

Abstract
Targeted transcriptional modulation in the central nervous system (CNS) can be achieved by adeno-associated virus (AAV) delivery of CRISPR activation (CRISPRa) and interference (CRISPRi) transgenes. To enable AAV packaging, we constructed minimal CRISPRa and CRISPRi transgenes by fusing catalytically inactive Staphylococcus aureus Cas9 (dSaCas9) to the transcriptional activator (VP64 and VP160) and repressor (KRAB and SID4X) domains along with truncated regulatory elements. We then evaluated the performance of these constructs in two reporter assays (bioluminescent and fluorescent), five endogenous genes (Camk2a, Mycn, Nrf2, Keap1, and PDGFRA), and two cell lines (neuro-2a [N2a] and U87) by targeting the promoter and/or enhancer regions. To enable systemic delivery of AAVs to the CNS, we have also generated an AAV1-PHP.B by inserting a 7-mer PHP.B peptide on AAV1 capsid. We showed that AAV1-PHP.B can efficiently cross the blood-brain barrier (BBB) and be taken up by the brain tissue upon lateral tail vein injection in mice. Importantly, a single-dose intravenous administration of AAV1-PHP.B expressing CRISPRa was shown to achieve targeted transgene activation in the mouse brain. This proof-of-concept study will contribute to the development of a non-invasive, specific and potent AAV-CRISPR system for correcting transcriptional misregulation in broad brain areas and multiple neuroanatomical structures.
Keywords: AAV capsid, adeno-associated virus, cis-regulatory elements, CRISPR activation, CRISPR interference, brain delivery
Graphical Abstract
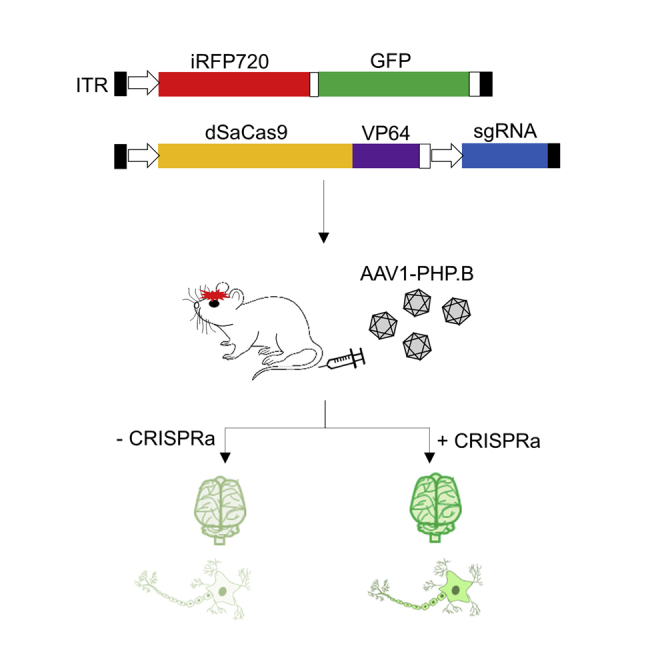
Introduction
CRISPR technology has become the preferred platform for modifications of the target DNA sequence such as gene knockout1, 2 and gene knockin3 in the genome of a living organism. A major addition to the CRISPR toolbox was the discovery of smaller-size Cas9 variants such as Staphylococcus aureus Cas9 (SaCas9) that has the capability to edit the genome with efficiency and specificity similar to the commonly used Streptococcus pyogenes Cas9 (SpCas9).4 Based on homology to nuclease-dead SpCas9 (also known as dCas9, contains D10A and H840A mutations), a catalytically inactive SaCas9 (dSaCas9) was derived by introducing two mutations, D10A and N580A, on the SaCas9.4 Similar to dSpCas9, dSaCas9 can be fused to a transcriptional activator (e.g., VP645) or repressor (e.g., KRAB6) domain for targeted modulation of gene expression. Owing to the large size of a dSpCas9 fusion protein, dSaCas9 can help to circumvent the issue of adeno-associated virus (AAV) applications by enabling delivery of a dSaCas9-fused effector domain and its single-guide RNA (sgRNA) in a single AAV vector. The development of an all-in-one AAV vector system can simplify the AAV production and improve gene modulation efficiency by ensuring delivery of all CRISPR components into individual cells.
AAV provides one of the most suitable viral vectors to package, deliver, and express CRISPR components in vivo owing to its good safety profile and promising therapeutic efficacy shown in a wide range of animal models and human clinical trials.7, 8, 9 In addition, AAV elicits a very mild immune response in vivo, and it rarely causes unwanted integration events in the host genome.7 However, most of the commonly used natural AAV serotypes (including AAV1) are inefficient in crossing blood-brain barriers and have low transduction efficiency in vivo. One of the possible ways to overcome these limitations is to de-target the native AAV tropism through genetic modification of the AAV capsids. For example, AAV9-PHP.B is one of the most promising AAV mutants engineered to date for systemic gene delivery to the CNS. AAV9-PHP.B was generated by inserting a 7-mer PHP.B peptide on the wild-type AAV9 capsid.10 Compared to the native AAV9, AAV9-PHP.B transduced human neurons and astrocytes more efficiently.10 It also enabled more efficient and widespread transduction in the adult mouse CNS after intravenous injection.10 Therefore, we envisioned to investigate whether the enhanced blood-brain barrier crossing of PHP.B modification could be extended to other AAV serotypes, such as AAV1. The capsid of AAV1 has been genetically modified to improve AAV1 transduction efficiency in cultured motor neurons11 and neuroblastoma12 and glioblastoma13 cells. AAV1 is also one of the most commonly used AAV serotypes for transduction of brain tissue via stereotactic injection.14, 15
In this study, we have developed a series of all-in-one CRISPR activation (CRISPRa) and interference (CRISPRi) expression cassettes, which have an accumulated transgene size within the 4.7-kb AAV-packaging capacity. We first validated the performance of these plasmid constructs using bicistronic infrared-GFP fluorescent and luciferase bioluminescence reporter assays. We then transfected these CRISPRa and CRISPRi constructs into two different cell lines for modulating the cis-regulatory elements (e.g., promoter and enhancer) of five endogenous genes (Camk2a, Mycn, Nrf2, Keap1, and PDGFRA). Next, we genetically modified native AAV1 to AAV1-PHP.B by inserting 7-mer PHP.B peptides on the AAV1 capsid. We then evaluated the transduction efficiency of this AAV1-PHP.B vector by using it to package the bicistronic infrared-GFP fluorescent reporters for delivery in vitro. These vectors were finally intravenously injected into the adult mice to track their localization and accumulation in the brain tissue. To demonstrate targeted transgene activation in the brain tissue, we also intravenously injected this AAV reporter vector together with another AAV1-PHP.B vector carrying CRISPRa into the mice.
Results
Construction of Minimal CRISPRa and CRISPRi Transgenes
Figure 1 illustrates the AAV-compatible CRISPRa and CRISPRi transgenes that have been constructed in this study. To achieve efficient packaging of the dSaCas9-fused effector domain and its sgRNA into a single AAV vector, a truncated version of the mouse Mecp2 promoter (235 bp, pMecp2); a minimal polyadenylation signal (48 bp, synthetic polyadenylation signal [spA]); and the smallest version of previously characterized chromatin catalytic domains such as VP64 activator (156 bp), VP160 activator (390 bp), KRAB repressor (227 bp), and SID4X repressor (432 bp) were used. The minimal pMecp2 promoter and the spA polyadenylation signal were recently shown capable of inducing robust transgene expression in primary mouse cortical neurons and mammalian brain.16 Several more potent transcriptional repressors (e.g., DNMT3A,17 histone deacetylase [HDAC],18 and LSD119) and activators (e.g., VP64-p65-Rta [VPR],20 TET1,21 and P30022) have been reported recently, however, their large size has ruled out the possibility of co-packaging them with dSaCas9 and its sgRNA into a single AAV vector.
Figure 1.

Constructions of Minimal CRISPRa and CRISPRi Transgenes into pAAV Vector Backbone
To achieve efficient packaging of dSaCas9 fusion proteins and its sgRNA in a single AAV vector, a truncated version of the mouse Mecp2 brain-specific promoter (235 bp, pMecp2); a minimal polyadenylation signal (48 bp, spA); and the smallest version of the transcriptional regulator domain, such as VP64 activator (156 bp), VP160 activator (390 bp), KRAB repressor (227 bp), and SID4X repressor (432 bp), were used.
Validation of CRISPRa and CRISPRi Functions Using Luciferase Reporter Assay
Super-enhancers are large clusters of transcriptional enhancers in close genomic proximity with unusually high levels of master regulators and mediator binding that play key roles in human cell identity, both in health and diseases.23 By analyzing the H3K27ac and MED1 enhancer marks of the major genes associated with the development of the nervous system and neuronal functions in the super-enhancer databases (dbSUPER and Super-Enhancer Archive [SEA]), we identified a putative super-enhancer region at the upstream of alpha CaMKII gene in mice. Therefore, the sgRNAs were designed to target the super-enhancer of mouse alpha CaMKII in the luciferase reporter assay. Alpha CaMKII is a neuronal-specific protein prominent in the cerebral cortex that plays key roles in neurotransmitter release, hippocampal synaptic plasticity, long-term potentiation, memory formation, learning, and cognitive function.24, 25 We classified the alpha CaMKII intergenic super-enhancer into proximal and distal super-enhancers based on two distinct clusters of transcriptional factories, DNaseI hypersensitivity and chromatin marks (H3K27ac and H3K4me1) in the mouse cerebral cortex, with the former closer to the alpha CaMKII promoter.
To examine the alpha CaMKII super-enhancer activity (Luc3), we inserted the full length of the intergenic super-enhancer upstream of the alpha CaMKII promoter and luciferase reporter gene (Figure 2A). As a positive control (Luc2), the well-characterized 1.3-kb alpha CaMKII promoter was inserted upstream of the luciferase vector. Three different sgRNAs of CRISPRa or CRISPRi transgenes were designed to target the alpha CaMKII promoter, and six sgRNAs were designed to target different regions of the intergenic super-enhancer. Multiple transcriptional regulator domains localized to the promoter were shown to synergistically enhance gene activation (Figure 2B). Similar results were obtained with the intergenic super-enhancer (Figure 2C). Simultaneous modulation of the promoter and super-enhancer by nine different CRISPRas resulted in the highest activation of luciferase activity. Importantly, the data from this luciferase reporter assay confirmed the functionality of our newly constructed dSaCas9-VP64, dSaCas9-VP160, dSaCas9-KRAB, and dSaCas9-SID4X.
