephrinBl (5’-CACTGTGCTTGATCCCAAT-3’), mouse ephrinB2 (5’-GCAGACAGATGCACAATTA-3’), mouse ephrinB3 (5’-GCCTTCGGAGAGTCGCCAC-3’), mouse ErbB4 (5’-CCAGACTACCTGCAGGAATAC-3’), and mouse DOCK7 (5’-GGTACAGTACACATTTACA-3’) were cloned into pSuper (Watabe-Uchida et al., 2006) via BglII and HindIII sites. The pCAG-miR vector backbone was made by inserting an EGFP-miR30 cassette (Stern et al., 2008) into the PacI and Nhel sites of pCAG-MCS. DNA fragments encoding miR30-based shRNAs against Renilla luciferase (5’-AGGAATT ATAATGCTTATCTA-3 ‘), mouse L1CAM (5’-GAGAATCAACGGAATGTCTAA-3’), mouse CHL1 (5’-TACCAGGATAGAGGAAATTAA- 3’), mouse ADAM22 (5’-ACATGGCAGATGTG ATCTATA-3’), mouse Neurofascin-186 (NF#1 5’-CACCAGTCAATGCCATCTATA-3 ‘; NF#2 5’-CACGATCTCGGTGAGAGTAAA- 3’), mouse SCN1B (5’-GTCTACCGTCTCCTCTTCTTA −3’), and mouse βIV-spectrin (βIV#1 5’-CAGGAGAAATTCTCAGAGTTA-3’; βIV#2 5’-GACCACGATCGAGAAACTCAA-3’) were cloned into the XhoI and EcoRI sites of pCAG-miR. The pCaMKII-miR vector backbone was constructed by replacing the CAG promoter of pCAG-miR with the CaMKII promoter via BamHI and PacI sites. pCaMKII-miR.LlCAM was constructed by inserting the miR30-based shRNA against mouse L1CAM into the XhoI and EcoRI sites of pCaMKII-miR. The pCAG-FLIP-miR vector backbone was described previously (Tai et al., 2014). pCAG-FLIP-miR.LlCAM was constructed by inserting the miR30-based shRNA against mouse L1CAM into the XhoI and EcoRI sites of pCAG-FLIP-miR. For lentiviral RNAi vectors (pTRIPΔU3-EF1α-EGFP + H1-shL1CAM#1, #2, and scramble control), DNA fragments encoding shRNAs against mouse L1CAM (shL1CAM#1 or shL1CAM#2) or scramble control were first cloned into the pSuper(EcoRI) vector (Janas et al., 2006) via BglII and HindIII sites. The H1 promoter and shRNA sequences were then isolated from pSuper(EcoRI) via EcoRI digestion and subcloned into the EcoRI site of pTRIPΔU3-EF1α-EGFP (Janas et al., 2006).
CRISPR construction
CRISPRs were designed at http://crispr.mit.edu provided by the Zhang laboratory and then cloned into pX330 CRISPR/Cas9 vector (pX330-U6-Chimeric_BB-CBh-hSpCas9; a gift from Dr. Feng Zhang (Addgene plasmid #42230)) following Zhang’s protocol (http://www.genome-engineering.org/crispr/?pageid=23). The target sequence of Nf186 is CCCCCGACGAGCAGTCCATT, and the control (LacZ) target sequence is GTGCGAATACGCGGACGCGAT.
In utero electroporation (IUE) and tamoxifen induction
To target/manipulate PyN gene expression and sparsely label ChCs in the same neocortical layer, ventricular zone (VZ)-directed IUE targeting neocortical progenitors was performed in Nkx2.1-CreER;Ai9 embryos. Specifically, timed-pregnant Swiss Webster females that were bred with Nkx2.1-CreER+/−;Ai9+/+ males were anesthetized at 15.5 d of gestation, the uterine horns were exposed, and approximately 1 μl of DNA solution (1 μg/ml) was injected manually into the lateral ventricle of their embryos using a beveled and calibrated glass micropipette (Drummond Scientific). After injection, five square 50 ms pulses of 45 V with 950 ms intervals were delivered across the uterus with two 5 mm electrode paddles (BTX, 45–0489) positioned on either side of the head (BTX, ECM830) (Fig. 1A1). After electroporation, the uterine horns were placed back in the abdominal cavity of the pregnant dam and the wound was surgically sutured. Tamoxifen (TMX; 3mg/30g of body weight) was administered to the pregnant dam by oral gavage at 18.5 d of gestation to induce CreER activity and excision of the STOP cassette, resulting in tdTomato red fluorescent protein (RFP) expression in a sparse population of nascent neocortical ChCs in their offspring. Pups were sacrificed at postnatal days as indicated.
For experiments investigating whether L1CAM is required for the maintenance of neocortical ChC/PyN AIS innervation, CD1 embryos were subjected to both E13.5 and E15.5 IUEs targeting nascent ChCs and PyNs, respectively (Fig. 5K). For E13.5 IUEs labeling nascent ChCs with tdTomato, 1 μl of pCAG-tdTomato plasmid (1μg/ml) was injected manually into the lateral ventricle of the embryos and custom-made tweezer electrodes were placed at about 60° from the brain’s horizontal plane so as to direct the current toward the ventral medial ganglionic eminence (vMGE) (Tai et al., 2014). Square electric pulses (35 V; 50 ms) were delivered five times at 950 ms intervals using an electroporator (BTX, ECM830). After electroporation, the uterine horns were placed back in the abdominal cavity of the pregnant dam and the wound was surgically sutured. Two days later, at E15.5, the same embryos were subjected to VZ-directed IUE with either an L1CAM-targeting or control Cre-dependent inducible RNAi expression vector (pCAG-FLIP-miR.L1CAM or pCAG-FLIP-miR.Ctrl, respectively) together with a vector expressing a TMX-inducible form of Cre recombinase (pCAG-ERT2CreERT2)(Matsuda and Cepko, 2007). TMX (3mg/30g of body weight) was administered to the doubly electroporated animals via IP injection at postnatal day 28 (P28) to induce CreER activity and expression of miR.LlCAM or miR.Ctrl. The animals were sacrificed at P40 for analysis.
Transfection of HEK293T, Neuro-2a, and primary cultured cortical neurons
HEK293T cells were transfected using the calcium phosphate co-precipitation method. Neuro-2a cells were transfected using FuGENE HD (Promega) according to the manufacturer’s instructions. Transfected cells were lysed 2 to 3 d after transfection and subjected to Western blot analysis. Dissociated primary cortical neurons were transfected using the Amaxa Nucleofection system (Amaxa, Lonza) (Yang et al., 2012). In brief, 4×106 cells for each experimental condition were resuspended in 110 μl electroporation buffer and transferred to an electroporation cuvette. Program O-005 was used to transfect the cells. After electroporation, cells were resuspended in MEM containing 10% FBS and plated at a density of 2×106 per well of a 6-well plate coated with 0.1 mg/ml poly-D-lysine. 2–4 h after plating, the medium was replaced with Neurobasal medium supplemented with 2% B27, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 4 mM L-glutamine. Cells were lysed 7 d (or 14 d in the case of AnkG and ADAM22) after transfection and subjected to Western blot analysis. For immunofluorescence experiments, electroporated neurons were plated on 12 mm glass coverslips (coated with 0.1 mg/ml poly-D-lysine) at a density of 1×102/cm2 and immunostained at days in vitro 10 (DIV10).
