

Abstract
Elevated shear force caused by an anastomotic stenosis is a common complication at the blood vessel-vascular implant interface. Although elevated shear forces were found to cause platelet aggregation around a stenotic region, transient platelet exposure to elevated shear forces and subsequent downstream events occurring under lower shear force were not extensively studied. We hypothesize that effects of elevated shear forces on pre-activation of platelets for downstream adhesion and activation are relevant in understanding the increased thrombotic risk associated with blood-contacting devices. We designed a microfluidic flow system to mimic the hemodynamic environment of vasculature with an upstream anastomotic stenosis with five wall shear strain rates ranging from 1620 s−1 to 11560 s−1. Under shear flow conditions, transient exposure of whole blood to elevated shear forces resulted in higher downstream platelet adhesion onto three different immobilized platelet agonists: fibrinogen, collagen, or von Willebrand factor. Platelet expression of four activation markers (P-selectin, GPIIb/IIIa, lysosomal glycoprotein, and phosphatidylserine) significantly increased after transient exposure to higher upstream wall shear strain rates of 2975 – 11560 s−1. A significant lysis was observed when platelets were primed by upstream wall shear strain rate of 11560 s−1. These experimental results could be helpful to understand how altered hemodynamics around an anastomotic stenosis promotes thrombus formation downstream.
Keywords: platelet adhesion and activation, platelet lysis, anastomotic stenosis, microfluidics, flow cytometry
Graphical Abstract
1. Introduction
Cardiovascular disease is often treated through the surgical implantation of vascular devices. When a device such as a synthetic small-diameter vascular graft is implanted into the vasculature, there are two anastomotic regions where the native vessel is joined with the synthetic material. Due to intimal fibrous hyperplasia, vascular grafts sometimes become stenotic (narrow) either at the anastomotic regions or along the inner wall of the grafts [1–3], leading to elevated shear forces on flowing platelets. Such higher shear forces have been shown to increase platelet concentration near the wall [4], and to induce intracellular signaling cascades characterized by binding of von Willebrand factor (vWF) to glycoprotein GPIbα [5], Ca2+ mobilization [6,7], actin polymerization and cytoskeletal rearrangements [8], protein kinase C expression [9], and GPIIb/IIIa activation [10]. Mechanical damage of platelets due to elevated shear forces may cause lysis [11], resulting in the release of procoagulant factors. In the case of damaged vessel endothelium, platelets will interact with collagen of exposed subendothelium through their surface receptors GPVI and GPIa/IIa. If the surface of a vascular device is not perfectly passivated, plasma proteins, including fibrinogen and vWF, will adsorb to the surface within few seconds upon exposure to blood [12,13]. Platelets will interact with these surface adsorbed agonists using their receptors GPIIb/IIIa and GPIb-IX-V. Binding to these three agonists, as well as shear induced platelet lysis will trigger the activation of platelets. During the activation platelets undergo a series of morphological changes [14] and release procoagulant factors from their granules [15,16], which stimulate further platelet recruitments, activation, aggregation and ultimately a thrombus formation.
Various techniques have been used to assess platelet function in vitro. To assess the effects of elevated shear forces on platelet function, research labs have typically used a cone- and-plate viscometer or closed-loop recirculation system [17–19]. In such platelet tests, a steady- state shear flow condition is created, i.e., there is zero shear gradient across the flowing blood. However, it has been shown that the majority of platelet interactions at a flowing blood-vascular implant interface are transient [20,21], similar to the transient platelet exposure to an elevated shear force caused by an anastomotic stenosis. Even though several studies applied single-pass perfusion system using microfluidic devices [22–27], it is the transient platelet exposure to elevated shear forces and subsequent downstream events occurring under normal shear exposure that have not been thoroughly investigated.
Shear gradients generated by transient flow acceleration and deceleration induce mechanical sensing in circulating platelets which has a significant role in causing their aggregation around a stenotic region [23,26,27]. Here, we address another fundamental question: how platelets, after transient passage through an upstream stenotic region, interact with an imperfect vascular implant surface placed downstream. We hypothesize that the transient exposure to an elevated shear force (i.e., shear gradient) pre-activates (a.k.a. “primes”) platelets for enhanced downstream adhesion and activation, the events that are dependent on the hemodynamic conditions around a stenotic region. We have previously found that platelets can be primed even by moderately increased shear forces (i.e., wall shear strain rates of 500 – 1000 s−1) for increased adhesion to the surface-immobilized capture agonists [28]. However, at these moderately increased shear forces the expression levels of GPIIb/IIIa and lysosomal glycoprotein in perfused blood were not found to be significantly increased [28]. Therefore, it remained to be investigated how highly elevated shear forces, typically found in severe stenosis, prime platelets for downstream adhesion and activation.
The present study was designed to provide the answers to these questions by designing a parallel array of flow channels in a microfluidic device that mimicked changes in hemodynamic flow conditions experienced in a stenotic vascular graft anastomosis. Anticoagulated whole blood was perfused through these flow channels where at the downstream position one of the three platelet agonists (fibrinogen, collagen, or vWF) was covalently immobilized to capture platelets primed upstream. Three different platelet agonists, which served as a mimic of an exposed subendothelium or a procoagulant surface of an implanted vascular graft, captured platelets by interacting with different platelet receptors. Platelet adhesion to these capture regions was quantified by counting adhered platelets using phase contrast microscopy. Downstream platelet activation was characterized by flow cytometry of perfused blood in the absence of any capture regions using four different activation markers (P-selectin, GPIIb/IIIa, lysosomal glycoprotein, and phosphatidylserine). The effects of elevated shear forces on platelet lysis were determined by a lactate dehydrogenase (LDH) assay. The results suggested that transient exposure to elevated upstream wall shear strain rates (2975 – 11560 s−1) primed platelets for significantly higher downstream adhesion and activation, and caused significant platelet lysis at the highest wall shear strain rate.