Figure 2.

Activation and Repression of Luciferase Activity in Mouse N2a Cells Mediated by Various Minimal CRISPRa and CRISPRi Transgenes
(A) Luciferase reporter vector construction. Luc1 is the promoterless pGL4.10[luc2] background control. Luc2 is a modified Luc1 vector that has been inserted with 1.3-kb mouse alpha CaMKII promoter amplified from the genomic DNA of N2a cells. Luc3 is a modified Luc1 vector that has been inserted with 6.8-kb mouse alpha CaMKII promoter and intergenic super-enhancer regions amplified from the genomic DNA of N2a cells. TK-Renilla was used as an internal normalization control in all luciferase reporter assay experiments. (B) Luciferase activity with dSaCas9-fused activators and repressors targeting mouse alpha CaMKII promoter. The CRISPRa or CRISPRi transgenes targeting the mouse alpha CaMKII promoter were co-transfected with Luc2 vector and TK-Renilla control vector. Three sgRNAs were designed to target three different binding sites on the promoter (Pm). Luciferase activities were measured when the CRISPRa or CRISPRi transgenes were used singly (site 1, 2, or 3) or in combination (sites 1, 2, and 3). (C) Luciferase activity with dSaCas9-fused activators and repressors targeting mouse alpha CaMKII promoter and intergenic super-enhancer. The sgRNAs targeting mouse alpha CaMKII promoter and intergenic super-enhancer were co-transfected with the Luc3 vector and TK-Renilla control vector. A total of nine sgRNAs was designed to target three different binding sites on the promoters (Pm), three on proximal (SEP) and three on distal intergenic super-enhancer sites (SED). All luciferase activities were normalized to the corresponding empty sgRNA. The statistical significance levels from control (empty sgRNA) are indicated as *p < 0.05, **p < 0.01, and ***p < 0.001. All data are presented as mean ± SD (n = 4).
Modulation of Endogenous Alpha CaMKII Promoter and Super-Enhancer by CRISPRa and CRISPRi
The same set of sgRNAs from the luciferase reporter assay was used to modulate the endogenous alpha CaMKII promoter and super-enhancer in mouse neuro-2a (N2a) cells (Figure 3A). In this case, only dSaCas9-VP160 activator and dSaCas9-KRAB repressor were tested. Plasmid transfection with dSaCas9 fusion proteins targeting a single DNA site (only one sgRNA) had no effect on alpha CaMKII transcriptional activity, which was similar to the results using GFP control plasmid and dSaCas9 fusion proteins with empty sgRNA. Nevertheless, co-transfection of multiple dSaCas9-VP160 at different sites could effectively upregulate alpha CaMKII expression levels in N2a cells (Figure 3B). The activation effect was strongest on the promoter, followed by the proximal and distal super-enhancers.
Figure 3.

Modulation of Endogenous Alpha CaMKII Expressions by Using Minimal CRISPRa and CRISPRi Transgenes Targeting Super-Enhancer and Promoter in Mouse N2a Cells
(A) sgRNA target sites on super-enhancer and promoter of mouse alpha CaMKII. The locations of nine different sgRNA target sites designed to target the promoter (Pm), proximal super-enhancer (SEP), and distal super-enhancer (SED) are shown. Red arrows indicate the sense or antisense orientation of sgRNAs designed to recognize target DNA sequences. (B) dSaCas9-VP160-mediated activation of endogenous alpha CaMKII mRNA level in mouse N2a cells. Nine dSaCas9-VP160 activators were designed to target the Pm, SEP, and SED. Expression levels of alpha CaMKII in N2a cells are shown. (C) dSaCas9-KRAB-mediated repression of endogenous alpha CaMKII mRNA level in mouse N2a cells. Nine dSaCas9-KRAB repressors were designed to target the Pm, SEP, and SED. Expression levels of alpha CaMKII in N2a cells are shown. (D) dSaCas9-mediated transcriptional interference of mouse alpha CaMKII. (E) Western blot for protein lysate of alpha CaMKII in mouse N2a cells. The monoclonal mouse alpha CaMKII- (54 kDa) specific antibody was used. Beta-actin (Actb, 42 kDa) was used as an internal control. Protein expression levels of alpha CaMKII were measured for wild-type (WT), knockout (KO), and under the effects of dSaCas9-VP160 and dSaCas9-KRAB in N2a cells. Right panel shows the normalized protein expression level of alpha CaMKII in N2a cells. (B-D) The statistical significance levels from control (wild-type N2a) are indicated as *p < 0.05, **p < 0.01, and ***p < 0.001. All data are presented as mean ± SD (n = 3).
Similar to the luciferase reporter data, the highest activation effect was observed by simultaneously targeting the promoter and super-enhancer regions. However, dSaCas9-KRAB downregulated (14%) the alpha CaMKII expression level only by designing sgRNAs to target the promoter region (Figure 3C). Therefore, we designed the sgRNAs of dSaCas9-KRAB to target the gene body (downstream of the promoter) in the subsequent experiments. In addition, the dSaCas9 alone had no impact on the alpha CaMKII expression (Figure 3D). The protein expression data of alpha CaMKII in western blot was highly consistent with the mRNA expression data (Figure 3E), confirming the modulation effect on cis-regulatory elements mediated by our minimal CRISPRa and CRISPRi transgenes. We also used SaCas9s to knock out the protein-coding region of alpha CaMKII in order to validate alpha CaMKII antibody specificity (Figure 3E). A strong downregulation of protein level was detected by western blot after the knockout of alpha CaMKII in N2a cells.
Modulation of Endogenous Promoters of PDGFRA, Mycn, Nrf2, and Keap1
We further validated the effectiveness of dSaCas9-VP64 activator and dSaCas9-KRAB repressor by designing sgRNAs to target the promoter of four additional endogenous genes (PDGFRA in human U87 cells; Mycn, Nrf2, and Keap1 in mouse N2a cells) (Figure 4). PDGFRA is a receptor tyrosine kinase gene and prominent glioma oncogene that is frequently mutated, amplified, or overexpressed in glioblastomas.26 Mycn is a proto-oncogene that is frequently amplified and overexpressed in neuroblastoma.27 Also, poor prognosis and treatment failure in patients with neuroblastoma were often associated with the amplification of MYCN.27 Nrf2 transcription factor is encoded by NFE2L2, which can regulate hundreds of genes involved in the cytoprotective response against oxidative stress by binding to the antioxidant response element (ARE) in their upstream promoter region.28 As a major regulator of oxidative stress defense in the human body, many pharmacological drugs in pre-clinical testing have been developed to target and activate NFE2L2 for the treatment of human neurodegenerative diseases, including Alzheimer disease29 and Parkinson’s disease.30 Under normal condition, Nrf2 gene expression is repressed by a negative regulator Keap1. When cells are exposed to oxidative stress, Nrf2 escapes Keap1-mediated repression and activates ARE-dependent detoxifying and antioxidant gene cells.31 Therefore, activating Nrf2 or repressing Keap1 to activate neuroprotective pathways can ameliorate the pathogenesis of neurodegenerative diseases.
Figure 4.

Modulation of Other Endogenous Genes in Mouse N2a and Human U87 Cells
(A) Modulation of endogenous Mycn gene in N2a cells. The locations of different sgRNA target sites designed to target the promoter (C10, C11, C12, and E13) and gene body (E14 and E15) are shown. Right panel shows the relative expression levels of Mycn in N2a cells. (B) Modulation of endogenous Nrf2 gene in N2a cells. The locations of different sgRNA target sites designed to target the promoter (C16, C17, and C18) and gene body (E19, E20 and E21) are shown. Right panel shows the relative expression levels of Nrf2 in N2a cells. (C) Modulation of endogenous Keap1 gene in N2a cells. The locations of different sgRNA target sites designed to target the promoter (C22, C23, and C24) and gene body (E25, E26, and E27) are shown. Right panel shows the relative expression levels of Keap1 in N2a cells. (D) Modulation of endogenous PDGFRA gene in U87 cells. The locations of different sgRNA target sites designed to target the promoter (C28, C29, and C30) and gene body (E31, E32, and E33) are shown. Right panel shows the relative expression levels of PDGFRA in U87 cells. Red arrows indicate the sense or antisense orientation of sgRNAs designed to recognize target DNA sequences. The statistical significance levels from control (wild-type N2a or U87) are indicated as *p < 0.05, **p < 0.01, and ***p < 0.001. All data are presented as mean ± SD (n = 3).
Three different sgRNAs of dSaCas9-VP64 activator or dSaCas9-KRAB repressor were designed to target the promoter of each gene. Designing sgRNAs of dSaCas9-VP64 to target the Mycn promoter effectively upregulated the Mycn expression level (Figure 4A). However, dSaCas9-KRAB had no significant effect on the Mycn expression, probably due to the amplification of this gene in neuroblastoma cells. In addition, we successfully used dSaCas9-VP64 and dSaCas9-KRAB to robustly upregulate Nrf2 (Figure 4B) and downregulate Keap1 (Figure 4C) in N2a cells, respectively. Weaker but statistically significant repression of Nrf2 (by 26%) and activation of Keap1 (by 31%), respectively, were also observed. We also designed sgRNAs to target the PDGFRA gene in human U87 glioblastoma cells (Figure 4D). dSaCas9-VP64 and dSaCas9-KRAB effectively upregulated and downregulated PDGFRA expression level in U87 cells, respectively. Taken together, dSaCas9-VP64 activator and dSaCas9-KRAB repressor could effectively modulate endogenous expression levels of these four genes, albeit to varying extents.
