

Abstract
Adolescent cannabis use has been implicated as a risk factor for schizophrenia; however, it is neither necessary nor sufficient. Previous studies examining this association have focused primarily on the role of the cannabinoid receptor 1 (CB1R) with relatively little known about a key regulatory protein, the cannabinoid receptor interacting protein 1 (CNRIP1). CNRIP1 is an intracellular protein that interacts with the C-terminal tail of CB1R and regulates its intrinsic activity. Previous studies have demonstrated aberrant CNRIP1 DNA promoter methylation in post-mortem in human patients with schizophrenia, and we have recently reported decreased methylation of the CNRIP1 DNA promoter in the ventral hippocampus (vHipp) of a rodent model of schizophrenia susceptibility. To examine whether augmented CNRIP1 expression could contribute to the pathology of schizophrenia, we performed viral-mediated overexpression of CNRIP1 in the vHipp of Sprague Dawley rats. We then tested these rats for behavioral correlates of schizophrenia symptoms, followed by electrophysiology to determine the effects on the dopamine system, known to underlie psychosis. Here, we report that overexpression of vHipp CNRIP1 induces impairments in latent inhibition and social interaction, similar to those observed in individuals with schizophrenia and in rodent models of the disease. Furthermore, rats overexpressing vHipp CNRIP1 displayed a significant increase in ventral tegmental area (VTA) dopamine neuron population activity, a putative correlate of psychosis. These data provide evidence that alterations in CNRIP1 may contribute to the pathophysiology of schizophrenia, as overexpression is sufficient to produce neurophysiological and behavioral correlates consistently observed in rodent models of the disease.
Keywords: Schizophrenia, cannabinoids, CB1, CNRIP1, ventral hippocampus
1. Introduction
Cannabis use has been suggested to increase the risk of developing schizophrenia in individuals with an underlying genetic predisposition (Andreasson et al., 1989; Arseneault et al., 2002; Henquet et al., 2005). Likewise, animal models of the disease display an increased susceptibility to exhibiting a schizophrenia-like phenotype when exposed to synthetic cannabinoids (Aguilar et al., 2018). Moreover, individuals with schizophrenia have a higher tendency to consume cannabis (Kovasznay et al., 1997), which can precipitate symptoms of the disease (Linszen et al., 1994). The cannabinoid hypothesis of schizophrenia speculates that alterations in the endocannabinoid system may contribute to the pathogenesis of schizophrenia (Emrich et al., 1997; Fernandez-Espejo et al., 2009). Additional support for this hypothesis stems from the observation that high densities of cannabinoid receptors type 1 (CB1R) are found in regions commonly implicated in schizophrenia (i.e. prefrontal cortex, hippocampus, basal ganglia, anterior cingulate cortex) (Berding et al., 2006; Glass et al., 1997; Zavitsanou et al., 2004). Furthermore, individuals with schizophrenia display increases in CB1R binding in these key brain regions (Dalton et al., 2011; Dean et al., 2001; Zavitsanou et al., 2004); however, it remains unclear as to how, or whether, these alterations contribute to symptoms associated with schizophrenia. Indeed, it is important to note that CB1R antagonists have failed to produce beneficial effects in clinical trials (Meltzer et al., 2004).
We have recently discovered alterations in the cannabinoid receptor interacting protein 1 (CNRIP1) in the MAM (methylazoxymethanol acetate) model, and in F2 MAM rats, a novel rodent model of schizophrenia susceptibility (Neary et al., 2017; Perez et al., 2016). F2 MAM rats were obtained by mating MAM-treated rats with control rats, resulting in a proportion of the offspring (~40%) displaying a schizophrenia-like phenotype (Perez et al., 2016). Further, evidence that this is a model of susceptibility was based on observations that exposure to schizophrenia risk factors, such as adolescent cannabinoids or stress, increases the proportion of F2 MAM rats displaying a schizophrenia-like phenotype (Aguilar et al., 2018). In our characterization of this model, we observed a decrease in the methylation of the DNA promoter for CNRIP1 (Perez et al., 2016), which is predicted to translate to an increase in CNRIP1 expression. Similarly, aberrant CNRIP1 DNA promoter methylation has been observed post-mortem in human patients with schizophrenia (Wockner et al., 2014). As its name suggests, CNRIP1 interacts with the C-terminal tail of CB1R (Nie and Lewis, 2001; Niehaus et al., 2007) and is reported to alter the constitutive activity of this receptor (Niehaus et al., 2007). Thus, an increase in CNRIP1 expression is hypothesized to lead to augmented constitutive activity at the CB1R, without overt changes in CB1R expression (Smith et al., 2010). The consequences of this potential increase in CNRIP1 expression is not currently known, and will thus be investigated in this study.
Aberrant hippocampal activity has been reported in individuals with schizophrenia (Heckers, 2001, 2004; Schobel et al., 2009), as well as in rodent models of the disease (Lodge and Grace, 2007; Perez and Lodge, 2013). Additionally, increases in the activity of ventral tegmental area (VTA) dopamine neurons have been attributed to hyperactivity of the vHipp (Lodge and Grace, 2007); furthermore, the positive symptoms associated with schizophrenia are thought to arise from dysfunction of the mesolimbic dopamine system (Abi-Dargham, 2004; Laruelle and Abi-Dargham, 1999). Indeed, interactions between the cannabinoid system and the dopaminergic system are suggested to lead to psychosis, as cannabinoids transiently increase the activity of dopamine neurons (Voruganti et al., 2001). Based on these data, we posit that alterations in vHipp CNRIP1 expression may contribute to a schizophrenia-like phenotype.
2. Materials and Methods
All experiments were performed in accordance with the guidelines outlined in the USPH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of UT Health San Antonio.
2.1. Western Blot
To verify that the alterations in CNRIP1 DNA promoter methylation (reported in F2 MAM-treated rats (Perez et al., 2016)) actually alters CNRIP1 protein expression, western blot was utilized. The vHipp was dissected from F2 MAM- and saline-treated rats (n= 9 per group), homogenized in ice-cold buffer (750μL) containing a protease inhibitor cocktail, centrifuged (14,000 r.p.m. for 2 minutes) and supernatant containing protein fractions was collected. Protein concentrations were determined using the Bradford method before incubation with Laemmli Sample Buffer containing 5% dithiothreitol (10 minutes at 90°C) and separated 150-200mA on a 10% acrylamide gel. Proteins were transferred (120 minutes at 250mA) to PVDF membranes. Membranes were then blocked for 1 hour (5% BSA in TBST) before incubation with primary antibody for CNRIP1 and CB1R (anti-CNRIP, 1:2000, Abeam, abl67087; anti-CNRI, 1:2000, Synaptic Systems, AA151-200; anti-β-actin, 1:20,000) for 1 hour at 4°C. Membranes were washed three times in TBST (10 minutes each), followed by incubation in secondary antibody (goat anti-rabbit IgG-HRP, 1:500, Santa Cruz Biotechnology, SC-2004 or goat anti-mouse antibody IgG-HRP, 1:500, Santa Cruz Biotechnology, SC-2005) at room temperature (1 hour).
