

Abstract
Patient derived organoids have emerged as robust preclinical models for screening anti-cancer therapeutics. Current 2D culturing methods do not provide physiological responses to therapeutics, therefore 3D models are being developed to better reproduce physiological responses. 3D culturing however often requires large initial cell populations and one week to one month to grow tumors ready for therapeutic testing. As a solution a 3D culturing system has been developed capable of producing physiologically relevant tumors in an expedited fashion while only requiring a small number of initial cancer cells. A bi-layer microfluidic system capable of facilitating active convective nutrient supply to populations of cancer cells facilitates expedited growth of cancer cells when starting with populations as small as 8 cells. The system has been shown to function well with adherent and non-adherent cell types by expediting cell growth by a factor ranging from 1.27 to 4.76 greater than growth under static conditions. Utilizing such an approach has enable to formation of tumors ready for therapeutic screening within 3 days and the ability to perform therapeutic screening within the microfluidic system is demonstrated. A mathematical model has been developed which allows for adjustments to be made to the dynamic delivery of nutrients in order to efficiently use culture media without excessive waste. We believe this work to be the first attempt to grow cancers in an expedited fashion utilizing only a convective nutrient supply within a microfluidic system which also faciliates on-device therapeutic screening. The developed microfluidic system and cancer growth method have the potential to offer improved drug screening for patients in clinical settings.
Graphical Abstract
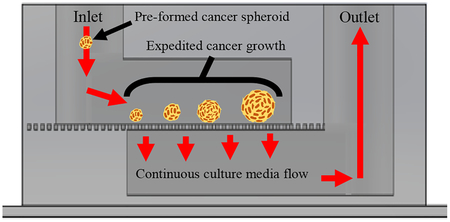
Expedited cancer growth technique to generate cancer populations for therapeutic screening within 3 days utilizing media flow and few cells.
Introduction
Throughout the world 14.1 million people suffer from cancers of various types as of 2012, with an estimated increase to around 21.3 million by 20301–3. The average survival rate after diagnosis varies based on cancer type and the stage at which the cancer is identified2,3. Improving the likelihood of patient survival requires early diagnosis along with rapid development and implementation of treatment plans2,3.
Current in vitro growth of cancer models and therapeutic performance tests are run using two-dimensional monolayers or sheet cultures of cancer cells4,5. While this approach reduces the time required to identify a viable candidate, it makes use of a model system which does not match what is experienced in vivo4–9. As such this method can result in the selection of a therapeutic treatment plan which is not ideal for use against the patient’s tumor mass due to misrepresentation of cell-cell interactions and differences in drug kinetics between 2D and 3D models4–9. In order to better identify viable therapeutic treatment options for patients a need exists for methods capable of generating three-dimensional tumor spheroids or masses in a short period of time4–9. Current in vitro approaches such as liquid-overlay10, hanging-drop11, magnetic levitation12, bioreactors13 and others are capable of producing tumor spheroids however suffer from issues of low spheroid quantities, inability to refresh culture media, difficulty with direct tumor imaging, difficulty with introduction of therapeutic for drug testing, high cost, large initial quantities of cancer cell require for growth and long periods of time as indicated in SF. 114–20. Ideally, such in vitro growth systems should be capable of generating tumor models without such issues for testing10–13,21. In addition, the tumor models should be produced quickly in order to reach an exponential growth phase, where treatment of in vivo tumors typically occurs4,5.
Current 3D growth models require 1 week to 1 month in order to fully establish cancer spheroids before any drug screening can occur4,22. Additionally, several hundreds to thousands of initial cancer cells are often required in order to form such 3D tumor models14–20. As an example, in vivo injection of cancer cells into animal models such as mice and rats often requires large populations (1×105 or more) of cancer cells for injection and can typically require a several weeks or more of growth before treatment testing can begin23–25. The acquisition of such a large population of cells from patient samples can often be difficult due to the rarity of cells in certain cancer types where fewer than 50 cells may be collected in biopsies14–20. Following the establishment of the tumor models, simultaneous therapeutic analysis is required in order to identify and viable therapeutic treatments14–20. As such, being able to perform tumor growth to a predetermined size in an expedited manner within a platform capable of facilitating tests with dozens of potential therapeutic options will aid to shorten the drug screening process14–20. However, performing multiple drug tests simultaneously increases the time required for data acquisition and analysis.
The overall pool of potential therapeutics capable of being utilized as treatment options continues to grow as new drugs are approved by the FDA. From the year 2013 to February of 2018, a total of nearly 160 new drugs for treatment of various cancers have been approved and released on the market26. For some specific cancers, such as leukemia, there exist multiple drug choices which could potentially serve as the best treatment option for patients, with BCR-ABLI T315I-positive cell leukemia alone having 11 potential options27. More substantial are the number of drug candidates which are made available to patients in clinical trials, where it is not uncommon to have upwards of 50 or more candidates available15. Such clinical trials typically pose more risk to patients as a full response profile has not been completely established and could serve to be a waste of patient’s time if the chosen treatment is not effective15. Current practices typically utilize trial dosages on patience to test drug candidate responses until an effective drug is found, which a time-consuming and painful process28–30. Such large quantities of potential therapeutic options is promising for patients, however, identifying which is best suited can serve to be an insurmountable challenge15–20. In order to better sample a larger range of the total available clinical trial and marketed drug options, a system must be developed which is capable of running tens to hundreds of potential options simultaneously in order to facilitate the timely selection of a viable treatment31,32.
Previous microfluidic platforms have been developed to help address the issues faced in cancer therapeutic testing33–35. All of these systems involve the production of devices which have specialty chambers in which cancer cells can be grown and exposed to therapeutic agents33–35. However, most of the systems designed to handle three dimensional tumor spheroids require that the spheroids be pre-formed outside of the device and later introduced upon reaching a suitable size or that the spheroids be grown slowly within the device33–37. In both cases, the time required to produce the tumor spheroids is long, large initial quantities of cancer cells are required and there is the need for an additional handling step when spheroids are formed outside of the microfluidics33–37. The previous approaches which form spheroids directly within microfluidic systems often rely on the use of hydrogel or ECM materials to facilitate the three dimensional growth of the tumors34,36,37. While the use of hydrogels or ECM is beneficial in providing structural support to the growing tumors, their presence around the spheroids limits the ability to deliver nutrients to the tumor34,36,37. Such growth models require the nutrients to undergo diffusion through the hydrogel materials before reaching the spheroids34–37. As such, the rate of growth for the spheroids is limited when compared to a system where nutrient delivery to the periphery of the tumor occurs unhindered. Therefore, when attempting to grow cancer spheroids as quickly as possible for use in therapeutic testing, the ability to avoid nutrient diffusion through hydrogels is important33,35. Based on such previous work, expedited growth and improved drug screening require methods for manipulating and processing cancer cells.
