

Abstract
Alternative splicing is commonly believed to be a major source of cellular protein diversity. However, although many thousands of alternatively spliced transcripts are routinely detected in RNAseq studies, reliable large-scale mass spectrometry-based proteomics studies identify only a small fraction of annotated alternative isoforms. The clearest finding from proteomics experiments is that most human genes have a single main protein isoform, while those alternative isoforms that are identified tend to be the most biologically plausible: those with the most cross-species conservation and those that do not compromise functional domains. Indeed, most alternative exons do not seem to be under selective pressure, suggesting that a large majority of predicted alternative transcripts may not even be translated into proteins.
Keywords: Alternative splicing, proteomics, RNAseq, homology, functional isoforms, dominant isoforms
One gene, one protein or one gene, many proteins?
Alternative splicing of messenger RNA produces a wide variety of differently spliced RNA transcripts that may be translated into diverse protein products. The presence of alternatively spliced transcripts is unequivocally supported by EST and cDNA sequence evidence [1], by microarray [2] and RNAseq data [3,4]. It has been estimated that most multi-exon human genes can undergo alternative splicing [5].
Manual genome annotation projects [1,6,7] have added substantial numbers of alternatively spliced transcripts to reference databases in recent years; the current version of the GENCODE human gene set (v24) [1] contains 82,141 CDS distinct protein-coding transcripts. Many estimates for the number of transcripts expressed in human cells are even higher; a recent large-scale RNAseq analysis [3] found multiple splice variants for 72% of annotated human genes, while another predicted that 205,000 transcripts had protein-coding potential, which would mean more than 10 variants per annotated gene [8].
The breadth of alternative splicing detectable at the transcript level has lead to claims that alternative protein isoforms could be the key to mammalian complexity [9]. How much of this alternative splicing is functional at the protein level is a long-standing open question of great importance for understanding eukaryotic biology [Box 1].
Box 1 – the role of alternative isoforms.
The functional role of alternative protein isoforms has been the subject of considerable debate. One strongly supported theory is that alternative splicing exists to allow the tissue-specific rewiring of protein-protein interaction networks [10,11]. This hypothesis is based on the tissue-specific expression of alternative transcripts, the loss of functional domains and the prevalence of disordered protein regions in alternative isoforms [12]. At the other extreme it has been suggested that stochastic models explain alternative splicing and that most alternative transcripts will not code for proteins [13].
Although there are 26,000 publications with the phrase “alternative splicing” in PubMed, very few alternative protein isoforms have well-characterised cellular function. The difficulty of determining molecular function means that even when alternative transcripts are found in tissues, what we know about their cellular role is incomplete [14,15]. A review of the role of more than 250 alternative isoforms [16] found that most alternative isoforms either sort into different cellular compartments or have a net negative effect on the function of the reference isoform. The review included 15 examples of modulation of function brought about by homologous exon substitution. In general the conclusion was that changes brought about by alterative splicing were hard to detect.
A major large-scale yeast 2-hybrid experiment with cloned alternative isoforms came to a contrasting conclusion. The authors found large functional differences between reference and alternative isoforms and showed that many alternative isoforms would indeed interact with different protein partners in vitro [17], in support of the tissue-specific rewiring hypothesis. This contrasting result was almost certainly due to the fact that 70% of the alternative isoforms that they expressed had lost more than 60 residues, greatly increasing the chances of affecting protein domains and impacting reference interactions.
Large-scale RNAseq experiments have shown that gene expression levels have strong tissue dependence that is conserved across both individuals [16] and different species [19]. However, alternative splicing levels are not conserved. For example, the GTex Consortium found that 84% of the variance between human tissues was due to gene expression, while splicing variation was much more pronounced between individuals [18], leading them to conclude that much alternative splicing is stochastic. Alternative exon usage also varies more between species [20,21] than it does between tissues. Meanwhile, Reyes et al. [22] found that a “sizeable minority” of exons, enriched in exons from 3’ and 5’ untranslated regions, had expression that was strongly tissue-specific across species.
Alternative splice isoforms
From the protein point of view there are two broad classes of alternative splicing: those that result in insertions or deletions (indels) and those that result in exon substitutions [figure 1]. The majority of annotated splice events involve the loss or gain of exons, or parts of exons [23]. These splice events generate alternative proteins with indels of widely different sizes as long as they do not cause a shift in the reading frame. Another common splice event is the substitution of one or more exons; this happens most often at the 3’ and 5’ ends of the transcripts [23,24]. Most of the resulting alternative proteins will have completely different N- or C-terminal sequences [figure 1]. However, a small proportion of these substituted exons have detectable homology, and mutually exclusive splicing of these exons [24,25] will result in alternative homologous protein sequences [figure 1].
