

Abstract
In mammals, the iron masterswitch hepcidin efficiently controls iron recycling by the macrophage-liver axis but the exact interplay between macrophages and hepatocytes remains poorly understood. We here study hepcidin response during macrophage differentiation as well as the macrophage-hepatocyte crosstalk and its subsequent effects on hepatocyte hepcidin using an in vitro co-culture model that mimics the physiological liver microenvironment. We show that macrophage differentiation strongly induces hepcidin by 60-fold both in THP1 macrophages and primary isolated monocyte-derived macrophages. Removal of H2O2 by catalase or inhibition of NOX2 efficiently blocked hepcidin induction. After differentiation, macrophage hepcidin accounted for 10% of total hepatocyte hepcidin and did not respond to low oxygen levels. In contrast, co-culture of differentiated macrophages with Huh7 cells significantly induced hepatocyte hepcidin, which was further potentiated under low oxygen levels. Hepatocyte hepcidin was also upregulated when Huh7 cells were solely exposed to macrophage-conditioned hypoxic medium. A cytokine screen identified macrophage secreted IL-1β as major inducer of hepcidin in hepatocytes. In confirmation, treatment of Huh7 cells with the IL-1 receptor antagonist (anakinra) completely blunted macrophage-mediated hepcidin transcription in hepatocytes. Finally, detailed analysis of potentially involved signaling pathways points toward STAT3 and CEBPδ-mediated hepcidin induction independent of IL-6. In conclusion, our study demonstrates a strong NOX2-mediated hepcidin induction during macrophage differentiation. These differentiated macrophages are able to efficiently induce hepatocyte hepcidin mainly through secretion of IL-1β. Our data highlight a hitherto unrecognized role of macrophage-hepatocyte crosstalk for a joint and oxygen-dependent hepcidin production through STAT3 and CEBPδ.
Keywords: Iron metabolism/hepcidin, Hypoxia, Hydrogen peroxide, NADPH oxidase, Cytokines, STAT3
Abbreviations: β2mg, β2-microglobulin; BMP, Bone morphogenetic protein; CAT, Catalase; CEBPδ, CCAAT/enhancer-binding protein delta; DHFDA, dichlorodihydrofluorescein diacetate; FPN1, ferroportin1; IL-1β, Interleukin 1 beta; IL-6, Interleukin 6; MCHM, macrophage conditioned hypoxic medium; MDM, Monocyte-derived macrophages; Mo, Monocytes; NOX, NADPH oxidase; PKC, Protein kinase C; PMA, phorbol myristate acetate; PBMC, peripheral blood mononuclear cells; ROS, Reactive oxygen species; STAT3, Signal transducer and activator of transcription 3
Graphical abstract
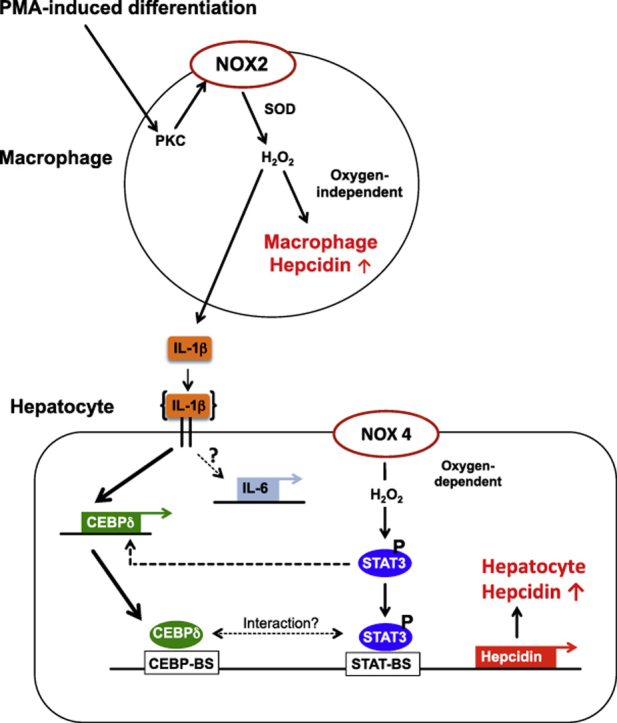
Highlights
-
•
Hepcidin is strongly induced during NOX2-mediated macrophage differentiation in a H2O2-dependent manner.
-
•
In contrast to hepatocyte hepcidin, macrophage hepcidin transcription is not modulated by low O2 level.
-
•
Macrophage released IL1-β strongly induces hepatocyte hepcidin via STAT3 signaling.
-
•
IL1-β mediated hepatocyte hepcidin induction is independent of IL-6.
-
•
Despite the mandatory requirement of STAT3, CEBPδ also involved in IL1-β induced hepatocyte hepcidin transcription.