Prior research in the field of cancer cell processing and manipulation has demonstrated the utility of microfluidics for producing tumors and performing therapeutic screening38–42. The ability to isolate cancer cell and precisely control their fate after isolation serves as the framework on which all down-stream therapeutic screening rest38,41,42. Many sorting, processing and manipulation techniques rely on well-characterized device geometries and flow conditions38,41,42. Utilizing the foundational work performed by other research groups, we have developed a system capable of manipulating cancer cells and actively delivering key growth nutrients38–42. The developed system and growth technique are designed to facilitate fast tumor growth directly within the microfluidic environment and coupled therapeutic testing.
Current methods for producing cancer cell populations for therapeutic testing require extended periods of time31,32,43,44. Such delays typically experienced when growing cancer populations result in lost time when attempting to identify viable therapeutic treatments31,32,43,44. In order to shorten the time delay experienced during cancer cell growth and drug screening from one or more weeks, down to just a few days requires a method of expedited growth. In order to address these issues, a microfluidic system will be developed which is capable of generating tumor spheroids in an expedited fashion utilizing convection driven nutrient delivery. The microfluidic system will be tested with adherent (spheroid forming) and non-adherent (suspension) cancer cell to verify performance across different cancer types. In addition, a mathematical model will be utilized to verify and predict the growth of the cancer within the microfluidic system. Application of mathematical model and direct imaging of the cancer populations will serve as a feedback loop to facilitate expedited cancer growth in an adaptive manner. Overall, outperforming current cancer growth techniques will serve to provide a platform better suited for therapeutic testing as shown in SF. 1.
Experimental
Device Fabrication
Bi-layer microfluidic platforms were produced following similar techniques outlined in previous microfluidic works from the Liu group45–49. The devices produced for expedited tumor spheroid growth contained a semi-permeable polycarbonate membrane which functioned to separate the apical and basal halves of the microfluidics. The circular chamber where the cancer cells were grown measured 3mm in diameter and 1.5mm in height. The straight sections of channel in the microfluidic devices measured 600μm in height and width. The use of the semi-permeable membrane within the system functioned to prevent the cancer cells introduced into the system from being washed downstream and out of the channel, which had an average pore diameter of 800nm. A depiction of the microfluidic setup can be viewed in Fig. 1.
Figure. 1.

Microfluidic device schema depicting device structure and cancer cell capture function utilizing semi-permeable membrane. Flow direction indicated by red arrows. (a), Device schema cross-section. (b), Device schema overview. (c), Image of sample microfluidic device utilized in experimental testing (white scale bar is 3mm long).
Leukemia Cell Growth
Leukemia cancer cells were flown into the system using a syringe pump (PHD 2000, Harvard Apparatus) with culture media (DMEM(1x) + GlutaMAX – I (gibco, life technologies), 10% HI-FBS (Sigma-Aldrich), 1% Penn/Strep Antibiotic(1X) (gibco, life technologies)). Due to the small pores of the semi-permeable membrane, the cancer cells were captured by the membrane and all held in the same viewing plane within the devices. The leukemia cells used for testing were purchased from ATCC and all tests were carried out with cells within 5 passages from the initial frozen stock provided by ATCC. The initial count of cells introduced into all devices was held constant at 1×105, which corresponds to the lower end of the range of leukemia cells per milliliter typically drawn in patient samples50–53. A hemocytometer was used to verify the concentration of cells in a stock suspension from which appropriate volumes of the suspension were separated to ensure that 1X105 cells were introduced into the microfluidic devices. Due to the large number of cells used in each test, visual confirmation was not possible. Culture media was continuously flown within the devices for the flow based test cases. Static test cases, only received an initial influx of culture media during the cell seeding process. For flow based tests, specific flow rates of 3.4×103μL/hr and 3.4×104μL/hr were generated in order to introduce a large quantity of nutrients into the growth system. Nutrient consumption by the cells was visualized over time using culture media containing phenol red that changes color from red to yellow as the pH of the culture media becomes more acidic. The visual observation of color change was used as an indirect measure of nutrient availability on the basis that cells utilizing nutrients undergo the Kreb Cycle and produce CO2 as a by-product that builds up in culture media. Increased CO2 concentration in the culture media leads to a more acidic environment and subsequent color change from red to yellow. Utilizing the visual culture media color change and nutrient consumption rates reported in literature for the cells allows for the estimation of nutrient consumption over time based on the size of the cancer cell populations. The leukemia cells were maintained and grown under standard culturing conditions of 37°C and 5% CO2 in an incubator. Lastly, due to the non-adherent nature of the leukemia cells no cancer cell spheroid can be produced with the leukemia cells. Instead of growing as a spheroid, the leukemia cells grow separate from one another.
HCT116 Spheroid Growth
Tumors were grown using HCT116 human colorectal cancer cells. The HCT116 cells used for testing were purchased from ATCC and all tests were carried out with cells within 5 passages from the initial frozen stock provided by ATCC. All spheroids utilized in this work were grown from an initial suspensions of 30 cells which were pre-formed into loose tumor spheroids via an 8 hour incubation in a low adhesion round bottom well plate under static conditions. The Ultra-Low Attachment Multiple Well Plates were manufactured by Corning that feature a covalently bound hydrogel layer that effectively inhibits cellular attachment. No additional coatings were required to facilitate spheroid formation in the round bottom wells. A hemocytometer was used to verify the concentration of cells in a stock suspension from which appropriate volumes of the suspension were separated to ensure that exactly 30 cells were introduced into each well. Due to the small number of cells used in each test, visual confirmation was performed for each well to ensure that 30 cells were present. Well containing greater or fewer than 30 cells were discarded and not used for growth testing. The pre-formed spheroids were then collected and flown into their respective microfluidic devices. Once inside the microfluidic devices, the semi-permeable membrane functioned to capture the pre-spheroids and hold them in place over the duration of the experimentation. The same process of an 8 hour incubation in a low adhesion well plate to pre-form spheroids was utilized for the static based tests. After pre-forming the initial static test case spheroids, Matrigel was added to the well plate in order to further facilitate spheroid growth over the course of 120 hours. Tumor spheroids grown under flow conditions were established as noted in the leukemia growth section at specific flow rates of 3.4×103μL/hr and 3.4×104μL/hr. Nutrient consumption for the HCT116 cells was also visualized and estimated as described for the leukemia cancer cells. Likewise, the HCT116 cells were maintained and grown under the same standard culturing conditions of 37°C and 5% CO2 in an incubator as the leukemia cells.