2.2. CB1R/CNRIP1 Co-lmmunoprecipitation
To demonstrate that there is indeed a direct association between CB1R and CNRIP1 in the vHipp we performed co-immunoprecipitation. Male Sprague Dawley rats (n=4; 400-550g) were anesthetized with Fluriso™ (2-5% Isoflurane, USP with oxygen flow at 1L/min; VetOne; #502017) and quickly decapitated. The vHipp were extracted, homogenized in ice-cold buffer (750μL; 50mM Tris-HCl, 150mL NaCl, 0.1% Triton X-100, pH 7.4) containing a protease inhibitor cocktail (Sigma; P8340), centrifuged (14,000 r.p.m. for 2 minutes) and supernatant was collected. SureBeads™ Protein G Magnetic beads (BioRad; #1614013) were incubated for 10 minutes with anti-CB1R antibody (2μg; anti-CB1; Abcam; ab23703) diluted in 200μL TBST (15mM Tris-HCl, 137mM NaCl and 0.1% Tween 20) at room temperature; the SureBeads™-antibody complexes were washed and incubated with 500μL of protein lysate for 15 minutes. The beads were then magnetized and washed 3 times using 500 μL TBST. Laemmli Sample Buffer (50 μL; BioRad; #161-0737) containing 5% dithiothreitol was added to the beads and heated for 10 minutes at 90°C. The immunoprecipitated protein (25μl per well) was separated (45 minutes at 150V) on Mini-Protean TGX Any kD gels (BioRad; #456-9035) in Tris/glycine/SDS buffer (Thermo Fisher Scientific; BP1341-1) and subsequently transferred to nitrocellulose/filter paper sandwiches (0.2μm; BioRad; #162-0213) with transfer buffer (25mM Tris-HCl, 192mM glycine, 20%w/v ethanol, pH 8.3) for one hour at 100V. Membranes were then blocked for 30 minutes (5% BSA in TBST) before incubation with primary antibody (anti-CB1R or anti-CNRIP1; 1:2000; Abcam; abl67087) for 1 hour at 4°C. Next, membranes were washed three times in TBST (10 minutes each), followed by incubation in Protein A - HRP (1:5000; BD Biosciences; #610438) at room temperature (1 hour). Lastly, membranes were treated with Pierce ECL Western Blotting substrate (1 minute; Thermo Fisher Scientific; #32106) and protein signal was captured with a G:BOX-XT4 Chemi system (Syngene).
2.3. Lentivirus overexpression of CNRIP1
Adult male Sprague Dawley rats (~350-375 g) were obtained from Envigo. All survival surgical procedures were performed under general anesthesia in a semi-sterile environment. Rats were anesthetized with sodium pentobarbital (60 mg/kg, i.p.) and placed in a stereotaxic apparatus. Anesthesia was maintained by supplemental administration of sodium pentobarbital as required to maintain suppression of limb compression withdrawal reflex. A core body temperature of 37°C was sustained by a thermostatically controlled heating pad. The skin was reflected and bore holes drilled bilaterally overlying the vHipp (A/P ± 5.3, M/L ± 5.0 from bregma and D/V −7.5 mm ventral to the brain surface). Rats were bilaterally injected with high-titer (0.75 μL/side; ~1×109 TU/mL) lentivirus particles containing vectors expressing CNRIP1 or a GFP expressing control (see Figure 2A for vector map: obtained from VectorBuilder). The skin was sutured closed and rats housed under ABSL2 conditions for 72 hours before being transferred to standard housing conditions. Rats were housed for a period of 6 weeks prior to any behavioral or electrophysiological examinations to ensure stable transgene expression.
Figure 2. Vector map for cannabinoid receptor interacting protein 1 (CNRIP1) over-expression (A) and validation of ventral hippocampal (vHipp) CNRIP1 overexpression.
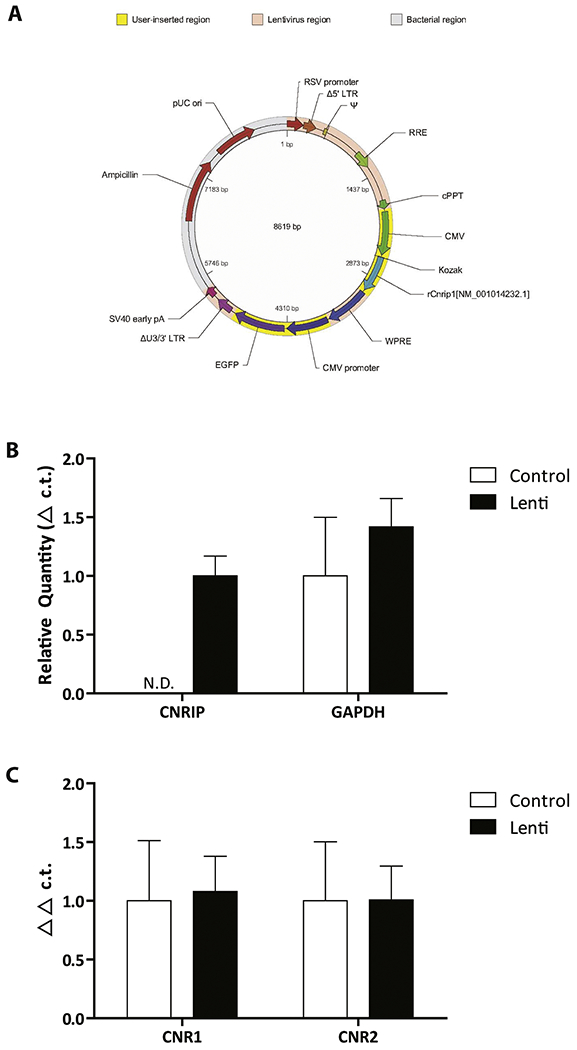
Over-expression of CNRIP1 in the vHipp was confirmed using RT-PCR (B; n= 6 rats per group). Since the levels of CNRIP1 in control rats were not detectable (N.D.), no statistical analyses were performed. Importantly, the expression of GAPDH was not different between groups. mRNA expression levels of the cannabinoid 1 receptor (CNR1) and the cannabinoid 2 receptor (CNR2) were not affected by the over expression of CNRIP1 (C; n= 6 rats per group).