Figure 1. Types of alternative isoforms.

This figure presents three types of alternative variants defined using the gene SLC25A3, a mitochondrial phosphate carrier protein. In each case we show the effect at the transcript level and at the protein level. (A) Homologous exons. Above, schema of variant SCL25A3–005, which is generated from variant SCL25A3–001 via the substitution of exon 2a (black) by exon 2b (orange). The differing protein sequences are shown in the alignment below the transcript level comparison. Middle, example spectra for the two peptides that identify the two different alternative isoforms. Below, the likely effect on protein structure (shown in two views) for the similar gene SLC25A4 (PDB code: 1okc); residues that differ between the two isoforms are shown as orange sticks. The change to the structure and function is likely to be comparatively subtle: no residues are lost and most of the changes are found on the outside of the pore. (B) Non-homologous substitution. Above, schema of variant SCL25A3–015, which is generated from variant SCL25A3–001 via the substitution of exon 3 (the longer alternative exon is in red). Below; the likely effect on protein structure shown in two different views; residues that would be lost in the alternative isoforms are shown in red. (C) Indels. Above, schema of variant SCL25A3–002, which is generated from variant SCL25A3–001 via the skipping of exon 6 (green). Below, the likely structural effect of this loss of 28 amino acids is shown in two different views; residues that would be lost in the alternative isoforms are shown in green. The deletion would remove the base of the pore and parts of two different trans-membrane helices meaning that the trans-membrane sections would have to completely refold. Images generated with the PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC.
Proteomics experiments find little evidence of alternatively spliced proteins
Recent advances have made tandem mass spectrometry (MS)-based proteomics experiments an increasingly important tool for validating the translation of protein coding genes [26,27] and large-scale mass spectroscopy experiments are now the main source of evidence of alternative splicing at the protein level.
We recently carried out a reanalysis of the peptides and spectra from eight large-scale experiments and databases [24]. In order to generate as reliable a set of peptides as possible we implemented a series of stringent filters [Box 2]. The rigorous quality controls allowed us to be confident that the vast majority of identified peptides and splice events were present in the individual studies. While relaxing quality controls would have allowed us to detect more alternative peptides, it would also have increased the proportion of false positive identifications based on marginally valid peptide spectrum evidence [Box 3].
Box 2 – stringent filters on large-scale proteomics data improves reliability .
The numbers of alternative splice events reported by large-scale proteomics experiments varies by many orders of magnitude [28–33]. However, those experiments with the highest numbers of alternative splice isoforms overestimate the number of alternative proteins [24]. Alternative isoforms should only be identified when peptides map to both sides of a splicing event (Figure I), but many studies report alternative isoforms when peptides identify just just one of the two splice isoforms.
Other large-scale proteomics experiments correctly identify splice isoforms [29,30], but then substantially underestimate the false positive rates of their experiments [34,35]. High false positive rates will artificially inflate the number of alternative isoforms detected; 11% of the theoretical peptides from the human reference annotation [1] map to alternative isoforms, so 1 in every 9 false positive peptide matches will “identify” a peptide that maps to an alternative isoform.
In our study we brought together peptides from eight large-scale studies. Combining many sources of data comes at a cost [26, 35], so it is vital to control false positive rates. We implemented a series of stringent filters on the eight individual experiments to remove as many false positive peptides as possible [24, 36].
Where two or more search engines were used to detect peptides we required that at least two search engines agreed on the peptide identified in each spectrum. All non-tryptic and semi-tryptic peptides were filtered out and missed cleavages were allowed only when they were also supported by one of the fully-cleaved tryptic peptides. Residues identified as leucine or isoleucine were allowed to map to both leucine and isoleucine in the GENCODE20 gene set. Peptides that mapped to more than one gene were removed.
We removed all peptides that were only identified in one of the eight studies. While some peptides that appear in a single study may be tissue-specific, or detected in just one study for technical reasons, peptides that are identified in just one experiment are also highly enriched in false positive identifications [35]. In this experiment we chose to sacrifice coverage for reliability. In order to detect a biological signal, we first had to remove as much noise as possible.
Further details can be found in two papers, Abascal et al [24] and Ezkurdia et al [36].
Box 2, Figure I. Identifying alternative splice events.
Part of an alignment between two splice isoforms of the gene EEF1D. Identified peptides are in red font and vertical lines mark the position of exon boundaries. The two regions that distinguish the isoforms are marked as A and B and the extent of the differences between the two regions are marked by a blue line. Region A differs by an indel; peptides that map to both sides of the indel confirm the translation of this splice isoform. By contrast, peptides map to just one side of the splice event in region B (a C-terminal substitution), so the translation of an alternative isoform with the alternative C terminus is not confirmed.