Minimum Cell Growth Requirements
In order to determine the minimum number of HCT116 cells required to produce tumor spheroids, a range of initial cell counts were tested within the microfluidic device. HCT116 cells were pre-clustered for 8 hours in low-adhesion 96 well plates in order to form loosely associated pre-spheroids as described in the previous section. The cells were maintained under standard culturing conditions coupled with slow swirling of the entire plate on a shaker for 4 hours followed by static culturing for the remaining 4 hours. Once the pre-spheroids were formed and stable enough to be transported out of the wells, a syringe was used to transfer the spheroids into the microfluidic devices as described above. Further culturing of the spheroids occurred within the microfluidic devices following the protocol described above for the HCT116 cells. The total range of initial cell counts tested was: 1, 3, 5, 8, 10, 13, 15, 18, 20, 23, 25, 28, 30, and 33. Three rounds of testing were performed with 8 replicates of each initial cell count run per round. The frequency of successfully forming pre-spheroids for each initial cell count can be seen in SF. 6.
Cancer Growth Measurements
Cancer growth was monitored daily over the course of five days with brightfield and fluorescence imaging. All data collected from the leukemia growth studies was accomplished using CellTracker™ Stain (10μM red, Thermo Fisher Scientific) and standard fluorescence imaging (Olympus IX70, Hamamatsu C9300, Plan Fluor 10x & 20x, NA: 0.3, RI: 1). Data collection for spheroid growth was accomplished utilizing CellTracker™ Stain (10μM red, Thermo Fisher Scientific) and confocal microscopy (Nikon C2+, Apo 4X). Images of each cancer growth set-up were captured every 12 hours over the course of 5 days. All images were processed with FIGI (ImageJ) in order to measure HCT116 tumor sizes and cell counts for leukemia tests.
Statistical Analysis
Statistical analysis of all obtained results were run utilizing IBM’s SPSS statistical software package (IBM Corp.). All of the figures have significant differences indicated above elements within the plots. One way ANOVA tests were run for each data set with confidence levels of 95% held throughout all plots. All analyses were carried out under conditions of Tukey equal variances assumed, along with tests of homogeneity of variance further verified by both Brown-Forsythe and Welch analyses. All line plots show statistically significant differences in means compared against the baseline tests for the statically grown cancer cells, indicated by “*”. Significance between 3.4×103μL/hr and 3.4×104μL/hr test data is indicated by “ ** “, noting that all differences are given at a confidence level of 95%. Sample sizes for all experimental testing were determined by performing estimation for multiple-sample one-way ANOVA pairwise comparison based on pilot studies utilizing the standard sample size approximation of: . All statistical comparisons are run under assumptions of equal variance between groups. This assumption of equal variance is verified via the Levene’s test where all p values must be greater than 0.05 in order to verify the equal variance assumption across groups. All data sets presented in this work pass the Levene’s test with p values greater than 0.05 when applicable.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding authors upon reasonable request.
Cell Line Authentication
The cell lines utilized in this work do not appear on the “List of Commonly Misidentified Cell Lines” maintained by the International Cell Line Authentication Committee (ICLAC). Cell line authentications (AR230-r: ATCC CRL-3346 & HCT116: ATCC CCL-247) have been provided directly from cell line source.
Results
Microfluidic Device Fabrication
A microfluidic platform has been developed which is capable of delivering a continuous nutrition supply to growing cells. The microfluidic device channels are made of Polydimethylsiloxane (PDMS) through standard photolithography47–49,54. Both the upper and lower channels are comprised of a short straight section of channel which leads into a large circular portion. The width of the straight sections for both channels are 600μm and the diameter of the circular portion is 3mm. The height of the both upper and lower channels is also 1.5mm over the entire length of the straight and circular channel regions of the device. Fig. #1 shows the overall design and layout of the microfluidic platform. The upper and lower channels of the device are separated by a polycarbonate semi-permeable membrane which contains uniformly distributed pores with 800nm diameters. The presence of the semi-permeable membrane functions to retain cancer cells and tumor spheroids inside of the device and also serves to keep all cells in the same viewing plane. The pores in the semi-permeable membrane also allow transport of culture media and cellular waste products through and out of device. Using the microfluidic system, non-adherent cancer cells and small preformed tumor spheroids can be grown and monitored over long time durations of a week or more.
Cancer Cell Culturing and Imaging
Adherent cancer cells (HCT116) grown within the devices were first aggregated together under static culturing conditions within low adhesion well-plates. The cells were added to the wells and given 8 hours to adhere into loose spheroids before being introduced into the microfluidic devices. Both leukemia and HCT116 cancer cells were successfully cultured within the microfluidic devices. Fig. 2c depicts representative images of leukemia cells grown within the devices when subjected to a flow rate of 3.4×104 μL/hr over the course of 120 hours. Fig. 3c depicts representative images of HCT116 cells grown when subjected to a flow rate of 3.4×104 μL/hr over the course of 120 hours. Both representative figures show overall increases in cell counts as time progresses as is indicated by the presented growth data in Figs. 2a and 3a. Additionally, control cancer populations were grown under static conditions in well plates which can be seen in SF. 2 (leukemia) and SF. 3 (HCT116).
Figure 2.

Leukemia cell growth over time based on flow conditions (105 cell initial count). a, Normalized leukemia cell count over time for various flow conditions and theoretical model predictions. b, Normalized leukemia nutrient availability within microfluidic system over time for various flow conditions. Equilibrium point indicates when nutrient availability within the system can no longer satisfy the requirements of the entire cancer cell population. Inset figure shows magnified view of the static test case crossing over equilibrium point. c, Representative fluorescence images of leukemia cell growth over the course of 120 hours under a flow rate of 3.4×104 μL/hr (stained with CellTracker Red™) (scale bars are 150μm). Statistical significance indicated by “*” between flow based tests and static conditions and “**” between flow based tests at p≤0.05.
Figure 3.

HCT116 cell growth over time based on flow conditions (30 cell initial count). a, Normalized HCT116 cell count over time for various flow conditions and theoretical model predictions. b, Normalized nutrient availability within microfluidic system over time for various flow conditions. Equilibrium point indicates when nutrient availability within the system can no longer satisfy the requirements of the entire cancer cell population. c, Representative fluorescence images of HCT116 tumor spheroid growth over the course of 120 hours under a flow rate of 3.4×104 μL/hr (scale bars are 60μm). Statistical significance indicated by “*” between flow based tests and static conditions and “**” between flow based tests at p≤0.05.