2.4. Latent Inhibition
As a model of positive symptoms associated with schizophrenia we examined latent inhibition. Rats over expressing vHipp CNRIP1 or controls were randomly assigned into two groups (tone pre-exposure or no tone exposure) and placed in a conditioning chamber (30.5 × 25.4 × 30.5 cm3) with metal walls and a stainless steel grid shock floor (Coulbourn Instruments, Holliston, MA). A subset of rats were presented with a 20 second tone over 16 trials with a pseudorandom inter-trial interval (ITI) averaging two minutes, while the remaining rats remained in their home cage. All rats then underwent an established fear conditioning procedure (Green et al., 2011) in which a 20 second tone co-terminated with a mild foot shock (0.8 mA, 0.5 sec). The tone-shock pairing was presented four times with a pseudorandom ITI averaging two minutes. Twenty-four hours after conditioning, rats were returned to the conditioning chamber and re-exposed to the 20 second tone over four trials. Behavior was video recorded at each stage and freezing behavior was analyzed offline using FreezeView (Coulbourn Instruments). Freezing behavior was defined as movement below a motion index threshold of ten, lasting at least one second. The percentage of latent inhibition was determined using the following formula: 100 − percent freezing in response to the conditioned tone (in tone pre-exposed rats / average non pre-exposed rats).
2.5. Social Interaction
To evaluate negative symptoms associated with schizophrenia we examined social behavior (as described previously (Cecchi et al., 2002)) in rats over expressing vHipp CNRIP1 or controls. In brief, rats were placed singly in a testing arena (100 × 100 × 40 cm) for 15 minutes per day for a period of two days prior to testing to habituate to the arena. On the testing day, experimental rats were placed in the arena with a weight matched “stimulus” rat. Testing was video recorded for five minutes by video camera for offline analysis by a blind experimenter. The dependent measure was the time the test animal spent actively engaged in social activity (sniffing, grooming, climbing on, following, or wrestling) with the stimulus rat.
2.6. In Vivo Extracellular Recordings
Rats over expressing vHipp CNRIP1 or controls were anesthetized with 8% chloral hydrate (400 mg/kg, i.p.), as this anesthetic does not significantly depress dopamine neuron activity (Hyland et al., 2002), and placed in a stereotaxic apparatus. Anesthesia was maintained by supplemental administration of chloral hydrate as required to maintain suppression of limb compression withdrawal reflex. A core body temperature of 37°C was sustained by a thermostatically controlled heating pad. Extracellular glass microelectrodes (impedance 6-14 MΩ) were lowered into the VTA (A/P ±5.3, M/L ±0.6 from bregma and D/V −6.5 to −9.0 mm from the surface of the brain) using a hydraulic micropositioner to measure dopamine neuron activity. Spontaneously active dopamine neurons were identified with open filter settings (low pass: 30 Hz; high pass: 30 kHz) using previously established electrophysiological criteria (Grace and Bunney, 1983), which have recently been reviewed and evaluated to support the reliability of these types of recordings (Ungless and Grace, 2012). Once isolated, dopamine neuron activity was recorded for 2-3 minutes. Three parameters of dopamine neuron activity were measured: population activity (defined as the number of spontaneously active dopamine neurons encountered while making six to nine vertical passes, separated by 200 μm in a predetermined pattern to sample equivalent regions of the VTA); basal firing rate; and burst firing (proportion of action potentials occurring in bursts).
2.7. Quantitative PCR
To verify CNRIP1 overexpression we performed qPCR. vHipp tissue was taken from a subset of rats following electrophysiology and processed using an RNA isolation kit (RNAqueous®-4PCR Total RNA Isolation Kit; Thermo Fisher Scientific; AM1914). Briefly, tissue samples were homogenized in Lysis/Binding solution and RNA was precipitated and separated by filtration. The concentration of RNA was determined by absorbance at 260 nm with a NanoDrop Spectrometer (ThermoFisher Scientific) and converted to single stranded cDNA by using a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific; #4368814) and thermal cycler. Real-time PCR was performed with FAM-labeled TaqMan primers (Thermo Fisher Scientific) targeting either CNRIP1 (Rn01434074_ml), CB1R (CNR1; Rn02758689_sl), CB2R (CNR2; Rn03993699_sl) or GAPDH (Rn01775763_gl).
2.8. Data Analysis
Electrophysiological analysis of dopamine neuron activity was performed using commercially available computer software (LabChart version 7.1; ADInstruments, Chalgrove, Oxford, UK). Detection of FAM-labeled DNA was performed by a CFX384 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). Co-lmmunoprecipitation blots were captured with a G:BOX-XT4 Chemi system (Syngene; Frederick, MD, USA). Western blots of F2 tissue were scanned and optical density measured using ImageJ. Data are represented as the mean ± s.e.m. unless otherwise stated, with n values representing the number of neurons recorded or number of animals per experimental group where indicated. All statistics were calculated using Prism software (GraphPad Software, San Diego, CA, USA) or SigmaPlot (Systat Software, Chicago, IL, USA). Data was analyzed by Unpaired-t test, one-way ANOVA or two-way ANOVA and significance determined at P<0.05.
2.9. Materials
Fluriso™ was purchased from VetOne (Boise, ID, USA). Chloral hydrate, sodium pentobarbital and protease inhibitor cocktail were purchased from Sigma-Aldrich (St. Louis, MO, USA). Lentiviral vectors were produced and packaged by VectorBuilder (Santa Clara, CA, USA). The Pierce ECL Western Blotting Substrate, Tris/glycine/SDS buffer RNAqueous®, 4PCR Total RNA Isolation Kit and High Capacity cDNA Reverse Transcriptase Kits, as well as FAM-labeled TaqMan Primers and Gene Expression Master Mix were purchased from ThermoFischer Scientific (Waltham, MA, USA). Anti-CB1R and anti-CNRIP1 antibodies were purchased from Abeam (Cambridge, MA, USA). Anti-CNR1 antibody (used in western blot) was purchased from Synaptic Systems (Goettingen, Germany). HRP Protein A was purchased from BD Biosciences (San Jose, CA, USA). Anti-β-actin was purchased from Chemicon. Goat anti-rabbit IgG-HRP and goat anti-mouse IgG-HRP were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). SureBeads™ Protein G Magnetic beads, Laemmli Sample Buffer, Mini-Protean TGX Any kD gels and nitrocellulose/filter paper sandwiches were supplied from BioRad (Hercules, CA, USA). All other chemicals and reagents were of either analytical or laboratory grade, and were purchased from various suppliers.
3. Results
3.1. Western Blot and Co-lmmunoprecipitation
We have previously shown that F2 MAM-treated rats display a decrease in CNRIP1 DNA promoter methylation (Perez et al., 2016) that likely translate to an increase in CNRIP1 expression. To examine whether F2 generation MAM- and saline-treated rats exhibit altered CNRIP1 and CB1R expression in the vHipp, we performed western blot analysis. Indeed, CNRIP1 expression in the vHipp is elevated in F2 MAM rats (~20% increase compared to F2 saline rats; n= 9 rats per group) without a change in the expression of CB1R (Figure 1A). In addition, further data analysis showed a significant positive correlation between CNRIP1 and CB1R expression in control rats (R2= 0.5254; P< 0.05) that was completely lost in F2 MAM rats (R2= 0.0074; P= 0.83; Figure 1B). In order to verify an association between CB1R and CNRIP1 in the vHipp, we performed co-immunoprecipitation (Figure 1C,D) and found that CNRIP1 was co-immunoprecipitated with CB1R, as determined by the presence of a 17kD band, consistent with previous reports of an association between CNRIP1 and the CB1R.