2. Materials and Methods
2.1. Flow channel preparation
Each flow cell, consisting of two parallel rectangular profile flow channels, was manufactured by placing a silicone gasket (Medical Grade, 0.007″, BioPlexus, Ventura, CA, USA) between a plasma-cleaned microscope slide (VWR International, Radnor, PA, USA) and a Nexterion-H slide (Schott, Tempe, AZ, USA) (Fig 1). Flow channel features were patterned on the silicone gasket using a laser cutter (VLS3.60, Universal Laser Systems, Scottsdale, AZ, USA). Flow channel dimensions were 1 mm wide, 0.18 mm high, and 70 mm long. Each flow channel contained an upstream stenotic region and a downstream capture region. Stenotic regions were 10 mm in length. Capture regions were 25 mm downstream from the end of the stenotic regions. The width of stenotic regions was varied from 1.0 mm to 0.2 mm to create five different stenosis levels, from 0 to 80% stenosis defined as:
where winlet was the width of inlet of the flow channels (1 mm) and wstenosis was the width of the stenotic regions. Control flow channel, a representative of small artery, did not contain any upstream stenotic region (0% stenosis). Wall shear strain rates were calculated at a constant volumetric flow rate by generating a three-dimensional model of each flow channel in COMSOL Multiphysics (COMSOL, Inc., Burlington, MA, USA).
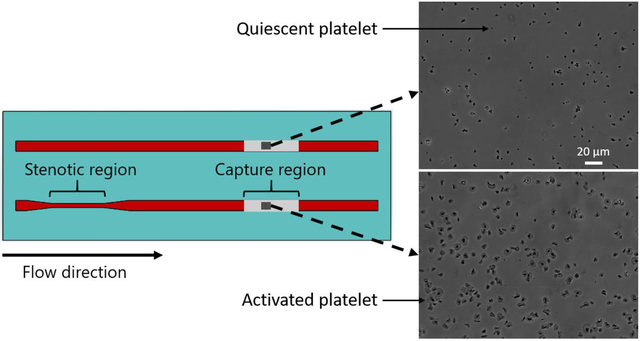
Figure 1.

Schematic diagram of the flow cell used in the flow-based platelet adhesion assay. Upstream stenotic regions were created by changing the width of the flow channels. Protein agonists were covalently immobilized downstream on the chemically reactive glass substrates by microcontact printing.
2.2. Preparation of downstream capture regions
The downstream capture regions (10 mm in length) were prepared to capture platelets primed upstream by stenosis. Fibrinogen and vWF (Haematologic Technologies, Essex Junction, VT, USA), and collagen I (Ibidi, Fitchburg, WI, USA) were covalently immobilized on the surface of Nexterion-H slides. Nexterion-H slides are coated with a poly(ethylene glycol) (PEG)-based polymer containing reactive N-hydroxysuccinimide (NHS) esters, thus providing means for covalent protein immobilization via their –NH2 moieties [29]. The proteins were deposited to the capture area by microcontact printing described in detail elsewhere [30]. Briefly, a flat polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning, Midland, MI, USA) stamp was coated with a protein solution (fibrinogen 1 mg/mL, collagen 3 mg/mL, or vWF 10 μg/mL) for 10 min, allowed to dry under a stream of nitrogen, and placed in contact with Nexterion-H slides for 1 hr. After that transfer time, the capture region was rinsed in deionized water and dried under the stream of nitrogen.
2.3. Flow channel operation
Flow channels were connected to a syringe pump (Kent Scientific, Torrington, CT, USA) using an Ismatec E-LFL tubing (wall shear strain rate of 30 s−1, Cole Parmer, Vernon Hills, IL, USA). Connecting tubing was kept as short as possible to minimize any possible priming effect. A solution of human serum albumin (HSA, 1 mg/mL in phosphate-buffered saline, Sigma Aldrich, St. Louis, MO, USA) was perfused through each flow channel and incubated for 1 hr at room temperature prior to blood perfusion. Covalently attached HSA served to passivate the nonreacted regions of the glass substrates as well as the walls of each flow channel and connecting tubing by physical adsorption.
Fresh whole blood was drawn via venipuncture from healthy human donors according to the protocol approved by the University of Utah Institutional Review Board. Blood was drawn into buffered 3.2% (0.105 M) sodium citrate and was treated with Phe-Pro-Arg- chloromethylketone (PPACK, 80 μM, Haematologic Technologies, Essex Junction, VT, USA) immediately after drawing blood to inhibit thrombin-induced coagulation. Blood sample was kept at 37°C in a water bath until being perfused through the flow channels at a constant flow rate of 0.5 mL/min in all experiments. Flow was sustained for 5 min, after which the flow channels were rinsed with prewarmed Tyrode’s buffer (37 °C, pH 7.4) and fixed in 4% paraformaldehyde solution. The attached cells were imaged using a phase contrast microscope (Diaphot 200, Nikon, Tokyo, Japan), and platelet adhesion density was quantified by counting adhered platelets in ten selected fields (180 × 236 μm) along centerline of the downstream capture regions.
2.4. Flow cytometry
Flow cytometry was used to measure the expression levels of CD62P (P-selectin), GPIIb/IIIa (integrin αIIbβ3), CD63 (lysosomal glycoprotein), and phosphatidylserine on perfused platelets.