Cancer Growth Measurements and Nutrient Availability
Successful culturing of both adherent and non-adherent cancer cell lines was accomplished, as demonstrated by the growth of human colorectal (HCT116) and human leukemia (AR230-r), respectively, in Fig. 2a and 3a. Growth of the cancer populations within the devices was measured directly through confocal imaging with CellTracker™ red stain. Through the application of various flow rates within the microfluidic devices, the rate of growth for the cancer cell populations was controlled, as shown in Fig. 2b and 3b. The use of convective flow allowed for a maximum of 1.27 to 4.76 times faster cancer cell growth when compared to the static growth cases for AR230-r and HCT116, respectively. When compared to the static test cases, introducing 3.4×103μL/hr nutrient flow into the systems containing leukemia cells results in an overall increase in the cell population by a factor of 1.27 at the end of 120 hours. Similarly, the normalized number of leukemia cells present after 120 hours of growth under flow conditions of 3.4×104μL/hr, was improved by a factor of 1.38. More significant were the observed increases in HCT116 growth with the 3.4×103μL/hr test case resulting in a 2.47 factor increase in the cell population. Even more impressive was the observed increase in HCT116 growth when supplied with a nutrient flow of 3.4×104μL/hr, which resulted in a 4.76 factor increase in the cell population. Representative images collected for static culturing conditions of both cell types can be seen in SF. 2 and SF. 3, respectively. The observed growth of both cancer types had good agreement with the theoretical predications made based on the mathematical growth model developed based on the transport of nutrients throughout tumors, as shown below. The model is a combination of equations for static cell growth that have been modified to include components of fluid transport in tissues specific for our microfluidic model and cell types utilized55–61. First the growth of the cell lines was verified under static conditions. The growth of either cell population is described by
| (1) |
where Np is the number of cancer cells after a growth time t, No is the initial number of cells in the population, ro is the static growth rate constant for the particular cell type (Leuk=0.00330 & HCT=0.01937), and t is time. As time increases, the population of cells increases based on a growth rate that is specific for each cell type. Once the static growth estimations are verified to agree well with experimental results, the consumption of the entire population of cells under static conditions can be determined by
| (2) |
where Cp is the nutrient consumption rate by the whole population of cells and Cc is the nutrient consumption rate of an individual cell based on type. Using equations 1 and 2 provide the ability to predict the growth and rate of nutrient consumption for static growth conditions however are not suitable for make predictions when convective nutrient transport is applied to the system. In order to model growth in the system, the convective transport, diffusion and consumption of nutrients must be considered. To start, the rate of nutrient convection to the cancer population is defined differently for adherent and non-adherent cells. In the case of non-adherent cells, the rate of nutrient convection (CTnad) simply takes the form
| (3) |
where [Nuo] is the stock media nutrient concentration and Qch is the flow rate established in the microfluidic channel. However when considering adherent cells growing in a tumor spheroid, the model requires additional considerations. Specifically, the influences of interstitial fluid pressure, channel flow rate, tumor surface area, tumor permeability, and nutrient concentration gradient within the tumor must be taken into consideration. The flow rate of interstitial fluid out of the tumor is defined as
| (4) |
where QIFP is the flow rate out the tumor resulting from interstitial fluid pressure, p is the tumor density, qif is the net fluid loss from the tumor periphery, aT is the radius of the tumor, AT is the cross sectional area of the tumor (πa2). The calculated flow rate of interstitial fluid leaving the tumor is subtracted from the flow rate in the channel to obtain the flow rate of nutrients interacting with the tumors surface as
| (5) |
where QT is the flow rate interacting with the tumor. We next determine the volume of culture media interacting with the tumor (vT) as
| (6) |
In order to determine the rate of nutrient convection into the tumor, the convective flux into the tumor is determined by
| (7) |
| (8) |
where SVT is the ratio of tumor surface area to tumor volume, SAT is the surface area of the tumor, VT is the volume of the tumor, cflux is the convective flux of nutrients into the tumor, P is the permeability of the tumor tissue, and [Nui] is the interstitial nutrient concentration. Finally, the rate of nutrient convection into an adherent cell tumor (cTad) is determined by
| (9) |
Beyond convection in the growth system, the amount of nutrients within the tumor relies on diffusion of nutrients. While the contribution of diffusion is relatively small when compared to convection, it is still important to consider in order to have a well developed model. It should also be noted that diffusion of nutrients is only considered for the adherent tumor spheroids and not the leukemia cancer. This is because there is no tissue for nutrient to diffuse through in the leukemia model and as such is disregarded. For the tumor spheroids, the diffusion of nutrients is defined by the concentration gradient across the tumor, the rate of nutrient diffusion in the tumor tissue, the diffusive flux of nutrients within the tissue and the surface area of the tumor as follows
| (10) |
| (11) |
| (12) |
where ∇nu is the concentration gradient of nutrients within the tumor, Dflux is the diffusive flux of nutrients into the tumor, D is the diffusion coefficient of nutrients into the tumor, and DT is the rate of nutrient diffusion into the tumor. Once the rates of nutrient convection and diffusion into the tumor have been determined, the change in nutrient availability within the tumor (ΔNu) at any given time point can be calculated as
| (13) |
Then in order to determine an updated growth rate to use in the cell growth equation, the average nutrient availability (Nuavg) within the tumor is calculated over the entire duration of the flow-assisted growth (120 hours in our tests).
| (14) |
where tn is the total number of time points during time-course t. From the average nutrient availability within the tumors over the growth time-course, an updated growth rate can be determined based on the specific nutrient flow rate used within the device. The equations governing the conversion of average nutrient availability to an updated growth rate are as follows
| (15) |
where a, b, c and d are constants determined based on a fit of the experimental growth rates observed and the average nutrient availability, which are cell type specific with HCT:(a=102.4, b=1000, c=0.0229, d=3.25) and Leuk:(a=1500, b=10, c=0.1046, d=8.4). rN is the adjusted growth rate constant for each cell type. Lastly, the updated growth rate constant that is determined can be used to predict the growth curve of the cancer cells using the following equation
| (16) |
It should be noted that the growth model is exactly the same as the one for the static growth cases, with the exception of the updated growth rate to reflect the changes introduced as a result of nutrient flow within the system. Values which were set based on literature values include: Cc=7.7×10−9 and 6.5×10−6 g/hr/cell for leukemia and HCT116 respectively, p=1.08 g/mL, qif=0.18mL/hr, P=57.34×10−7cm/sec), and D=2.57×10−8cm2/sec55–61. All other values were determined experimentally or specifically chosen based on desired experimental testing conditions.
The above set of equations were used to model the growth of both cell types under static and flow based conditions. The method of generating updated growth rate values for the flow cases has been shown to produce predicted growth curves that closely match the experimental results for the slow and fast flow rates used on both cell types. The close agreement of the models predictions and experimental results can be seen in Figs. 2a and 3a. In addition to predicting cell growth, the same governing model was used to predict the time point at which the rate of nutrient delivery into the tumor balances with rate of nutrient consumption by cells within the system, referred to as an equilibrium point. The predicted timeframes in Figs. 2b and 3b for these equilibrium points, corresponds to the timeframes when the cancer growth curves in Figs. 2a and 2b fall behind the maximum growth curve (red curves). As an example, the nutrient availability graph in Fig 3b for the HCT116 spheroids, predicts that the amount of available nutrients in the tumor will drop below the total nutrient demand of the tumor for the 3.4×103μL/hr test case (blue curve) around hour 72. This prediction of insufficient nutrient supply to feed the entire tumor around hour 72 corresponds well with the point in time when the 3.4×103μL/hr test case (blue curve) in Fig. 3a drops away from the 3.4×104μL/hr test case (red curve) which still has a sufficient supply of nutrients around hour 72. The ability to make such predicts along with the close predictions of both static and unrestricted nutrient supply (3.4×104μL/hr test case) cell growth for both cell types indicates that the model developed to describe the nutrient delivery within the microfluidic system closely mimics what occurs during experimental testing.