Lentivirus production and infection of primary neuronal cultures
Lentiviral supernatants were prepared and concentrated largely as described (Janas et al., 2006). Briefly, lentiviruses were produced by co-transfecting the transfer vector (pTRIPΔU3-EF1α-EGFP + H1-shL1CAM#1, #2, or scramble control), the HIV-1 packaging vector Δ8.9 (pCMV-Δ8.9), and the VSVG envelope glycoprotein vector (pVSVG) into HEK293T cells using the calcium phosphate co-precipitation method (Janas et al., 2006). Supernatants of culture media were collected 48 h after transfection and concentrated via ultracentrifugation at 100,000 × g for 2 h. The resulting pellet was resuspended in PBS, flash-frozen, and stored at −80°C. Primary mouse cortical neurons prepared at E15.5 (see above) were infected at DIV3, harvested at DIV10, and subsequently processed for Western blot analysis.
Western blotting
Cells were washed with ice-cold PBS and lysed in RIPA lysis buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 5% glycerol, 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF, and 1× cOmplete, EDTA-free protease inhibitor cocktail (Sigma-Aldrich). Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Cell lysates were resolved by SDS-PAGE and transferred to an Immobilon-P PVDF membrane (Millipore). Membranes were blocked in Tris-buffered saline with 0.5% Tween 20 (VWR) (TBST) containing 5% fat-free milk for 30 min at RT and then incubated in TBST with 5% bovine serum albumin (BSA) (Equitech-Bio, Inc.) and 0.05% NaN3 containing primary antibodies overnight at 4°C. The following day, the membranes were incubated in blocking buffer containing horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at RT. The following primary antibodies were used: anti-L1CAM (mouse mAb, 1:1000, Abcam ab24704), anti-TAG-1 (goat pAb, 1:1000, R&D Systems AF4439), anti-CHL1 (goat pAb, 1:1000, R&D Systems AF2147), anti-γ-tubulin (mouse mAb IgG1, 1:5000, Sigma-Aldrich T6557), anti-β-actin (mouse mAb IgG1, 1:9000, Sigma A5441), anti-vinculin (rabbit mAb, 1:2000, Cell Signaling Technology 13901T), anti-Myc (mouse mAb, 1:5000, EMD Millipore 05–724), anti-HA.11 (mouse mAb IgG1,κ, 1:1000, BioLegend 901514), anti-NrCAM (rabbit pAb, 1:1000, Abcam ab24344), anti-ADAM22 (mouse mAb IgG2b, 1:200, NeuroMab 75–083), and anti-AnkG (mouse mAb IgG1, 1:200, NeuroMab 75–187). HRP-conjugated anti-rabbit (Cell Signaling Technology 7047), anti-mouse (Dako P044701–2), and anti-goat (Santa Cruz Biotechnology sc-2350) secondary antibodies were used at 1:5,000. Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific) was used for detection of HRP activity.
Immunohistochemistry and immunocytochemistry
For immunostaining of tissue sections, animals were deeply anesthetized with isoflurane and perfused transcardially with PBS and 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer. Brains were post-fixed in 4% PFA in 0.1 M phosphate buffer overnight at 4°C and then cryoprotected with 30% sucrose in PBS. Of note, for neurofascin-186, NrCAM, and Nav1.6 immunostaining, animals were perfused and post-fixed in 1% PFA in 0.1 M phosphate buffer prior to cryoprotection. 50 μm thick coronal sections were subsequently generated using a Vibratome (Leica VT1000S). For gephyrin, TAG-1, and SCN1B immunostaining, antigen retrieval was performed prior to the application of primary antibodies. Namely, brain sections were incubated at 95°C in antigen retrieval buffer (25 mM Tris-HCl, pH 8.5; 1 mM EDTA, 0.05% SDS) for 15 min. For immunostaining of endogenous L1CAM in brain slices, fresh brains were flash frozen in liquid nitrogen, cryosectioned at 12 μm thickness using a cryostat (Leica CM1850), and mounted onto Superfrost Plus microscope slides (Fisherbrand). Mounted brain sections were then thawed at RT and briefly fixed in 2% PFA in PBS for 90 seconds. Following the aforementioned methods of tissue fixation and sectioning described for each immunohistochemistry (IHC) experiment, brain slices were blocked and permeabilized with 10% normal goat serum (NGS) or normal donkey serum (NDS) and 0.3% Triton X-100 in PBS at RT for 1 h and then incubated with primary antibodies diluted in 3% NGS or NDS and 0.3% Triton X-100 in PBS overnight at 4°C. Fluorescently-conjugated secondary antibodies diluted in 3% NGS or NDS and 0.3% Triton X-100 in PBS were applied for 1 h at RT the following day. For live cell surface labeling of L1CAM on DIV10 cortical neurons, cortical neuron cultures were washed twice with PBS and live-labeled with an anti-L1CAM antibody (1:150) diluted in PBS for 15 min at 37°C. The cultures were then fixed with 4% PFA in 0.1 M phosphate buffer for 15 min at 4°C, permeabilized with 0.1% Triton X-100 in PBS for 5 min, blocked with 10% NGS in PBS for 1 h at RT, and subsequently incubated with anti-GFP (1:1000) and anti-AnkG (1:1000) antibodies diluted in 3% NGS in PBS overnight at 4°C. The next day the cultures were incubated with fluorescently-conjugated secondary antibodies (1:2000) diluted in 3% NGS in PBS for 1 h at RT. The following primary antibodies were used: anti-GFP (chicken pAB, 1:1000, Aves Labs GFP 1020), anti-AnkG (mouse mAb IgG2a, 1:500 (IHC) or 1:1000 (immunocytochemistry), NeuroMab 75–146), anti-Neurofascin-186 (rabbit pAb, 1:500, gift from P. Brophy or rabbit mAb, 1:500, Cell Signaling Technology 15034), anti-RFP (rabbit pAb, 1:1000, Rockland 600–401-379), anti-tdTomato (goat pAb, 1:100, Sicgen Antibodies AB8181–200), anti-βIV-spectrin (rabbit pAb, 1:500, gift from M. Rasband), anti-NrCAM (rabbit pAb, 1:500, Abcam ab24344), anti-Gephyrin (mouse mAb IgG1, 1:500, Synaptic Systems 147011), anti-VGAT (guinea pig pAb, 1:500, Synaptic Systems 131004), anti-Nav1.6 (rabbit pAb, 1:100, Alomone Labs asc-009), anti-TAG-1 (goat pAb, 1:200, R&D Systems AF4439), anti-SCN1B (rabbit pAb, 1:100, Abcam ab107370), anti-HA (rabbit mAb, 1:500, Cell Signaling Technology 3724), and anti-L1CAM (rat mAb, 1:150, EMD Millipore MAB5272). The following secondary antibodies were used: Alexa Fluor (AF) 488 goat anti-chicken IgY (1:1000, Thermo Fisher Scientific A-11039), AF Plus 555 goat anti-rabbit (1:1000, Thermo Fisher Scientific A32732), AF 647 goat anti-mouse IgG2a (1:1000, Thermo Fisher Scientific A-21241), AF 488 goat anti-mouse IgG1 (1:1000, Thermo Fisher Scientific A-21121), AF 555 donkey anti-goat (1:1000, Thermo Fisher Scientific A-21432), AF 647 donkey anti-mouse (1:1000, Thermo Fisher Scientific A-31571), AF 594 donkey anti-rabbit (1:1000, Thermo Fisher Scientific R37119), and AF 488 donkey anti-chicken (1:1000, Jackson ImmunoResearch 703–545-155).