Validation of CRISPRa Function Using Bicistronic Fluorescent Reporter Vectors
To enable in vivo and ex vivo imaging of brain tissue, we have constructed plasmid AAV (pAAV)-based bicistronic fluorescent reporter vectors that can co-express iRFP720 and GFP trangenes driven by three different promoters, including mouse methyl CpG-binding protein 2 promoter (pMecp2), human synapsin 1 promoter (pSyn1), and cytomegalovirus promoter (pCMV) (Figure S1A). Near-infrared fluorescent proteins (iRFPs) derived from bacterial phytochrome photoreceptors are potentially useful for in vivo whole-body imaging due to their deep tissue penetration capacity, low autofluorescence, and light-scattering effects.32, 33 In contrast to the widely used luciferase reporter, iRFP does not require an exogenous supply of the chromophore biliverdin to fluoresce in mammalian tissues.32, 33 Flow cytometry analysis revealed no fluorescence spillover between iRFP720 and GFP proteins; thus, both of these fluorescent proteins could be co-expressed for detection (Figure S1B). The neuron-specific pMecp2 was used to confirm the functionality of this promoter in driving the expression of CRISPR components in mouse neuroblastoma cells and brain tissue.
To further confirm the luciferase reporter data of dSaCas9-VP64, we designed sgRNAs to target three different sites on pSyn1 (Figure S2A). To avoid targeting the mouse genome in N2a cells and brain tissue, the sgRNAs were designed to target the human pSyn1 instead of the mouse neuron-specific promoters, such as pMecp2 and pCaMKIIα. Again, the data from fluorescent microscope imaging of cells confirmed the functionality of dSaCas9-VP64 in activating gene expression (Figure S2B). Co-transfection of dSaCas9-VP64s with three different sgRNAs resulted in the highest increase in iRFP720 and GFP expressions.
Validation of AAV1-PHP.B Transduction Efficiency in Brain Tissue Using Bicistronic Fluorescent Reporter Vectors
To enable blood-brain barrier (BBB) crossing and improve the transduction efficiency in brain tissue, we genetically modified the AAV1 capsid to AAV1-PHP.B by inserting a 7-mer PHP.B peptide between S588 and T589 (Figure 5A). This modification site of AAV1 capsid was equivalent to the insertion site between N587 and R588 of the AAV2 capsid.34, 35 Real-time PCR showed similar viral yields in AAV1-wild-type (WT) and AAV1-PHP.B, indicating insertion of a 7-mer peptide between S588 and T589 of AAV1 capsid did not affect AAV1 packaging and production. We then used this AAV1-PHP.B to package the CMV-driven bicistronic fluorescent reporter transgene for delivery (Figure 5B). Using an MOI of 40,000 viral genomes/cell, we compared the transduction efficiency between AAV1-WT and AAV1-PHP.B in HEK293FT (Figure 5C) and U87 cells (Figure 5D). Both fluorescent microscopy imaging and flow cytometry analysis of iRFP720 and GFP fluorescence-expressing cells revealed a higher transduction efficiency in AAV1-PHP.B than in AAV1-WT. A similar pattern of data was obtained for both cell lines, as well as the experiments performed with single and double transductions. Taken together, these data supported that displaying a PHP.B peptide between S588 and T589 of AAV1 capsid can improve AAV1 transduction efficiency while preserving vector genome-packaging capability, capsid assembly, infectivity, and gene transfer.
Figure 5.

Efficient Transduction of Bicistronic Fluorescent Reporter Vectors Using AAV1-PHP.B
(A) Inserting a 7-mer PHP.B peptide sequence in between S588 and T589 of the AAV1 capsid to generate AAV1-PHP.B. (B) pCMV-iRFP720-GFP construct fusing iRFP720 infrared to the GFP fluorescent transgenes via the T2a bicistronic element. CMV promoter is used to drive the expression of bicistronic fluorescent reporter vectors. (C) AAV1-WT and AAV1-PHP.B transductions of HEK293FT cells. (D) AAV1-WT and AAV1-PHP.B transductions of U87 cells. After 24 h of incubation with 0.1× ViraDuctin, AAVs were added to the U87 cells. GFP fluorescent images were taken 3 days after the first transduction (for single transduction) or 2 days after the second transduction (for double transduction). Second transduction was performed 24 h after the first transduction. In the flow cytometry analysis (right), the fluorescent channels APC-H7 and FITC were used to detect the iRFP720- and GFP-expressing cells, respectively. The percentage of cells expressing both iRFP720 and GFP are indicated in the density plot.
AAV1-PHP.B-Mediated Transgene Activation in the Brain Tissue
To track and compare the accumulation of AAV1-WT and AAV1-PHP.B in the brain tissue, pSyn1-driven bicistronic fluorescent reporter transgene was packaged into these viruses for delivery (Figure 6A). These viruses were then introduced into the mice by lateral tail vein injection. This bicistronic fluorescent reporter vector enabled iRFP720 imaging in vivo to detect the accumulation of AAV particles in the brain of living mice (Figure 6B). At 7 weeks after AAV injection, the brains were harvested and both iRFP720 and GFP fluorescent intensities were quantified ex vivo. Our ex vivo imaging data of the brain tissue revealed a stronger fluorescent intensity from both iRFP720 and GFP in AAV1-PHP.B than in AAV1-WT, indicating a higher blood-brain barrier-crossing ability and accumulation of AAV1-PHP.B particles in the brain (Figure 6C; Figure S3). Importantly, co-injection of two different AAV1-PHP.B vectors, one vector carried pSyn1-iRFP720-GFP transgene and the other vector carried dSaCas9-VP64 that targets the Syn1 promoter, generated the strongest fluorescent signals. These data demonstrate that a single-dose intravenous administration of AAV1-PHP.B expressing dSaCas9-VP64s was sufficient to achieve targeted transgene activation in the mouse brain.
Figure 6.

Targeted Transgene Activation in the Brain Tissue by Systemic Delivery of AAV1-PHP.B Expressing CRISPRa
(A) pSyn1-iRFP720-GFP construct. The red arrows (34, 35, and 36) on the hSyn1 promoter indicate the locations of three different sgRNA target sites for dSaCas9-VP64. (B) Example of in vivo imaging of the mouse brains. AAV1-WT (left) or AAV1-PHP.B (right) expressing iRFP720 and GFP fluorescent proteins was intravenously injected into the mice. In vivo imaging was carried out 5 weeks after injection. iRFP720 signal was detected only at the head of the AAV1-PHP.B-injected mouse. (C) Ex vivo imaging of the dissected ventral brain tissues. AAV1-PHP.B expressing dSaCas9-VP64 was injected together with AAV1-PHP.B expressing iRFP720 and GFP fluorescent proteins. After seven weeks of AAVs injection, the mouse brains were harvested. Ex vivo imaging was then carried out to detect and quantify the GFP (left) and iRFP720 (right) fluorescent signals on the mouse ventral brains. The statistical significance levels from control (uninjected brain) are indicated as *p < 0.05, **p < 0.01, and ***p < 0.001. All data are presented as mean ± SD.
Discussion
Here we have successfully developed a potent AAV-CRISPR system for targeted transgene activation in the mouse brain by engineering both AAV1-PHP.B and CRISPRa vectors. The recently developed dSpCas9-based CRISPRa and CRISPRi were not feasible for AAV packaging, though these transgenes can be easily packaged into the lentiviral vector for delivery.36, 37, 38 Therefore, we developed all-in-one CRISPRa and CRISPRi vectors that each has a transgene size fall within the AAV-packaging capacity. Despite these vectors being minimal in size, their transgene expression and functionality were not compromised. Transcriptional modulation offers several advantages over genome editing, such as restoring normal gene expression without inducing DNA damage and off-target mutagenesis. With the exception of gene knockout,39 no study yet has reported the use of an all-in-one AAV system for the delivery of CRISPR fusion protein and its sgRNA into the adult mouse brain.40 To our knowledge, the existing strategies to enable transcriptional modulation in vivo were based on the use of a dual-vector AAV system,41 a split dCas9 AAV system,42, 43 or Cre-inducible dCas9-expressing mice.44, 45, 46 In the future, it remains possible to improve the potency of our CRISPRa and CRISPRi vectors using the recently in vitro-validated strategies such as truncation of small dCas9 orthologs47 (e.g., mini-dSaCas9) and highly potent effector domains47, 48 (e.g., truncated VPR transactivation domain).
In addition, we show that the enhanced blood-brain barrier crossing of PHP.B modification can be extended to AAV1. Importantly, AAV1-PHP.B enabled the delivery of CRISPRa to the CNS, and it mediated transgene activation in the mouse brain tissue upon lateral tail vein injection. Based on amino acid sequence alignment analysis, we engineered our AAV1-PHP.B by inserting a PHP.B peptide sequence between S588 and T589 of the AAV1 capsid, which is equivalent to the common insertion site between N587 and R588 of the AAV2 capsid.34, 35 S588 and T589 amino acid residues are located within surface region VIII (588-STDPATGDVH-597) of the AAV1 capsid.49 This surface region VIII has been shown to be mutable and tolerate small peptide insertions without impairing the AAV1 production and infectivity.12, 13 Modification on this AAV1 capsid site also allows effective displaying of foreign peptide epitopes on all VPI, VP2, and VP3 capsid proteins.12, 50 Because the sialic acid-binding site for AAV1 is located on N447, S472, V473, N500, T502, and W503,51 modification at S588 or T589 of the AAV1 capsid is unlikely to disrupt its primary native glycan receptor for cell entry. Therefore, AAV1-PHP.B can be an additional tool besides AAV9-PHP.B for the efficient delivery of transgene throughout the CNS.
While brain-targeting specificity can be improved using a neuron-specific promoter, it will be interesting to investigate whether the enhanced blood-brain barrier crossing of AAV1-PHP.B is restricted to specific mouse strains and animal models just like AAV9-PHP.B. For example, the enhanced tropism in the CNS was restricted to mouse strain C57BL/6J, and AAV9-PHP.B may perform poorly in other mouse strains.52 Furthermore, a recent study revealed a similar transduction efficiency between AAV9-PHP.B and native AAV9 in the marmoset brain.53
We have shown that co-injection of the two AAV1-PHP.B vectors was able to achieve targeted transgene activation in the mouse brain. In contrast, separate injection of these two AAV1-PHP.B vectors, with a 24-h time interval, decreased the fluorescent signals (Figure S4). This observation suggests that intravenous injection of AAV1-PHP.B can trigger the production of neutralizing antibodies against the later AAV1-PHP.B administration. In fact, a recent study revealed that second time administration of another AAV9-PHP.B vector after 24-h exposure to AAV9-PHP.B could pronouncedly decrease the transgene’s expression in the mouse brain.54 Furthermore, the production of neutralizing antibodies became detectable 2 days after intravenous injection of AAV9-PHP.B into mice.54
Materials and Methods
Cell Line Cultures
Mouse N2a, human HEK293FT, and U87 glioblastoma cell lines were cultured in DMEM (high glucose) (Gibco 11965092) with 10% fetal bovine serum (Gibco 10270106). The cells were cultured under a standard cell culture condition (37°C, 5% CO2) in a humidified incubator.