Box 3 – The difficulty of correctly identifying peptide spectrum matches.
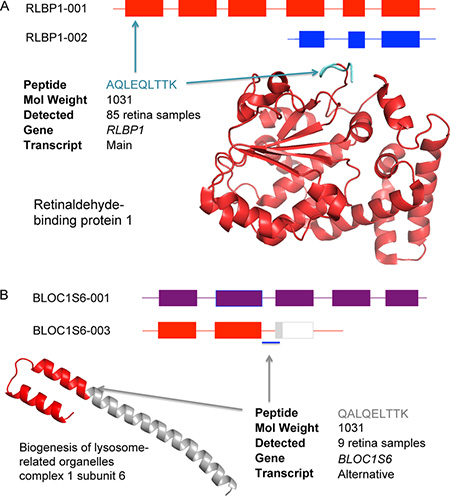
It is easy to misidentify peptides in proteomics experiments (Figure I). Here two similar peptides with the same amino acid composition and molecular weight (AQLEQLTTK and QALQELTTK) were identified from a single spectrum during a reanalysis of the Kim et al. [29] experiment (Figure I). This was not an isolated spectrum; many of the spectra from Kim analysis retina samples did not have enough information for search engines to distinguish one peptide from the other. While peptide AQLEQLTTK is from Retinaldehyde-binding protein 1 (RLBP1), a retina-specific protein for which 80% of the sequence was identified by peptides found in retina samples, the peptide QALQELTTK maps to BLOC1S6, a gene that the Kim analysis places almost entirely in hematopoietic cells. We did not identify QALQELTTK in any tissue other than retina.
The spectrum can only belong to one of the two peptides and AQLEQLTTK clearly fits the tissue specificity of the experiments much better than QALQELTTK. Further support for peptide AQLEQLTTK comes from the reliable PeptideAtlas database [24] where the peptide has been identified 51 times, all in retina-specific experiments. QALQELTTK has never previously been identified in PeptideAtlas.
Search engines performing the reanalysis identified AQLEQLTTK 85 times peptide QALQELTTK 9 times in spectra from retina samples. Given the tissue-specificity of BLOC1S6, this is 9 times too many, and to make matters worse the identification of QALQELTTK was determined to be significant in 3 cases. This is important because QALQELTTK would be used to identify an alternative isoform of BLOC1S6. In large-scale analyses researchers cannot carry out similar in-depth investigations into all peptides and spectra, so the BLOC1S6 alternative variant would be identified as being expressed in retina. This isoform was not detected in our pipeline because of the rigorous quality controls we had in place.
This case is based on the misidentification of a good spectrum with multiple assigned peaks. If the spectra are poor or if the peptide identifications are borderline, the chances of misidentification will multiply. Post-translational modifications complicate the identifications still further; if post-translational modifications are taken into account, correctly identifying peptide-spectrum matches becomes even more complex [24]. These problems complicate the identification of novel coding regions and alternative isoforms in large-scale proteomics studies [35] and are currently not being addressed.
Box 3, Figure I. Identifying two peptides from the same spectrum.
(A) The peptide AQLEQLTTK is from the main isoform of RLBP1 (Retinaldehyde-binding protein 1), a protein expressed in retina. The structure of RLBP1 has been resolved and is shown bottom right; the position of peptide AQLEQLTTK is marked in blue. (B) Peptide QALQELTTK supports the presence of an alternative isoform of BLOC1S6 that would cause the loss of the large coiled coil region shown in grey in the figure.
After applying these stringent filters we still found peptides for the majority of protein coding genes (12,716), but few genes (246) had reliable evidence for more than one isoform. This strongly suggests that alternative variants are not abundant at the protein level. The low number of protein splice isoforms is in stark contrast to the abundance of alternative transcripts in microarray and RNAseq experiments and is especially surprising in light of the fact that the eight large-scale experiments interrogated more than 100 different tissues, cell lines and developmental stages [24].
We carried out simulations to test whether the number was smaller than expected [Box 4]. Simulations that assumed that all isoforms in a gene were equally likely detected alternative isoforms for over 3,500 genes, while we found alternative splicing for more than 1,250 genes in simulations where reference isoforms were 50 times more abundant.
Box 4 – Estimating the expected number of alternative splice isoforms.
We estimated the numbers of alternative splice isoforms we would expect to detect in the experiments via simulations. For the first simulation we assumed that all transcripts were expressed equally. We carried out an in silico lysis of the GENCODE20 database [1] to produce tryptic peptides and selected at random the same number of peptides for each gene as were identified in the experiments. We mapped these peptides to the database, repeated the experiment 100 times and took the average values.
If we had only used tryptic peptides in our analysis we would have found alternative splicing for 226 genes instead of 246 (20 splice isoforms were identified via missed cleavages), and 14 genes would have had evidence of two or more alternative isoforms.