In vivo single-cell labeling of endogenous L1CAM by CRISPR-Cas9-mediated homology-directed repair (SLENDR)
As a complementary approach to in vivo endogenous L1CAM immunostaining, we took advantage of SLENDR, a CRISPR-Cas9-mediated homology-directed repair genome editing strategy (Mikuni et al., 2016), to insert an HA epitope tag into the genomic locus of L1CAM and subsequently visualize the protein’s subcellular localization via HA immunostaining at single-cell resolution in vivo. Specifically, a single-guide RNA (sgRNA) (5’-TGGTGGACCTTGCTATTCT-3’) targeting the vicinity of L1CAM’s stop codon was designed and cloned into the pX330 CRISPR/Cas9 vector following Zhang’s protocol (http://www.genome-engineering.org/crispr/7pageid=23) (pX330-crL1CAM). In addition, a corresponding single-stranded oligodeoxynucleotide (ssODN-L1CAM) (5’-ggcaagaaagagaaggaggcagcaggaggcaatgacagttcaggggctacctctcctatcaatcctgcagtagccctagaaTACCCATACGATGTTCCAGATTACGCTtagcaaggtccagccatgtgaggcagggccaagctgggcccagggccagaggtgcaggagagcccaggggccaagacacctggcc-3’; HA sequence underlined) was designed to mediate the integration of the HA epitope tag sequence into the genome immediately upstream of L1CAM’s stop codon. A cocktail of pX330-crL1CAM (1μg/μl), ssODN-L1CAM (20pM), and pCAG-YFP (1μg/μl) was then used in E13.5 IUEs targeting PyN progenitors, as described above. Given that L1CAM is located on the X chromosome, only electroporated male pups were analyzed to avoid any complications arising from differences in the number of alleles edited. Male pups were sacrificed at P10 and P13 and brain slices from such animals were immunostained (as described above) with anti-HA and anti-AnkG antibodies.
Preparation of acute brain slices and electrophysiology
CD1 mice (P27–30) were anaesthetized with isoflurane, decapitated, and their brains quickly removed and chilled in ice-cold dissection buffer (110 mM choline chloride, 25 mM NaHCO3, 1.25 mM NH2PO4, 2.5 mM KCl, 0.5 mM CaCl2, 7 mM MgCl2, 25 mM glucose, 11.6 mM ascorbic acid, and 3.1 mM pyruvic acid; gassed with 95% O2 and 5% CO2). Coronal slices (300 pm) containing the somatosensory cortex were cut in dissection buffer using a Vibratome (Leica VT1000S) and subsequently transferred to a storage chamber containing artificial cerebrospinal fluid (ACSF; 118 mM NaCl, 2.5 mM KCl, 26.2 mM NaHCO3, 1 mM NH2PO4, 20 mM glucose, 2 mM MgCl2, and 2 mM CaCl2 at 34 °C, pH 7.4; gassed with 95% O2 and 5% CO2). After at least 40 min recovery time, slices were transferred to RT and constantly perfused with ACSF.
Whole-cell patch-clamp recordings were performed on electroporated GFP+ PyNs in layer II/III in the somatosensory cortex. Recordings were obtained with Multiclamp 700B amplifiers (Molecular Devices) under visual guidance using a Zeiss Axioskop microscope equipped with both transmitted light illumination and epifluorescence illumination. Miniature inhibitory postsynaptic currents (mIPSCs) were recorded in voltage-clamp mode with borosilicate pipettes (3.5–5 MΩ). The internal solution (ACSF) contained the following: 115 mM cesium methanesulphonate, 20 mM CsCl, 10 mM HEPES, 2.5 mM MgCl2, 4 mM Na2-ATP, 0.4 mM Na3GTP, 10 mM Na-phosphocreatine, and 0.6 mM EGTA (pH 7.2). mIPSCs were recorded at a holding potential of 0 mV with 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX, 20 μM), DL-2-amino-5-phosphonopentanoic acid (DL-AP5, 100 μM), and tetrodotoxin (TTX; 1 μM) added to the ACSF. Electrophysiological data were acquired using Axon pCLAMP 10 software (Molecular Devices) and analyzed using Mini Analysis Program (Synaptosoft).
Confocal image acquisition and analysis of ChC/PyN percent innervation and inhibitory synapse number
For analysis of ChC/PyN AIS percent innervation, images of coronal brain slices (50 μm thickness) were acquired using an LSM 800 confocal laser-scanning microscope (Zeiss) with a 63x oil-immersion objective and sequential acquisition settings applied at a resolution of 1024×1024 pixels. 200 μm × 200 μm images of single RFP+ ChCs and neighboring GFP+ electroporated PyNs in layer II of the somatosensory cortex were collected from E18.5 TMX-induced Nkx2.J-CreER+/−;Ai9+/− mice (at postnatal ages as indicated) using a z-series of 40–50 images with a depth interval of 1 μm. ChC/PyN AIS percent innervation was calculated by dividing the number of GFP+ PyNs innervated by a single RFP+ ChC by the total number of GFP+ PyNs present in the entire 200 μm × 200 μm image z-stack. Average ChC cartridge number was determined for PyN L1CAM and control knockdown conditions by quantifying the number of RFP+ cartridges from single RFP+ ChCs in 200 μm × 200 μm confocal images acquired from Nkx2.J-CreER+/−;Ai9+/− coronal brain slices (50 μm thickness) as described above. Importantly, TMX administration at E18.5 was essential for standardizing our analyses of ChC/PyN AIS percent innervation to single ChCs, since such an induction protocol enables the sparse labeling of spatially isolated neocortical ChCs in Nkx2.J-CreER+/−;Ai9+/− animals. Of note, TMX induction even 24 hours earlier (at E17.5) causes a significantly larger population of overlapping and non-homogenously distributed ChCs to be labeled in the neocortex (Fig. S5A), which causes a marked increase in variability of ChC/PyN AIS percent innervation values across different images analyzed (Fig. S5B). Notably, variations in the number of electroporated GFP+ PyNs present in each field of view (FOV) did not impact ChC/PyN AIS percent innervation (Fig S5C). Moreover, “convex hull” analysis (Inan et al., 2013) of ChC/PyN AIS percent innervation was performed on serial reconstructions of entire individual RFP+ ChCs and all neighboring GFP+ PyNs within the domain of the ChC axonal arbor. Such reconstructions were generated from serial 320 μm × 320 μm z-stack images acquired from four to six 50 μm thick brain slices using an LSM 800 confocal laser-scanning microscope (Zeiss) equipped with an 40x oil-immersion objective. Importantly, this analysis recapitulated the PyN L1CAM knockdown innervation phenotype relative to the control condition obtained in the 200 μm × 200 μm FOV (Fig. S5D). Given this, all analyses of ChC/PyN AIS percent innervation were performed on 200 μm × 200 μm z-stacks of 40–50 images.
Representative maximum projection images of ChC/PyN AIS innervation with gephyrin or VGAT staining were generated using a z-series of 4–8 images with a depth interval of 0.37 μm. The average number of gephyrin or VGAT puncta per PyN AIS was determined manually. The percentage of RFP+ ChC boutons overlapping with AIS gephyrin puncta was calculated by dividing the number of RFP+ ChC boutons overlapping with gephyrin puncta by the total number of RFP+ ChC boutons per AIS. The average number of gephyrin or VGAT puncta per μm on PyN dendrites was quantified by manually counting the number of gephyrin or VGAT puncta within 30 μm of the cell body on the apical dendrite and dividing this sum by the length of the dendrite analyzed. To quantify the average number of VGAT puncta per μm on PyN cell bodies, single z-plane images were acquired using an LSM 800 confocal laser-scanning microscope (Zeiss) with a 63x oil-immersion objective and acquisition settings applied at a resolution of 1024 × 1024 pixels. The number of VGAT puncta overlapping with GFP+ PyN cell bodies was manually counted and the circumference of the cell bodies was measured using Zeiss Zen (Blue Edition) imaging software. The average number of VGAT puncta per μm on PyN cell bodies was then calculated by dividing the number of PyN cell body VGAT puncta by the circumference of the cell body. For quantification of the average fluorescence intensity (AU) of PyN perisomatic gephyrin signal, the “integrated fluorescence intensity” (the sum of the fluorescence intensity values of all pixels in a region of interest (ROI)) was calculated in a perisomatic ring (an intracellular ring ROI spanning 2 μm from the cell membrane) in a single z-plane image using ImageJ software. The “mean fluorescence background” was also measured by selecting an area(s) outside of the cell and next to each ROI that has no fluorescence. The average corrected fluorescence intensity of perisomatic gephyrin was then calculated as: ((Integrated fluorescence intensity of perisomatic gephyrin) – (Area of ROI × Mean fluorescence intensity of background readings))/Area of ROI.