Figure 1. F2 MAM-treated rats display an increase (~20%) in the expression of ventral hippocampal (vHipp) cannabinoid receptor interacting protein 1 (CNRIP1).
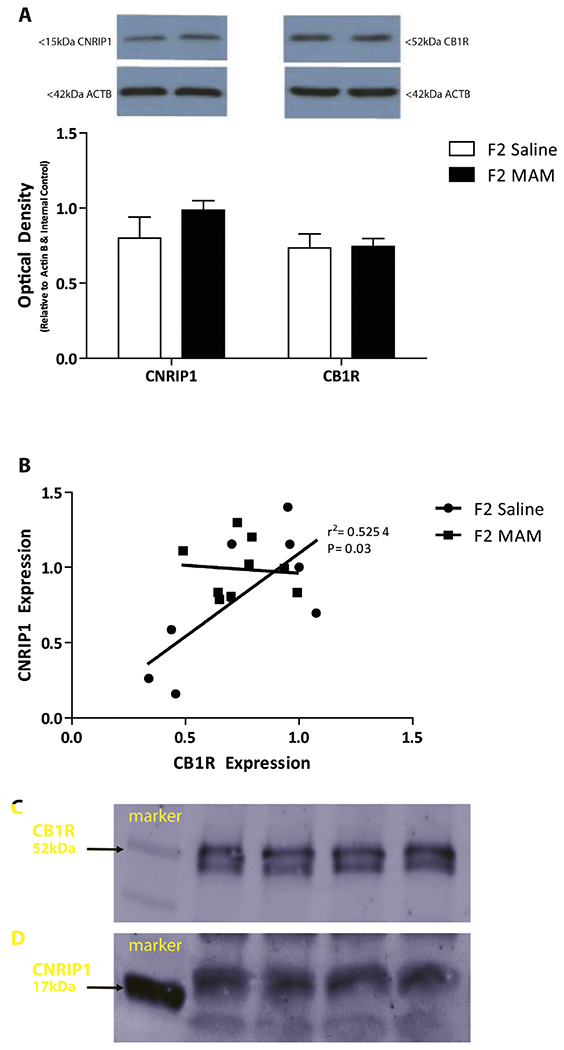
Quantification of CNRIP1 and cannabinoid 1 receptor (CB1R) protein expression in the vHipp of F2 MAM- and saline-treated rats (A). There is a significant correlation between CNRIP1 and CB1R in the vHipp of control rats and not in F2 MAM- treated rats (B; Pearson correlation; R2= 0.5254). In normal rats, co-immunoprecipitation revealed an association between CB1R and CNRIP1 (n= 4 rats). Representative films demonstrating CNRIP1 (C) and CB1R (D) expression.
3.2. Quantitative PCR.
The over-expression of CNRIP1 in the vHipp was confirmed post-mortem by quantitative PCR. Rats that received lentivirus particles containing vectors expressing CNRIP1 (n= 6; Figure 2A) displayed robust expression of vHipp CNRIP1 mRNA (Figure 2B). Statistical analysis was not performed, as CNRIP1 expression levels in control rats were below the limits of detection. Importantly, GAPDH mRNA expression was observed in equivalent amounts in both groups (relative quantity; control: 1.00±0.50; Lenti: 1.42±0.24: Unpaired t test; t= 0.75; P= 0.47). In addition, there was not a significant difference between the levels of CB1R mRNA (CNR1; control: 1.00±0.51; Lenti: 1.08±0.30) and CB2R mRNA (Figure 2C; CNR2; control: 1.00±0.50; Lenti: 1.01±0.29; two-way ANOVA; P= 0.93).
3.3. Latent Inhibition
Latent inhibition can be measured across species and deficits are observed in individuals with schizophrenia (Swerdlow et al., 2003). Latent inhibition is the observation that a conditioned or familiar stimulus takes more time to become meaningful when compared to a new stimulus. Individuals with schizophrenia display decreases in latent inhibition, which is thought to reflect an inability to ignore irrelevant stimuli (Weiner and Arad, 2009). Here we demonstrate that rats overexpressing CNRIP1 in the vHipp displayed a trend (p=0.05) for a deficit in latent inhibition (n= 6; −57.54 ± 51.71% latent inhibition) when compared to control rats (n=6; bilaterally injected with lentivirus particles containing vectors expressing GFP; +64.42 ± 20.37 % latent inhibition; Unpaired t test; t=2.19, p=0.05; Figure 3A).
Figure 3. Cannabinoid receptor interacting protein 1 (CNRIP1) over-expression in the ventral hippocampus (vHipp) impairs latent inhibition and causes deficits in social interaction.

Rats in which CNRIP1 is over-expressed in the vHipp display a deficit in latent inhibition, a behavioral correlate of positive symptoms, when compared to control rats (A; n= 6 rats per group). *p=0.05. CNRIP1 overexpression in the vHipp reduced time spent interacting time when compared to control rats (B; n= 11-12 rats per group), a marker of negative associated symptoms. *p<0.05.
3.4. Social Interaction
Individuals with schizophrenia often display negative symptoms, such as social avoidance (Hansen et al., 2009). Rodents display a relatively consistent social behavior that is disrupted in putative models of the disease (Donegan et al., 2017; Sams-Dodd, 1995, 1996). Thus in this study we used the social interaction test as a correlate of these symptoms. CNRIP1 over-expression in the vHipp reduced social interaction time (n=11; 74.45 ± 6.88 sec) when compared to control rats (n=12; 95.00± 5.70 sec; Unpaired t test; t=2.32, p<0.05; Figure 3B), which is a consistent observation in various other rodent models of schizophrenia (Donegan et al., 2017; Flagstad et al., 2004; Seillier and Giuffrida, 2009).
3.5. In Vivo Extracellular Recordings
Over-expression of CNRIP1 in the vHipp caused downstream changes in dopamine neuron activity within the VTA. Control rats (n= 6;) displayed an average of 1.00 ±0.06 spontaneously active dopamine neurons per track, consistent with previous findings in untreated rats (Boley et al., 2014; Lodge and Grace, 2007; Perez et al., 2014; Perez and Lodge, 2012; Perez et al., 2013; Shah and Lodge, 2013). Rats over-expressing CNRIP1 in the vHipp (n= 10) had a significant increase in the number of spontaneously active dopamine neurons (1.98±0.11) when compared to control rats (one-way ANOVA; P< 0.001; Figure 4A). No significant differences were observed in the firing rate or percent burst firing of VTA dopamine neurons (n= 36-118; one-way ANOVA; Figure 4B,C).
Figure 4. Ventral hippocampal (vHipp) over-expression of cannabinoid receptor interacting protein 1 (CNRIP1) causes changes in ventral tegmental area (VTA) dopamine neuron activity.