Following blood perfusion through a flow channel with a stenotic region but with no capture region, a 5 μL aliquot of blood supernatant was incubated for 20 minutes with anti-human CD62P, PAC−1, anti-human CD63, or annexin V (BD Biosciences, San Jose, CA, USA) to label P-selectin, active GPIIb/IIIa, lysosomal glycoprotein, and phosphatidylserine, respectively. Two 5 μL blood aliquots were obtained and labeled prior to perfusion. Platelet activation in one aliquot was achieved by addition of thrombin immediately prior to labeling (c = 0.1 units/mL, EMD Millipore, Billerica, MA, USA) and other aliquot was left unstimulated to serve as positive and negative activation controls, respectively. Blood was not treated with PPACK for thrombin activated positive control. To locate platelets in flow cytometry analysis, another 5 μL blood aliquot was labeled with anti-human CD41b (BD Biosciences, San Jose, CA, USA), which binds to the GPIIb subunit of GPIIb/IIIa regardless of the activation state of the receptor. Following labeling, platelets were fixed in 1% paraformaldehyde and stored at 4 °C. Analysis of 100,000 events was conducted on a FACScanto analyzer (BD Biosciences, San Jose, CA, USA).
2.5. LDH assay
The lactate dehydrogenase (LDH) assay was performed using perfused platelet rich plasma (PRP) instead of whole blood using Pierce LDH Cytotoxicity Assay Kit (Thermo Fisher Scientific (Rockford, IL, USA). PRP was prepared by centrifugation of anticoagulated whole blood at 275 g for 15 min. Platelet concentration was adjusted to 4.0×105 platelets/mL using a prewarmed HEPES-buffered Tyrode’s solution (37 °C, pH 7.4). Following PRP perfusion over an albumin coated flow channel in the absence of capture region, a 50 μL aliquot of perfused PRP was collected and incubated with a reaction mixture at room temperature. After 30 min incubation, the reactions were stopped by adding a stop solution. Absorbance at 490 nm and 680 nm was measured using a Synergy HTX microplate reader (Biotek, Winooski, VT, USA) to determine the LDH activity. Two 50 μL aliquots of PRP were obtained and labeled prior to perfusion. One aliquot lysis was stimulated by addition of lysis buffer immediately prior to labeling and the other was left unstimulated to serve as positive and negative controls, respectively. Platelets were also stimulated by addition of thrombin (c = 0.1 units/mL) to evaluate the influence of thrombin on lysis of platelets.
2.6. Statistical analysis
All experiments were carried out independently in minimum four replicates. Box and whisker plots presented the distribution of platelet adhesion density through their quartiles and outliers. Bar plots were presented as mean ± standard deviation of platelet activation and lysis data. Statistical significance was established using a paired t-test at p-values < 0.05. The significance level was presented as *p < 0.05, **p < 0.005, or ***p < 0.0005. Data were analyzed for significance with OriginPro 9.0 software (OriginLab, Northampton, MA, USA).
3. Results
The present study investigated how brief transient platelet exposures to elevated shear forces in a microfluidic device affect downstream platelet adhesion and activation. Anticoagulated whole blood was perfused through the stenosed flow channels where platelets entered the upstream region of elevated shear forces and subsequently exited to a lower shear force region. In the lower shear force region, the platelets were captured by platelet binding proteins immobilized downstream (Fig 1). Figure 2a shows the design of stenotic region in the microfluidic flow channels and the control flow channel. Stenotic regions were created 5 mm downstream from the channel inlet to gradually increase wall shear strain rates. After transient exposure to elevated wall shear strain rates in the stenotic regions, platelets were gradually exposed to lower wall shear strain rate of 1620 s−1. The stenotic chamber design ensured laminar flow throughout the chambers, where recirculation and stagnation flow regions were avoided. Computational fluid dynamics indicated that blood flow will be highly affected by shear forces in the upstream stenosis, especially for 60% and 80% stenosis chambers (Fig 2b). The centerline wall shear strain rates (γc) increased linearly, for example for 80% stenosis from 1620 s−1 to 11560 s−1 with a gradient of 2.0 × 104 s−1/cm. Centerline wall shear strain rates remained constant within each stenotic region and decreased linearly along the 5 mm long expanding region back to 1620 s−1 (Fig 2b). Table 1 lists the flow parameters in all five microfluidic chambers used. Maximum fluid velocity (vmax) increased sharply over the 5 mm long region before the stenosis resulting in the maximum elongation rate (εmax) ranging from 0 to 316 s−1. Capture region, 10 mm long was positioned 25 mm downstream of the stenotic region (Fig 1). Reynolds numbers within the stenotic regions were 4.3, 5.2, 6.5, 8.8 and 13.4 for 0%, 20%, 40%, 60% and 80% stenotic levels, respectively. These Reynolds numbers were in the typical range of arterial blood flow [31].
Figure 2.

Wall shear strain rate (γw) distribution in the microfluidic flow channels. (A) The first 20 mm of each experimental flow channel was simulated using COMSOL Multiphysics to evaluate wall shear strain rates in the anastomotic stenosis. The colors indicate local wall shear strain rates at the channel bottom wall. Centerline wall shear strain rate (γc) represents wall shear strain rate across the centerline (i.e., dotted white line in A) of the channel bottom wall. (B) Centerline wall shear strain rates indicate a steep gradient, particularly for 60% and 80% stenotic models. Centerline wall shear strain rates within the stenotic regions were 1620, 2145, 2975, 4860 and 11560 s−1 for 0%, 20%, 40%, 60% and 80% stenotic models, respectively. Downstream centerline wall shear strain rate (1620 s−1) was kept constant in all experiments.
Table 1.
The flow parameters in five microfluidic chambers used in experiments.
| Stenosis (%) | Width of the stenotic region wstenosis (mm) | Centerline wall shear strain rate, γc (s−1) | Maximum velocity in the stenotic region, vmax (mm s−1) | Maximum elongation rate, εmax (s−1) |
|---|---|---|---|---|
| 0 | 1 | 1620 | 81.9 | 0 |
| 20 | 0.8 | 2145 | 104.2 | 7 |
| 40 | 0.6 | 2975 | 145.6 | 23 |
| 60 | 0.4 | 4860 | 234.6 | 86 |
| 80 | 0.2 | 11560 | 496.7 | 316 |
Figure 3 shows the surface density of adhered platelets (number/area) on the downstream capture regions with one of the three platelet binding proteins: fibrinogen, collagen and vWF. The finding that the platelets adhered to three different capture proteins in a shear strain rate- dependent manner suggested that the transient exposure to elevated upstream shear forces primed platelets for the downstream adhesion occurring under low shear conditions. Overall adhesion to each capture region was found to be significantly higher after the platelets passed through upstream 40%, 60% and 80% stenotic regions (transient exposure to 2975, 4800 and 11560 s−1 wall shear strain rates) than the adhesion in the cases of 0% stenosis control. Up to 4860 s−1, we found indistinguishable platelet adhesion density among all three capture proteins for each wall shear strain rate. However, platelet adhesion density at 11560 s−1 shows a larger number of platelets adhered to vWF relative to fibrinogen and collagen.