1. Introduction
Hepcidin is the master regulator of systemic iron homeostasis in mammals [1,2]. It is mainly expressed in hepatocytes and secreted from the liver. Although other tissues can express hepcidin at low levels, such as heart, kidney, brain it is also found in activated macrophages [3,4]. It was first described as 25 amino acid antimicrobial peptide and family member of the defensins in ultrafiltrates of human blood and urine. Circulating hepcidin binds to the iron exporter ferroportin 1 (FPN1) on the cell surface of duodenal enterocytes, macrophages and hepatocytes leading to its internalization and subsequent degradation thereby reducing iron absorption, iron recycling and mobilization from liver iron stores [[5], [6], [7]].
Corresponding to its role as antimicrobial peptide, hepcidin is strongly induced by inflammation namely by IL-6 but also by microbial molecules such as lipopolysaccharide (LPS) rapidly leading to a decrease of serum iron. These are important evolutionary conserved mechanisms to induce hepcidin during infections causing the so-called anemia of chronic disease and limiting iron availability for e.g. bacterial growth [8]. Beginning with technical problems to accurately detect hepcidin in human samples, several methods of quantifying mature hepcidin-25 have been developed. Different kind of strategies have been applied for hepcidin quantification; MS based (including LC-MS/MS, SELDI-TOF and MALDI-TOF), based on immunochemical principles (ELISA and radioimmunoassay) or by using PCR [9]. In addition, it is now established and widely accepted that hepcidin mRNA levels correspond well to peptide levels [10].
Macrophages play a major role in iron homeostasis by recycling hemoglobin bound iron (approximately 20–25 mg per day) from senescent or damaged erythrocytes in the liver, spleen and bone marrow but can additionally take up hemoglobin and heme from the circulatory system [11]. Furthermore, liver resident macrophages, the Kupffer cells may also play a pivotal role in the uptake and clearance of damaged hepatocytes including the recycling of iron stored in these cells [12]. Thereby macrophages provide the majority of iron for new incorporation into heme in red blood cell hemoglobin in the bone marrow. Kupffer cells represent a cellular key component of the human liver in mediating inflammatory and innate immune responses and are the first sensors of initial liver injury [13]. These cells are activated in response to hepatocyte injury and inflammation e.g. induced by alcohol and have been shown to further recruit pro-inflammatory monocytes, which mature into monocyte-derived macrophages secreting even more inflammatory mediators (such as IL-6, IL-1β, TNFα and reactive oxygen species (ROS)) thereby eventually aggravating liver damage [14]. These pro-inflammatory mediators and cytokines directly influence hepatocytes and other non-parenchymal cells thereby regulating hepatic hepcidin secretion and changing systemic iron homeostasis as well as storage in the liver.
Hepcidin expression has been shown to increase with elevated hepatic iron levels [15]; hepatic iron induces BMP6 expression, which stimulates hepcidin transcription via BMP-responsive elements (BMP-RE) 1 and 2 in the promoter region. In contrast, inflammatory cytokines such as IL-6 and inflammatory conditions such as hypoxia and H2O2 upregulate hepcidin expression through activation of the signal transducer and activator of transcription (STAT) 3 binding site (STAT-BS) in the hepcidin gene [[16], [17], [18]].
In view of many trans-activating factors influencing hepcidin regulation, we investigated in detail the cellular crosstalk between macrophages and hepatocytes in regulating hepatic hepcidin under physiological low oxygen levels and cell ratios. We first show a strong and NOX2-dependent induction of macrophage hepcidin during the differentiation process. Moreover, macrophage differentiation causes hepatocyte hepcidin induction mainly through IL-1β involving STAT3 and CEBPδ mediated mechanisms.
2. Materials and methods
2.1. Cell culture
Huh7 cells from the Japanese Cancer Research Resources Bank (JCRB, Tokyo, Japan) were grown under standard conditions using Dulbecco's modified Eagle medium (Sigma-Aldrich, Taufkirchen, Germany), 25 mM glucose and 10% fetal calf serum under 21% oxygen and 5% CO2 [18]. The immortalized human monocyte THP-1 cells from the American Type Culture Collection (ATCC, Manassas, VA, USA) were grown in RPMI-1640 medium with 25 mM glucose (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum. THP-1 cells were seeded in 12-well plates and treated with phorbol myristate acetate (PMA) at 100 ng/ml for 24 h to induce differentiation. After differentiation, cells were washed and incubated in fresh media for 24 h before experimentation.
2.2. Isolation of blood monocytes and differentiation into MDM
Human primary monocytes were isolated and purified from peripheral blood mononuclear cells (PBMCs) of buffy coats obtained from healthy blood donors using a Biocoll gradient (Biochrom, Berlin, Germany). After isolation, PBMCs were resuspended in RPMI medium and seeded at a density of 2 × 106 cells/well in non-coated 12-well plates. The monocytes were obtained by selective adherence after 1 h of incubation and cultured over 7 days in RPMI medium supplemented with 10% FBS to allow the differentiation into monocyte-derived macrophages (MDM).