Confocal image acquisition and analysis of endogenous and surface L1CAM and other AIS proteins
For quantification of endogenous L1CAM or L1CAM-HA expression levels in vivo, images of brain slices immunostained for L1CAM or HA (see above) were acquired using an LSM 800 confocal laser-scanning microscope (Zeiss) with a 63x oil-immersion objective and sequential acquisition settings applied at a resolution of 1024×1024 pixels. Representative maximum projection images of PyN AISs were generated using a z-series of 4–8 images with a depth interval of 0.37 μm. Relative fluorescence intensity of AIS-localized L1CAM or L1CAM-HA was calculated using ImageJ software. Namely, the relative fluorescence intensity was calculated by first measuring the “integrated fluorescence intensity” of L1CAM on the entire AIS in spatially isolated PyNs in addition to the “mean fluorescence background” (as described above). The relative corrected fluorescence intensity was then calculated as: ((Integrated fluorescence intensity of L1CAM) - (Area of the AIS × Mean fluorescence intensity of background readings))/Area of AIS normalized to P10. The relative fluorescence intensities of AIS-enriched proteins under PyN L1CAM knockdown conditions were calculated as described above and normalized to control conditions.
For surface L1CAM staining in cultured primary cortical neurons, images of randomly chosen GFP+ transfected cells were acquired using an LSM 800 confocal laser-scanning microscope (Zeiss) with a 40x oil-immersion objective. Sequential acquisition settings were applied at a resolution of 1024×1024 pixels. For representative images of GFP+ cultured cortical neurons, maximum projections of 4–6 sequential z-stack images at a 0.6 μm depth interval were generated. To calculate average surface L1CAM fluorescence intensity (AU), ImageJ software was used to measure the “integrated fluorescence intensity” of surface L1CAM on the AIS or distal axon (within a region of the axon spanning approximately 40 μm from the AIS) of an isolated GFP+ neuron as well as the “mean fluorescence background” (as described above). The average corrected fluorescence intensity of surface L1CAM was then calculated as: ((Integrated fluorescence intensity of surface L1CAM) – (Area of ROI × Mean fluorescence intensity of background readings))/Area of ROI. The average corrected fluorescence intensity of GFP for each ROI was also calculated in the same manner and the average corrected fluorescence intensity of surface L1CAM was then normalized to the average corrected fluorescence intensity of GFP for each ROI by dividing the average corrected fluorescence intensity of surface L1CAM by the average corrected fluorescence intensity of GFP. In addition, the ratio of GFP-normalized surface L1CAM fluorescence intensity at the AIS to GFP-normalized surface L1CAM fluorescence intensity at the distal axon was calculated per cell and averaged for each condition. AIS length and area were measured using the appropriate measurement tools in Zeiss Zen (Blue Edition) imaging software and ImageJ, respectively.
ChC axonal 3D reconstruction and morphometric analyses
For immunostaining of tissue sections for ChC axonal 3D reconstructions and morphometric analyses, brains from E18.5 TMX-induced Nkx2.J-CreER+/−;Ai9+/− animals were collected at the postnatal ages indicated and processed/sectioned and immunostained as described above. Immunostained brain sections were subsequently imaged using an LSM 800 confocal laser-scanning microscope (Zeiss) with a 63x oil-immersion objective and sequential acquisition settings applied at a resolution of 1024×1024 pixels. Of note, for all 3D reconstructions, 200 μm × 200 μm images of single RFP+ ChCs, which were spatially isolated from other RFP+ ChCs and processes, were collected in the somatosensory cortex using a z-series of 40–50 images with a depth interval of 1 μm. ChC axonal arbors were reconstructed using the User-Guided tracing mode of Neurolucida 360 (Microbrightfield Bioscience). Morphometric analyses of ChC axonal complexity, length, and the number of branch points as well as ChC axonal Sholl analysis were carried out using Neurolucida Explorer (Microbrightfield Bioscience). ChC axonal complexity index was defined as: (∑ terminal orders + number of terminals) × (total axonal length) where the number of “terminal orders” for each terminal point is calculated as the number of branches that appear in proceeding backward from the defined terminal to the ChC soma. ChC “terminals” (or axonal endings) were defined as the smallest RFP+ ChC axonal processes clearly identifiable from the last branching point in high resolution confocal images.
QUANTIFICATION AND STATISTICAL ANALYSIS
For quantifications/analyses of ChC/PyN percent innervation, 5–29 ChCs and 4–630 PyNs per ChC from at least 3 animals were analyzed. For immunostaining and SLENDR-based fluorescence intensity experiments, 14–155 AISs from 3 animals (in vivo) or 35–43 neurons from 3 independent cultures (from at least 3 individual animals) (in vitro) were analyzed. Of note, for in vivo and in vitro immunostaining experiments, all depicted images are representative. For quantifications/analyses of PyN AIS length and area, 14–40 AISs from 3 animals were analyzed and for average cartridge number per ChC, 10 ChCs from 3 animals were analyzed. Moreover, for gephyrin and VGAT immunostaining analysis, 8–49 AISs, 30–34 cell bodies, or 24–51 dendrites from at least 3 animals were analyzed. 16 GFP+ PyNs from 4 animals were analyzed for electrophysiological measurements of mIPSC amplitude and frequency. For ChC soma and axon 3D reconstructions and morphometric analyses, maximum projection renderings are representative and 4–7 ChCs from 3 animals were analyzed. Lastly, all western blot experiments were performed in triplicate and depicted images are representative. Exact sample sizes for all experiments are indicated in their respective figure legends.