Figure 3.

Platelet adhesion to three downstream capture proteins as a function of centerline wall shear strain rates in the upstream stenosis compared with control sample containing no stenotic region (γc = 1620 s−1). Statistical significance was obtained using a paired t-test (n = 40, *p < 0.05, **p < 0.005 and ***p < 0.0005).
Transiently elevated wall shear strain rate did not only result in an increasing number of adhered platelets, but also affected the morphology of adhered platelets. Figures 4a–d show the phase contrast images of adhered platelets onto fibrinogen coated capture region after passing through the upstream stenotic region with centerline wall shear strain rates of 1620 – 11560 s−1.
Figure 4.

Representative phase contrast images (A-D) show morphological changes of adhered platelets onto fibrinogen coated capture region after passing through the chamber containing no stenotic region (centerline wall shear strain rate of 1620 s−1) and through the chambers containing upstream stenotic region (centerline wall shear strain rates of 2975 – 11560 s−1). Representative images E and F show morphology of platelets adhered onto collagen and vWF coated capture regions, respectively, after passing through the upstream stenotic region with centerline wall shear strain rate of 11560 s−1.
The shape of adhered platelets can be taken as evidence of platelet activation; after passing through elevated wall shear strain rate regions, adhered platelets changed from ellipsoidal inactivated state to a fully spread activated state. Very few platelets primed by 1620 s−1 spread fully on fibrinogen capture region (Fig 4a). However, the spreading area increased with increasing wall shear strain rates from 1620 to 2975 s−1 (Fig 4b). A significant number of platelets primed by 4860 and 11560 s−1 upstream wall shear strain rates were fully spread on each capture region (Figs 4c–d). After transient exposure to 11560 s−1 upstream wall shear strain rate typical morphologies of adhered platelets on all three capture regions were similar showing a large fraction of platelets fully spread and activated (Figs 4e–f).
The interactions between the platelets and elevated upstream shear forces primed a fraction of platelets which formed stable adhesion on the capture region. Figures 5 and 6 show the result of the flow cytometry analysis of perfusate, a whole blood volume that flowed through the albumin coated stenotic chambers without the capture regions. Four activation markers: P- selectin, GPIIb/IIIa (Fig 5), and lysosomal glycoprotein, and phosphatidylserine (Fig 6) were quantified in the perfused blood. The percent of expression events (out of 100,000 events for each marker) was compared to blood with no prior perfusion (negative control) and to blood with no prior perfusion after thrombin stimulation (positive control). All four platelet activation markers were significantly higher when platelets were transiently exposed to wall shear strain rates ranging from 2975 s−1 to 11560 s−1. In addition to the activation of P-selectin and lysosomal glycoprotein (secretory products of granules and lysosomes), transiently elevated shear strain rates (γc ≥ of 2975 s−1) also activated GPIIb/IIIa (receptor of fibrinogen) and phosphatidylserine (a procoagulant factor), indicating an underlying role of elevated shear forces in eventual thrombus formation downstream. These findings supported the published observation which documented that a threshold shear strain rate of 2200 s−1 is necessary to induce platelet activation in whole blood [19]. The expression levels of activation markers increased with wall shear strain rates in a roughly proportional manner (Figs 5–6). The flow cytometry results generally supported the conclusion derived from the data shown in Figure 3, indicating that the transient exposure to higher shear forces was sufficient to activate the platelets for enhanced downstream adhesion.
Figure 5.

Flow cytometry results of platelet activation. Expression levels of (A) P-selectin and (B) activated GPIIb/IIIa (PAC−1 receptor) in perfused blood samples were compared with unstimulated (negative control) and thrombin stimulated (positive control) blood samples collected prior to perfusion. Analysis of 100,000 events for each sample was conducted, and events of platelets expressing each marker were recorded. Statistical significance was obtained using a paired t-test (n = 4, *p < 0.05, **p < 0.005 and ***p < 0.0005 relative to “no stimulation” control).
Figure 6.

Flow cytometry results of platelet activation. Expression levels of (A) annexin V receptor (phosphatidylserine) and (B) CD63 (lysosomal glycoprotein) in perfused blood samples were compared with unstimulated (negative control) and thrombin stimulated (positive control) samples collected prior to perfusion. Analysis of 100,000 events for each sample was conducted, and events of platelets expressing each marker were recorded. Statistical significance was obtained using a paired t-test (n = 4, *p < 0.05 and ***p < 0.0005 relative to “no stimulation” control).
To determine whether transient exposure to elevated shear forces also caused platelet lysis, the release of cytoplasmic LDH was measured in the perfused platelet rich plasma (PRP) (Fig 7). Platelets exposed low levels of wall shear strain rates (1620 – 2975 s−1) showed no notable effect on platelet lysis. Wall shear strain rate of 4860 s−1 slightly increased the release of LDH, but that increase was statistically insignificant when compared with the control (p > 0.05). Only the transient exposure to 11560 s−1 wall shear strain rate resulted in statistically significant platelet lysis, suggesting that priming effect observed at this high wall shear strain rate could have been caused by both the lysis and sublytic activation of platelets. However, that overall LDH release was significantly lower compared to lysis buffer stimulated positive control (p < 0.0005). It was also found that thrombin stimulation of PRP did not significantly increase the release of LDH, indicating that platelets can be activated without being lysed.