In contrast the numbers from the in silico analysis were substantially larger. We identified alternative splicing for 3,508 genes (15.5 times greater than the experiments), and two or more alternative isoforms for 937 genes (67 times greater than the experiments). This clearly suggests that one protein isoform per gene is dominant.
We repeated the experiment simulating a model where one isoform had 50-fold dominance over the other isoforms. We generated 50 times more peptides for the principal isoform of each gene via the in silico lysis (principal isoforms taken from the APPRIS database [43]) and repeated the simulation with this larger database. This time the peptides identified 1,289 genes with evidence of alternative isoforms and 152 genes with two or more alternative isoforms. The numbers from the 50-fold dominant model are still much larger than the experiments, implying that alternative isoforms are expressed at a much lower level than the main isoforms. The simulations demonstrate that we ought to detect many more alternative isoforms than we did, so the lack of alternative isoforms in the experiments is not solely the result of poor coverage.
In fact the proteomics experiments also find many fewer alternative peptides than expected. While more than 11% of the tryptic peptides from GENCODE20 map to alternative isoforms, alternative peptides are just 0.376% of the peptides identified in proteomics experiments.
Almost all coding genes seem to have a main protein isoform
The question of whether or not genes have dominant variants has become increasingly important as the numbers of annotated transcripts has grown. Large-scale transcriptomics studies [37–39] have shown that genes have dominant transcripts, even if a proportion of them are non-coding or subject to nonsense-mediated decay [37]. Most genes have a single dominant transcript across all cell lines [37,38], but as many as a third of genes have tissue-dependent dominant transcripts [39].
By contrast, proteomics studies strongly suggest that most genes have a single main protein isoform; 99.63% of the peptides we detected mapped to just one isoform in each gene [24]. This evidence motivated us to determine a “main” experimental isoform. We summed up the peptides detected for each isoform across the eight studies and the unique CDS with the most peptides was the main isoform. We determined a main isoform for 5,011 of the 12,716 genes and compared these to known reference variants [36].
“Dominant” RNAseq transcripts are those that are expressed at least 5-fold more than other transcripts across all tissues or cell lines [37]. We found that the agreement between dominant variants from the two experimental procedures was just 77–78% [fig 2]. The main reason for the disagreement is likely to be technical rather than biological: transcript reconstruction from short RNAseq reads is a complex problem and algorithms for reconstructing and quantifying full-length mRNA transcripts are inaccurate [40].
Figure 2. Coincidence between main proteomics isoforms and other reference isoforms.

The percentage of genes in which there was agreement between the reference isoform for a gene and the main proteomics isoform calculated from the proteomics experiments [36]. The comparison was made over all 5,011 genes from the same proteomics study for the longest isoform, over a subset of 3,331 genes with CCDS unique isoforms [41] for the CCDS comparison, over a subset of 4,186 genes with principal isoforms for the APPRIS comparison [43] and over a subset of 1,038 genes with five-fold dominant transcripts across all tissues for the RNAseq comparison [37]. The Highest Connected Isoform comparison was made using data from the paper that introduced the method [42]. A random selection of isoforms would have agreed with the main proteomics isoform 46% of the time.
The longest isoform is chosen as the reference isoform for technical reasons in practically all studies and databases. Although it has no biological basis, the longest isoform still agreed with the main experimental proteomics isoform across 89.6% of genes [fig 2], suggesting that this is a reasonable but far from perfect strategy.
Consensus coding DNA sequence (CCDS) variants [41] are transcript models agreed on by independent teams of manual annotators using genomic evidence including the presence of cDNAs. When there is just one CCDS variant per gene these can be used as a proxy for the reference variant. The agreement between the main experimental isoforms and unique CCDS variants was an impressive 98.6%.
In addition to the experiment-based methods, there are also two recently developed computational methods that predict reference isoforms. Highest Connected Isoforms [42] predict reference isoforms based on transcript expression data, amino acid composition, and protein-protein docking. APPRIS [43] determines “principal” isoforms using cross-species conservation and the conservation of protein structure and functional features. The agreement between the Highest Connected Isoforms and the main experimental isoforms was just 78% [fig 2]. By contrast, the APPRIS principal isoforms coincided with the main experimental isoform over 97.6% of comparable genes.
Remarkably, the agreement between the main proteomics isoform, the APPRIS principal isoforms and the unique CCDS variants was almost perfect (99.4%) over the 3,015 genes where all three methods had a single reference isoform [37]. The fact that three entirely orthogonal sources of reference isoforms have such an outstanding agreement highlights the biological significance of the results from the proteomics experiments and significantly reinforces the likelihood that the main proteomics isoform is the dominant protein isoform in the cell.