CRISPRa and CRISPRi pAAV Vector Backbone Construction
To construct pAAV-pMecp2-SaCas9-spA-pU6-sgRNA (vector A), the truncated version of the mouse Mecp2 promoter (235 bp, pMecp2) was amplified from the plasmid PX551 (Addgene 60957) and replaced the CMV promoter in the plasmid backbone pX601 (Addgene 61591), using XbaI and AgeI. A short oligonucleotide containing a minimal polyadenylation signal (48 bp, spA) was digested with EcoRI and KpnI to replace the bGHpA in plasmid pX601. To construct pAAV-pMecp2-dSaCas9-spA-pU6-sgRNA (vector B), the SaCas9 from vector A was replaced with the catalytically inactive SaCas9 (dSaCas9) from plasmid PX603 (Addgene 61594), using AgeI and EcoRI. The minimal catalytic domain of the transcriptional regulators (156-bp VP64, 390-bp VP160, 227-bp KRAB, and 432-bp SID4X) was amplified from the plasmid backbones dCAS9-VP6436 (Addgene 61422), dCas9-VP16055 (Addgene 48240), dCas9-KRAB56 (Addgene 50919), and SID4X domain57 (Addgene 43882), respectively. To construct the dSaCas9-based transcriptional modulator, the amplified transcriptional regulator domain was inserted into the previously constructed vector B using BamHI and EcoRI. The newly constructed CRISPRa and CRISPRi included pAAV-pMecp2-dSaCas9-VP64-spA-pU6-sgRNA (vector C), pAAV-pMecp2-dSaCas9-VP160-spA-pU6-sgRNA (vector D), pAAV-pMecp2-dSaCas9-KRAB-spA-pU6-sgRNA (vector E), and pAAV-pMecp2-dSaCas9-SID4X-spA-pU6-sgRNA (vector F) (Figure 1). Primers used for vector construction are listed in Table S1.
Design of sgRNA Target Sites
To construct the sgRNA guide sequence, a pair of annealed oligonucleotides was cloned into a CRISPR expression cassette bearing the sgRNA scaffold backbone, tracrRNA and SaCas9 or dSaCas9 nuclease using BsaI (New England Biolabs R3535). The oligonucleotides were designed based on the target site sequence (21 or 22 bp), and they were flanked on the 3′ end by a 6-bp 5′-NNGRRT-3′ (R = A/G) PAM sequence. The first nucleotide of the transcribed gRNA was a guanine nucleotide (G) to maximize U6 promoter activity. The CRISPR RGEN (RNA-guided endonuclease) Tool Cas-Designer (http://www.rgenome.net/cas-designer/) was used to identify the target sequence of sgRNAs. The selected sgRNA target sequences had no potential off-target sites of RNA-guided endonucleases within 2-nt mismatches, as determined by blasting the genome of Homo sapiens (GRCh38/hg38) or Mus musculus (mm10) in Cas-Designer. By inferring to Common SNP Track Settings for mouse (dbSNP 142) and human (dbSNP 146), the BLAT Search Genome tool in UCSC Genome Browser was used to confirm the absence of common genetic variants (≥1%) for each selected sgRNA target site. BsaI and XbaI were used to screen the clones with target sequence insertion. Two DNA fragment sizes (3 and ∼4 kb) were observed for the sgRNA vector with no insertion, while a single DNA band (∼7 kb) was observed when digested with BsaI and XbaI for the modified sgRNA. Then, the pU6-seq primer was used for DNA sequencing to confirm successful target sequence insertion in the sgRNA. All primers used for designing sgRNAs are listed in Table S2.
Luciferase Bioluminescence Reporter Assays
For the construction of pCaMKIIα-Luc vector (Luc2), the 1,316 bp of mouse alpha CaMKII promoter was amplified from the genomic DNA of N2a cells and inserted into the promoterless pGL4.10[luc2] (Promega E6651), using KpnI and HindIII. Nested PCR was used to amplify the mouse alpha CaMKII promoter and super-enhancer at intergenic Camk2a-Arsi from the genomic DNA of N2a cells. Then, the amplified 6.8-kb mouse alpha CaMKII promoter and intergenic super-enhancer (Luc3) were inserted into promoterless pGL4.10[luc2] with SacI and HindIII. Platinum PCR SuperMix High Fidelity (Invitrogen 12532016) was used for the PCR amplification. For the amplification of 6.8-kb DNA fragments, PCR was carried out as follows: an initial denaturation step at 98°C for 30 s followed by 40 cycles at 98°C for 10 s, 65°C for 30 s, and 68°C for 7 min, with a final extension step at 68°C for 10 min. Primers used for luciferase vector construction are listed in Table S3.
Using Lipofectamine 3000 Transfection Reagent (Invitrogen L3000001), the luciferase reporter (50 ng/well) and CRISPR (50 ng/well) constructs were co-transfected with the TK-Renilla control (Promega E2241) (10 ng/well) at a ratio of 10:1 into a 96-well plate. For co-transfection of multiple CRISPRs, an equal amount of each CRISPR was used. After 48 h of transfection, cells were lysed and quantified for the luciferase activity using the Dual-Luciferase Reporter (DLR) Assay System (Promega E1910). The firefly luminescence on a 96-well white polystyrene plate was measured with the SpectraMax M5e Microplate Reader (Molecular Devices). Four replicates for each experimental group were performed.
Plasmid Transfection of CRISPRa and CRISPRi
At 1 day prior to plasmid transfection, 3 × 105 N2a or U87 cells was seeded into each well of a 6-well plate. Briefly, 2,500 ng DNA was mixed with 5 μL P3000 Reagent and 5 μL Lipofectamine 3000 (Invitrogen) in 1 mL Opti-MEM I Reduced Serum Medium (Gibco 31985062). For co-transfection of multiple CRISPRs, an equal amount of each CRISPR was used. The mRNA and protein were harvested from the cells after 2 and 3 days, respectively.
qRT-PCR and Melt Curve Analysis
Total RNA was extracted from the N2a and U87 cells with the RNeasy Mini Kit (QIAGEN 74104) and treated with DNase I to digest genomic DNA (QIAGEN 79254). The concentration and purity of total RNAs were then determined with a Biochrom spectrophotometer. The total harvested RNA was reverse transcribed using SuperScript III First-Strand Synthesis System (Invitrogen 18080051), according to the manufacturer’s protocol. For qRT-PCR, the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad 1725271) was used to amplify the synthesized cDNA. The mixture for each qPCR reaction was 10 μL 2X SsoAdvanced Universal SYBR Green Supermix, 0.2 μL 10 μM forward primer, 0.2 μL 10 μM reverse primer, 2 μL 2.5 ng/μL cDNA sample, and 7.6 μL nuclease-free water. qPCR was done by using Thermal Cycling (Bio-Rad CFX96 Connect System).
qPCR was carried out as follows: an initial denaturation step at 95°C for 3 min followed by 40 cycles at 95°C for 15 s and 60°C for 60 s. The housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (Gapdh) was used for internal normalization. Relative quantification of gene expression was calculated using the 2-ΔΔCt method. Technical triplicates for each sample were performed. Melt curve analysis was done at 65°C –95°C with 0.5°C increments and 2–5 s/step to check specificity of the primers and to ensure no primer-dimer formation during the qPCR. Melt curve analysis was also used to confirm the absence of longer qPCR products to ensure no genomic DNA amplification. An exon-exon junction primer was designed to avoid genomic DNA amplification during the qPCR. Primers used for qRT-PCR are listed in Table S4.
Protein Extraction and Western Blot Analysis
Ice-cold radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling Technology 9806) with added Protease Inhibitor Cocktail (Cell Signaling Technology 5871) was used to lyse cultured cells for protein extraction. Protein concentration was determined with a Pierce BCA Protein Assay Kit (Thermo Scientific 23227) using the SpectraMax M5e Microplate Reader (Molecular Devices). Then, 50 μg protein lysate was isolated by NuPAGE Novex 4%–12% Bis-Tris Protein Gels (Invitrogen NP0335BOX) and transferred to 0.45-μm Invitrolon polyvinylidene fluoride (PVDF) membranes (Invitrogen LC2005). NuPAGE LDS Sample Buffer (Invitrogen NP0007) and NuPAGE Sample Reducing Agent (Invitrogen NP0004) were added to reduce the protein samples prior to electrophoresis. The proteins were separated by electrophoresis at 200 V constant for 35 min in NuPAGE MES SDS Running Buffer (Invitrogen NP0002) supplemented with NuPAGE Antioxidant (Invitrogen NP0005). The membrane transfer was performed at 40 V for 90 min in ice-cold NuPAGE Transfer Buffer (Invitrogen NP0006) supplemented with NuPAGE Antioxidant and 10% methanol.
The membranes were blocked for 1 h at room temperature with 5% blocking buffer (Bio-Rad 170-6404), then incubated with 1,000×-diluted CaMKII-α (6G9) mouse monoclonal antibody (Cell Signaling Technology 50049) in 5% BSA (Cell Signaling Technology 9998) at 4°C overnight. The membrane was then incubated with anti-mouse immunoglobulin G (IgG), horseradish peroxidase (HRP)-linked antibody (Cell Signaling Technology 7076). CaMKII-α (6G9) mouse monoclonal antibody (mAb) recognizes endogenous levels of total CaMKII-α protein and does not cross-react with CaMKII-β or CaMKII-δ proteins. All the membrane-washing steps were performed with Tris-buffered saline supplemented with Tween 20 (TBST) (Cell Signaling Technology 9997). Immunoreactive bands were visualized using Pierce ECL Western Blotting Substrate (Thermo Scientific 32109). The alpha CaMKII-staining membrane was stripped with Restore Western Blot Stripping Buffer (Thermo Scientific 21062) and reprobed with β-actin (8H10D10) mouse monoclonal antibody (Cell Signaling Technology 3700) to confirm equal loading. Precision Plus Protein Kaleidoscope Prestained Protein Standards (Bio-Rad 1610375) was used for band referencing and blot orientation during electrophoresis and membrane transfer. MagicMark XP Western Protein Standard (Invitrogen LC5603) was used to determine the protein band size during western blot detection with Luminescent Image Analyzer LAS-4000 (Fujifilm). The protein band intensities were analyzed and quantified with ImageJ (NIH, USA).