All data are presented as mean ± SEM. Differences between experimental conditions were analyzed with GraphPad Prism 7 software (GraphPad). Comparisons of two groups were made using unpaired two-tailed Student’s t-tests. For comparisons of more than two groups, one-way analysis of variance (ANOVA) with post hoc Tukey-Kramer multiple comparison tests were performed. Statistical significance was defined as p < 0.05, p < 0.01, or p < 0.001 (indicated as *, **, or ***, respectively). p values ≥ 0.05 were considered not significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken polyclonal anti-GFP | Aves Labs | Cat# GFP-1020; RRID: AB_10000240 |
| Mouse monoclonal anti-AnkG (IgG2a) | NeuroMab | Cat# 75–146; RRID: AB_10673030 |
| Mouse monoclonal anti-AnkG (IgG1) | NeuroMab | Cat# 75–187; RRID: AB_2057718 |
| Rabbit polyclonal anti-Neurofascin-186 | Gift from P.J. Brophy | N/A |
| Rabbit monoclonal anti-Neurofascin-186 | Cell Signaling Technology | Cat# 15034S; RRID:AB_2773024 |
| Rabbit polyclonal anti-RFP | Rockland | Cat# 600–401-379; RRID: AB_2209751 |
| Goat polyclonal anti-tdTomato | Sicgen Antibodies | Cat# AB8181–200; RRID:AB_2722750 |
| Rabbit polyclonal anti-βIV-Spectrin | Gift from M. Rasband | N/A |
| Rabbit polyclonal anti-NrCAM | Abcam | Cat# ab24344; RRID: AB_448024 |
| Mouse monoclonal anti-Gephyrin (IgG1) | Synaptic Systems | Cat# 147011; RRID: AB_887717 |
| Guinea pig polyclonal anti-VGAT | Synaptic Systems | Cat# 131004; RRID: AB_887873 |
| Rabbit polyclonal anti-Nav1.6 | Alomone Labs | Cat# asc-009; RRID: AB_2040202 |
| Rat monoclonal anti-L1CAM | EMD Millipore | Cat# MAB5272; RRID: AB_2133200 |
| Mouse monoclonal anti-L1CAM | Abcam | Cat# ab24704; RRID: AB_448241 |
| Goat polyclonal anti-TAG-1 | R&D Systems | Cat# AF4439; RRID: AB_2044647 |
| Goat polyclonal anti-CHL1 | R&D Systems | Cat# AF2147; RRID: AB_2079332 |
| Mouse monoclonal anti-ADAM22 | NeuroMab | Cat# 75–083; RRID:AB_10675128 |
| Rabbit polyclonal anti-SCN1B | Abcam | Cat# ab107370; RRID: AB_10861904 |
| Mouse monoclonal anti-γ-Tubulin | Sigma-Aldrich | Cat# T6557; RRID: AB_477584 |
| Rabbit monoclonal anti-Vinculin | Cell Signaling Technology | Cat# 13901; RRID:AB_2728768 |
| Mouse monoclonal anti-β-Actin | Sigma-Aldrich | Cat# A5441; RRID: AB_476744 |
| Mouse monoclonal anti-Myc | EMD Millipore | Cat# 05–724; RRID: AB_11211891 |
| Mouse monoclonal anti-HA.11 | BioLegend | Cat# 901514; RRID: AB_2565336 |
| Rabbit monoclonal anti-HA | Cell Signaling Technology | Cat# 3724; RRID: AB_1549585 |
| Goat anti-chicken IgYAlexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11039; RRID: AB_142924 |
| Goat anti-rabbit Alexa Fluor Plus 555 | Thermo Fisher Scientific | Cat# A32732; RRID: AB_2633281 |
| Goat anti-mouse IgG2a Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21241; RRID: AB_141698 |
| Goat anti-mouse IgG1 Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21121; RRID: AB_141514 |
| Donkey anti-goat Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21432; RRID: AB_141788 |
| Donkey anti-mouse Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31571; RRID: AB_162542 |
| Donkey anti-rabbit Alexa Fluor 594 | Thermo Fisher Scientific | Cat# R37119; RRID: AB_2556547 |
| Donkey anti-chicken IgY Alexa Fluor 488 | Jackson ImmunoResearch | Cat# 703–545-155; RRID: AB_2340375 |
| Anti-rabbit-HRP | Cell Signaling Technology | Cat# 7047; RRID: AB_2099233 |
| Anti-mouse-HRP | Dako | Cat# P044701–2; RRID: AB_2617137 |
| Anti-goat-HRP | Santa Cruz Biotechnology | Cat# sc-2350; RRID: AB_634811 |
| Bacterial and Virus Strains | ||
| One Shot Stbl3 Chemically Competent E. Coli | Thermo Fisher Scientific | Cat# C737303 |
| Lentivirus: TRIPΔU3-EF1α-EGFP | (Janas et al., 2006) | N/A |
| Lentivirus: TRIPΔU3-EF1α-EGFP + H1-shL1CAM#1 | This paper | N/A |
| Lentivirus: TRIPΔU3-EF1α-EGFP + H1-shL1CAM#2 | This paper | N/A |
| Lentivirus: TRIPΔU3-EF1α-EGFP + H1-scramble control | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma-Aldrich | Cat# T5648; CAS: 10540–29-1 |
| cOmplete, EDTA-free protease inhibitor cocktail | Sigma-Aldrich | Cat# 11873580001 |
| Poly-D-Lysine | Sigma-Aldrich | Cat# A-003-E |
| Bovine Serum Albumin | Equitech-Bio, Inc. | Cat# BAH65 |
| Tetrodotoxin | Tocris | Cat# 1078 |
| 6-cyano-7-nitroquinoxaline-2, 3-dione | Sigma-Aldrich | Cat# C127 |
| DL-2-amino-5-phosphonopentanoic acid | Sigma-Aldrich | Cat# 074K38051 |
| Critical Commercial Assays | ||
| FuGENE HD | Roche | Cat# 4709691001 |
| AMAXA Nucleofector Kit for mouse neurons | Lonza | Cat# VPG1001 |
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | Cat# M0530S |
| Takara DNA Ligation Kit Ver.2.1 | Takara | Cat# 6022 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Pierce™ ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat# 32106 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| Neuro-2a | ATCC | Cat# CCL-131; RRID: CVCL_0470 |
| Experimental Models: Organisms/Strains | ||
| Mouse: CD1 | Charles River | Cat# CRL:22; RRID: IMSR_CRL:22 |
| Mouse: CFW(Swiss Webster) | Charles River | Cat# CRL:24; RRID: IMSR_CRL:24 |
| Mouse: Nkx2–1tm1.1(Cre/ERT2)Zjh/J | Gift from Z.J. Huang | JAX: 014552; RRID: IMSR_JAX:014552 |
| Mouse: B6;129S6-Gt(Rosa)26Sortm9(CAG-tdTomato)Hze/J | Gift from Z.J. Huang | JAX: 007905; RRID: IMSR_JAX:007905 |
| Oligonucleotides | ||
| LacZ sgRNA: GTGCGAATACGCGGACGCGAT | This paper | N/A |
| Nf186 sgRNA, targeting exon 26: CCCCCGACGAGCAGTCCATT | This paper | N/A |
|
L1CAM sgRNA, targeting exon 29 (C-terminal): TGGTGGACCTTGCTATTCT |
This paper | N/A |
| ssODN sequence to integrate HA sequence into the genome just upstream of L1CAM’s stop codon (HA sequence; underlined): ggcaagaaagagaaggaggcagcaggaggcaatgacagttcaggggctacctctcctatcaatcctgcagtagccctagaaTACCCATACGATGTTCCAGATTACGCT tagcaaggtccagccatgtgaggcagggccaagctgggcccagggccagaggtgcaggagagcccaggggccaagacacctggcc |
This paper | N/A |
| For all shRNA sequences, see Table S2 | N/A | N/A |
| Recombinant DNA | ||
| Plasmid: pX330-U6-Chimeric_BB_CBh_hSpCas9 | Gift from F. Zhang | Addgene plasmid #42230 (Cong et al., 2013) |
| Plasmid: pX330-crNF186 | This paper | N/A |
| Plasmid: pX330-crLacZ | This paper | N/A |
| Plasmid: pX330-crL1CAM | This paper | N/A |
| Plasmid: pSuper | OligoEngine | Cat# VEC-IND-0002 |
| Plasmid: pSuper-shL1CAM#1 | This paper | N/A |
| Plasmid: pSuper-shL1CAM#2 | This paper | N/A |
| Plasmid: pSuper-shNrCAM | This paper | N/A |
| Plasmid: pSuper-shTAG-1 | This paper | N/A |
| Plasmid: pSuper-shCASPR2 | This paper | N/A |
| Plasmid: pSuper-shEphA3 | This paper | N/A |
| Plasmid: pSuper-shEphA4 | This paper | N/A |
| Plasmid: pSuper-shEphB2 | This paper | N/A |
| Plasmid: pSuper-shEfnB1 | This paper | N/A |
| Plasmid: pSuper-shEfnB2 | This paper | N/A |
| Plasmid: pSuper-shEfnB3 | This paper | N/A |
| Plasmid: pSuper-shCtrl | This paper | N/A |
| Plasmid: pCAGGS (pCAG) | (Watabe-Uchida et al., 2006) | N/A |
| Plasmid: pCAG-MCS | This paper | N/A |
| Plasmid: pCAG-MCS-4 | This paper | N/A |
| Plasmid: pCAG-miR | (Tai et al., 2014) | N/A |
| Plasmid: pCAG-miR.L1CAM | This paper | N/A |
| Plasmid: pCAG-miR.ADAM22 | This paper | N/A |
| Plasmid: pCAG-miR.NF#1 | This paper | N/A |
| Plasmid: pCAG-miR.NF#2 | This paper | N/A |
| Plasmid: pCAG-miR.SCN1B | This paper | N/A |