Based on the observed results and predictions made by the developed mathematical model, the 3.4×104μL/hr test case for both cell types showed continuous expedited cell growth even at 120 hours, while the static and 3.4×103μL/hr test cases entered into conditions of insufficient nutrient supply for the entire cell populations prior to reaching the 120 hour time point. Additionally for the 3.4×104μL/hr test cases, sufficient cancer cell populations were reached around hour 72, indicating that therapeutic screening could commence after 72 hours of culturing within the device.
In order to further verify the formation of spherical tumor clusters, representative confocal scans were periodically run. SF. 4 depicts one of the representative confocal scans showing a typical spherical tumor cluster produced within the microfluidic system. Additionally, the expression of E-Cadherin under static and flow conditions were tested in 2D and 3D cultures as shown in SF. 5. The level of expression between all 4 groups tested were very similar indicating that no major changes in cell-cell junction protein expression were occurring as a result of the expedited 3D growth system. Lastly, in order to test the systems capability to grow tumor spheroid from limited amount of cells, a set of tumor spheroid growth experiments were run which utilized a range of initial cell numbers in the devices to grow tumors. A process of pre-clustering the cells into loose spheroids was utilized which involved a slow swirling of the cancer cells in a low adhesion well plate for 4 hours followed by 4 hours of static culture, both under standard culturing conditions. The resulting loosely associated spheroids after 8 hours could be transferred into the microfluidic devices for further culturing. The results in SF. 6 indicate that a minimum of 8 cells are required for this growth technique with a likelihood of success around 12.5%. Further testing indicated that the initial use of 30 cells or more almost guaranteed the formation of tumor spheroids for growth within the device.
Discussion
From this work, a platform for the expedited growth of cancers is offered which can function with both adherent and non-adherent cell lines. The overall goal of the study was to demonstrate the influence of active convective flow on the growth rate of cancer cell populations within the microfluidic system. The system has been shown to facilitate a range of culturing conditions from static up to flow rates of 3.4×104μL/hr which influence the rate at which cancer cells grow over the course of 120 hours. Additionally, the design of the device allows for direct observations and measurements of the HCT116 and leukemia cancer population to be made via brightfield and fluorescence microscopy over the entire time-course of the study.
The outcomes from this work indicate that the growth of both cell types are assisted by the active convective flow of nutrients through the devices. The degree of growth improvement has been shown to be controlled by the type of cell being used and the flow rate at which nutrients are introduced into the devices. Overall, the improved growth of the leukemia cells with the addition of nutrient flow was less than the improvement observed when flow was applied to the HCT116 cells. This difference in growth improvement is contributed to a couple of factors. The first is that the leukemia cells are non-adherent cells and as such, do not grow in a tightly associated spheroid. The leukemia cells spread out, growing in the same plane of the device and as such nutrient transport to a given cell in the population is not hindered by any surrounding cells. The HCT116 cells grow as a single mass making it difficult to deliver nutrients to cells within the tumor without the aid of convection. Therefore when convection driven nutrient transport is applied, the HCT116 tumor mass receives more benefit when compared to the individually growing leukemia cells as nutrients are forced further into the core of the HCT116 spheroids. The second reason why a more significant improvement in HCT116 growth was observed when compared to the leukemia cells is because the HCT116 cells have a naturally faster growth rate even under static conditions. The faster growth rate for the HCT116 cells allows the cells to turn over new generations of cells in a shorter period of time, when then can go on to further take advantage of the excess nutrients supplied by the culture media flow. When compared to the static test cases, introducing 3.4×103μL/hr nutrient flow into the systems containing leukemia cells results in an overall increase in the cell population by a factor of 1.27 at the end of 120 hours. Similarly, the normalized number of leukemia cells present after 120 hours of growth under flow conditions of 3.4×104μL/hr, was improved by a factor of 1.38. More significant were the observed increases in HCT116 growth with the 3.4×103μL/hr test case resulting in a 2.47 factor increase in the cell population. Even more impressive was the observed increase in HCT116 growth when supplied with a nutrient flow of 3.4×104μL/hr, which resulted in a 4.76 factor increase in the cell population.
The increased growth observed under flow conditions for both cancer types is contributed to the convective flow of nutrients to the cancer cells. While static culturing conditions rely purely on diffusion of nutrients within the system, the application of convective flow provides a continuously refreshed source of nutrients while also removing cellular waste excreted by the cancer cells. This two-fold effect in turn can be used to ensure that the cells growing in the population are never lacking nutrients. The function of the convective nutrient flow for the spheroid based tests also facilitates deeper penetration of fresh nutrients into the core of the tumors because the flowing nutrients are forced between intercellular gaps in the tumor tissue. When compared to static cultures, the nutrient transport through intercellular gaps can only occur via diffusion which requires long periods of time for transport over such long distances to reach the cores of the growing tumors. As such, static culturing conditions often result in necrotic tumor spheroid cores as demonstrated heavily in literature. The application of nutrient flow in our system instead functions to accelerate the rate of tumor growth over time. In addition, comparisons to literature data for tumor growth in animal models and hydrogel based models have shown that comparable increases in cell number or tumor volume can take one week to two months39,40,62–64,64. Therefore, these growth techniques are comparable to static tumor growth4,39,40,62–64,64. Application of the flow assisted growth method, has the ability to outperform these other common types of tumor growth while facilitating easier data collection and on-device high throughput therapeutic screening.
As a note, the experimental setup was designed to observe changes in tumor growth as the culture media flow rate was varied. As such, a conclusion was made that the addition of culture media flow assisted in the growth of the cancer populations. However, this is not to say that the convective delivery of nutrients was the only factor that facilitated the improved growth. In turn, continually expedited growth of the tumors is not as simple as providing nutrients at greater and greater flow rates. Instead, factors such as the tumor size, surface area to volume ratio, diffusion coefficient, rate of waste production, concentration of nutrients in the stock solutions, as well as cell and cell type specific characteristics are all also key factors that can influence the delivery of nutrients throughout the tumor and ultimately the growth of the tumor as a whole65–72. Previous work by others have identified the importance of key factors such as the depth of nutrient penetration and cellular growth state that heavily influence the rate of overall tumor growth65–72. Nutrient penetration has been shown to rely on the degree of tumor vascularization and the overall structural architecture of the growing cells65–69. Further, pervious work has indicated the state which various populations of cells exist in within the tumor greatly influence the overall growth70–72. Larger populations of quiescent and necrotic cells leads to reduced tumor growth as the total number of actively proliferating cells becomes reduced70–72. In order to better understand the degree to which each of these factors influences the rate of growth for the entire tumor, more complex experimental setups and computational simulations are required. Such experimental setups and convection-diffusion-reaction simulations will need to provide a high degree of control over each factor to tease out how the interconnected system as a whole functions to improve tumor growth. Future work along these lines is planned in an attempt to further push the speed at which tumors can be produced while facilitate a high degree of control over the entire process.