Detected splice events have comparatively subtle effects on the protein
Standard MS proteomics experiments only identify a proportion of the peptide ions present in protease digests [44]. The peptide coverage for highly expressed proteins is rarely complete and proteins expressed in low quantities are often not detected at all [44]. This means that alternative splice isoforms present in low quantities in the cell may not be picked up by proteomics experiments, which could partly explain why so few alternative isoforms are detected in proteomics experiments.
It is also possible that the low numbers of alternative peptides is in part due to limited sampling depth. Although the combined large-scale experiments covered more than 100 tissues and developmental stages, the low coverage typical of proteomics experiments would make tissue-specific splice isoforms harder to detect.
Despite these technical issues, the patterns evident in the set of alternative isoforms identified in the proteomics experiments clearly show that some alternative variants are more important than others. These patterns are further strong indications that limited sampling depth and low coverage are not the only reason for not finding larger numbers of alternative peptides [Box 4].
Alternative splice isoforms identified in the experiments were highly enriched in duplicated homologous exon substitutions, both in the human proteomics experiments and in parallel analyses carried out with mouse [24]. Sixty of the 282 events that were detected in the human study were generated from homologous exons, a number that was substantially greater than expected (21% of identifiable homologous exon substitutions were identified in the proteomics analysis, compared to just 0.01% of other annotated splice events). Analysis of other studies backs this up: proteomics studies detect a high proportion of alternative isoforms generated by swapping one homologous exon for another [28–31].
There was evidence for all 60 homologous substitutions in the genomes of bony fish, suggesting that all these splice events had ancient origins, evolving at least 460 millions years ago. While alternative isoforms generated from homologous exons were highly conserved, we found that just 19% of alternative exons annotated in the human reference set were conserved in mouse [24].
These homologous exon splice events will hav e only subtle effects on structure and function (fig 3). One way of measuring the effect on structure and function is to analyse the composition of conserved Pfam functional domains [46] in the predicted protein product. Alternative isoforms identified in the proteomics experiments were highly enriched in splice events that did not affect Pfam functional domain composition. Only 15% of the alternative splice events would damage or cause the loss of a Pfam domain, whereas 68% of the annotated alternative splice events in CDS regions would break or cause the loss of one or more Pfam domains.
Figure 3. Solved crystal structures for two pairs of MS-detected alternative isoforms.

Solved protein structures for alternative isoforms that differ by substitution of homologous exons. In each figure one isoform is coloured orange and the other blue. The region coded by the homologous exons is shown in light blue and light orange. (A) Pyruvate kinase isoforms M1 and M2 [53], those residues that differ in the alternative isoform are shown as sticks. The two structures (PDB codes 1srf and 1srd) are practically identical, the largest differences are in a loop from the substituted region (bottom right) and in the loop region when the M2 isoform binds the fructose biphosphate substrate and the M1 isoform does not (top right). (B) “central” and “peripheral” isoforms of ketohexokinse [54]. Both isoforms bind the substrate fructose; the homologous exon substitution affects the substrate-binding site; the two residues that differ in the site are shown as blue and grey sticks. The peripheral isoform does not bind fructose as strongly as the central isoform; the change in binding residues may mean that the peripheral isoform has a different substrate.
The preservation of functional domains, the enrichment in homologous exon substitutions and the cross species conservation, clearly demonstrate that alternative isoforms with the most conservative changes tend to be the most prevalent in the cell.
Most alternative exons are not under selective pressure
Most annotated alternative isoforms are not supported by proteomics evidence and have limited cross-species conservation. However, these isoforms may be lineage-specific innovations [10]. Variation within human populations could provide support for this hypothesis; if recently evolved exons code for functionally relevant proteins, then they should be evolving under purifying selection.
A recent analysis of data from healthy patients in the 1,000 genomes project [45] demonstrated that alternative exons from the reference annotation had proportionally more predicted high impact variants than the APPRIS principal isoforms [48]. This result indicates that alternative exons are under weaker purifying selection than the APPRIS principal isoforms.
Our own in-house investigation of the same data supports these results. Exons from APPRIS principal isoforms have a substantially lower proportion of high Impact variants than exons from alternative isoforms (Fig 4). Not only are alternative exons evolving under weaker purifying selection, but the patterns observed for rare and common variants suggest that most alternative exons evolve neutrally; even though alternative sites represented only 5% of our data, they contributed 29% of the high impact variants across all allele frequencies and 57% of high impact variants for the most common allele frequencies.
Figure 4. Genome-wide distribution of sequence variants in principal and alternative isoforms.