| Plasmid: pCAG-miR.βIV#1 | This paper | N/A |
| Plasmid: pCAG-miR.βIV#2 | This paper | N/A |
| Plasmid: pCAG-miR.CHL1 | This paper | N/A |
| Plasmid: pCAG-miR.Ctrl | This paper | N/A |
| Plasmid: pCAG-FLIP-miR | (Tai et al., 2014) | N/A |
| Plasmid: pCAG-FLIP-miR.L1CAM | This paper | N/A |
| Plasmid: pCAG-FLIP-miR.Ctrl | This paper | N/A |
| Plasmid: pCAG-Cre | (Tai et al., 2014) | N/A |
| Plasmid: pCAG-ERT2CreERT2 | Gift from C. Cepko | Addgene plasmid #13777(Matsuda and Cepko, 2007) |
| Plasmid: pCaMKII-miR | This paper | N/A |
| Plasmid: pCaMKII-miR.L1CAM | This paper | N/A |
| Plasmid: pCaMKII-miR.Ctrl | This paper | N/A |
| Plasmid: pcDNA3.1 mouse L1CAM | Gift from K. Kaibuchi | N/A |
| Plasmid: pcDNA3.1 human L1CAM | Gift from V. Lemmon | Addgene plasmid #12307(Hlavin and Lemmon, 1991) |
| Plasmid: pcDNA3.1 human L1CAM Y1229H | Gift from P. Maness | N/A |
| Plasmid: pCAG human L1CAM | This paper | N/A |
| Plasmid: pCAG human L1CAM-Y1229H | This paper | N/A |
| Plasmid: HA-NF186 | Gift from M. Rasband | N/A |
| Plasmid: pcDNA3.1 mouse CHL1 | Gift from K. Kaibuchi | N/A |
| Plasmid: pcDNA3.1 rat SCN1B-Myc | This paper | N/A |
| Plasmid: pCMV6-XL4-NrCAM-FLAG | Gift from D. Montag | N/A |
| pCMV-Δ8.9 | (Janas et al., 2006) | N/A |
| pVSVG | (Janas et al., 2006) | N/A |
| Plasmid: pTRIPΔU3-EF1α-EGFP | Gift from B. Verhasselt | N/A |
| Plasmid: pTRIPΔU3-EF1α-EGFP + H1- shL1CAM#1 | This paper | N/A |
| Plasmid: pTRIPΔU3-EF1α-EGFP + H1-shL1CAM#2 | This paper | N/A |
| Plasmid: pTRIPΔU3-EF1α-EGFP + H1-scramble control | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | http://imagej.nih.gov/ij/; RRID: SCR_003070 |
| LSM 5 Image Browser | Zeiss | http://www.embl.de/eamnet/html/body_image_browser.html; RRID: SCR_014344 |
| Zeiss Zen (Blue Edition) Imaging Software | Zeiss | http://www.zeiss.com/microscopy/en_us/products/microscope-software/zen.html#introduction; RRID: SCR_013672 |
| Axon pCLAMP 10 Electrophysiological Data Acquisition & Analysis Software | Molecular Devices | http://mdc.custhelp.com/app/answers/detail/a_id/18779/~/axon%E2%84%A2pclamp%E2%84%A2–10-electrophysiology-data-acquisition-%26-analysis-software-download; RRID:SCR_011323 |
| Mini Analysis Program | Synaptosoft | http://www.synaptosoft.com/MiniAnalysis/; RRID:SCR_002184 |
| GraphPad Prism 7 | GraphPad | http://www.graphpad.com/scientific-software/prism; RRID: SCR_002798 |
| Neurolucida | Microbrightfield Bioscience | https://www.mbfbioscience.com/neurolucida; RRID: SCR_001775 |
Highlights.
NF186 is dispensable for neocortical PyN AIS innervation by ChCs
Postsynaptic PyN-expressed L1CAM regulates PyN AIS synaptic innervation by ChCs
L1CAM is required for the establishment and maintenance of ChC/PyN AIS innervation
AnkG-mediated L1CAM anchoring at the AIS is necessary for ChC/PyN AIS innervation
ACKNOWLEDGMENTS
We thank members of the Van Aelst lab, Z.J. Huang, J. Skowronski, and E.-E. Govek for helpful discussions and critical reading of the manuscript. We also thank P. Brophy, M. Rasband, P. Maness, V. Lemmon, K. Kaibuchi, C. Cepko, D. Montag, F. Zhang, and B. Verhasselt for providing critical reagents and Z.J. Huang for providing Nkx2.1-CreER and Ai9 mice. This work was supported by US National Institutes of Health (NIH) grants R01MH082808 and R01NS082266 to L. Van Aelst, US NIH fellowship F31MH117871 to N.B. Gallo, and NARSAD Young Investigator grant to M. Wang.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information includes six figures and two tables.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Anderson GR, Galfin T, Xu W, Aoto J, Malenka RC, and Sudhof TC (2012). Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc Natl Acad Sci U S A 109, 18120–18125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ango F, di Cristo G, Higashiyama H, Bennett V, Wu P, and Huang ZJ (2004). Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at purkinje axon initial segment. Cell 119, 257–272. [DOI] [PubMed] [Google Scholar]
- Appel F, Holm J, Conscience JF, and Schachner M (1993). Several extracellular domains of the neural cell adhesion molecule L1 are involved in neurite outgrowth and cell body adhesion. J Neurosci 13, 4764–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariza J, Rogers H, Hashemi E, Noctor SC, and Martinez-Cerdeno V (2018). The Number of Chandelier and Basket Cells Are Differentially Decreased in Prefrontal Cortex in Autism. Cereb Cortex 28, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi S, Betley JN, Comer JD, Brenner-Morton S, Bar V, Shimoda Y, Watanabe K, Peles E, Jessell TM, and Kaltschmidt JA (2014). Neuronal Ig/Caspr recognition promotes the formation of axoaxonic synapses in mouse spinal cord. Neuron 81, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baohan A, Ikrar T, Tring E, Xu X, and Trachtenberg JT (2016). Pten and EphB4 regulate the establishment of perisomatic inhibition in mouse visual cortex. Nat Commun 7, 12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolini G, Ciceri G, and Marin O (2013). Integration of GABAergic interneurons into cortical cell assemblies: lessons from embryos and adults. Neuron 79, 849–864. [DOI] [PubMed] [Google Scholar]
- Blaess S, Kammerer RA, and Hall H (1998). Structural analysis of the sixth immunoglobulin-like domain of mouse neural cell adhesion molecule L1 and its interactions with alpha(v)beta3, alpha(IIb)beta3, and alpha5beta1 integrins. J Neurochem 71, 2615–2625. [DOI] [PubMed] [Google Scholar]
- Boiko T, Vakulenko M, Ewers H, Yap CC, Norden C, and Winckler B (2007). Ankyrin-dependent and -independent mechanisms orchestrate axonal compartmentalization of L1 family members neurofascin and L1/neuron-glia cell adhesion molecule. J Neurosci 27, 590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennaman LH, Zhang X, Guan H, Triplett JW, Brown A, Demyanenko GP, Manis PB, Landmesser L, and Maness PF (2013). Polysialylated NCAM and ephrinA/EphA regulate synaptic development of GABAergic interneurons in prefrontal cortex. Cereb Cortex 23, 162–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani V, Chedotal A, Schachner M, Faivre-Sarrailh C, and Rougon G (2000). Analysis of the L1-deficient mouse phenotype reveals cross-talk between Sema3A and L1 signaling pathways in axonal guidance. Neuron 27, 237–249. [DOI] [PubMed] [Google Scholar]
- Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, Orr A, Lotz G, Kim DH, Hamto P, et al. (2011). Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen NR, Taylor JS, Scott LB, Guillery RW, Soriano P, and Furley AJ (1998). Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr Biol 8, 26–33. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JQ, and Bennett V (1994). Ankyrin binding activity shared by the neurofascin/Ll/NrCAM family of nervous system cell adhesion molecules. J Biol Chem 269, 27163–27166. [PubMed] [Google Scholar]
- DeBernardo AP, and Chang S (1996). Heterophilic interactions of DM-GRASP: GRASP-NgCAM interactions involved in neurite extension. J Cell Biol 133, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelipe J, Hendry SH, Jones EG, and Schmechel D (1985). Variability in the terminations of GABAergic chandelier cell axons on initial segments of pyramidal cell axons in the monkey sensory-motor cortex. J Comp Neurol 231, 364–384. [DOI] [PubMed] [Google Scholar]