Figure 7.

Effect of wall shear strain rates on the release of LDH from platelets in platelet rich plasma after 5 min perfusion through the albumin coated flow channels. Statistical significance was obtained using a paired t-test (n = 4, *p < 0.05 and ***p < 0.0005 relative to “no stimulation” control).
4. Discussion
Blood clotting on biomedical devices is the result of a multistep process that involves plasma protein adsorption, platelet adhesion and activation, formation of thrombin, and embolization. Evaluation of platelet responses is essential for the management of patients who are treated with vascular implants and remain on anticoagulation therapy. Understanding the mechanisms regulating platelet adhesion and activation is important, since the majority of platelets adhering to blood contacting devices undergo a variable period of surface translocation prior to forming stable adhesive contacts [32,33]. The hemodynamic environment plays a key role in thrombus formation. Blood flow is highly disturbed by severe stenosis, where recirculation and stagnation flow regions often develop [25]. Even though shear-induced platelet aggregation is a well- studied phenomenon, the relationship between altered hemodynamics around an anastomotic stenosis and downstream platelet response remains to be thoroughly investigated to minimize potential thrombotic risk. Hence, the mechanisms by which platelets respond to elevated shear forces and subsequent downstream response need to be fully characterized.
Previous in vitro studies on transient exposure of platelets to high shear stress followed by a longer exposure to low shear stress indeed showed enhanced platelet aggregation and activation [34,35]. However, a cone-and-plate viscometer used in these studies did not mimic the hemodynamic environment that platelets experience upon passage through a stenotic vascular device. In addition, in a cone-and-plate viscometer, platelets were continuously exposed to elevated shear forces for many seconds. In contrast, the passage through a stenotic region in circulation is typically lasting a few milliseconds. Therefore, a physiologically relevant microfluidic device is required for assessment of downstream platelet response under transient upstream shear gradient conditions. Several studies that used microfluidic devices confirmed shear-dependent platelet aggregation at the site of stenosis or in close proximity to the stenotic region [23,26,36], however, subsequent downstream interactions of platelets after they returned to normal shear conditions and had a chance to interact with biomaterial surfaces, have not been experimentally addressed.
Platelet adhesion is mediated by a number of hemodynamic, rheological and biological factors. Shear strain rate is a key factor in the platelet transport through a stenotic region and subsequent adherence, either to exposed subendothelium at downstream anastomosis, or to a procoagulant surface of an implanted vascular device. Shear strain rate is highest at the vessel wall and under flow condition, wall shear strain rates typically range 20–200 s−1 in veins, 300–800 s−1 in large arteries, and 800−10000 s−1 in stenotic vessels [37]. We have previously found that moderately increased wall shear strain rates of 500−1000 s−1 pre-activated platelets for enhanced adhesion to the surface-immobilized capture agonists downstream at low shear condition [28]. The platelets primed by an upstream shear strain rate of 1000 s−1 expressed larger number of P- selectin and annexin V in perfused blood, however, the expression levels of activated GPIIb/IIIa and CD63 were found insignificant. It is known that the formation of platelet thrombi is regulated by the binding of fibrinogen to activated GPIIb/IIIa glycoprotein and that CD63 modulates spreading and tyrosine phosphorylation of platelets by recruiting signaling enzymes on their surface through phosphatidylinositol 4-kinase [16]. Since platelets adhered to the surface-immobilized agonists in a wall shear strain rate-dependent manner, the previous study indicated that platelets primed by even moderately increased wall shear strain rates became partly activated. It was therefore important to investigate how platelets respond to transient exposure to elevated wall shear strain rates that are much larger than 1000 s−1 and how this exposure affects their downstream adhesion to surface-immobilized platelet binding proteins. It was reported that wall shear strain rates can exceed 10000 s−1 in severe stenotic vessels such as in stenosed coronary arteries [38,39]. Previous studies indicated that intimal hyperplasia can cause mild to severe stenosis at the anastomotic regions of implanted arterial grafts [3,40]. In the present study, wall shear strain rates from 2145 s−1 to 11560 s−1 represented mild to severe arterial graft stenosis [41].
Platelet adhesion to biomaterials coated by blood plasma proteins is a critical initial event determining hemocompatibility of blood contacting devices, often leading to irreversible platelet adhesion, aggregation and subsequent thrombus formation. The substrates used in the present study to mimic protein coated biomaterials were glass slides coated with a PEG-based polymer with reactive NHS ester groups, which allowed selected proteins to be covalently attached. The PEG-based polymer background minimized non-specific protein adsorption and platelet adhesion during the blood perfusion [42]. The three platelet binding proteins were selected to differentiate between different platelet receptors and different modes of adhesion. The results of the platelets adhesion to fibrinogen or vWF implied the activation of GPIIb/IIIa (integrin αIIbβ3), or GPIbα and GPIIb/IIIa, respectively (Figs 3a and 3c). Platelets adherence to collagen coated capture region implied the activation of surface receptors GPVI and GPIa/IIa (Fig 3b) [43]. The surface receptors GPVI and GPIa/IIa require initial attachment (tethering) and rolling onto immobilized collagen to form stable adhesive contacts. Plasma fibrinogen bound to activated GPIIb/IIIa is the predominant adhesive bridge required for platelet aggregation [44]. Several studies demonstrated the function of vWF in initial platelet adhesion, in which the binding of subunit GPIbα to vWF, particularly at high shear forces, leads to the transient capture of flowing platelets on the surface [32,45]. Since the binding of GPIbα to vWF is an early stage event strongly influenced by highly elevated shear forces, platelet adhesion after exposure to 11560 s−1 wall shear strain rate was expected to be higher on vWF coated surface as compared to fibrinogen or collagen coated capture regions, a result that was indeed found here (Fig 3). In addition to enhanced platelet adhesion, elevated upstream wall shear strain rates were also found to induce morphological changes in primed platelets (Figure 6a–f) serving as another evidence of platelet activation. A significant number of platelets primed by 4860 s−1 and 11560 s−1 wall shear strain rates spread onto all three immobilized proteins, indicating the propagation of GPIIb/IIIa- mediated outside-in signaling [46], actin polymerization and cytoskeletal rearrangements [8].