The ratio of non-synonymous to synonymous variants (A) and the percentage of high-impact variants (B) shown for three sets of protein-coding sites: Alternative, those sites that fall inside exons belonging exclusively to alternative variants (895,887 sites in total); APPRIS, those sites from exons that code for APPRIS main isoforms [43] and not for alternative isoforms (4,732,523 sites); and Intersection, those sites that fall inside exons that code for both alternative variants and APPRIS main isoforms (10,792,735 sites). Each ratio was calculated both for rare and common allele frequencies identified from phase3 of the 1000 Genomes project [47] (the boundary between rare and common was set at an allele count of 25, corresponding to an allele frequency of 0.005). High impact variants defined by VEP [55] were splice acceptor variants, splice donor variants, stop gains, stop losses and frameshift variants.
The fact that exons from alternative isoforms have a substantially greater proportion of high impact and missense variants shows that most alternative isoforms are not under selective pressure. This underscores the importance of the main protein isoforms and suggests that most alternative isoforms, if translated, will have little or no functional relevance as proteins.
Concluding remarks
Alternative splicing is well documented at the transcript level, and microarray and RNAseq experiments routinely detect evidence for many thousands of splice variants. However, large-scale proteomics experiments identify few alternative isoforms. The gap between the numbers of alternative variants detected in large-scale transcriptomics experiments and proteomics analyses is real and is difficult to explain away as a purely technical phenomenon. While alternative splicing clearly does contribute to the cellular proteome, the proteomics evidence indicates that it is not as widespread a phenomenon as suggested by transcript data. In particular the popular view that alternative splicing can somehow compensate for the perceived lack of complexity in the human proteome [9,17] is manifestly wrong.
Those isoforms detected in proteomics experiments are highly conserved and significantly enriched in mutually exclusively spliced homologous exons and subtle splice events that do not disrupt functional domains. This is highly suggestive of a model in which splice isoforms with small variations can more easily gain a functional role in the cell, and in which those alternative isoforms with changes leading to loss of structure and function (such as the damage or loss of a functional domain) are less likely to acquire functional importance.
What happens to alternative transcripts that are not translated in detectable quantities is not clear. Some may be expressed in small quantities, in limited tissues or under special circumstances, some may be regulated by cellular quality control pathways [49,50], ensuring that isoforms with damaged domains are not present in the cell, and some may have functions other than generating a protein product [51]. Resolving the fate of these missing isoforms will be of great importance to help understand the cellular machinery.
The agreement between the main experimental proteomics isoform, the CCDS variants from genomic information and the APPRIS principal isoforms from conservation demonstrates that the vast majority of genes have a single dominant splice isoform. The fact that the main isoforms detected at the protein level agree with the APPRIS principal isoforms is an important detail, because it means that dominant cellular isoforms can be predicted for any well annotated genome.
The importance of this main cellular isoform, especially in large-scale experiments and biomedical applications, can be appreciated from the remarkable results from the variant calling experiments (Figure 4). These results show that most alternative exons are evolving neutrally, suggesting that most alternative splice events are not evolutionary innovations. Of course, this also suggests that many alternative transcripts will not be translated into functional proteins.
This has important practical implications for predicting the effect of genetic variants. High-impact variants are usually the most interesting in clinical studies, but they are also the variants most enriched in false positives [52] and those that most frequently associate to alternative transcripts. Variant effects should be predicted for main isoforms rather than, as frequently done, choosing the transcript with the highest predicted impact.
The results from large-scale proteomics experiments [24,36] are in line with evidence from cross-species conservation [24], human population variation studies [48] and investigations into the relative effect of gene expression and alternative splicing [18,22]. Gene expression levels, not alternative splicing, seem to be the key to tissue specificity [18]. While a small number of alternative isoforms are conserved across species, have strong tissue dependence and are translated in detectable quantities, most have variable tissue specificities and appear to be evolving neutrally. This suggests that most annotated alternative variants are unlikely to have a functional cellular role as proteins.
Box 4 – genes with strong evidence for alternative splice isoforms.
Analysis of the alternative isoforms identified in large-scale proteomics experiments [24] shows that many of them are well characterized in the literature, appear in certain cellular processes, are conserved in distant species or are generated from small changes in amino acid sequence. Many of the splice isoforms are detected across multiple proteomics studies and/ or in different species.
High-throughput proteomics studies would be expected to detect peptide evidence for specific splice isoforms from the following genes. A proteomics study that did not detect splice isoforms for a high proportion of these genes would be exceptional.