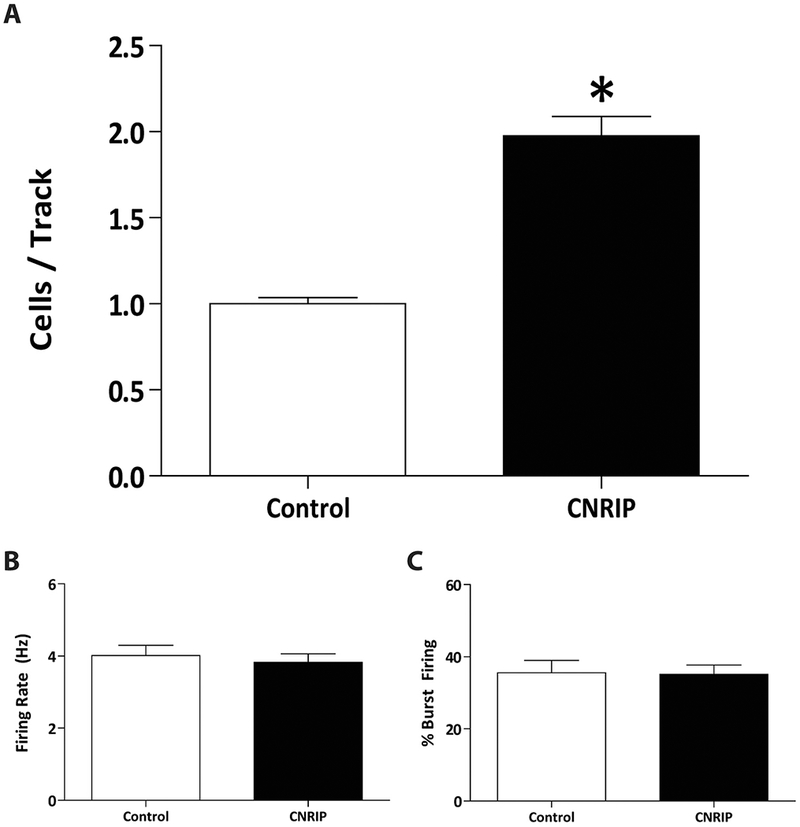
Specifically, we observed significantly increases in dopamine neuron population activity (A; n= 6-10 rats per group) without altering the average firing rate (B; n= 36-118 cells per group) or percent burst firing (C; n= 36-118 cells per group). *p<0.001.
4. Discussion
Recent post-mortem studies of individuals with schizophrenia suggest that epigenetic modifications, such as differential DNA methylation, are important in the etiology of the disease (Connor and Akbarian, 2008; Nishioka et al., 2012; Wockner et al., 2014). Aberrant DNA methylation of the promoter for the CB1 receptor interacting protein, CNRIP1, has been observed in individuals with schizophrenia (Wockner et al., 2014) and in F2-MAM rats (Perez et al., 2016), a rodent model of heritable susceptibility. Surprisingly, there is very little information about the role of CNRIP1, which is particularly striking given the extensive literature on the cannabinoid system. Here, we demonstrate that overexpression of CNRIP1 in the vHipp of rodents leads to downstream changes in dopamine neuron activity and behavioral deficits analogous to positive and negative symptoms commonly observed in individuals with schizophrenia.
Cannabinoid receptors (i.e. CB1R) are G-protein-coupled receptors (GPCRs) that play a role in central nervous system function by mediating the effects of tetrahydrocannabinol (THC; the principal psychoactive constituent of cannabis) and endocannabinoids (Howlett et al., 2002). Expression levels of CNRIP1 do not appear to influence the expression of CB1R or ligand binding affinity (Niehaus et al., 2007; Smith et al., 2015). Similarly, here we demonstrate that the overexpression of CNRIP1 mRNA does not affect the expression of CB1 or CB2 receptors, as measured by qPCR. Western blot analysis performed in vHipp tissue revealed a 20% increase in CNRIP1 protein expression in F2 MAM-treated rats when compared to controls, with no change in CB1R protein expression. Additionally, evidence supports an interaction between CNRIP1 and CB1R (Blume et al., 2013; Ludanyi et al., 2008), such that CNRIP1 may play a regulatory role in the cannabinoid system by modulating CB1R activation (Smith et al., 2015). Whether CB1R activity contributes to, or is affected by schizophrenia, is not currently known and was the focus of this study. Here we confirm an association between CB1R and CNRIP1 in the vHipp by co-immunoprecipitation and demonstrate a significant correlation between CB1R and CNRIP1 protein expression. This correlation was completely lost in F2 MAM rats, where increases in CNRIP1 were independent of CB1 levels, suggesting a possible dysregulation of CB1R function in these animals.
In rats that over expressed CNRIP1 in the vHipp, we performed the latent inhibition behavioral assay to model the positive symptoms associated with schizophrenia. Latent inhibition is disrupted in patients with schizophrenia (Gray et al., 1995b), as patients typically have difficulty ignoring irrelevant stimuli. Similarly, rodents with increased mesolimbic dopaminergic activity display deficits in latent inhibition (Moore et al., 2006; Young et al., 1993). Typical antipsychotics have been shown to enhance latent inhibition preclinically, as well as in patients (Gray et al., 1995a; Gray et al., 1995b; Leumann et al., 2002; Young et al., 1993). Rats that over-expressed CNRIP1 in the vHipp exhibited a deficit in latent inhibition when compared to their respective controls, mimicking positive symptoms associated with schizophrenia. We were not able to directly correlate the latent inhibition with our readout of dopamine neuron activity (population activity); however, studies have demonstrated that latent inhibition relies on dopamine system function (Gray et al., 1995a; Young et al., 1993). Additionally, as a marker of negative symptoms, we measured social interaction time. Individuals with schizophrenia often display deficits in social functioning or behaviors (Blanchard et al., 1998). Rodent models of the disease display similar deficits in the social interaction task, as they spend less time interacting when introduced to a novel rat (Flagstad et al., 2004; Sams-Dodd, 1995; Seillier and Giuffrida, 2009). Likewise, we observed a decrease in the time spent interacting in rats with over-expression of CNRIP1 in the vHipp. Most commonly associated with negative symptoms is the medial prefrontal cortex (mPFC), which is innervated by the vHipp (Carr and Sesack, 1996; Jay et al., 1989; Wolkin et al., 1992). Indeed, we have previously demonstrated that rodent behavioral correlates of negative symptoms can be reversed by normalizing activity in the vHipp (Donegan et al., 2017). Thus, we posit that increases in vHipp CNRIP1 may cause deficits in social interaction due to aberrant innervating of the mPFC.