- DeFelipe J, Lopez-Cruz PL, Benavides-Piccione R, Bielza C, Larranaga P, Anderson S, Burkhalter A, Cauli B, Fairen A, Feldmeyer D, et al. (2013). New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat Rev Neurosci 14, 202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Pino I, Garcia-Frigola C, Dehorter N, Brotons-Mas JR, Alvarez-Salvado E, Martinez de Lagran M, Ciceri G, Gabaldon MV, Moratal D, Dierssen M, et al. (2013). Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron 79, 1152–1168. [DOI] [PubMed] [Google Scholar]
- Demyanenko GP, Siesser PF, Wright AG, Brennaman LH, Bartsch U, Schachner M, and Maness PF (2011). L1 and CHL1 Cooperate in Thalamocortical Axon Targeting. Cereb Cortex 21, 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristo G, Wu C, Chattopadhyaya B, Ango F, Knott G, Welker E, Svoboda K, and Huang ZJ (2004). Subcellular domain-restricted GABAergic innervation in primary visual cortex in the absence of sensory and thalamic inputs. Nat Neurosci 7, 1184–1186. [DOI] [PubMed] [Google Scholar]
- Enneking EM, Kudumala SR, Moreno E, Stephan R, Boerner J, Godenschwege TA, and Pielage J (2013). Transsynaptic coordination of synaptic growth, function, and stability by the L1-type CAM Neuroglian. PLoS Biol 11, e1001537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzari P, Paternain AV, Valiente M, Pla R, Lujan R, Lloyd K, Lerma J, Marin O, and Rico B (2010). Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature 464, 1376–1380. [DOI] [PubMed] [Google Scholar]
- Friedlander DR, Milev P, Karthikeyan L, Margolis RK, Margolis RU, and Grumet M (1994). The neuronal chondroitin sulfate proteoglycan neurocan binds to the neural cell adhesion molecules Ng-CAM/L1/NILE and N-CAM, and inhibits neuronal adhesion and neurite outgrowth. J Cell Biol 125, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickfeld LL, Roberts JD, Somogyi P, and Scanziani M (2009). Interneurons hyperpolarize pyramidal cells along their entire somatodendritic axis. Nat Neurosci 12, 21–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greferath U, Canty AJ, Messenger J, and Murphy M (2002). Developmental expression of EphA4-tyrosine kinase receptor in the mouse brain and spinal cord. Mech Dev 119, S231–238. [DOI] [PubMed] [Google Scholar]
- Guan H, and Maness PF (2010). Perisomatic GABAergic innervation in prefrontal cortex is regulated by ankyrin interaction with the L1 cell adhesion molecule. Cereb Cortex 20, 2684–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom KL, Xu X, Ogawa Y, Frischknecht R, Seidenbecher CI, Shrager P, and Rasband MN (2007). Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J Cell Biol 178, 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlavin ML, and Lemmon V (1991). Molecular structure and functional testing of human L1CAM: an interspecies comparison. Genomics 11, 416–423. [DOI] [PubMed] [Google Scholar]
- Hortsch M, Nagaraj K, and Godenschwege TA (2009). The interaction between L1-type proteins and ankyrins--a master switch for L1-type CAM function. Cell Mol Biol Lett 14, 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard A, Tamas G, and Soltesz I (2005). Lighting the chandelier: new vistas for axoaxonic cells. Trends Neurosci 28, 310–316. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Di Cristo G, and Ango F (2007). Development of GABA innervation in the cerebral and cerebellar cortices. Nat Rev Neurosci 8, 673–686. [DOI] [PubMed] [Google Scholar]
- Inan M, and Anderson SA (2014). The chandelier cell, form and function. Curr Opin Neurobiol 26, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inan M, Blazquez-Llorca L, Merchan-Perez A, Anderson SA, DeFelipe J, and Yuste R (2013). Dense and overlapping innervation of pyramidal neurons by chandelier cells. J Neurosci 33, 1907–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inan M, Welagen J, and Anderson SA (2012). Spatial and temporal bias in the mitotic origins of somatostatin- and parvalbumin-expressing interneuron subgroups and the chandelier subtype in the medial ganglionic eminence. Cereb Cortex 22, 820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inda MC, DeFelipe J, and Munoz A (2006). Voltage-gated ion channels in the axon initial segment of human cortical pyramidal cells and their relationship with chandelier cells. Proc Natl Acad Sci U S A 103, 2920–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inda MC, Defelipe J, and Munoz A (2007). The distribution of chandelier cell axon terminals that express the GABA plasma membrane transporter GAT-1 in the human neocortex. Cereb Cortex 17, 2060–2071. [DOI] [PubMed] [Google Scholar]
- Janas J, Skowronski J, and Van Aelst L (2006). Lentiviral delivery of RNAi in hippocampal neurons. Methods Enzymol 406, 593–605. [DOI] [PubMed] [Google Scholar]
- Jones EG (1975). Varieties and distribution of non-pyramidal cells in the somatic sensory cortex of the squirrel monkey. J Comp Neurol 160, 205–267. [DOI] [PubMed] [Google Scholar]
- Jouet M, Rosenthal A, Armstrong G, MacFarlane J, Stevenson R, Paterson J, Metzenberg A, Ionasescu V, Temple K, and Kenwrick S (1994). X-linked spastic paraplegia (SPG1), MASA syndrome and X-linked hydrocephalus result from mutations in the L1 gene. Nat Genet 7, 402–407. [DOI] [PubMed] [Google Scholar]
- Kania A, and Klein R (2016). Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat Rev Mol Cell Biol 17, 240–256. [DOI] [PubMed] [Google Scholar]
- Kenwrick S, Jouet M, and Donnai D (1996). X linked hydrocephalus and MASA syndrome. J Med Genet 33, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs A, and Fishell G (2014). Interneuron cell types are fit to function. Nature 505, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodosevich K, Watanabe Y, and Monyer H (2011). EphA4 preserves postnatal and adult neural stem cells in an undifferentiated state in vivo. J Cell Sci 124, 1268–1279. [DOI] [PubMed] [Google Scholar]
- Kriebel M, Metzger J, Trinks S, Chugh D, Harvey RJ, Harvey K, and Volkmer H (2011). The cell adhesion molecule neurofascin stabilizes axo-axonic GABAergic terminals at the axon initial segment. J Biol Chem 286, 24385–24393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurumaji A, Nomoto H, Okano T, and Toru M (2001). An association study between polymorphism of L1CAM gene and schizophrenia in a Japanese sample. Am J Med Genet 105, 99–104. [PubMed] [Google Scholar]
- Lemmon V, Farr KL, and Lagenaur C (1989). Ll-mediated axon outgrowth occurs via a homophilic binding mechanism. Neuron 2, 1597–1603. [DOI] [PubMed] [Google Scholar]
- Leterrier C (2016). The Axon Initial Segment, 50Years Later: A Nexus for Neuronal Organization and Function. Curr Top Membr 77, 185–233. [DOI] [PubMed] [Google Scholar]
- Lewis DA (2011). The chandelier neuron in schizophrenia. Dev Neurobiol 71, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner J, Rathjen FG, and Schachner M (1983). L1 mono- and polyclonal antibodies modify cell migration in early postnatal mouse cerebellum. Nature 305, 427–430. [DOI] [PubMed] [Google Scholar]