2.3. Co-culture, treatments and hypoxia studies
THP-1 monocytes were seeded for differentiation with PMA at 0.2 × 105 or 0.5 × 105 cells/well in 12-well plates for a ratio hepatocyte to macrophage of 10:1 or 4:1, respectively. After 48 h of differentiation, Huh7 cells were added to macrophages at a density of 1.3 × 105 cells/well and incubated over night for attachment. The co-culture was conditioned to low O2 levels by keeping the culture plates in a sealed chamber flushed with 5% O2, 5% CO2 and 90% N2 or with 1% O2, 5% CO2 and 94% N2 at 37 °C for 24 h. Aiming at studying the effects of macrophage-conditioned medium, differentiated THP-1 macrophages were conditioned for 24 h to normoxia or to hypoxia using the sealed chamber flushed with 1% O2 [17,19]. Huh7 cells were exposed to this macrophage-conditioned medium and kept under 21% or 1% O2 for 24 h. For inhibition experiments, IL-1β receptor antagonist (Anakinra - Kineret®) or IL-8 receptor inhibitor (Reparixin) was added during the incubation of Huh7 cells with the conditioned medium at 0.5 μg/ml or 10 μg/ml respectively.
2.4. Chemicals and reagents
PMA (100 ng/ml), VAS2870 (5 μM), luminol, horseradish peroxidase and catalase were all purchased from Sigma-Aldrich (Taufkirchen, Germany). Dihydrofluorescein diacetate (DHFDA) was supplied by Molecular Probes Europe BV (Leiden, Holland) and Reparixin by Cayman Chemical (Ann Arbor, MI, USA). Kineret® (Sobi, Stockholm, Sweden) was kindly provided by Prof. U Merle (University of Heidelberg, Germany). Effectiveness of target inhibition by chemical inhibitors is shown in Fig. 2 and suppl. Fig. 1. Stock solutions prepared in DMSO were further diluted in culture medium with a final concentration of 0.1% DMSO. Human recombinant IL-1β was purchased from Enzo Lifesciences (Lörrach, Germany).
Fig. 2.

Differentiation-induced NOX2 leads to enhanced ROS production, IL-1β release and hepcidin upregulation. A) The release of H2O2 by THP-1 cells was highly increased within 60 min of PMA-induced differentiation and was efficiently blocked by co-incubation with the pan NOX inhibitor VAS2870. Extracellular H2O2 levels were measured by HRPO-enhanced luminescence (relative light units; RLU). B) Increased intracellular ROS accumulation could be lowered by VAS2870 during the first 24 h of differentiation with PMA. THP-1 monocytes were pre-loaded with dihydrofluorescein diacetate (DHF) and differentiated with PMA for 24 h. Intracellular ROS were detected by measuring the fluorescence at 540 nm and the mean fluorescence intensity is given in relative fluorescence units (RFU) after subtraction of the control in the absence of PMA (RFU ± SD). C) Treatment of THP-1 cells with VAS2870 while PMA-mediated differentiation significantly inhibited hepcidin, and D) IL-1β mRNA induction during 24 h. E) siRNA-mediated silencing of NOX2 while differentiation significantly decreased hepcidin transcription but had no effect on IL-1β mRNA expression. NOX2, IL-1β and hepcidin mRNA was quantified by quantitative real-time PCR and the results are the mean of mRNA levels normalized to β2-microglobulin ± SD. Significant differences are marked by asterisks (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
2.5. Extracellular release of H2O2
The extracellular release of H2O2 by enzymes such as NOXs was assessed using a horseradish peroxidase (HRP)-luminol based reaction [20,21]. Briefly, THP-1 monocytes were seeded in white 96-well plate at a density of 5 × 104 cells/well in HBSS (Sigma-Aldrich, Taufkirchen, Germany) without FBS. Luminol and HRP were added to each well at a final concentration of 10 μM and a 1:1000 dilution. After addition of PMA (100 ng/ml) the luminescence signal was measured every 2 min for 1 h with the fluostar device (BMG Labtech, Ortenberg, Germany).
2.6. Intracellular detection of H2O2
THP-1 monocytes (1 × 106 cells/ml) were pre-loaded with DHFDA at a concentration of 10 μM in PBS [21]. After 30 min, the cells were washed with PBS and seeded in a black 96-well plate at 1 × 105 cells/well. During 24 h the cells were incubated with PMA (100 ng/ml) in RPMI-1640 medium without phenol red (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum. Fluorescence was determined (excitation 492 nm; emission 540 nm) using a fluorometer (Fluostar, BMG Labtech, Ortenberg, Germany) and the results are presented in fluorescence intensity normalized to the control.