Besides simply observing expedited growth of cancer populations under such convective flows of nutrients, a mathematical model has been developed to help describe and predict the outcomes of such growth experiments. The mathematical model has been based on the active convection of nutrients resulting from flow, the passive diffusion of nutrients within the spheroids, and the consumption of nutrients by the cancer cell populations over time. The developed model has been simplified for ease of use, with the major factors resulting in the growth of cancer populations being taken into consideration. Despite the simplified nature of the model, good agreements with experimental testing has been achieved which indicates that the model can be used to predict the growth of cancer cells within the microfluidic system. The application of the model in a predictive manner in conjunction with the microfluidic growth system can result in expedited cancer growth of adherent and non-adherent cancer cells for therapeutic screening or genetic analysis. In addition, the models ability to predict nutrient availability within the growth system, allows for a control feedback loop to be established in order to vary the nutrient supply rate to maximized cell growth while limiting waste. An example of such a feedback loop would involve the continuous monitoring of cell growth which in turn would be used to update the mathematical model. The initial nutrient supply rate can be very low and slowly increased as the cell population grows over time. By doing so, expensive reagents can be used sparingly while not limiting the total potential growth of the cancer cells. As a final note, the predictive nature of the developed model shows good agreement with the timeframes when variations in experimental cell growth were expected. By predicting the availability of nutrients within the system, it is possible to identify how long it will take before the cell population runs into a deficit of nutrients. The predicted times to reach conditions of nutrient deficit for each testing condition (static and two flow cases) correspond well with the time points in the experimental data where the static and slow flow rate growth curves fall behind the growth of the faster flow rate. This correlation between the predicted and observed results indicates that the developed model is sufficient enough to represent the experimental conditions being tested.
Additionally, the minimum number of required HCT116 cancer cells required to successfully form tumor spheroids was tested. The conclusions indicate that at least 8 cells must be present in order to successfully form a spheroid with a success rate of around 12.5%. However, when 30 cells or more are used within the device to produce a spheroid, the successful formation of such a spheroid is almost guaranteed. These results are important to understand if such a device is to be used in a clinical setting with very limited number of cells from patient samples. The number of such cells collected from patient samples are typically very low and as such, having a method to successfully form spheroids for drug screening will need to function under such conditions. It should be noted however that the cells used in this study were well established cell lines routinely grown in a laboratory setting. Future work along these lines will include tests run with patient derived cancer cells in order to more fully validate the system for potential future use in the clinical space.
As a final analysis, the developed microfluidic system was tested to demonstrate the capability of performing on-device therapeutic screening. The resulting APH assay comparison of drug response by 2D and 3D cultures of HCT116 cells to (a) Paclitaxel, (b) Doxorubicin, and (c) Capecitabine can be seen in Fig. 4. The system has shown good agreement with literature findings that 3D cancer cultures possess a greater resistance to therapeutic treatment when compared to 2D cultures. Additionally, a high throughput version of the microfluidic system is demonstrated and prototyped to enable the screening of multiple drugs in parallel in an effort to expedite the screening process. The high throughput prototype and initial benchmarking data can be seen in Fig. 5.
Figure. 4.

The drug effect on HCT116 tumor spheroids grown within the bi-layer microfluidic system by APH assay. Drug effects of Paclitaxel (a), Doxorubicin (b), and Capecitabine (c) on spheroids cultures, with comparison to monolayer cell cultures performed in flat bottomed well plates. Dose-response curves plotted exponentially over drug concentration range of 1nM to 1μM for all three drugs. All data points shown were run in replicates of four (n=4) for each testing condition and drug.
Figure. 5.

Representative prototype of high-throughput array microfluidic system allowing 16 therapeutic screening tests to be run simultaneously. a, 3D model of high-throughput array microfluidic well plate designed for use with high-throughput imaging and data collection systems. b, Prototype well plate system manufactured via 3D printing being used to test data collection from a fluorescent microscope. c, Prototype well plate system being tested with a plate reader to verify data collection. d, Normalized fluorescence intensity data of FITC dye flown through the prototype high-throughput array system. Fluorescence intensity was measured at various dye concentrations while passing through the device and compared to fluorescence intensities at the same concentrations in standard well plates. The data collected by both methods are in good agreement, indicating that the prototyped system functions well plate reader data collection. Data collection tests were run five times (n=5) to achieve statistical significance.
Overall, the developed microfluidic system has been shown to facilitate the growth of adherent and non-adherent cancer cell types in an expedited fashion through the application of nutrient flow. The improvement in cancer cell growth is contributed to the continuous convective delivery of fresh nutrients to the cells while also actively removing cellular waste. The expression of cell-cell adhesion molecule E-Cadherin has also been examined to show that the growth method does not interfere or influence the normal phenotypic expression of such extracellular proteins. The developed mathematical model has been shown to serve as a good predictor of cancer growth within the system and can be used as a feedback loop to adjust the rate at which nutrients are supplied to cancer cell populations. The minimum require number of HCT116 cells has been determined in order to successfully form tumor spheroids with the microfluidic system. Lastly, the microfluidic system as a whole has been demonstrated to function for on-device therapeutic screening applications and a high throughput prototype of the system has been demonstrated to allow 16 drugs to be screened simultaneously. Still there exists many routes of future work along the lines of the developed cancer growth device.