Well-studied splice variants:
Prelamin-A/C (LMNA*), pyruvate kinase (PKM*), actinins (ACTN1*,ACTN4*), Microtubule-associated protein tau (MAPT), dystrophin (DMD), cyclin-dependent kinase inhibitor 2A (CDKN2A)
The most highly expressed splice variants:
LAP2alpha (TMPO), Inhibitor of nuclear factor kappa-B kinase-interacting protein (IKBIP*), plectin (PLEC1), tropomyosins (TPM1*1,TPM3*1,TPM4*), pyruvate kinase (PKM*), glutaminase kidney isoform (GLS), fibulin 1 (FBLN1*)
Highly conserved splice variants:
plasma membrane calcium-transporting ATPases (ATP2B1*,ATP2B4*), mannan-binding lectin serine protease 1 (MASP1*), LIM domain-binding protein 3 (LDB3*1)
Splice isoforms that swap one set of Pfam domains for another:
nebulin (NEBL), homeobox protein cut-like 1 (CUX1), dystonin (DST)
Splice variants linked to disease:
cyclin-dependent kinase inhibitor 2A (CDKN2A), annexin A6 (ANXA6), calumenin (CALU*), cell division control protein 42 homolog (CDC42*), pyruvate kinase (PKM*)
Heart and skeletal muscle specific splice isoforms:
LIM domain-binding protein 3 (LDB3)*1, tropomyosins (TPM1*1, TPM2*1), titin (TTN*), PDZ and LIM domain protein 5 (PDLIM5), PDZ and LIM domain protein 3 (PDLIM3*),
Splicing factors:
splicing factor 1 (SF1), heterogeneous nuclear ribonucleoproteins (HNRNPC, HNRNPD, HNRNPK, HNRNPR), polypyrimidine tract-binding protein 2 (PTBP2), poly(U)-binding-splicing factor PUF60 (PUF60)
Splicing variants generated from tandem alternative splice sites [43]:
drebrin-like protein (DBNL), cellular nucleic acid-binding protein (CNBP), translation initiation factor eIF-2B subunit delta (EIF2B4), heterogeneous nuclear ribonucleoprotein (HNRNPR)
* - splice variant generated from homologous exons
1 – more than one distinct variant detected for this gene
Acknowledgements
The authors would like to thank Iakes Ezkurdia for his input on the paper. This work was supported by the National Institutes of Health (NIH, grant number U41 HG007234).
References
- 1.Harrow J et al. (2012) GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 760–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sánchez-Pla A et al. (2012) Transcriptomics: mRNA and alternative splicing. J Neuroimmunol. 248, 23–31 [DOI] [PubMed] [Google Scholar]
- 3.Uhlén M et al. (2015) Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 4.Juntawong P et al. (2012) Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A 111:E203–E212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mollet IG et al. (2010) Unconstrained mining of transcript data reveals increased alternative splicing complexity in the human transcriptome. Nucleic Acids Res. 38, 4740–4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pruitt KD et al. (2013) RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 42, D756–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pundir S et al. (2015) Searching and navigating UniProt databases. Curr. Protoc. Bioinformatics 50, 1.27.1–1.27.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Z et al. (2015) Revealing missing human protein isoforms based on ab initio prediction, RNA-seq and proteomics. Sci. Rep 5, 10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nilsen TW and Graveley BR (2010) Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buljan M et al. (2012) Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mole. Cell 46, 871–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellis JD et al. (2012) Tissue-specific alternative splicing remodels protein-protein interaction networks. Mole. Cell 46, 884–892 [DOI] [PubMed] [Google Scholar]
- 12.Colak R et al. (2013) Distinct types of disorder in the human proteome: functional implications for alternative splicing. PLoS Comput. Biol 9, e1003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melamud E and Moult J (2009) Stochastic noise in splicing machinery. Nucleic Acids Res. 37, 4873–4886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weeland CJ et al. (2015) Insights into alternative splicing of sarcomeric genes in the heart. J. Mol. Cell. Cardiol 81, 107–113 [DOI] [PubMed] [Google Scholar]
- 15.Foley KS and Young PW (2013) An analysis of splicing, actin-binding properties, heterodimerization and molecular interactions of the non-muscle α-actinins. Biochem J. 452, 477–488 [DOI] [PubMed] [Google Scholar]
- 16.Kelemen O et al. (2013) Function of alternative splicing. Gene 514, 1–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X et al. (2016) Widespread expansion of protein interaction capabilities by alternative splicing. Cell 164, 805–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melé M et al. (2015) Human genomics. The human transcriptome across tissues and individuals. Science 348, 660–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brawand D et al. (2011) The evolution of gene expression levels in mammalian organs. Nature 478, 343–348 [DOI] [PubMed] [Google Scholar]
- 20.Merkin J et al. (2012) Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 338, 1593–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbosa-Morais NL et al. (2012) The evolutionary landscape of alternative splicing in vertebrate species. Science 338, 1587–1593 [DOI] [PubMed] [Google Scholar]