Flow Cytometry Analysis
The BD FACSVerse flow cytometer and BD FACSuite software (BD Biosciences) were used to quantify iRFP720- and GFP-expressing cells. 100,000 cells were analyzed for each sample. The fluorescent channels allophycocyanin (APC)-H7 and fluorescein isothiocyanate (FITC) were used to detect the iRFP720- and GFP-expressing cells, respectively.
Construction of iRFP720 and GFP Fusion Vectors
To construct pAAV-pMecp2-iRFP720-spA, the 948-bp iRFP720 was amplified from the plasmid piRFP720-N1 (Addgene 45461) and replaced the SpCas9 in the plasmid backbone PX551 (Addgene 60957), using AgeI and EcoRI. Then, 774-bp T2A-GFP was amplified from the plasmid pX601-GFP (Addgene 84040) and we inserted the T2A-GFP into the pAAV-pMecp2-iRFP720-spA using a single EcoRI digestion. BamHI was used to confirm correct orientation of the inserted T2A-GFP. A stop codon was added to the downstream sequence of GFP in the pAAV-pMecp2-iRFP720-T2A-GFP-spA vector.
To construct the pAAV-pCMV-iRFP720-T2A-GFP-spA vector, the 553-bp CMV promoter was amplified from the plasmid piRFP720-N1 (Addgene 45461) and replaced the pMecp2 promoter in the pAAV-pMecp2-iRFP720-T2A-GFP-spA vector, using XbaI and AgeI. To construct the pAAV-pSyn1-iRFP720-T2A-GFP-spA vector, the 476-bp human Syn1 promoter was amplified from the plasmid pAAV-hSyn-hChR2(H134R)-mCherry (Addgene 26976) and replaced the pMecp2 promoter in the pAAV-pMecp2-iRFP720-T2A-GFP-spA vector, using XbaI and AgeI. These three newly modified constructs were subjected to DNA sequencing confirmation after being checked with the restriction enzymes. The other iRFP variant, iRFP713 (Addgene 31857), was also tested in this study. Primers used for vector construction are listed in Table S5.
AAV1 Capsid Modification
AAV-1 Helper Free Packaging System, consisting of pAAV-RC1 Vector, pHelper Vector, and pAAV-GFP Control Vector, was purchased from Cell Biolabs (VPK-401). The wild-type AAV1 capsid sequence on the pAAV-RC1 Vector was genetically modified by inserting a 7-mer PHP.B (TLAVPFK) in between S588 and T589. The 21-bp DNA oligonucleotide encoding PHP.B was introduced into the AAV1 capsid via MscI and SphI. The modified AAV1 capsid vector (AAV1-PHP.B) was subjected to DNA sequencing confirmation. DNA oligonucleotides used for AAV1 capsid modification are listed in Table S6.
Recombinant AAV Vector Production
At 1 day before plasmid transfection, ∼7 × 106 HEK293FT cells were seeded on each 100-mm dish. To generate recombinant AAV vectors, HEK293FT cells were transfected with transgene plasmid (iRFP720-GFP or CRISPR encoded), pAAV1 serotype vector (AAV1-WT or AAV1-PHP.B), and pHelper vector at a 1:1:1 ratio using Calcium Phosphate Transfection Kit (Invitrogen K278001), with some modifications. Briefly, 4 h prior to transfection, the culture media in each 100-mm dish were replaced with 12 mL fresh media consisting of 1% heat-inactivated fetal bovine serum (Gibco 10082139). Then, 600 μL transfection mixture that consisted of 36 μL 2M CaCl2, 20 µg DNA, 300 μL 2× HEPES-buffered saline (HBS), and H2O was added to the cells. After 72 h of plasmid transfection, cells from each 100-mm dish were collected into a single tube for AAV particle extraction. AAV particles within the cells were extracted using AAVpro Extraction Solution (Takara 6235). The extracted AAV particles in each tube (cells from a 100-mm dish) were suspended in 400 μL AAV Extraction Solution A, followed by 40 μL AAV Extraction Solution B. The mixture was then stored at -80°C.
Viral Genome Extraction and TaqMan qPCR
Viral nucleic acids from AAV vector preparations were extracted with the High Pure viral nucleic acid kit (Roche 11858874001), according to the manufacturer’s instructions. Briefly, 20 μL harvested viral supernatant was used for viral genome extraction. The purified viral genome was eluted into 50 μL elution buffer. The eluted viral DNA was diluted 100× for qPCR titering. SsoAdvanced Universal Probes Supermix (Bio-Rad 172-5280) and high-performance liquid chromatography (HPLC)-purified 6-FAM/ZEN/Iowa Black-labeled probes (Integrated DNA Technologies) were used to measure the titer of the AAV viruses. The probes and primers specific to dSaCas9 or iRFP720 were used (Table S7). The qPCR reaction mixtures consisted of 10 μL SsoAdvanced Universal Probes Supermix (2×), 0.2 μL of each 10 μM forward and reverse primers, 0.2 μL 10 μM FAM-labeled probe, 2 μL 100×-diluted viral genome, and 7.4 μL nuclease-free water. The thermal cycling of Bio-Rad CFX96 Connect System was settled at 95°C for 3 min for polymerase activation and DNA denaturation, followed by 40 cycles of denaturation at 95°C for 15 s and annealing at 60°C for 60 s. Plasmid pAAV-CMV-iRFP720-GFP (5,313 bp), pAAV-hSyn1-iRFP720-GFP (5,185 bp), or pAAV-Mecp2-dSaCas9-VP64 (6,992 bp) at 1 ng/μL was used to generate the DNA standard curve in 10-fold series dilution from 1 × 108 to 1 × 101 viral copies.
AAV Transduction
At 1 day prior to AAV transduction, 5 × 104 HEK293FT or U87 cells were seeded into each well of a 24-well plate. On the next day, the culture media in each well were replaced with 400 μL 1% heat-inactivated fetal bovine serum (FBS) media containing AAV particles. After 24 h of AAV infection, the transduction mixture was removed and the cells were maintained in 1 mL fresh culture media containing 1% heat-inactivated FBS (Gibco 10082139). For transduction of bicistronic fluorescent reporter vectors, AAV1-WT or AAV1-PHP.B particles were added at an MOI of 40,000 viral genomes/cell. A second transduction was performed 24 h after the first transduction. At 2 days after the second transduction of AAV viruses, the iRFP720- and GFP-expressing individual cells were analyzed with flow cytometry and imaged with fluorescent microscopy. To transduce U87 cells, ViraDuctin AAV Transduction Reagents (Cell Biolabs AAV-201) were added prior to incubating the cells with the AAV-containing media. Briefly, the ViraDuctin was added to the culture medium at a final concentration of 0.1×. After 24-h incubation of ViraDuctin, the media were removed and the cells were added to fresh media containing AAV particles. ViraDuctin can improve the transduction efficiency of AAVs by overcoming the rate-limiting step in the viral second-strand DNA synthesis.
Systemic Administration of AAV Particles into the Mice
All animal experiments were approved by the ethics committee of City University of Hong Kong and carried out according to the regulation in Hong Kong. The AAV particles were injected into the adult male C57BL/6J mice (∼20 g) intravenously via lateral tail vein. Each mouse was injected with 1.0 × 1010 copies of AAV particles. The AAV supernatant was diluted to 200 μL with PBS for injection into each mouse. Mice were divided into 4 different treatment groups. The first group of non-injected mice was a negative control. The second group of mice was injected with wild-type AAV1 expressing bicistronic fluorescent reporters (pSyn1-iRFP720-GFP). The third group of mice was injected with engineered AAV1-PHP.B expressing bicistronic fluorescent reporters. The last group of mice was co-injected with two different AAV1-PHP.B vectors, each 1.0 × 1010 copies: one of the AAV1-PHP.B vectors carried bicistronic fluorescent reporters, while the other AAV1-PHP.B vector carried dSaCas9-VP64s (targeting 34, 35, and 36 sites on the hSyn1 promoter). At 7 weeks after AAV injection, the mouse brains were harvested and fixed in 10% formalin overnight. The tissue was imaged ex vivo using the In-Vivo Xtreme Imaging System (Bruker) to measure the intensity of both GFP and iRFP720. Excitation at 700 nm and emission at 750 nm were used to detect the iRFP720 signals. Excitation at 480 nm and emission at 535 nm were used to detect the GFP signals.
Bioinformatics Analysis
Two super-enhancer databases, dbSUPER (http://bioinfo.au.tsinghua.edu.cn/dbsuper/) and SEA, were used to identify the potential super-enhancers implicated in neuronal and brain functions. Then, the Encyclopedia of DNA Elements (ENCODE) and Roadmap Epigenomics Integrative Analysis in the UCSC Genome Browser were used to get detailed epigenetic information on selected super-enhancers for designing target sites of sgRNAs.
Statistical Analysis
Using IBM SPSS Statistics Program, significant differences in continuous variables among subject groups were confirmed by ANOVA, followed by Dunnett (for comparing to a control) or Tukey (for all pairwise comparisons) post hoc multiple comparisons. All p values were two tailed. The statistical significance levels are indicated as *p < 0.05, **p < 0.01, and ***p < 0.001. All data are presented as mean ± SD.