2.7. RNA isolation, cDNA synthesis and real time quantitative PCR analysis
RNA was isolated with Trifast (Peqlab biotechnology GmbH, Erlangen, Germany) according to the manufacturer specifications. Reverse transcription and real time quantitative PCR reactions were performed as previously described [18]. Primers and probes were designed using the Probefinder software (Roche, Mannheim, Germany) and the sequences are provided in Table 1.
Table 1.
Primer list of the genes analyzed by quantitative RT-PCR.
| Gene | Primer sequence |
|---|---|
| human β2-MG | forward: 5′-tga ctt tgt cac agc cca aga ta-3′ |
| reverse: 5′-aat cca aat gcg gca tct tc-3′ | |
| probe: FAM-tga tgc tgc tta cat gtc tcg atc cca-TAM | |
| human hepcidin | forward 5′-cag gac aga gct gga gcc a -3′ |
| reverse: 5′-gca gca cat ccc aca ctt tg-3′ | |
| probe: FAM-ctg ctg cgg ctg ctg tca tcg a-TAM | |
| human NOX2 | forward: 5′- cat tca acc tct gcc acc at -3′ |
| reverse: 5′- ccc cag cca aac cag aat -3′ | |
| probe UPL # 26 (Roche) | |
| human IL-1β | forward: 5′- aaa gct tgg tga tgt ctg gtc -3′ |
| reverse: 5′- gga cat gga gaa cac cac ttg -3′ | |
| probe UPL # 39 (Roche) | |
| human IL-6 | forward: 5′- gcc cag cta tga act cct tct -3′ |
| reverse: 5′- ctt ctc ctg ggg gta ctg g -3′ | |
| probe UPL # 68 (Roche) | |
| human SMAD7 | forward: 5′- aaa cag ggg gaa cga att atc -3′ |
| reverse: 5′- acc acg cac cag tgt gac -3′ | |
| probe UPL # 50 (Roche) | |
| forward: 5′- gga cat agg agc gca aaa gaa -3′ | |
| human CEBPδ | reverse: 5′- gct tct ctc cgc agt tta gtg g -3′ |
| probe UPL # 64 (Roche) |
2.8. Transfection experiments
Huh7 cells were transfected with hepcidin promoter constructs (wild-type or mutated promoters) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Hepcidin promoter constructs containing firefly luciferase and a control plasmid containing renilla luciferase were co-transfected at a ratio of 1:16 for 48 h as published previously [18]. For hypoxia studies, cells were transferred for 24 h to 1% O2 using a hypoxia chamber. For the RNA silencing experiments Huh7 cells were transfected using Lipofectamine 2000 and 10 nM STAT3 siRNA or 40 nM C/EBPδ siRNA. 48 h after transfection, the cells were conditioned to 1% O2 in a hypoxia chamber for additional 24 h. Validated siRNA against STAT3 was purchased from Ambion (Foster City, CA, USA) and the double-stranded siRNA against CEBPδ was synthesized by Eurofins Genomics (Ebersberg, Germany) with the following nucleotide sequence: 5′-aca gcc ugg acu uac cac cac uaa a-3’. Equimolar concentrations of universal negative siRNA (Sigma-Aldrich, Taufkirchen, Germany) were used as control.
THP-1 cells were transfected with HiPerfect (Qiagen, Hilden, Germany) according to the supplier's protocol. Briefly, the cells were seeded at 2 × 105 cells/well in a 12-well plate and transfected with 10 nM siRNA against NOX2 (Ambion, Foster City, CA, USA) for 30 h. During the last 24 h the cells were incubated in the presence of PMA (100 ng/ml).
2.9. Immunoblotting
Cells were washed in ice-cold 1xPBS and harvested in RIPA buffer plus 1 × Complete protease inhibitor with EDTA (Roche Applied Sciences, Penzberg, Germany) on ice. Western Blotting was performed as described previously [18]. Following the transfer, the proteins immobilized on nitrocellulose membranes were incubated over night with the antibodies anti-pSTAT3, anti-STAT3 (1:1000 dilution; Cell Signaling Technology, Frankfurt am Main, Germany) or anti-β-actin (1:5000 dilution; Sigma-Aldrich, Taufkirchen, Germany). After incubation with the IRDye-conjugated 680 anti-mouse or 800 anti-rabbit antibody (1:10 000 dilution; LI-COR, Inc., Lincoln, NE, USA) the membranes were scanned using an infrared imaging system (Odyssey CLx; LI-COR, Inc., Lincoln, NE, USA).
2.10. Statistical analysis
All the data are expressed as mean ± SD. Significant differences (P < 0.05) between means of data sets were assessed by one-way ANOVA with Tukey's test or two-way ANOVA with Sidak's test using GraphPad Prism 6 software.