Interestingly, CNRIP1 over-expression in the vHipp was sufficient, in and of itself, to induce downstream changes in VTA dopamine neuron population activity, such that the number of spontaneously active dopamine neurons was significantly increased, without any change to the average firing rate, or burst firing pattern. This not surprising as previous studies have demonstrated that the vHipp can regulate VTA dopamine neuron activity, such that aberrant activity in the vHipp leads to a significant increase in population activity (Lodge and Grace, 2007). Increases in dopamine neuron population activity are commonly observed in rodent models of schizophrenia [i.e. MAM, chronic PCP, and F2-MAM models (Aguilar, 2014; Lodge and Grace, 2007; Perez et al., 2016; Perez et al., 2014; Perez and Lodge, 2012, 2013; Perez et al., 2013)] and attributed to aberrant regulation of the mesolimbic dopamine system by the vHipp (Aguilar, 2014; Lodge and Grace, 2007). Further, studies have shown that the firing rates and burst firing activity of individual dopamine neurons are affected by manipulations to the pedunculopontine tegmental nucleus, medial prefrontal cortex, and lateral dorsal tegmentum, without altering the population activity of dopamine neurons (Floresco et al., 2003; Murase et al., 1993). Thus, CNRIP1 over-expression accurately recapitulates the schizophrenia-like phenotype of increased population activity observed in various rodent models of the disease (Aguilar, 2014; Perez et al., 2016; Perez et al., 2014; Perez and Lodge, 2012, 2013; Perez et al., 2013). Measuring cells per track has been shown to be a reliable method to study in vivo changes in dopamine neuron population activity, as well as differences in average firing rate and burst firing patterns (Grace and Bunney, 1983; Ungless and Grace, 2012). Although it has been suggested that ‘silent’ dopamine neurons may not be detected using this method because of low firing rates and/or low electrotonic length (Dai and Tepper, 1998), evidence from more recent studies suggests that this is a reliable measure of the activity of the population of dopamine neurons in the VTA (Choong and Shen, 2004; Shen and Choong, 2006; Ungless and Grace, 2012).
The exact mechanism as to how this over-expression of vHipp CNRIP1 yields a schizophrenia-like phenotype is unclear; however, as mentioned previously alterations in cannabinoid signaling have been observed in individuals with schizophrenia (De Marchi et al., 2003; Leweke et al., 1999). Pre-clinically, studies have demonstrated that administration of CB1R agonists, in F2 MAM susceptible rats, can significantly increase the proportion of rats that exhibit a schizophrenia-like phenotype (Aguilar et al., 2018). Specifically, those that develop the phenotype display increases in population activity and an increased sensitivity to psychomotor stimulants (a marker of positive symptoms) (Aguilar et al., 2018). A class of fast-spiking containing PV, a calcium binding protein, have been associated with aberrant activity in the vHipp and downstream dysfunction of VTA dopamine neurons (Boley et al., 2014; Perez and Lodge, 2014; Shah and Lodge, 2013). Further, protein expression of parvalbumin (PV) in the vHipp was significantly decreased by adolescent cannabinoid exposure and negatively correlated with VTA dopamine neuron population activity, such that those rats that had lower levels of PV expression displayed a higher population activity (Aguilar et al., 2018). It is possible that over-expressing CNRIP1, leads to aberrant constitutive activity of the CB1R, which may alter the function of PV interneurons; however, this remains to be established.
In general, modeling symptoms of schizophrenia is challenging, as this disease truly is human in nature. However, given that we don’t completely understand the pathophysiology of the disease, rodent models are essential to provide information to guide novel therapies. Here, we demonstrate that alterations in CNRIP1 expression could contribute to the pathophysiology of schizophrenia. Indeed, altered CNRIP1 expression may increases the susceptibility of an individual to develop schizophrenia. This is supported by preclinical data demonstrating that adolescent cannabinoid agonist administration increases the proportion of F2 MAM rats displaying a schizophrenia-like phenotype (Aguilar et al., 2018). In our model of vHipp CNRIP1 overexpression, there were downstream changes in dopamine neuron population activity, which contribute to positive symptoms of the disease, as well as behaviors analogous to positive and negative symptoms associated with the disease. These data are consistent with observations made in various other rodent models of schizophrenia. Further, this model will serve as a useful tool to further investigate interactions between CNRIP1 and CB1R and the role of hippocampal CNRIP1 in developing schizophrenia; thus, allowing us to develop new targets for therapeutic interventions for the treatment of schizophrenia.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abi-Dargham A, 2004. Do we still believe in the dopamine hypothesis? New data bring new evidence. The international journal of neuropsychopharmacology / official scientific journal of the Collegium Internationale Neuropsychopharmacologicum 7 Suppl 1, S1–5. [DOI] [PubMed] [Google Scholar]
- Aguilar DD, Chen L & Lodge DJ, 2014. Increasing Endocannabinoid Levels in the Ventral Pallidum Restores Aberrant Dopamine Neuron Activity in the Subchronic PCP Rodent Model of Schizophrenia. The international journal of neuropsychopharmacology / official scientific journal of the Collegium Internationale Neuropsychopharmacologicum. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar DD, Giuffrida A, Lodge DJ, 2018. Adolescent Synthetic Cannabinoid Exposure Produces Enduring Changes in Dopamine Neuron Activity in a Rodent Model of Schizophrenia Susceptibility. The international journal of neuropsychopharmacology / official scientific journal of the Collegium Internationale Neuropsychopharmacologicum 21(4), 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasson S, Allebeck P, Rydberg U, 1989. Schizophrenia in users and nonusers of cannabis. A longitudinal study in Stockholm County. Acta psychiatrica Scandinavica 79(5), 505–510. [DOI] [PubMed] [Google Scholar]
- Arseneault L, Cannon M, Poulton R, Murray R, Caspi A, Moffitt TE, 2002. Cannabis use in adolescence and risk for adult psychosis: longitudinal prospective study. Bmj 325(7374), 1212–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berding G, Schneider U, Gielow P, Buchert R, Donnerstag F, Brandau W, Knapp WH, Emrich HM, Muller-Vahl K, 2006. Feasibility of central cannabinoid CB1 receptor imaging with [124I]AM281 PET demonstrated in a schizophrenic patient. Psychiatry research 147(2-3), 249–256. [DOI] [PubMed] [Google Scholar]
- Blanchard JJ, Mueser KT, Bellack AS, 1998. Anhedonia, positive and negative affect, and social functioning in schizophrenia. Schizophrenia bulletin 24(3), 413–424. [DOI] [PubMed] [Google Scholar]