Such future work will include further expediting the growth of cancer cell populations through the introduction of growth factors into the nutrient flow. However, changes in cell phenotype and protein expression will need to be closely monitored to ensure that the system is still representative of phenotypically normal conditions. Additionally, further testing of additional cancer cell types, both established and patient derived, is needed to expand the scope of the model as a whole. Tests run with patient derived cancer cells will also serve to identify the minimum number of cells required for successful formation of spheroids for potential clinical use in drug screening assays and genetic analyses.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health (NIH) grants R01HL131750, National Science Foundation (NSF) grants PFI:AIR-TT 1701136, DMS 1516236, and the Pennsylvania Infrastructure Technology Alliance (PITA) program.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Krebber AMH, Buffart LM, Kleijn G, Riepma IC, De Bree R, Leemans CR, Becker A, Brug J, Van Straten A, Cuijpers P, Verdonck-De Leeuw IM, Psychooncology, 2014, 23, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-tieulent J, Jemal A, CA a cancer J Clin, 2015, 65, 87. [DOI] [PubMed] [Google Scholar]
- 3.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A, CA Cancer J Clin, 2016, 66, 271. [DOI] [PubMed] [Google Scholar]
- 4.Imamura Y, Mukohara T, Shimono Y, Funakoshi Y, Chayahara N, Toyoda M, Kiyota N, Takao S, Kono S, Nakatsura T, Minami H, Oncol Rep, 2015, 33, 1837. [DOI] [PubMed] [Google Scholar]
- 5.Stock K, Estrada MF, Vidic S, Gjerde K, Rudisch A, Santo VE, Barbier M, Blom S, Arundkar SC, Selvam I, Osswald A, Stain Y, Gruenewald S, Brito C, Van Weerden W, Rotter V, Boghaert E, Oren M, Sommergruber W, Chong Y, De Hoogt R, Graeser R, Sci Rep., 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Duinen V, Trietsch SJ, Joore J, Vulto P, Hankemeier T, Curr Opin Biotechnol, 2015, 35, 118. [DOI] [PubMed] [Google Scholar]
- 7.Hsiao AY, suke Torisawa Y, Tung YC, Sud S, Taichman RS, Pienta KJ, Takayama S, Biomaterials, 2009, 30, 3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toh Y-C, Zhang C, Zhang J, Khong YM, Chang S, Samper VD, van Noort D, Hutmacher DW, Yu H, Lab Chip, 2007, 7, 302. [DOI] [PubMed] [Google Scholar]
- 9.Shin Y, Han S, Jeon JS, Yamamoto K, Zervantonakis IK, Sudo R, Kamm RD, Chung S, Nat Protoc., 2012, 7, 1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costǎchel O, Fadei L, Badea E, Z Krebsforsch, 1969, 72, 24. [DOI] [PubMed] [Google Scholar]
- 11.Lin RZ, Chang HY, Biotechnol J., 2008, 3, 1172. [DOI] [PubMed] [Google Scholar]
- 12.Ho VHB, Barcza A, Chen R, Müller KH, Darton NJ, Slater NKH, Biomaterials, 2009,30, 6548. [DOI] [PubMed] [Google Scholar]
- 13.Hammond TG, Hammond JM, Am J Physiol Physiol., 2001, 281, F12. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Z, Wang H, Ding Q, Xing Y, Xu Z, Lu C, Luo D, Xu L, Xia W, Zhou C, Shi M, PLoS One, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, Lampis A, Eason K, Huntingford I, Burke R, Rata M, Koh DM, Tunariu N, Collins D, Hulkki-Wilson S, Ragulan C, Spiteri I, Moorcraft SY, Chau I, Rao S, Watkins D, Fotiadis N, Bali M, Darvish-Damavandi M, Lote H, Eltahir Z, Smyth EC, Begum R, Clarke PA, Hahne JC, Dowsett M, De Bono J, Workman P, Sadanandam A, Fassan M, Sansom OJ, Eccles S, Starling N, Braconi C, Sottoriva A, Robinson SP, Cunningham D, Valeri N, Science (80-), 2018, 359, 920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, Balgobind AV, Wind K, Gracanin A, Begthel H, Korving J, van Boxtel R, Duarte AA, Lelieveld D, van Hoeck A, Ernst RF, Blokzijl F, Nijman IJ, Hoogstraat M, van de Ven M, Egan DA, Zinzalla V, Moll J, Boj SF, Voest EE, Wessels L, Van Diest PJ, Rottenberg S, Vries RGJ, Cuppen E, Clevers H, Cell, 2018, 172, 373. [DOI] [PubMed] [Google Scholar]
- 17.HAGEMANN J, JACOBI C, HAHN M, SCHMID V, WELZ C, SCHWENK-ZIEGER S, STAUBER R, BAUMEISTER P, BECKER S, Anticancer Res, 2017, 37, 2201. [DOI] [PubMed] [Google Scholar]
- 18.Wang R, Chu GCY, Mrdenovic S, Annamalai AA, Hendifar AE, Nissen NN, Tomlinson JS, Lewis M, Palanisamy N, Tseng HR, Posadas EM, Freeman MR, Pandol SJ, Zhau HE, Chung LWK, Asian J Urol, 2016, 3, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jabs J, Zickgraf FM, Park J, Wagner S, Jiang X, Jechow K, Kleinheinz K, Toprak UH, Schneider MA, Meister M, Spaich S, Sutterlin M, Schlesner M, Trumpp A, Sprick M, Conrad C, Mol Syst Biol, 2017, 13, 955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van De Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, Van Houdt W, Van Gorp J, Taylor-Weiner A, Kester L, McLaren-Douglas A, Blokker J, Jaksani S, Bartfeld S, Volckman R, Van Sluis P, Li VSW, Seepo S, Sekhar Pedamallu C, Cibulskis K, Carter SL, McKenna A, Lawrence MS, Lichtenstein L, Stewart C, Koster J, Versteeg R, Van Oudenaarden A, Saez-Rodrigues J, Vries RGJ, Getz G, Wessels L, Sratton MR, McDermott U, Meyerson M, Garnett MJ, Clevers H, Cell, 2015, 161, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zanoni M, Piccinini F, Arienti C, Zamagni A, Santi S, Polico R, Bevilacqua A, Tesei A, Sci Rep, 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garralda E, Paz K, López-Casas PP, Jones S, Katz A, Kann LM, Lopez-Rios F, Sarno F, Al-Shahrour F, Vasquez D, Bruckheimer E, Angiuoli SV, Calles A, Diaz LA, Velculescu VE, Valencia A, Sidransky D, Hidalgo M, Clin Cancer Res, 2014, 20, 2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Habu S, Fukui H, Shimamura K, Kasai M, Nagai Y, Okumura K, Tamaoki N, J Immunol, 1981, 127, 34. [PubMed] [Google Scholar]
- 24.Sun WH, Burkholder JK, Sun J, Culp J, Turner J, Lu XG, Pugh TD, Ershler WB, Yang NS, ProcNatlAcadSciUSA., 1995, 92, 2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Brien CA, Pollett A, Gallinger S, Dick JE, Nature, 2007, 445, 106. [DOI] [PubMed] [Google Scholar]
- 26.Food and Drug Administration. Approved Drug Products with Therapeutic Equivalence Evaluations.; 2017.