The downstream platelet adhesion was found to be strongly dependent on upstream wall shear strain rates, suggesting that elevated shear strain rates at the sites of stenosis are responsible for subsequent platelet activation, downstream adhesion, and eventual thrombus formation. Another plausible explanation is that the elongation rate in the middle of the chamber is causing platelet activation and perhaps lysis. However, even at moderately high wall shear strain rates, platelets are concentrated near the walls and red blood cells are in the center of flowing blood [47]. Elevated shear strain rates have been known to enhance such platelet margination [4,48], which is presumed here to be an essential factor in allowing platelet adherence to the surface-immobilized platelet binding proteins downstream. The margination of platelets to the surface increases when wall shear strain rate is increased [49]. That alone may be interpreted as one of the reasons for increasing number of adhered platelets, however, the increased adhesion observed in the present study actually occurred at the low shear strain rate conditions after the transient exposure to higher wall shear strain rates.
Platelet activation was assessed quantitatively using flow cytometry. The relationship between platelet activation and the magnitude of wall shear strain rate agreed with previously reported in vitro studies [22,50]. The increase in expression levels of all four activation markers were significantly lower when compared to thrombin stimulated positive controls (p < 0.05). Such difference was likely due inhomogeneous shear rate distribution in a rectangular flow channel where shear rate is highest at the walls. A fraction of platelets that was not marginated towards the channel walls simply passed through the stenotic region retaining their quiescent state.
The lysis results were consistent with the previous findings. Previously it was observed that shear stresses below about 150 dyne/cm2 (or shear strain rate of ~4000 s−1) do not cause LDH release for 5 min PRP perfusion [11]. To avoid difficulties of measuring absorbance in whole blood, we used PRP for studying platelet lysis, which does not exhibit platelet margination due to absence of red blood cells. The lack of margination means that a smaller fraction of platelets is exposed to the high wall shear rate than in the whole blood. In other words, platelet lysis in whole blood could be even larger than observed here. The duration of platelet exposure to elevated shear forces is also an important factor in causing lysis [11], which was not investigated in the present study. The distance between the upstream stenotic region and the downstream capture region is another important parameter, which remains to be further investigated for understanding how long upstream shear-induced platelet priming persists downstream. In addition to understanding the interactions between platelets and stenotic vascular devices, the present study could help to understand the mechanism of thrombus formation when blood vessels become stenotic as a result of atherosclerosis.
5. Conclusions
Using microfluidic stenotic chambers, the present study was designed to mimic in vitro the flow conditions encountered in a stenosed vascular graft anastomosis at a range of 1620 – 11560 s−1 centerline wall shear strain rates. We provided evidence that upstream stenosed geometries resulted in altered fluid mechanical conditions that promoted priming of platelets for enhanced downstream adhesion and activation. We also found that highly elevated wall shear strain rates (4860 s−1 and 11560 s−1) were able to induce fully spread and activated platelets as well as platelet lysis. The overall results suggested that the initiation of intracellular signaling and morphological changes, followed by the propagation of signals and the release of coagulation factors from granules could eventually lead to thrombus formation downstream. The present assay could be also utilized to study how an actual biomaterial placed downstream interacts with platelets primed by upstream agonists under different flow conditions. Although all experiments were performed in vitro with anticoagulated whole blood or PRP, the findings may be relevant to understanding why patients with implanted vascular devices are at thrombotic risk, even with anticoagulation therapy. Since vascular devices are sometimes characterized by anastomotic stenosis, the findings of the present study may also have implications for the re-design of vascular devices as well improving the design of in vitro platelet adhesion and activation assays.
Statement of Significance.
Studying the downstream response of platelets following transient exposure to an upstream agonist is important because of significant clinical implications to the implantation of vascular devices. Due to intimal fibrous hyperplasia, vascular biomaterials such as synthetic small-diameter vascular grafts sometimes become stenotic (narrow), leading to transient platelet exposure to elevated shear forces. In this study, a microfluidic flow system was developed to mimic a stenosed vascular graft and to investigate how highly elevated, transient upstream shear forces, typically found in severe stenosis, results in the pre-activation of platelets for downstream adhesion and activation. The findings of the present study have implications for optimizing the design of blood-contacting biomaterials in order to minimize thrombotic risk associated with transiently elevated shear forces. The findings also provide additional insights into the mechanisms of thrombus formation at the post-stenotic regions of vascular implants.