Author Contributions
C.T. conceived and supervised the project. C.-H.L. designed and performed the experiments. C.-H.L. collected and analyzed the data. J.W.-T.H., P.K.L., and C.T. contributed reagents, materials, and analysis tools. C.-H.L. and C.T. wrote and revised the manuscript. All authors read, corrected, and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We would like to thank Prof. Chi-Ming Che and Dr. Chun-Nam Lok (The University of Hong Kong) for providing U87 cells and Dr. Regina Cheuk-Lam Lo (The University of Hong Kong) for providing HEK293FT cells. We would also like to thank Dr. Wenjun Xiong (City University of Hong Kong) for her advice on AAV production. This work was supported by the National Natural Science Foundation of China (NSFC), China (31671103), the Research Grants Council of Hong Kong (project CityU11213717), the Health and Medical Research Fund (HMRF), Food and Health Bureau, Hong Kong (projects 03141076 and 05160336), and the City University of Hong Kong (project CityU7005088).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2019.04.015.
Supplemental Information
References
- 1.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]; Shalem, O., Sanjana, N.E., Hartenian, E., Shi, X., Scott, D.A., Mikkelson, T., Heckl, D., Ebert, B.L., Root, D.E., Doench, J.G., and Zhang, F. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84-87. [DOI] [PMC free article] [PubMed]
- 2.Cho S.W., Kim S., Kim J.M., Kim J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]; Cho, S.W., Kim, S., Kim, J.M., and Kim, J.S. (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230-232. [DOI] [PubMed]
- 3.Paquet D., Kwart D., Chen A., Sproul A., Jacob S., Teo S., Olsen K.M., Gregg A., Noggle S., Tessier-Lavigne M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]; Paquet, D., Kwart, D., Chen, A., Sproul, A., Jacob, S., Teo, S., Olsen, K.M., Gregg, A., Noggle, S., and Tessier-Lavigne, M. (2016). Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125-129. [DOI] [PubMed]
- 4.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ran, F.A., Cong, L., Yan, W.X., Scott, D.A., Gootenberg, J.S., Kriz, A.J., Zetsche, B., Shalem, O., Wu, X., Makarova, K.S., et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186-191. [DOI] [PMC free article] [PubMed]
- 5.Maeder M.L., Linder S.J., Cascio V.M., Fu Y., Ho Q.H., Joung J.K. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods. 2013;10:977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]; Maeder, M.L., Linder, S.J., Cascio, V.M., Fu, Y., Ho, Q.H., and Joung, J.K. (2013). CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 10, 977-979. [DOI] [PMC free article] [PubMed]
- 6.Thakore P.I., D’Ippolito A.M., Song L., Safi A., Shivakumar N.K., Kabadi A.M., Reddy T.E., Crawford G.E., Gersbach C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods. 2015;12:1143–1149. doi: 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]; Thakore, P.I., D’Ippolito, A.M., Song, L., Safi, A., Shivakumar, N.K., Kabadi, A.M., Reddy, T.E., Crawford, G.E., and Gersbach, C.A. (2015). Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143-1149. [DOI] [PMC free article] [PubMed]
- 7.Samulski R.J., Muzyczka N. AAV-mediated gene therapy for research and therapeutic purposes. Annu. Rev. Virol. 2014;1:427–451. doi: 10.1146/annurev-virology-031413-085355. [DOI] [PubMed] [Google Scholar]; Samulski, R.J., and Muzyczka, N. (2014). AAV-mediated gene therapy for research and therapeutic purposes. Annu. Rev. Virol. 1, 427-451. [DOI] [PubMed]
- 8.Kotterman M.A., Schaffer D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014;15:445–451. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kotterman, M.A., and Schaffer, D.V. (2014). Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 15, 445-451. [DOI] [PMC free article] [PubMed]
- 9.Daya S., Berns K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008;21:583–593. doi: 10.1128/CMR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; Daya, S., and Berns, K.I. (2008). Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 21, 583-593. [DOI] [PMC free article] [PubMed]
- 10.Deverman B.E., Pravdo P.L., Simpson B.P., Kumar S.R., Chan K.Y., Banerjee A., Wu W.L., Yang B., Huber N., Pasca S.P., Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]; Deverman, B.E., Pravdo, P.L., Simpson, B.P., Kumar, S.R., Chan, K.Y., Banerjee, A., Wu, W.L., Yang, B., Huber, N., Pasca, S.P., and Gradinaru, V. (2016). Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 34, 204-209. [DOI] [PMC free article] [PubMed]
- 11.Davis A.S., Federici T., Ray W.C., Boulis N.M., OʼConnor D., Clark K.R., Bartlett J.S. Rational design and engineering of a modified adeno-associated virus (AAV1)-based vector system for enhanced retrograde gene delivery. Neurosurgery. 2015;76:216–225, discussion 225. doi: 10.1227/NEU.0000000000000589. [DOI] [PubMed] [Google Scholar]; Davis, A.S., Federici, T., Ray, W.C., Boulis, N.M., OʼConnor, D., Clark, K.R., and Bartlett, J.S. (2015). Rational design and engineering of a modified adeno-associated virus (AAV1)-based vector system for enhanced retrograde gene delivery. Neurosurgery 76, 216-225, discussion 225. [DOI] [PubMed]
- 12.Federici T., Davis A., Teng Q., Riley J., Bartlett J.S., Boulis N.M. Genetic modification of AAV1 capsid for WN targeted gene delivery. Mol. Ther. 2007;15(Suppl 1):S31. [Google Scholar]; Federici, T., Davis, A., Teng, Q., Riley, J., Bartlett, J.S., and Boulis, N.M. (2007). Genetic modification of AAV1 capsid for WN targeted gene delivery. Mol. Ther. 15 (Suppl 1), S31.
- 13.Adachi K., Nakai H. A new recombinant adeno-associated virus (Aav)-based random peptide display library system: infection-defective Aav1.9-3 as a novel detargeted platform for vector evolution. Gene Ther. Regul. 2010;5:31–55. doi: 10.1142/S1568558610000197. [DOI] [PMC free article] [PubMed] [Google Scholar]; Adachi, K., and Nakai, H. (2010). A new recombinant adeno-associated virus (Aav)-based random peptide display library system: infection-defective Aav1.9-3 as a novel detargeted platform for vector evolution. Gene Ther. Regul. 5, 31-55. [DOI] [PMC free article] [PubMed]
- 14.Hadaczek P., Stanek L., Ciesielska A., Sudhakar V., Samaranch L., Pivirotto P., Bringas J., O’Riordan C., Mastis B., San Sebastian W. Widespread AAV1- and AAV2-mediated transgene expression in the nonhuman primate brain: implications for Huntington’s disease. Mol. Ther. Methods Clin. Dev. 2016;3:16037. doi: 10.1038/mtm.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hadaczek, P., Stanek, L., Ciesielska, A., Sudhakar, V., Samaranch, L., Pivirotto, P., Bringas, J., O’Riordan, C., Mastis, B., San Sebastian, W., et al. (2016). Widespread AAV1- and AAV2-mediated transgene expression in the nonhuman primate brain: implications for Huntington’s disease. Mol. Ther. Methods Clin. Dev. 3, 16037. [DOI] [PMC free article] [PubMed]
- 15.Hadaczek P., Forsayeth J., Mirek H., Munson K., Bringas J., Pivirotto P., McBride J.L., Davidson B.L., Bankiewicz K.S. Transduction of nonhuman primate brain with adeno-associated virus serotype 1: vector trafficking and immune response. Hum. Gene Ther. 2009;20:225–237. doi: 10.1089/hum.2008.151. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hadaczek, P., Forsayeth, J., Mirek, H., Munson, K., Bringas, J., Pivirotto, P., McBride, J.L., Davidson, B.L., and Bankiewicz, K.S. (2009). Transduction of nonhuman primate brain with adeno-associated virus serotype 1: vector trafficking and immune response. Hum. Gene Ther. 20, 225-237. [DOI] [PMC free article] [PubMed]
- 16.Swiech L., Heidenreich M., Banerjee A., Habib N., Li Y., Trombetta J., Sur M., Zhang F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015;33:102–106. doi: 10.1038/nbt.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]; Swiech, L., Heidenreich, M., Banerjee, A., Habib, N., Li, Y., Trombetta, J., Sur, M., and Zhang, F. (2015). In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 33, 102-106. [DOI] [PMC free article] [PubMed]
- 17.Vojta A., Dobrinić P., Tadić V., Bočkor L., Korać P., Julg B., Klasić M., Zoldoš V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016;44:5615–5628. doi: 10.1093/nar/gkw159. [DOI] [PMC free article] [PubMed] [Google Scholar]; Vojta, A., Dobrinić, P., Tadić, V., Bočkor, L., Korać, P., Julg, B., Klasić, M., and Zoldoš, V. (2016). Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 44, 5615-5628. [DOI] [PMC free article] [PubMed]
- 18.Kwon D.Y., Zhao Y.T., Lamonica J.M., Zhou Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017;8:15315. doi: 10.1038/ncomms15315. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kwon, D.Y., Zhao, Y.T., Lamonica, J.M., and Zhou, Z. (2017). Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 8, 15315. [DOI] [PMC free article] [PubMed]
- 19.Kearns N.A., Pham H., Tabak B., Genga R.M., Silverstein N.J., Garber M., Maehr R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods. 2015;12:401–403. doi: 10.1038/nmeth.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kearns, N.A., Pham, H., Tabak, B., Genga, R.M., Silverstein, N.J., Garber, M., and Maehr, R. (2015). Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 12, 401-403. [DOI] [PMC free article] [PubMed]