- Lu J, Tucciarone J, Padilla-Coreano N, He M, Gordon JA, and Huang ZJ (2017). Selective inhibitory control of pyramidal neuron ensembles and cortical subnetworks by chandelier cells. Nat Neurosci 20, 1377–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maness PF, and Schachner M (2007). Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci 10, 19–26. [DOI] [PubMed] [Google Scholar]
- Matsuda T, and Cepko CL (2007). Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A 104, 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland AC, Sheffler-Collins SI, Kayser MS, and Dalva MB (2009). Ephrin-B1 and ephrin-B2 mediate EphB-dependent presynaptic development via syntenin-1. Proc Natl Acad Sci U S A 106, 20487–20492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikuni T, Nishiyama J, Sun Y, Kamasawa N, and Yasuda R (2016). High-Throughput, High-Resolution Mapping of Protein Localization in Mammalian Brain by In Vivo Genome Editing. Cell 165, 1803–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham LK, Thelen K, and Maness PF (2001). Cytoplasmic domain mutations of the L1 cell adhesion molecule reduce L1-ankyrin interactions. J Neurosci 21, 1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikimi M, Oishi K, Tabata H, Torii K, and Nakajima K (2011). Segregation and pathfinding of callosal axons through EphA3 signaling. J Neurosci 31, 16251–16260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Horresh I, Trimmer JS, Bredt DS, Peles E, and Rasband MN (2008). Postsynaptic density-93 clusters Kv1 channels at axon initial segments independently of Caspr2. J Neurosci 28, 5731–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Oses-Prieto J, Kim MY, Horresh I, Peles E, Burlingame AL, Trimmer JS, Meijer D, and Rasband MN (2010). ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. J Neurosci 30, 1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleszewski M, Beer S, Katich S, Geiger C, Zeller Y, Rauch U, and Altevogt P (1999). Integrin and neurocan binding to L1 involves distinct Ig domains. J Biol Chem 274, 24602–24610. [DOI] [PubMed] [Google Scholar]
- Poltorak M, Wright R, Hemperly JJ, Torrey EF, Issa F, Wyatt RJ, and Freed WJ (1997). Monozygotic twins discordant for schizophrenia are discordant for N-CAM and L1 in CSF. Brain Res 751, 152–154. [DOI] [PubMed] [Google Scholar]
- Rathjen FG, and Schachner M (1984). Immunocytological and biochemical characterization of a new neuronal cell surface component (L1 antigen) which is involved in cell adhesion. EMBO J 3, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribak CE (1985). Axon terminals of GABAergic chandelier cells are lost at epileptic foci. Brain Res 326, 251–260. [DOI] [PubMed] [Google Scholar]
- Rocco BR, DeDionisio AM, Lewis DA, and Fish KN (2017). Alterations in a Unique Class of Cortical Chandelier Cell Axon Cartridges in Schizophrenia. Biol Psychiatry 82, 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotland P, Zhou D, Benveniste H, and Bennett V (1998). Nervous system defects of AnkyrinB (−/−) mice suggest functional overlap between the cell adhesion molecule L1 and 440-kD AnkyrinB in premyelinated axons. J Cell Biol 143, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P (1977). A specific ‘axo-axonal’ interneuron in the visual cortex of the rat. Brain Res 136, 345–350. [DOI] [PubMed] [Google Scholar]
- Steinecke A, Hozhabri E, Tapanes S, Ishino Y, Zeng H, Kamasawa N, and Taniguchi H (2017). Neocortical Chandelier Cells Developmentally Shape Axonal Arbors through Reorganization but Establish Subcellular Synapse Specificity without Refinement. eNeuro 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern P, Astrof S, Erkeland SJ, Schustak J, Sharp PA, and Hynes RO (2008). A system for Cre-regulated RNA interference in vivo. Proc Natl Acad Sci U S A 105, 13895–13900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentagothai J, and Arbib MA (1974). Conceptual models of neural organization. Neurosci Res Program Bull 12, 305–510. [PubMed] [Google Scholar]
- Tai Y, Janas JA, Wang CL, and Van Aelst L (2014). Regulation of chandelier cell cartridge and bouton development via DOCK7-mediated ErbB4 activation. Cell Rep 6, 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, Lu J, and Huang ZJ (2013). The spatial and temporal origin of chandelier cells in mouse neocortex. Science 339, 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telley L, Cadilhac C, Cioni JM, Saywell V, Jahannault-Talignani C, Huettl RE, Sarrailh-Faivre C, Dayer A, Huber AB, and Ango F (2016). Dual Function of NRP1 in Axon Guidance and Subcellular Target Recognition in Cerebellum. Neuron 91, 1276–1291. [DOI] [PubMed] [Google Scholar]
- Tremblay R, Lee S, and Rudy B (2016). GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 91, 260–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viney TJ, Lasztoczi B, Katona L, Crump MG, Tukker JJ, Klausberger T, and Somogyi P (2013). Network state-dependent inhibition of identified hippocampal CA3 axo-axonic cells in vivo. Nat Neurosci 16, 1802–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida M, John KA, Janas JA, Newey SE, and Van Aelst L (2006). The Rac activator DOCK7 regulates neuronal polarity through local phosphorylation of stathmin/Op18. Neuron 51, 727–739. [DOI] [PubMed] [Google Scholar]
- Weller S, and Gartner J (2001). Genetic and clinical aspects of X-linked hydrocephalus (L1 disease): Mutations in the L1CAM gene. Hum Mutat 18, 1–12. [DOI] [PubMed] [Google Scholar]
- Wimmer VC, Harty RC, Richards KL, Phillips AM, Miyazaki H, Nukina N, and Petrou S (2015). Sodium channel beta1 subunit localizes to axon initial segments of excitatory and inhibitory neurons and shows regional heterogeneity in mouse brain. J Comp Neurol 523, 814–830. [DOI] [PubMed] [Google Scholar]
- Winckler B, Forscher P, and Mellman I (1999). A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature 397, 698–701. [DOI] [PubMed] [Google Scholar]
- Woodruff AR, Anderson SA, and Yuste R (2010). The enigmatic function of chandelier cells. Front Neurosci 4, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff AR, McGarry LM, Vogels TP, Inan M, Anderson SA, and Yuste R (2011). State-dependent function of neocortical chandelier cells. J Neurosci 31, 17872–17886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu NJ, and Henkemeyer M (2009). Ephrin-B3 reverse signaling through Grb4 and cytoskeletal regulators mediates axon pruning. Nat Neurosci 12, 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Tam M, and Anderson SA (2008). Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J Comp Neurol 506, 16–29. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ogawa Y, Hedstrom KL, and Rasband MN (2007). betaIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J Cell Biol 176, 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YT, Wang CL, and Van Aelst L (2012). DOCK7 interacts with TACC3 to regulate interkinetic nuclear migration and cortical neurogenesis. Nat Neurosci 15, 1201–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Stornetta RL, and Zhu JJ (2004). Chandelier cells control excessive cortical excitation: characteristics of whisker-evoked synaptic responses of layer 2/3 nonpyramidal and pyramidal neurons. J Neurosci 24, 5101–5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.