Acknowledgements
This work was supported by the National Institutes of Health (R01 HL126864). The authors would like to thank Drs. Jules Magda and Aaron Fogelson for their advices and discussions, and Dr. Robert Campbell from Weyrich’s lab for providing human donors blood necessary to complete the experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Swedberg SH, Brown BG, Sigley R, Wight TN, Gordon D, Nicholls SC, Intimal fibromuscular hyperplasia at the venous anastomosis of PTFE grafts in hemodialysis patients: clinical, immunocytochemical, light and electron microscopic assessment, Circulation 80 (1989) 1726–1736. [DOI] [PubMed] [Google Scholar]
- [2].Wilson YG, Davies AH, Currie IC, Morgan M, Vein graft stenosis: incidence and intervention, Eur. J. Vasc. Endovasc. Surg 11 (1996) 164–169. [DOI] [PubMed] [Google Scholar]
- [3].Huo Y, Luo T, Guccione JM, Teague SD, Tan W, Navia JA, Kassab GS, Mild anastomotic stenosis in patient-specific CABG model may enhance graft patency: a new hypothesis, PLoS One 8 (2013) e73769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhao R, Kameneva MV, Antaki JF, Investigation of platelet margination phenomena at elevated shear stress, Biorheology 44 (2007) 161–177. [PubMed] [Google Scholar]
- [5].Sugimoto M, Tsuji S, Kuwahara M, Matsui H, Shear-dependent functions of the interaction between soluble von Willebrand factor and platelet glycoprotein Ib in mural thrombus formation on a collagen surface, J. Hematol 69 (1999) 48–53. [PubMed] [Google Scholar]
- [6].Chow T, Hellums J, Moake J, Kroll M, Shear stress-induced von Willebrand factor binding to platelet glycoprotein Ib initiates calcium influx associated with aggregation, Blood 80 (1992) 113–120. [PubMed] [Google Scholar]
- [7].Nesbitt WS, Kulkarni S, Giuliano S, Goncalves I, Dopheide SM, Yap CL, Salem HH, Jackson SP, Distinct glycoprotein Ib/V/IX and integrin IIb 3-dependent calcium signals cooperatively regulate platelet adhesion under flow, J. Biol. Chem 277 (2001) 2965–1972. [DOI] [PubMed] [Google Scholar]
- [8].Yuan Y, Kulkarni S, Ulsemer P, Cranmer SL, The von Willebrand factor-glycoprotein Ib/V/IX interaction induces actin polymerization and cytoskeletal reorganization in rolling platelets and glycoprotein Ib/V/IX-transfected cells, J. Biol. Chem 274 (1999) 36241–36251. [DOI] [PubMed] [Google Scholar]
- [9].Kroll MH, Hellums JD, Guo Z, Durante W, Razdan K, Hrbolich JK, Schafer AI, Protein kinase C is activated in platelets subjected to pathological shear stress, J. Biol. Chem 268 (1993) 3520–3524. [PubMed] [Google Scholar]
- [10].Chen Z, Mondal NK, Ding J, Koenig SC, Slaughter MS, Griffith BP, Wu ZJ, Activation and shedding of platelet glycoprotein IIb/IIIa under non-physiological shear stress, Mol. Cell. Biochem 409 (2015) 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Anderson GH GH, Hellums JD, Moake JL, Alfrey CP, Platelet lysis and aggregation in shear fields, Blood Cells 4 (1977) 499–511. [PubMed] [Google Scholar]
- [12].Vroman L, Adams AL, Identification of rapid changes at plasma-solid interfaces, J. Biomed. Mater. Res. A 3 (1969) 43–67. [DOI] [PubMed] [Google Scholar]
- [13].Andrade JD, Hlady V, Protein adsorption and materials biocompatibility: a tutorial review and suggested hypotheses, Adv. Polym. Sci 79 (1986) 1–63. [Google Scholar]
- [14].Maxwell MJ, Dopheide SM, Turner SJ, Jackson SP, Shear induces a unique series of morphological changes in translocating platelets: effects of morphology on translocation dynamics, Arterioscler. Thromb. Vasc. Biol 26 (2006) 663–669. [DOI] [PubMed] [Google Scholar]
- [15].Lentz BR, Exposure of platelet membrane phosphatidylserine regulates blood coagulation, Prog. Lipid Res. 42 (2003) 423–438. [DOI] [PubMed] [Google Scholar]
- [16].Israels SJ, McMillan-Ward EM, CD63 modulates spreading and tyrosine phosphorylation of platelets on immobilized fibrinogen, Thromb. Haemost 93 (2005) 311–318. [DOI] [PubMed] [Google Scholar]
- [17].Peterson DM, Stathopoulos NA, Giorgio TD, Hellums JD, Moake JL, Shear-induced platelet aggregation requires von Willebrand factor and platelet membrane glycoproteins Ib and IIb-IIIa, Blood 69 (1987) 625–628. [PubMed] [Google Scholar]
- [18].Miyazaki Y, Nomura S, Miyake T, Kagawa H, Kitada C, Taniguchi H, Komiyama Y, Fujimura Y, Ikeda Y, Fukuhara S, High shear stress can initiate both platelet aggregation and shedding of procoagulant containing microparticles, Blood 88 (1996) 3456–3464. [PubMed] [Google Scholar]
- [19].Shankaran H, Alexandridis P, Neelamegham S, Aspects of hydrodynamic shear regulating shear-induced platelet activation and self-association of von Willebrand factor in suspension, Blood 101 (2003) 2637–2645. [DOI] [PubMed] [Google Scholar]
- [20].Godo MN, Sefton MV, Characterization of transient platelet contacts on a polyvinyl alcohol hydrogel by video microscopy, Biomaterials 20 (1999) 1117–1126. [DOI] [PubMed] [Google Scholar]
- [21].Corum LE, Hlady V, The effect of upstream platelet–fibrinogen interactions on downstream adhesion and activation, Biomaterials 33 (2012) 1255–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Holme PA, Orvim U, Hamers MJ, Solum NO, Brosstad FR, Barstad RM, Sakariassen KS, Shear-induced platelet activation and platelet microparticle formation at blood flow conditions as in arteries with a severe stenosis, Arterioscler. Thromb. Vasc. Biol 17 (1997) 646–653. [DOI] [PubMed] [Google Scholar]
- [23].Nesbitt WS, Westein E, Tovar FJ, A shear gradient–dependent platelet aggregation mechanism drives thrombus formation, Nat. Med 15 (2009) 665–673. [DOI] [PubMed] [Google Scholar]
- [24].Li M, Ku DN, Forest CR, Microfluidic system for simultaneous optical measurement of platelet aggregation at multiple shear rates in whole blood, Lab Chip 12 (2012) 1355–1362. [DOI] [PubMed] [Google Scholar]