- Blume LC, Bass CE, Childers SR, Dalton GD, Roberts DC, Richardson JM, Xiao R, Selley DE, Howlett AC, 2013. Striatal CB1 and D2 receptors regulate expression of each other, CRIP1A and delta opioid systems. Journal of neurochemistry 124(6), 808–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boley AM, Perez SM, Lodge DJ, 2014. A fundamental role for hippocampal parvalbumin in the dopamine hyperfunction associated with schizophrenia. Schizophrenia research 157(1-3), 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DB, Sesack SR, 1996. Hippocampal afferents to the rat prefrontal cortex: synaptic targets and relation to dopamine terminals. The Journal of comparative neurology 369(1), 1–15. [DOI] [PubMed] [Google Scholar]
- Cecchi M, Khoshbouei H, Morilak DA, 2002. Modulatory effects of norepinephrine, acting on alpha 1 receptors in the central nucleus of the amygdala, on behavioral and neuroendocrine responses to acute immobilization stress. Neuropharmacology 43(7), 1139–1147. [DOI] [PubMed] [Google Scholar]
- Choong K, Shen R, 2004. Prenatal ethanol exposure alters the postnatal development of the spontaneous electrical activity of dopamine neurons in the ventral tegmental area. Neuroscience 126(4), 1083–1091. [DOI] [PubMed] [Google Scholar]
- Connor CM, Akbarian S, 2008. DNA methylation changes in schizophrenia and bipolar disorder. Epigenetics 3(2), 55–58. [DOI] [PubMed] [Google Scholar]
- Dai M, Tepper JM, 1998. Do silent dopaminergic neurons exist in rat substantia nigra in vivo? Neuroscience 85(4), 1089–1099. [DOI] [PubMed] [Google Scholar]
- Dalton VS, Long LE, Weickert CS, Zavitsanou K, 2011. Paranoid schizophrenia is characterized by increased CB1 receptor binding in the dorsolateral prefrontal cortex. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 36(8), 1620–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marchi N, De Petrocellis L, Orlando P, Daniele F, Fezza F, Di Marzo V, 2003. Endocannabinoid signalling in the blood of patients with schizophrenia. Lipids Health Dis 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean B, Sundram S, Bradbury R, Scarr E, Copolov D, 2001. Studies on [3H]CP-55940 binding in the human central nervous system: regional specific changes in density of cannabinoid-1 receptors associated with schizophrenia and cannabis use. Neuroscience 103(1), 9–15. [DOI] [PubMed] [Google Scholar]
- Donegan JJ, Tyson JA, Branch SY, Beckstead MJ, Anderson SA, Lodge DJ, 2017. Stem cell-derived interneuron transplants as a treatment for schizophrenia: preclinical validation in a rodent model. Molecular psychiatry 22(10), 1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emrich HM, Leweke FM, Schneider U, 1997. Towards a cannabinoid hypothesis of schizophrenia: cognitive impairments due to dysregulation of the endogenous cannabinoid system. Pharmacol Biochem Behav 56(4), 803–807. [DOI] [PubMed] [Google Scholar]
- Fernandez-Espejo E, Viveros MP, Nunez L, Ellenbroek BA, Rodriguez de Fonseca F, 2009. Role of cannabis and endocannabinoids in the genesis of schizophrenia. Psychopharmacology 206(4), 531–549. [DOI] [PubMed] [Google Scholar]
- Flagstad P, Mork A, Glenthoj BY, van Beek J, Michael-Titus AT, Didriksen M, 2004. Disruption of neurogenesis on gestational day 17 in the rat causes behavioral changes relevant to positive and negative schizophrenia symptoms and alters amphetamine-induced dopamine release in nucleus accumbens. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 29(11), 2052–2064. [DOI] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA, 2003. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nature neuroscience 6(9), 968–973. [DOI] [PubMed] [Google Scholar]
- Glass M, Dragunow M, Faull RL, 1997. Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 77(2), 299–318. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS, 1983. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons--1. Identification and characterization. Neuroscience 10(2), 301–315. [DOI] [PubMed] [Google Scholar]
- Gray JA, Joseph MH, Hemsley DR, Young AM, Warburton EC, Boulenguez P, Grigoryan GA, Peters SL, Rawlins JN, Taib CT, et al. , 1995a. The role of mesolimbic dopaminergic and retrohippocampal afferents to the nucleus accumbens in latent inhibition: implications for schizophrenia. Behavioural brain research 71(1-2), 19–31. [DOI] [PubMed] [Google Scholar]
- Gray NS, Pilowsky LS, Gray JA, Kerwin RW, 1995b. Latent inhibition in drug naive schizophrenics: relationship to duration of illness and dopamine D2 binding using SPET. Schizophrenia research 17(1), 95–107. [DOI] [PubMed] [Google Scholar]
- Green MK, Rani CS, Joshi A, Soto-Pina AE, Martinez PA, Frazer A, Strong R, Morilak DA, 2011. Prenatal stress induces long term stress vulnerability, compromising stress response systems in the brain and impairing extinction of conditioned fear after adult stress. Neuroscience 192, 438–451. [DOI] [PubMed] [Google Scholar]
- Hansen CF, Torgalsboen AK, Melle I, Bell MD, 2009. Passive/apathetic social withdrawal and active social avoidance in schizophrenia: difference in underlying psychological processes. The Journal of nervous and mental disease 197(4), 274–277. [DOI] [PubMed] [Google Scholar]
- Heckers S, 2001. Neuroimaging studies of the hippocampus in schizophrenia. Hippocampus 11(5), 520–528. [DOI] [PubMed] [Google Scholar]
- Heckers S, 2004. The hippocampus in schizophrenia. American Journal of Psychiatry 161(11), 2138–2139. [DOI] [PubMed] [Google Scholar]
- Henquet C, Murray R, Linszen D, van Os J, 2005. The environment and schizophrenia: the role of cannabis use. Schizophrenia bulletin 31(3), 608–612. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG, 2002. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 54(2), 161–202. [DOI] [PubMed] [Google Scholar]
- Hyland BI, Reynolds JN, Hay J, Perk CG, Miller R, 2002. Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience 114(2), 475–492. [DOI] [PubMed] [Google Scholar]
- Jay TM, Glowinski J, Thierry AM, 1989. Selectivity of the hippocampal projection to the prelimbic area of the prefrontal cortex in the rat. Brain research 505(2), 337–340. [DOI] [PubMed] [Google Scholar]
- Kovasznay B, Fleischer J, Tanenberg-Karant M, Jandorf L, Miller AD, Bromet E, 1997. Substance use disorder and the early course of illness in schizophrenia and affective psychosis. Schizophrenia bulletin 23(2), 195–201. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, 1999. Dopamine as the wind of the psychotic fire: new evidence from brain imaging studies. Journal of psychopharmacology 13(4), 358–371. [DOI] [PubMed] [Google Scholar]
- Leumann L, Feldon J, Vollenweider FX, Ludewig K, 2002. Effects of typical and atypical antipsychotics on prepulse inhibition and latent inhibition in chronic schizophrenia. Biological psychiatry 52(7), 729–739. [DOI] [PubMed] [Google Scholar]
- Leweke FM, Giuffrida A, Wurster U, Emrich HM, Piomelli D, 1999. Elevated endogenous cannabinoids in schizophrenia. Neuroreport 10(8), 1665–1669. [DOI] [PubMed] [Google Scholar]