- 20.Chavez A., Scheiman J., Vora S., Pruitt B.W., Tuttle M., P R Iyer E., Lin S., Kiani S., Guzman C.D., Wiegand D.J. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods. 2015;12:326–328. doi: 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chavez, A., Scheiman, J., Vora, S., Pruitt, B.W., Tuttle, M., P R Iyer, E., Lin, S., Kiani, S., Guzman, C.D., Wiegand, D.J., et al. (2015). Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12, 326-328. [DOI] [PMC free article] [PubMed]
- 21.Liu X.S., Wu H., Ji X., Stelzer Y., Wu X., Czauderna S., Shu J., Dadon D., Young R.A., Jaenisch R. Editing DNA methylation in the mammalian genome. Cell. 2016;167:233–247.e17. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu, X.S., Wu, H., Ji, X., Stelzer, Y., Wu, X., Czauderna, S., Shu, J., Dadon, D., Young, R.A., and Jaenisch, R. (2016). Editing DNA methylation in the mammalian genome. Cell 167, 233-247.e17. [DOI] [PMC free article] [PubMed]
- 22.Hilton I.B., D’Ippolito A.M., Vockley C.M., Thakore P.I., Crawford G.E., Reddy T.E., Gersbach C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hilton, I.B., D’Ippolito, A.M., Vockley, C.M., Thakore, P.I., Crawford, G.E., Reddy, T.E., and Gersbach, C.A. (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 33, 510-517. [DOI] [PMC free article] [PubMed]
- 23.Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hnisz, D., Abraham, B.J., Lee, T.I., Lau, A., Saint-Andre, V., Sigova, A.A., Hoke, H.A., and Young, R.A. (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934-947. [DOI] [PMC free article] [PubMed]
- 24.Yamagata Y., Kobayashi S., Umeda T., Inoue A., Sakagami H., Fukaya M., Watanabe M., Hatanaka N., Totsuka M., Yagi T. Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIalpha in dendritic spine enlargement, long-term potentiation, and learning. J. Neurosci. 2009;29:7607–7618. doi: 10.1523/JNEUROSCI.0707-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yamagata, Y., Kobayashi, S., Umeda, T., Inoue, A., Sakagami, H., Fukaya, M., Watanabe, M., Hatanaka, N., Totsuka, M., Yagi, T., et al. (2009). Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIalpha in dendritic spine enlargement, long-term potentiation, and learning. J. Neurosci. 29, 7607-7618. [DOI] [PMC free article] [PubMed]
- 25.Frankland P.W., O’Brien C., Ohno M., Kirkwood A., Silva A.J. Alpha-CaMKII-dependent plasticity in the cortex is required for permanent memory. Nature. 2001;411:309–313. doi: 10.1038/35077089. [DOI] [PubMed] [Google Scholar]; Frankland, P.W., O’Brien, C., Ohno, M., Kirkwood, A., and Silva, A.J. (2001). Alpha-CaMKII-dependent plasticity in the cortex is required for permanent memory. Nature 411, 309-313. [DOI] [PubMed]
- 26.Martinho O., Longatto-Filho A., Lambros M.B., Martins A., Pinheiro C., Silva A., Pardal F., Amorim J., Mackay A., Milanezi F. Expression, mutation and copy number analysis of platelet-derived growth factor receptor A (PDGFRA) and its ligand PDGFA in gliomas. Br. J. Cancer. 2009;101:973–982. doi: 10.1038/sj.bjc.6605225. [DOI] [PMC free article] [PubMed] [Google Scholar]; Martinho, O., Longatto-Filho, A., Lambros, M.B., Martins, A., Pinheiro, C., Silva, A., Pardal, F., Amorim, J., Mackay, A., Milanezi, F., et al. (2009). Expression, mutation and copy number analysis of platelet-derived growth factor receptor A (PDGFRA) and its ligand PDGFA in gliomas. Br. J. Cancer 101, 973-982. [DOI] [PMC free article] [PubMed]
- 27.Ruiz-Pérez M.V., Henley A.B., Arsenian-Henriksson M. The MYCN protein in health and disease. Genes (Basel) 2017;8:E113. doi: 10.3390/genes8040113. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ruiz-Perez, M.V., Henley, A.B., and Arsenian-Henriksson, M. (2017). The MYCN protein in health and disease. Genes (Basel) 8, E113. [DOI] [PMC free article] [PubMed]
- 28.Nguyen T., Nioi P., Pickett C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nguyen, T., Nioi, P., and Pickett, C.B. (2009). The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 284, 13291-13295. [DOI] [PMC free article] [PubMed]
- 29.Cui Y., Ma S., Zhang C., Li D., Yang B., Lv P., Xing Q., Huang T., Yang G.L., Cao W., Guan F. Pharmacological activation of the Nrf2 pathway by 3H-1, 2-dithiole-3-thione is neuroprotective in a mouse model of Alzheimer disease. Behav. Brain Res. 2018;336:219–226. doi: 10.1016/j.bbr.2017.09.011. [DOI] [PubMed] [Google Scholar]; Cui, Y., Ma, S., Zhang, C., Li, D., Yang, B., Lv, P., Xing, Q., Huang, T., Yang, G.L., Cao, W., and Guan, F. (2018). Pharmacological activation of the Nrf2 pathway by 3H-1, 2-dithiole-3-thione is neuroprotective in a mouse model of Alzheimer disease. Behav. Brain Res. 336, 219-226. [DOI] [PubMed]
- 30.Jing X., Wei X., Ren M., Wang L., Zhang X., Lou H. Neuroprotective effects of Tanshinone I against 6-OHDA-induced oxidative stress in cellular and mouse model of Parkinson’s disease through upregulating Nrf2. Neurochem. Res. 2016;41:779–786. doi: 10.1007/s11064-015-1751-6. [DOI] [PubMed] [Google Scholar]; Jing, X., Wei, X., Ren, M., Wang, L., Zhang, X., and Lou, H. (2016). Neuroprotective effects of Tanshinone I against 6-OHDA-induced oxidative stress in cellular and mouse model of Parkinson’s disease through upregulating Nrf2. Neurochem. Res. 41, 779-786. [DOI] [PubMed]
- 31.Zhang D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]; Zhang, D.D. (2006). Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 38, 769-789. [DOI] [PubMed]
- 32.Shcherbakova D.M., Verkhusha V.V. Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat. Methods. 2013;10:751–754. doi: 10.1038/nmeth.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]; Shcherbakova, D.M., and Verkhusha, V.V. (2013). Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat. Methods 10, 751-754. [DOI] [PMC free article] [PubMed]
- 33.Filonov G.S., Piatkevich K.D., Ting L.M., Zhang J., Kim K., Verkhusha V.V. Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat. Biotechnol. 2011;29:757–761. doi: 10.1038/nbt.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]; Filonov, G.S., Piatkevich, K.D., Ting, L.M., Zhang, J., Kim, K., and Verkhusha, V.V. (2011). Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat. Biotechnol. 29, 757-761. [DOI] [PMC free article] [PubMed]
- 34.Work L.M., Büning H., Hunt E., Nicklin S.A., Denby L., Britton N., Leike K., Odenthal M., Drebber U., Hallek M., Baker A.H. Vascular bed-targeted in vivo gene delivery using tropism-modified adeno-associated viruses. Mol. Ther. 2006;13:683–693. doi: 10.1016/j.ymthe.2005.11.013. [DOI] [PubMed] [Google Scholar]; Work, L.M., Buning, H., Hunt, E., Nicklin, S.A., Denby, L., Britton, N., Leike, K., Odenthal, M., Drebber, U., Hallek, M., and Baker, A.H. (2006). Vascular bed-targeted in vivo gene delivery using tropism-modified adeno-associated viruses. Mol. Ther. 13, 683-693. [DOI] [PubMed]
- 35.Geoghegan J.C., Keiser N.W., Okulist A., Martins I., Wilson M.S., Davidson B.L. Chondroitin sulfate is the primary receptor for a peptide-modified AAV that targets brain vascular endothelium in vivo. Mol. Ther. Nucleic Acids. 2014;3:e202. doi: 10.1038/mtna.2014.50. [DOI] [PMC free article] [PubMed] [Google Scholar]; Geoghegan, J.C., Keiser, N.W., Okulist, A., Martins, I., Wilson, M.S., and Davidson, B.L. (2014). Chondroitin sulfate is the primary receptor for a peptide-modified AAV that targets brain vascular endothelium in vivo. Mol. Ther. Nucleic Acids 3, e202. [DOI] [PMC free article] [PubMed]
- 36.Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]; Konermann, S., Brigham, M.D., Trevino, A.E., Joung, J., Abudayyeh, O.O., Barcena, C., Hsu, P.D., Habib, N., Gootenberg, J.S., Nishimasu, H., et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583-588. [DOI] [PMC free article] [PubMed]
- 37.Klann T.S., Black J.B., Chellappan M., Safi A., Song L., Hilton I.B., Crawford G.E., Reddy T.E., Gersbach C.A. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 2017;35:561–568. doi: 10.1038/nbt.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]; Klann, T.S., Black, J.B., Chellappan, M., Safi, A., Song, L., Hilton, I.B., Crawford, G.E., Reddy, T.E., and Gersbach, C.A. (2017). CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 35, 561-568. [DOI] [PMC free article] [PubMed]
- 38.Zheng Y., Shen W., Zhang J., Yang B., Liu Y.N., Qi H., Yu X., Lu S.Y., Chen Y., Xu Y.Z. CRISPR interference-based specific and efficient gene inactivation in the brain. Nat. Neurosci. 2018;21:447–454. doi: 10.1038/s41593-018-0077-5. [DOI] [PubMed] [Google Scholar]; Zheng, Y., Shen, W., Zhang, J., Yang, B., Liu, Y.N., Qi, H., Yu, X., Lu, S.Y., Chen, Y., Xu, Y.Z., et al. (2018). CRISPR interference-based specific and efficient gene inactivation in the brain. Nat. Neurosci. 21, 447-454. [DOI] [PubMed]
- 39.Lau C.H., Suh Y. In vivo genome editing in animals using AAV-CRISPR system: applications to translational research of human disease. F1000Res. 2017;6:2153. doi: 10.12688/f1000research.11243.1. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lau, C.H., and Suh, Y. (2017). In vivo genome editing in animals using AAV-CRISPR system: applications to translational research of human disease. F1000Res. 6, 2153. [DOI] [PMC free article] [PubMed]