3. Results
3.1. Monocyte to macrophage differentiation strongly induces macrophage hepcidin expression mediated by NOX2 and subsequent H2O2
THP1 monocytes typically grow in suspension and require PMA-mediated PKC activation for differentiation into macrophages [22,23]. This differentiation process is known to involve NOX2-mediated reactive oxygen species production mainly superoxide which is rapidly converted into H2O2 [24]. Interestingly, and in accordance with an increase of NOX2 mRNA by almost 4-fold, differentiation of THP1 cells led to 60-fold upregulation of macrophage hepcidin under 21% O2 during the first 24 h (Fig. 1A and B). Macrophage hepcidin levels decreased after 48 h, but were still significantly higher (about 30-fold increase) as compared to nearly absent monocyte baseline levels (Fig. 1B). In addition, ferroportin mRNA levels were decreased during PMA-mediated macrophage differentiation accompanied with increased ferritin mRNA levels (heavy and light chain) (see suppl. Fig. 2). Hepcidin induction could be recapitulated in primary human monocytes isolated from human blood and differentiated into monocyte-derived macrophages (MDM) using M-CSF for 7 days (Fig. 1C). Of note, PMA-induced differentiation of THP-1 cells into macrophages did not further induce NOX1 (suppl. Fig 3).
Fig. 1.

Differentiation of monocytes into macrophages leads to transcriptional activation of NOX2 and hepcidin. A) PMA-mediated differentiation of THP-1 monocytes into macrophages caused a significant increase of NOX2 and B) hepcidin mRNA expression during 24 h. C) Differentiation of primary isolated human monocytes into monocyte derived macrophages (MDM) using M-CSF for 7 days causes a significant increase of hepcidin mRNA expression. NOX2 and hepcidin mRNA was quantified by quantitative real-time PCR and the results are represented as mean of mRNA levels normalized to β2-microglobulin ± SD. Significant differences are marked by asterisks ( ***, P < 0.001).
Using horseradish peroxidase (HRP)-enhanced luminol chemiluminescence, we could demonstrate an efficient H2O2 release during PMA-induced differentiation of THP-1 monocytes into macrophages. Intracellular ROS accumulation was further confirmed by dihydrofluorescein diacetate (DHFDA) during 24 h (Fig. 2A and B). Based on our recent studies on peroxide-mediated hepcidin regulation in hepatocytes, we hypothesized that H2O2 may be responsible for differentiation-induced macrophage hepcidin expression. Indeed, the treatment of THP-1 cells during PMA-mediated differentiation with H2O2-degrading catalase or the pan-NOX inhibitor VAS2870 reduced ROS production as measured by DHFDA (suppl. Fig. 4A and 2A) and significantly blocked macrophage hepcidin induction (suppl. Fig. 4B and 2C). We further studied the effect of siRNA-mediated NOX2 inhibition in THP-1 cells. Transfection with NOX2 siRNA significantly decreased the amount of NOX2 mRNA (Fig. 2E) and caused a modest and significant inhibition of hepcidin but not back to baseline (compare Fig. 2E). These results suggest, that NOX2 enzyme activity is most important for hepcidin regulation whereas the amount of newly synthesized mRNA plays only a minor role. Furthermore, the transcription of IL-1β, an important inflammatory cytokine was also completely blocked by VAS2780 treatment, whereas siRNA-mediated NOX2 knockdown had no significant effect (compare Fig. 2D with E).
Taken together, we here demonstrate a strong and NOX2-peroxide dependent induction of hepcidin in macrophages. Of note and in comparison to the hepatoma cell line Huh7, an accepted model to study hepcidin regulation, basal hepcidin mRNA expression in PMA-differentiated THP-1 macrophages was 8-fold less than in Huh7 cells (suppl. Fig. 5).
3.2. Hypoxia has no effect on macrophage hepcidin
We could recently demonstrate that physiological low oxygen levels strongly enhance peroxide or oxidase-mediated upregulation of hepcidin in hepatocytes [17,18]. In contrast to these reports, macrophage hepcidin expression was not responsive to hypoxia. Moreover, neither NOX2 nor hepcidin mRNA levels are further induced in PMA-differentiated THP-1 macrophages cultivated under 1–5% O2 (suppl. Fig. 6A). In confirmation, the same results could be obtained in human primary monocyte derived macrophages (MDM cells) (suppl. Fig. 6B) although differentiation caused a drastic induction of NOX2 and hepcidin mRNA. Thus, contrary to hypoxia-induced NOX4 with subsequent NOX4-mediated hepcidin induction in hepatocytes, macrophage NOX2 and hepcidin were not further induced by low oxygen levels pointing towards a cell type-specific response to hypoxia.