- Linszen DH, Dingemans PM, Lenior ME, 1994. Cannabis abuse and the course of recent-onset schizophrenic disorders. Archives of general psychiatry 51(4), 273–279. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA, 2007. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 11424–11430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludanyi A, Eross L, Czirjak S, Vajda J, Halasz P, Watanabe M, Palkovits M, Magloczky Z, Freund TF, Katona I, 2008. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. The Journal of neuroscience : the official journal of the Society for Neuroscience 28(12), 2976–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer HY, Arvanitis L, Bauer D, Rein W, Meta-Trial Study, G., 2004. Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. The American journal of psychiatry 161(6), 975–984. [DOI] [PubMed] [Google Scholar]
- Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA, 2006. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biological psychiatry 60(3), 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase S, Grenhoff J, Chouvet G, Gonon FG, Svensson TH, 1993. Prefrontal cortex regulates burst firing and transmitter release in rat mesolimbic dopamine neurons studied in vivo. Neuroscience letters 157(1), 53–56. [DOI] [PubMed] [Google Scholar]
- Neary JL, Perez SM, Peterson K, Lodge DJ, Carless MA, 2017. Comparative analysis of MBD-seq and MeDIP-seq and estimation of gene expression changes in a rodent model of schizophrenia. Genomics 109(3-4), 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie J, Lewis DL, 2001. The proximal and distal C-terminal tail domains of the CB1 cannabinoid receptor mediate G protein coupling. Neuroscience 107(1), 161–167. [DOI] [PubMed] [Google Scholar]
- Niehaus JL, Liu Y, Wallis KT, Egertova M, Bhartur SG, Mukhopadhyay S, Shi S, He H, Selley DE, Howlett AC, Elphick MR, Lewis DL, 2007. CB1 cannabinoid receptor activity is modulated by the cannabinoid receptor interacting protein CRIP 1a. Molecular pharmacology 72(6), 1557–1566. [DOI] [PubMed] [Google Scholar]
- Nishioka M, Bundo M, Kasai K, Iwamoto K, 2012. DNA methylation in schizophrenia: progress and challenges of epigenetic studies. Genome Med 4(12), 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SM, Aguilar DD, Neary JL, Carless MA, Giuffrida A, Lodge DJ, 2016. Schizophrenia-Like Phenotype Inherited by the F2 Generation of a Gestational Disruption Model of Schizophrenia. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 41(2), 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SM, Carreno FR, Frazer A, Lodge DJ, 2014. Vagal nerve stimulation reverses aberrant dopamine system function in the methylazoxymethanol acetate rodent model of schizophrenia. The Journal of neuroscience : the official journal of the Society for Neuroscience 34(28), 9261–9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SM, Lodge DJ, 2012. Aberrant dopamine D2-like receptor function in a rodent model of schizophrenia. Journal of Pharmacology and Experimental Therapeutics 343(2), 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SM, Lodge DJ, 2013. Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Molecular psychiatry 18(11), 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SM, Lodge DJ, 2014. New approaches to the management of schizophrenia: focus on aberrant hippocampal drive of dopamine pathways. Drug Des Devel Ther 8, 887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SM, Shah A, Asher A, Lodge DJ, 2013. Hippocampal deep brain stimulation reverses physiological and behavioural deficits in a rodent model of schizophrenia. The international journal of neuropsychopharmacology / official scientific journal of the Collegium Internationale Neuropsychopharmacologicum 16(6), 1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sams-Dodd F, 1995. Distinct effects of d-amphetamine and phencyclidine on the social behaviour of rats. Behav Pharmacol 6(1), 55–65. [PubMed] [Google Scholar]
- Sams-Dodd F, 1996. Phencyclidine-induced stereotyped behaviour and social isolation in rats: a possible animal model of schizophrenia. Behav Pharmacol 7(1), 3–23. [PubMed] [Google Scholar]
- Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, Small SA, 2009. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Archives of general psychiatry 66(9), 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seillier A, Giuffrida A, 2009. Evaluation of NMDA receptor models of schizophrenia: divergences in the behavioral effects of sub-chronic PCP and MK-801. Behavioural brain research 204(2), 410–415. [DOI] [PubMed] [Google Scholar]
- Shah A, Lodge DJ, 2013. A loss of hippocampal perineuronal nets produces deficits in dopamine system function: relevance to the positive symptoms of schizophrenia. Translational psychiatry 3, e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen RY, Choong KC, 2006. Different adaptations in ventral tegmental area dopamine neurons in control and ethanol exposed rats after methylphenidate treatment. Biological psychiatry 59(7), 635–642. [DOI] [PubMed] [Google Scholar]
- Smith TH, Blume LC, Straiker A, Cox JO, David BG, McVoy JR, Sayers KW, Poklis JL, Abdullah RA, Egertova M, Chen CK, Mackie K, Elphick MR, Howlett AC, Selley DE, 2015. Cannabinoid receptor-interacting protein 1a modulates CB1 receptor signaling and regulation. Molecular pharmacology 87(4), 747–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TH, Sim-Selley LJ, Selley DE, 2010. Cannabinoid CB1 receptor-interacting proteins: novel targets for central nervous system drug discovery? British journal of pharmacology 160(3), 454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Stephany N, Wasserman LC, Talledo J, Sharp R, Auerbach PP, 2003. Dopamine agonists disrupt visual latent inhibition in normal males using a within-subject paradigm. Psychopharmacology 169(3–4), 314–320. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Grace AA, 2012. Are you or aren’t you? Challenges associated with physiologically identifying dopamine neurons. Trends in Neurosciences 35(7), 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voruganti LN, Slomka P, Zabel P, Mattar A, Awad AG, 2001. Cannabis induced dopamine release: an invivo SPECT study. Psychiatry research 107(3), 173–177. [DOI] [PubMed] [Google Scholar]
- Weiner I, Arad M, 2009. Using the pharmacology of latent inhibition to model domains of pathology in schizophrenia and their treatment. Behavioural brain research 204(2), 369–386. [DOI] [PubMed] [Google Scholar]
- Wockner LF, Noble EP, Lawford BR, Young RM, Morris CP, Whitehall VL, Voisey J, 2014. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Translational psychiatry 4, e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolkin A, Sanfilipo M, Wolf AP, Angrist B, Brodie JD, Rotrosen J, 1992. Negative symptoms and hypofrontality in chronic schizophrenia. Archives of general psychiatry 49(12), 959–965. [DOI] [PubMed] [Google Scholar]
- Young AM, Joseph MH, Gray JA, 1993. Latent inhibition of conditioned dopamine release in rat nucleus accumbens. Neuroscience 54(1), 5–9. [DOI] [PubMed] [Google Scholar]
- Zavitsanou K, Garrick T, Huang XF, 2004. Selective antagonist [3H]SR141716A binding to cannabinoid CB1 receptors is increased in the anterior cingulate cortex in schizophrenia. Progress in neuro-psychopharmacology & biological psychiatry 28(2), 355–360. [DOI] [PubMed] [Google Scholar]