- 40.Lau C.H., Suh Y. In vivo epigenome editing and transcriptional modulation using CRISPR technology. Transgenic Res. 2018;27:489–509. doi: 10.1007/s11248-018-0096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lau, C.H., and Suh, Y. (2018). In vivo epigenome editing and transcriptional modulation using CRISPR technology. Transgenic Res. 27, 489-509. [DOI] [PMC free article] [PubMed]
- 41.Thakore P.I., Kwon J.B., Nelson C.E., Rouse D.C., Gemberling M.P., Oliver M.L., Gersbach C.A. RNA-guided transcriptional silencing in vivo with S. aureus CRISPR-Cas9 repressors. Nat. Commun. 2018;9:1674. doi: 10.1038/s41467-018-04048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; Thakore, P.I., Kwon, J.B., Nelson, C.E., Rouse, D.C., Gemberling, M.P., Oliver, M.L., and Gersbach, C.A. (2018). RNA-guided transcriptional silencing in vivo with S. aureus CRISPR-Cas9 repressors. Nat. Commun. 9, 1674. [DOI] [PMC free article] [PubMed]
- 42.Chew W.L., Tabebordbar M., Cheng J.K., Mali P., Wu E.Y., Ng A.H., Zhu K., Wagers A.J., Church G.M. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chew, W.L., Tabebordbar, M., Cheng, J.K., Mali, P., Wu, E.Y., Ng, A.H., Zhu, K., Wagers, A.J., and Church, G.M. (2016). A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods 13, 868-874. [DOI] [PMC free article] [PubMed]
- 43.Moreno A.M., Fu X., Zhu J., Katrekar D., Shih Y.V., Marlett J., Cabotaje J., Tat J., Naughton J., Lisowski L. In situ gene therapy via AAV-CRISPR-Cas9-mediated targeted gene regulation. Mol. Ther. 2018;26:1818–1827. doi: 10.1016/j.ymthe.2018.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; Moreno, A.M., Fu, X., Zhu, J., Katrekar, D., Shih, Y.V., Marlett, J., Cabotaje, J., Tat, J., Naughton, J., Lisowski, L., et al. (2018). In situ gene therapy via AAV-CRISPR-Cas9-mediated targeted gene regulation. Mol. Ther. 26, 1818-1827. [DOI] [PMC free article] [PubMed]
- 44.Liao H.K., Hatanaka F., Araoka T., Reddy P., Wu M.Z., Sui Y., Yamauchi T., Sakurai M., O’Keefe D.D., Núñez-Delicado E. In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell. 2017;171:1495–1507.e15. doi: 10.1016/j.cell.2017.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liao, H.K., Hatanaka, F., Araoka, T., Reddy, P., Wu, M.Z., Sui, Y., Yamauchi, T., Sakurai, M., O’Keefe, D.D., Nuñez-Delicado, E., et al. (2017). In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell 171, 1495-1507.e15. [DOI] [PMC free article] [PubMed]
- 45.Zhou H., Liu J., Zhou C., Gao N., Rao Z., Li H., Hu X., Li C., Yao X., Shen X. In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nat. Neurosci. 2018;21:440–446. doi: 10.1038/s41593-017-0060-6. [DOI] [PubMed] [Google Scholar]; Zhou, H., Liu, J., Zhou, C., Gao, N., Rao, Z., Li, H., Hu, X., Li, C., Yao, X., Shen, X., et al. (2018). In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nat. Neurosci. 21, 440-446. [DOI] [PubMed]
- 46.Wangensteen K.J., Wang Y.J., Dou Z., Wang A.W., Mosleh-Shirazi E., Horlbeck M.A., Gilbert L.A., Weissman J.S., Berger S.L., Kaestner K.H. Combinatorial genetics in liver repopulation and carcinogenesis with a in vivo CRISPR activation platform. Hepatology. 2018;68:663–676. doi: 10.1002/hep.29626. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wangensteen, K.J., Wang, Y.J., Dou, Z., Wang, A.W., Mosleh-Shirazi, E., Horlbeck, M.A., Gilbert, L.A., Weissman, J.S., Berger, S.L., and Kaestner, K.H. (2018). Combinatorial genetics in liver repopulation and carcinogenesis with a in vivo CRISPR activation platform. Hepatology 68, 663-676. [DOI] [PMC free article] [PubMed]
- 47.Ma D., Peng S., Huang W., Cai Z., Xie Z. Rational design of mini-Cas9 for transcriptional activation. ACS Synth. Biol. 2018;7:978–985. doi: 10.1021/acssynbio.7b00404. [DOI] [PubMed] [Google Scholar]; Ma, D., Peng, S., Huang, W., Cai, Z., and Xie, Z. (2018). Rational design of mini-Cas9 for transcriptional activation. ACS Synth. Biol. 7, 978-985. [DOI] [PubMed]
- 48.Vora S., Cheng J., Xiao R., VanDusen N.J., Quintino L., Pu W.T., Vandenberghe L.H., Chavez A., Church G. Rational design of a compact CRISPR-Cas9 activator for AAV-mediated delivery. bioRxiv. 2018 [Google Scholar]; Vora, S., Cheng, J., Xiao, R., VanDusen, N.J., Quintino, L., Pu, W.T., Vandenberghe, L.H., Chavez, A., and Church, G. (2018). Rational design of a compact CRISPR-Cas9 activator for AAV-mediated delivery. bioRxiv, doi: 10.1101/007344.
- 49.Tse L.V., Klinc K.A., Madigan V.J., Castellanos Rivera R.M., Wells L.F., Havlik L.P., Smith J.K., Agbandje-McKenna M., Asokan A. Structure-guided evolution of antigenically distinct adeno-associated virus variants for immune evasion. Proc. Natl. Acad. Sci. USA. 2017;114:E4812–E4821. doi: 10.1073/pnas.1704766114. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tse, L.V., Klinc, K.A., Madigan, V.J., Castellanos Rivera, R.M., Wells, L.F., Havlik, L.P., Smith, J.K., Agbandje-McKenna, M., and Asokan, A. (2017). Structure-guided evolution of antigenically distinct adeno-associated virus variants for immune evasion. Proc. Natl. Acad. Sci. USA 114, E4812-E4821. [DOI] [PMC free article] [PubMed]
- 50.Miller E.B., Gurda-Whitaker B., Govindasamy L., McKenna R., Zolotukhin S., Muzyczka N., Agbandje-McKenna M. Production, purification and preliminary X-ray crystallographic studies of adeno-associated virus serotype 1. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006;62:1271–1274. doi: 10.1107/S1744309106048184. [DOI] [PMC free article] [PubMed] [Google Scholar]; Miller, E.B., Gurda-Whitaker, B., Govindasamy, L., McKenna, R., Zolotukhin, S., Muzyczka, N., and Agbandje-McKenna, M. (2006). Production, purification and preliminary X-ray crystallographic studies of adeno-associated virus serotype 1. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62, 1271-1274. [DOI] [PMC free article] [PubMed]
- 51.Huang L.Y., Patel A., Ng R., Miller E.B., Halder S., McKenna R., Asokan A., Agbandje-McKenna M. Characterization of the adeno-associated virus 1 and 6 sialic acid binding site. J. Virol. 2016;90:5219–5230. doi: 10.1128/JVI.00161-16. [DOI] [PMC free article] [PubMed] [Google Scholar]; Huang, L.Y., Patel, A., Ng, R., Miller, E.B., Halder, S., McKenna, R., Asokan, A., and Agbandje-McKenna, M. (2016). Characterization of the adeno-associated virus 1 and 6 sialic acid binding site. J. Virol. 90, 5219-5230. [DOI] [PMC free article] [PubMed]
- 52.Hordeaux J., Wang Q., Katz N., Buza E.L., Bell P., Wilson J.M. The neurotropic properties of AAV-PHP.B are limited to C57BL/6J mice. Mol. Ther. 2018;26:664–668. doi: 10.1016/j.ymthe.2018.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hordeaux, J., Wang, Q., Katz, N., Buza, E.L., Bell, P., and Wilson, J.M. (2018). The neurotropic properties of AAV-PHP.B are limited to C57BL/6J mice. Mol. Ther. 26, 664-668. [DOI] [PMC free article] [PubMed]
- 53.Matsuzaki Y., Konno A., Mochizuki R., Shinohara Y., Nitta K., Okada Y., Hirai H. Intravenous administration of the adeno-associated virus-PHP.B capsid fails to upregulate transduction efficiency in the marmoset brain. Neurosci. Lett. 2018;665:182–188. doi: 10.1016/j.neulet.2017.11.049. [DOI] [PubMed] [Google Scholar]; Matsuzaki, Y., Konno, A., Mochizuki, R., Shinohara, Y., Nitta, K., Okada, Y., and Hirai, H. (2018). Intravenous administration of the adeno-associated virus-PHP.B capsid fails to upregulate transduction efficiency in the marmoset brain. Neurosci. Lett. 665, 182-188. [DOI] [PubMed]
- 54.Shinohara Y., Konno A., Nitta K., Matsuzaki Y., Yasui H., Suwa J., Hiromura K., Hirai H. Effects of neutralizing antibody production on AAV-PHP.B-mediated transduction of the mouse central nervous system. Mol. Neurobiol. 2018 doi: 10.1007/s12035-018-1366-4. Published online October 5, 2018. [DOI] [PubMed] [Google Scholar]; Shinohara, Y., Konno, A., Nitta, K., Matsuzaki, Y., Yasui, H., Suwa, J., Hiromura, K., and Hirai, H. (2018). Effects of neutralizing antibody production on AAV-PHP.B-mediated transduction of the mouse central nervous system. Mol. Neurobiol. Published online October 5, 2018. 10.1007/s12035-018-1366-4. [DOI] [PubMed]
- 55.Cheng A.W., Wang H., Yang H., Shi L., Katz Y., Theunissen T.W., Rangarajan S., Shivalila C.S., Dadon D.B., Jaenisch R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23:1163–1171. doi: 10.1038/cr.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cheng, A.W., Wang, H., Yang, H., Shi, L., Katz, Y., Theunissen, T.W., Rangarajan, S., Shivalila, C.S., Dadon, D.B., and Jaenisch, R. (2013). Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 23, 1163-1171. [DOI] [PMC free article] [PubMed]
- 56.Kearns N.A., Genga R.M., Enuameh M.S., Garber M., Wolfe S.A., Maehr R. Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development. 2014;141:219–223. doi: 10.1242/dev.103341. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kearns, N.A., Genga, R.M., Enuameh, M.S., Garber, M., Wolfe, S.A., and Maehr, R. (2014). Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development 141, 219-223. [DOI] [PMC free article] [PubMed]
- 57.Cong L., Zhou R., Kuo Y.C., Cunniff M., Zhang F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat. Commun. 2012;3:968. doi: 10.1038/ncomms1962. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cong, L., Zhou, R., Kuo, Y.C., Cunniff, M., and Zhang, F. (2012). Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat. Commun. 3, 968. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.