3.3. Macrophage-hepatocyte crosstalk under physiological low oxygen levels induces hepcidin in hepatocytes
Since NOX2 activation seems to be crucial for the transcriptional activation of hepcidin during differentiation of monocytes into macrophages, we further analyzed the impact of macrophage differentiation, namely the activation of macrophage NOX2, on hepatocyte hepcidin expression by use of a co-culture model. In order to mimic liver conditions, we both explored 21% O2 as well as physiological low oxygen levels (1–5% O2) and (patho)physiological hepatocyte/macrophage cell ratios (10:1 vs. 4:1). In a normal experimental design, THP-1 cells were differentiated into macrophages for 24 h followed by co-cultivation for another 24 h with Huh7 cells under 21% O2 or 1–5% O2 using the hypoxia chamber for 24 h. Co-culture at both hepatocyte/macrophage ratios (10:1 and 4:1) under 21% O2 induced hepcidin mRNA levels, which was further enhanced under 1–5% O2 (Fig. 3A). Notably, cell ratios had no effect on hepcidin expression. In contrast and in confirmation of a recent study by Chaston et al. [25], hepcidin mRNA was suppressed under 1% O2 using a non-physiological hepatocyte/macrophage cell ratio of 1:1.6 (see suppl. Fig. 7). In addition, NOX2 mRNA was also significantly upregulated in both hepatocyte/macrophage co-cultures (4:1 and 10:1 ratio) under 1–5% O2. Of note, the increased number of macrophages in the 4:1 co-culture contributed to the elevated NOX2 mRNA expression (suppl. Fig. 8). We next studied the involvement of STAT3 and/or BMP signaling in macrophage-mediated hepatic hepcidin induction under physiological low oxygen levels. As reported recently [17], low oxygen tension per se strongly induced STAT3 phosphorylation in Huh7 monocultures. Co-cultivation enhanced pSTAT3 under normoxia, which was potentiated under low oxygen levels (1–5% O2) in line with significantly upregulated hepcidin mRNA levels (Fig. 3A). Notably, no significant contribution of the BMP signaling pathway was observed since hypoxia as well as hepatocyte/macrophage co-cultures had no effect on pSmad1/5/8 protein as well as inhibitory Smad7 mRNA levels (Fig. 3B). In summary, these data suggest that hepatocyte hepcidin is induced in hepatocyte/macrophage co-cultures via STAT3 but not the BMP/SMAD signaling.
Fig. 3.

Induction of hepatocellular hepcidin in macrophage/hepatocyte co-cultures under low oxygen levels is mediated by STAT3 but not BMP signaling pathway. A) Hepcidin mRNA as well as Stat3 protein levels are increased in hepatocyte/macrophage co-cultures using different (patho-)physiologic cell ratios and low oxygen levels. B) Smad7 mRNA as well as Smad1/5/8 protein levels are not significantly changed in hepatocyte/macrophage co-cultures using different (patho-)physiologic cell ratios and low oxygen levels. qRT-PCR results are presented as mean of target mRNAs normalized to β2-microglobulin ± SD. Western Blots are representatives of three independent experiments. Significant differences are marked by asterisks (***, P < 0.001).
3.4. Macrophage-secreted factors induce hepatocellular hepcidin via STAT3 signaling
We now used macrophage-conditioned medium instead of direct co-cultures to shed more light on potential soluble factors secreted by macrophages in modulating hepatocyte hepcidin expression. Macrophage-conditioned medium significantly increased hepcidin mRNA level in hepatocytes (see Fig. 4A). Medium from differentiated macrophages maintained under 1% O2 (macrophage conditioned hypoxic medium = MCHM) showed the highest induction of hepcidin expression in Huh7 cells cultured under 21% O2. Highest upregulation of hepcidin was obtained if both macrophages and hepatocytes were cultured under 1% O2. Notably, levels of pSTAT3 clearly reflect hepcidin mRNA levels (Fig. 4A). Spearman Rho correlation between densitometric pSTAT3/STAT3 expression levels and hepcidin mRNA levels showed a high and significant association (r = 0.943; P = 0.017, see suppl. Fig. 9). These data point towards a macrophage-secreted factor whose expression level is enhanced by low oxygen tension (1% O2) and is able to stimulate hepatocyte hepcidin. Of note, the macrophage-mediated effect was stronger if direct hepatocytes/macrophage co-cultures were used. To further narrow the search for the macrophage-secreted factor(s), we next studied the transcriptional mechanism of hepcidin induction by MCHM. Luciferase reporter assays were performed using full length wild type hepcidin promoter construct (WT 3 kb), three increasingly truncated wild type constructs (WT 1 kb, WT 385bp and WT 165bp) and full length hepcidin promoter constructs with selective deletion of the STAT3-binding site (STAT3-del) or the BMP-responsive elements 1, 2, and 3 (BMP-RE1-3 del). As shown in Fig. 4C and consistent with the mRNA results, hepcidin promoter activity was 4-fold increased when Huh7 cells were exposed to MCHM if compared to Huh7 monocultures under normoxic conditions. In addition, no change of promoter activity in response to MCHM was observed when truncating the wildtype hepcidin promoter construct starting from the 5′-end or the BMP-responsive elements were deleted. Of note, selective deletion of the BMP-RE2 still allowed for MCHM-dependent promoter activation but not reaching statistical significance. In contrast, only the deletion of the STAT3-binding site completely blocked macrophage-meditated induction of hepcidin promoter activity under hypoxia.