- 27.Quintás-Cardama A, Cortes J, Clin Cancer Res, 2008, 14, 4392. [DOI] [PubMed] [Google Scholar]
- 28.Serafini AN, Houston SJ, Resche I, Quick DP, Grund FM, Ell PJ, Bertrand A, Ahmann FR, Orihuela E, Reid RH, Lerski RA, Collier BD, McKillop JH, Purnell GL, Pecking AP, Thomas FD, Harrison KA, J Clin Oncol, 1998, 16, 1574. [DOI] [PubMed] [Google Scholar]
- 29.De Bono JS, Oudard S, Ozguroglu M, Hansen S, MacHiels JP, Kocak I, Gravis G, Bodrogi I, MacKenzie MJ, Shen L, Roessner M, Gupta S, Sartor AO, Lancet, 2010, 376, 1147. [DOI] [PubMed] [Google Scholar]
- 30.Aaronson NK, Ahmedzai S, Bergman B, Bullinger M, Cull A, Duez NJ, Filiberti A, Flechtner H, Fleishman SB, Haes JCJMDe., Kaasa S, Klee M, Osoba D, Razavi D, Rofe PB, Schraub S, Sneeuw K, Sullivan M, Takeda F, J Natl Cancer Inst, 1993, 85, 365. [DOI] [PubMed] [Google Scholar]
- 31.Ellis PM, Vandermeer R, J Thorac Dis, 2011, 3, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simunovic M, Gagliardi A, McCready D, Coates A, Levine M, DePetrillo D, CMAJ, 2001, 165, 421. [PMC free article] [PubMed] [Google Scholar]
- 33.Mehta G, Hsiao AY, Ingram M, Luke GD, Takayama S, J Control Release, 2012, 164, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Z, Gao Y, Hao Y, Li E, Wang Y, Zhang J, Wang W, Goa Z, Wang Q, Biomaterials, 2013, 34, 4109. [DOI] [PubMed] [Google Scholar]
- 35.Kwapiszewska K, Michalczuk A, Rybka M, Kwapiszewski R, Brzózka Z, Lab Chip, 2014, 14, 2096. [DOI] [PubMed] [Google Scholar]
- 36.Chen Y, Gao D, Liu H, Lin S, Jiang Y, Anal Chim Acta, 2015, 898, 85. [DOI] [PubMed] [Google Scholar]
- 37.Chen MCW, Gupta M, Cheung KC, Biomed Microdevices, 2010, 12, 647. [DOI] [PubMed] [Google Scholar]
- 38.Sajeesh P, Raj A, Doble M, Sen AK, RSC Adv., 2016, 6, 74704. [Google Scholar]
- 39.Alessandri K, Sarangi BR, Gurchenkov VV, Sinha B, Kiessling TR, Fetler L, Rico F, Scheuring S, Lamaze C, Simon A, Geraldo S, Vignjevic D, Domejean H, Rolland L, Funfak A, Bibetter J, Bremond N, Nassoy P, Proc Natl Acad Sci, 2013, 110, 14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Helmlinger G, Netti PA, Lichtenbeld HC, Melder RJ, Jain RK, Nat Biotechnol, 1997, 15, 778. [DOI] [PubMed] [Google Scholar]
- 41.Sajeesh P, Manasi S, Doble M, en AK, Lab Chip, 2015, 15, 3738. [DOI] [PubMed] [Google Scholar]
- 42.Raj A, Dixit M, Doble M, Sen AK, Lab Chip, 2017, 17, 3704. [DOI] [PubMed] [Google Scholar]
- 43.Bairati I, Fillion L, Meyer FA, Héry C, Larochelle M, Eur J Cancer Care (Engl), 2006, 15, 183. [DOI] [PubMed] [Google Scholar]
- 44.Bright K, Barghash M, Donach M, de la Barrera MG, Schneider RJ, Formenti SC, The Breast, 2011, 20, S54. [DOI] [PubMed] [Google Scholar]
- 45.Thomas A, Ou-Yang DH, Lowe-Krentz L, Muzykantov VR, Liu Y, Biomicrofluidics, 2016, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas A, Tan J, Liu Y, Microvasc Res, 2014, 94, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas A, Wang S, Sohrabi S, Orr C, He R, Shi W, Liu Y, Biomicrofluidics, 2017, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uhl CG, Gao Y, Zhou S, Liu Y, RSC Adv., 2018, 8, 8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uhl CG, Muzykantov VR, Liu Y, Biomicrofluidics, 2018, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Byun YJ, Park B-B, Lee ES, Choi KS, Lee DS, Blood Res, 2014, 49, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sawyers CL, N Engl J Med, 1999, 340, 1330. [DOI] [PubMed] [Google Scholar]
- 52.Sullivan C, Peng C, Chen Y, Li D, Li S, Biochem Pharmacol, 2010, 80, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumano K, Arai S, Hosoi M, Taoka K, Takayama N, Otsu M, Nagae G, Ueda K, Nakasaki K, Kamikubo Y, Eto K, Aburatani H, Nakauchi H, Kurokawa M, Blood, 2012, 119, 6234. [DOI] [PubMed] [Google Scholar]
- 54.Tan J, Shah S, Thomas A, Ou-Yang HD, Liu Y, Microfluid Nanofluidics, 2013, 14, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vaupel P, Kallinowski F, Okunieff P, Cancer Res, 1989, 49, 6449. [PubMed] [Google Scholar]
- 56.Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, di Tomaso E, Brown EB, Izumi Y, Campbell RB, Berk DA, Jain RK, Proc Natl Acad Sci, 2001, 98, 4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baxter LT, Jain RK, Microvasc Res., 1989, 37, 77. [DOI] [PubMed] [Google Scholar]
- 58.Ferreira SC, Martins ML, Vilela MJ, Phys Rev E Stat Nonlin Soft Matter Phys, 2002, 65, 21907. [DOI] [PubMed] [Google Scholar]
- 59.Thurber GM, Schmidt MM, Wittrup KD, Adv Drug Deliv Rev, 2008, 60, 1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meyskens FL, Thomson SP, Moon TE, Cancer Res, 1984, 44, 271. [PubMed] [Google Scholar]
- 61.Jain RK, Baxter LT, Cancer Res, 1988, 48, 7022. [PubMed] [Google Scholar]
- 62.Ovejera AA, Houchens DP, Barker AD, Ann Clin Lab Sci, 1978, 8, 50. [PubMed] [Google Scholar]
- 63.Wang JX, Feng YY, Li FF, Appl Mech Mater, 2011, 58, 1636. [Google Scholar]
- 64.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MTW, Factor R, Matsen C, Milash BA, Nelson E, Neumayer L, Randall RL, Stijleman IJ, Welm BE, Welm AL, Nat Med, 1997, 14, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ward J, Math Med Biol, 1997, 14, 39. [Google Scholar]
- 66.Sherratt JA, Chaplain MAJ, J Math Biol, 2001, 43, 291. [DOI] [PubMed] [Google Scholar]
- 67.Martins ML, Ferreira SC, Vilela MJ, Phys Life Rev, 2007, 4, 128. [Google Scholar]
- 68.Chaplain MAJ, Math Comput Model, 1996, 23, 47 [Google Scholar]
- 69.Ambrosi D, Preziosi L, Math Model Methods Appl Sci, 2002, 12, 737 [Google Scholar]
- 70.Roose T, Chapman SJ, Maini PK, SIAM Rev., 2007, 49, 179. [Google Scholar]
- 71.Dormann S, Deutsch A, In Silicon Biol, 2002, 2, 393. [PubMed] [Google Scholar]
- 72.Jiang Y, Pjesivac-Grbovic J, Cantrell C, Freyer JP, Biophys J, 2005, 89, 3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding authors upon reasonable request.