- [25].Ha H, Lee SJ, Hemodynamic features and platelet aggregation in a stenosed microchannel, Microvasc. Res 90 (2013) 96–105. [DOI] [PubMed] [Google Scholar]
- [26].Westein E, van der Meer AD, Kuijpers MJE, Frimat JP, van den Berg A, Heemskerk JWM, Atherosclerotic geometries exacerbate pathological thrombus formation poststenosis in a von Willebrand factor-dependent manner, Proc. Natl. Acad. Sci 110 (2013) 1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jain A, Graveline A, Waterhouse A, Vernet A, Flaumenhaft R, Ingber D, A shear gradient-activated microfluidic device for automated monitoring of whole blood hemostasis and platelet function, Nat. Commun 7 (2016) 10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rahman SM, Eichinger CD, Hlady V, Effects of upstream shear forces on priming of platelets for downstream adhesion and activation, Acta Biomater. 73 (2018) 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Takahashi H, Emoto K, Dubey M, Castner DG, Grainger DW, Imaging surface immobilization chemistry: correlation with cell patterning on non‐adhesive hydrogel thin films, Adv. Funct. Mater 18 (2008) 2079–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Corum LE, Eichinger CD, Hsiao TW, Hlady V, Using microcontact printing of fibrinogen to control surface-induced platelet adhesion and activation, Langmuir 27 (2011) 8316–8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ku DN, Blood flow in arteries, Annu. Rev. Fluid Mech 29 (1997) 399–434. [Google Scholar]
- [32].Savage B, Saldivar E, Ruggeri ZM, Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand Factor, Cell 84 (1996) 289–297. [DOI] [PubMed] [Google Scholar]
- [33].Dopheide SM, Maxwell MJ, Jackson SP SP, Shear-dependent tether formation during platelet translocation on von Willebrand factor, Blood 99 (2002) 159–167. [DOI] [PubMed] [Google Scholar]
- [34].Zhang J, Bergeron AL, Yu Q, Sun C, McIntire LV, Lopez JA, Dong J, Platelet aggregation and activation under complex patterns of shear stress, Thromb. Haemost 88 (2002) 817–821. [PubMed] [Google Scholar]
- [35].Sheriff J, Bluestein D, Girdhar G, Jesty J, High-shear stress sensitizes platelets to subsequent low-shear conditions, Ann. Biomed. Eng 38 (2010) 1442–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tovar-Lopez FJ, Rosengarten G, Nasabi M, Sivan V, Khoshmanesh K, Jackson SP, Mitchell A, Nesbitt WS, An investigation on platelet transport during thrombus formation at micro-scale stenosis, PLoS One 8 (2013) e74123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kao S, McIntire LV, Rheology, in: Loscalzo J, Schafer AI (Eds.), Thrombosis and Hemorrhage, Lippincott Williams & Wilkins, Philadelphia, 2002, pp. 294–314. [Google Scholar]
- [38].Strony J, Beaudoin A, Brands D, Adelman B, Analysis of shear stress and hemodynamic factors in a model of coronary artery stenosis and thrombosis, Am. J. Physiol 265 (1993) H1787–H1796. [DOI] [PubMed] [Google Scholar]
- [39].Siegel JM, Markou CP, Ku DN, Hanson SR, A scaling law for wall shear rate through an arterial stenosis, J. Biomech. Eng 116 (1994) 446–451. [DOI] [PubMed] [Google Scholar]
- [40].Mitchell RN, Blood vessels, in: Kumar V, Abbas AK, Aster JC (Eds.), Robbins and Cotran Pathologic Basis of Disease, Elsevier Saunders, Philadelphia, 2014, pp. 483–522. [Google Scholar]
- [41].Sakariassen KS, Orning L, Turitto VT, The impact of blood shear rate on arterial thrombus formation, Future Sc. OA 1 (2015) FSO30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Harbers GM, Emoto K, Greef C, Metzger SW, Woodward HN, Mascali JJ, Grainger DW, Lochhead MJ , A functionalized poly(ethylene glycol)-based bioassay surface chemistry that facilitates bio-immobilization and inhibits non-specific protein, bacterial, and mammalian cell adhesion, Chem. Mater 19 (2007) 4405–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jarvis GE, Atkinson BT, Snell DC, Watson SP, Distinct roles of GPVI and integrin α2β1 in platelet shape change and aggregation induced by different collagens, Br. J. Pharmacol 137 (2002) 107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Litvinov RI, Farrell DH, Weisel JW, Bennett JS, The platelet integrin αIIbβ3 differentially interacts with fibrin versus fibrinogen, J. Biol. Chem 291 (2016) 7858–7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Reininger AJ, Heijnen HFG, Schumann H, Specht HM, Schramm W, Ruggeri ZM, Mechanism of platelet adhesion to von Willebrand factor and microparticle formation under high shear stress, Blood 107 (2006) 3537–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Podolnikova NP, Yermolenko IS, Fuhrmann A, Lishko VK, Magonov S, Bowen B, Enderlein J, Podolnikov AV, Ros R, Ugarova TP, Control of integrin αIIbβ3 outside-in signaling and platelet adhesion by sensing the physical properties of fibrin(ogen) substrates, Biochemistry 49 (2010) 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Aarts P, van den Broek S, Prins GW, Kuiken GD, Sixma JJ, Heethaar RM, Blood platelets are concentrated near the wall and red blood cells, in the center in flowing blood, Arterioscler. Thromb. Vasc. Biol 8 (1988) 819–824. [DOI] [PubMed] [Google Scholar]
- [48].Yazdani A, Karniadakis GE, Sub-cellular modeling of platelet transport in blood flow through microchannels with constriction, Soft Matter 12 (2016) 4339–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bark DL, Ku DN, Platelet transport rates and binding kinetics at high shear over a thrombus, Biophys. J 105 (2013) 502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lu Q, Malinauskas RA, Comparison of two platelet activation markers using flow cytometry after in vitro shear stress exposure of whole human blood, Artif. Organs 35 (2011) 137–144. [DOI] [PubMed] [Google Scholar]