Fig. 4.

Macrophage-secreted factors induce hepatocellular hepcidin via STAT3 signaling pathway. A) Hepcidin mRNA as well as pStat3 protein levels were significantly increased in hepatocytes treated with medium of macrophages conditioned to 1% O2 and further cultivated under 21% and 1% O2. B) Macrophage-conditioned hypoxic medium induced hepatocellular hepcidin transcription as well as phosphorylation of Stat3 is completely blocked by co-treatment with the pan NOX inhibitor VAS2870. Hepcidin mRNA levels were quantified by qRT-PCR and the results are presented as mean of hepcidin mRNAs normalized to β2-microglobulin ± SD. C) The deletion of the STAT3-binding site completely blocked the induction on hepcidin promoter activity after incubation with macrophage-conditioned medium. Huh7 cells were transfected with hepcidin promoter constructs including the wild type promoter (WT), promoter regions with decreased length of the 5‘-flanking region (WT 1 kb and WT 165bp) or the WT with specific deletions of transcription factors binding sites (STAT3-binding site (STAT3bs del) and the BMP-responsive elements 1–3 (bmp-RE1 to -3 del)). Huh7 cells were transfected for 48 h and then cultivated with macrophage-conditioned hypoxic medium for 24 h. Renilla plasmid was used as control for expression and transfection. The results are expressed as fold induction ± SD of firefly/Renilla luciferase activity relatively to the normoxic control of each construct. (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Moreover, and in order to validate the important role of NOX2 activation during PMA-induced macrophage differentiation and hepatocellular hepcidin regulation, we analyzed the effect of NOX2 inhibition by using the pan NOX inhibitor VAS2870. We could demonstrate that VAS2870 treatment completely blocked the induction of hepatocellular hepcidin as well as pSTAT3 expression (Fig. 4B) during macrophage differentiation under hypoxia (1% O2) and sub-cultivation of Huh7 cells with the resulting medium. These data suggest that NOX2 and peroxide as well as other secreted factors are involved in the macrophage-mediated upregulation of hepatocyte hepcidin. Since removal of exogenous H2O2 by catalase was not able to block hepcidin induction in hepatocytes (data not shown), we suspect a macrophage-secreted factor as potential hepcidin inducer.
3.5. Identification of macrophage secreted factors responsible for hepcidin induction
We next analyzed the cytokine profile of PMA-differentiated THP-1 macrophages under 21% as well as 1% O2 and identified a large panel of cytokines including IL-1β, IL-8, Oncostatin M (OSM) and leukemia inhibitory factor (LIF), which are all known hepcidin inducers over the Jak/STAT signaling pathway. We focused on IL-1β, since its secretion was enhanced under 1% O2 and on IL-8 because of its high abundance (Fig. 5A). We next determined the effects of IL-1β receptor antagonist anakinra (IL-1RA) as well as Reparixin, an inhibitor of CXC chemokine receptors 1 and 2, which prevents CXCR activation by IL-8, on MCHM-induced hepatocyte hepcidin expression. As shown in Fig. 5B treatment of Huh7 cells with MCHM led to a significant increase of hepcidin mRNA, which was completely blocked by IL-1RA treatment but only slightly diminished by Reparixin. The combined treatment with both inhibitors did not show any additive effect (data not shown). The use of IL-1RA also completely inhibited the MCHM-induced phosphorylation of STAT3, whereas Reparixin had no effect (compare Fig. 5C, bar 1 and 4 with bar 3 and 6). These results clearly show, that physiological IL-1β levels secreted by macrophages under 1% O2 are able to induce hepatocyte hepcidin expression. In addition, this regulatory circuit seems to be more sensitive under low oxygen levels and involves STAT3 signaling.
Fig. 5.

IL-1β is the main cytokine involved in the macrophage-mediated induction of hepatocellular hepcidin transcription. A) The levels of 25 different cytokines were determined in the supernatant from PMA-differentiated THP-1 cells cultivated under 21% and 1% O2 using a cytokine protein array. The level of cytokines were quantified using ImageJ software and expressed as normalized density units. IL-1β was induced under hypoxic conditions and is known to regulate hepcidin. B) Induction of hepcidin transcription in hepatocytes by MCHM was completely abolished by IL-1 receptor antagonist treatment (IL-1RA; Anakinra), whereas IL-8 inhibition by Reparixin had no significant effect. C) IL-1RA treatment also inhibited the MCHM-induced phosphorylation of Stat3 in hepatocytes. Representative data of at least three independent experiments are shown. Hepcidin mRNA levels were quantified by RT-PCR and presented as mean Hepcidin mRNA normalized to β2-microglobulin ± SD. Significant differences in relation to the respective controls are marked by asterisks (*, P < 0.05; **, P < 0.01; ***, P < 0.001) and significant differences to the macrophage-conditioned hypoxic medium control by hash tags (#, P < 0.05; ##, P < 0.01; ###, P < 0.001).