- 22.Reyes A et al. (2013) Drift and conservation of differential exon usage across tissues in primate species. Proc. Natl. Acad. Sci. U.S.A 110, 15377–15382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mudge JM et al. (2011) The origins, evolution, and functional potential of alternative splicing in vertebrates. Mol. Biol. Evol 28, 2949–2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abascal F et al. (2015) Alternatively spliced homologous exons have ancient origins and are highly expressed at the protein level. PLoS Comput. Biol 11, e1004325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kondrashov FA and Koonin EV (2001) Origin of alternative splicing by tandem exon duplication. Hum. Mol. Genet 10, 2661–2669 [DOI] [PubMed] [Google Scholar]
- 26.Deutsch EW et al. (2012) State of the human proteome in 2014/2015 as viewed through PeptideAtlas: enhancing accuracy and coverage through the AtlasProphet. J. Proteome Res 14, 3461–3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ezkurdia I et al. (2014) Multiple evidence strands suggest that there may be as few as 19 000 human protein-coding genes. Hum. Mol. Genet 23, 5866–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ezkurdia I et al. (2012) Comparative proteomics reveals a significant bias toward alternative protein isoforms with conserved structure and function. Mol. Biol. Evol 29, 2265–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim MS et al. (2014) A draft map of the human proteome. Nature 509, 575–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilhelm M et al. (2014) Mass-spectrometry-based draft of the human proteome. Nature 509, 582–587. [DOI] [PubMed] [Google Scholar]
- 31.Tay AP et al. (2012) Proteomic validation of transcript isoforms, including those assembled from RNA-Seq data. J. Proteome Res 14, 3541–3554 [DOI] [PubMed] [Google Scholar]
- 32.Chang KY and Muddiman DC (2011) Identification of alternative splice variants in Aspergillus flavus through comparison of multiple tandem MS search algorithms. BMC Genomics 12, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ly T et al. (2014) A proteomic chronology of gene expression through the cell cycle in human myeloid leukemia cells. Elife 3, e01630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ezkurdia I et al. (2014) Analyzing the first drafts of the human proteome. J. Proteome Res 13, 3854–3855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ezkurdia I et al. (2015) The potential clinical impact of the release of two drafts of the human proteome. Expert Rev. Proteomics. 23, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ezkurdia I et al. (2015) Most highly expressed protein-coding genes have a single dominant isoform. J. Proteome Res 214, 1880–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gonzàlez-Porta M et al. (2013) Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome Biol. 14, R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taneri B et al. (2011) Distribution of alternatively spliced transcript isoforms within human and mouse transcriptomes. J OMICS Res 14, 1–5 [Google Scholar]
- 39.Djebali S et al. (2012) Landscape of transcription in human cells. Nature 14, 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayer KE et al. (2015) Benchmark analysis of algorithms for determining and quantifying full-length mRNA splice forms from RNA-seq data. Bioinformatics 31, 3938–3945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harte RA et al. (2012) Tracking and coordinating an international curation effort for the CCDS Project. Database 2012, bas008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li HD, et al. (2015) Functional networks of highest-connected splice isoforms: from the chromosome 17 Human Proteome Project. J. Proteome Res 14, 3484–3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez JM et al. (2013) APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res. 41, D110–D117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gstaiger M and Aebersold R (2009) Applying mass spectrometry-based proteomics to genetics, genomics and network biology. Nat. Review Genet 10, 617. [DOI] [PubMed] [Google Scholar]
- 45.Hiller M, et al. (2006) TassDB: a database of alternative tandem splice sites. Nucleic Acids Res. 35, D188–D192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Punta M et al. (2012) The Pfam protein families database. Nucleic Acids Res. 40, D290–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.1000 Genomes Project Consortium. (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu T and Lin K (2015) The distribution pattern of genetic variation in the transcript isoforms of the alternatively spliced protein-coding genes in the human genome. Mol. Biosyst 11, 1378–1388 [DOI] [PubMed] [Google Scholar]
- 49.Lykke-Andersen J and Bennett EJ (2014) Protecting the proteome: Eukaryotic cotranslational quality control pathways. J. Cell Biol 204, 467–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruggiano A et al. (2014) Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol 204, 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lareau LF and Brenner SE (2015) Regulation of splicing factors by alternative splicing and NMD is conserved between kingdoms yet evolutionarily flexible. Mol. Biol. Evol 32, 1072–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacArthur DG et al. (2012) A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mirtschink P et al. (2015) HIF-driven SF3B1 induces KHK-C to enforce fructolysis and heart disease. Nature 522, 444–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Israelsen WJ and Vander Heiden MG (2015) Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol 43, 43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yates A et al. (2016) Ensembl 2016. Nucleic Acids Res. 44, D710–D716 [DOI] [PMC free article] [PubMed] [Google Scholar]