3.6. MCHM-containing IL-1β induces hepcidin independently of IL-6
Since it has been shown that IL-1β induces IL-6 production in hepatocytes [26] and this may be responsible for hepcidin transcription, we determined IL-6 as well as hepcidin mRNA levels in Huh7 cells treated with commonly used IL-1β concentration of 10 ng/ml. As shown in Fig. 6A, nearly no IL-6 mRNA expression was detected in untreated Huh7 cells under 21% O2. However, and in confirmation with previous reports, Huh7 cells treated with commonly used high IL-1β concentrations (10 ng/ml) showed over 10 times induced expression of IL-6 along with twice as high hepcidin mRNA level under 21% O2 (Fig. 6A). Therefore, we next questioned, whether hepcidin upregulation found in our experimental setting using low oxygen tension was likewise mediated by IL-1β-induced IL-6 stimulation and subsequent hepcidin transcription. We therefore incubated Huh7 cells with increasing IL-1β concentrations under 1% O2 and found that hepcidin mRNA expression was significantly induced at concentrations higher than 0.1 ng/ml IL-1β under 1% O2 (Fig. 6B). Interestingly, none of the tested recombinant IL-1β concentrations as well as the MCHM-containing IL-1β was able to significantly induce IL-6 expression in Huh7 cells, but lead to significantly enhanced pSTAT3 levels (Fig. 6C and D). In addition, we also examined the effect of MCHM combined with NOX inhibition by VAS2870 or IL-1RA treatment on hepatocellular IL-6 expression. Both inhibitors had no significant impact on IL-6 mRNA levels in hepatocytes (Fig. 6E and F).
Fig. 6.

Low physiologic concentrations of IL-1β secreted by macrophages induce hepatocellular hepcidin independently of IL-6. A) Hepcidin as well as IL-6 mRNA are significantly induced in Huh7 cells using artificially high IL-1β concentrations (10 ng/ml recombinant IL-1β). B) Under 1% O2 IL-1β (>0.1 ng/ml) significantly increases hepcidin C) but not IL-6 mRNA expression. CEBPδ mRNA is highly upregulated under 1% O2 by IL-1β concentrations starting from 0.05 ng/ml. D) Under 1% O2 IL-1β also significantly increases STAT3 phosphorylation. E) MCHM also significantly induces CEBPδ mRNA expression without affecting IL-6 mRNA in Huh7 cells. Treatment of Huh7 cells with IL-1 receptor antagonist (IL-1RA; Anakinra) or F) co-treatment of macrophages with VAS2870 completely inhibited the effect of MCHM on CEBPδ transcription (###, P < 0.001). Hepcidin, IL-6 and CEBPδ mRNA levels were quantified by RT-PCR and presented as mean mRNA normalized to β2-microglobulin ± SD. Significant differences in relation to the respective controls are marked by asterisks (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Since the induction of hepcidin transcription by IL-6 stimulation was found to be dose dependent and only artificially high IL-1β concentrations led to IL-6 stimulation with hepcidin induction, while physiological IL-1β concentrations as secreted by macrophages upregulate hepcidin expression without affecting IL-6, we suggest an IL-6 independent mechanism for hepatocyte hepcidin induction by MCHM.
3.7. MCHM-containing IL-1β efficiently induces CEBPδ
Furthermore, and in agreement with recent findings by Kanamori et al. we could show that already very low IL-1β concentrations or MCHM-containing IL-1β highly and significantly induce the expression of CEBPδ (Fig. 6C and F), which might bind to the putative CEBP-BS on the hepcidin promoter to activate its transcription. We also examined the effect of MCHM combined with IL-1RA treatment and NOX inhibition by VAS2870 on hepatocyte CEBPδ mRNA expression. NOX2 as well as IL-1 receptor inhibition completely blocked MCHM-induced CEBPδ expression (Fig. 6E and F). Based on this data and the results of increased phosphorylated STAT3 after IL-1β treatment (Fig. 6D) we hypothesize that IL-1β secreted by macrophages under low oxygen levels induces hepatocellular hepcidin transcription involving STAT3 and CEBPδ signaling.
3.8. CEBPδ binding to the hepcidin promoter is involved in IL-1β-induced and STAT3-mediated hepcidin transcription under low oxygen levels
Provided that STAT3 is mandatory for MCHM-induced hepcidin transcription and to proof our postulated hypothesis, siRNA mediated knockdown of STAT3 and CEBPδ was explored using Huh7 cells exposed to MCHM. Silencing of STAT3 led to significantly reduced basal hepcidin mRNA levels in controls as previously shown [17] as well as in Huh7 cells exposed to MCHM (Fig. 7A). Efficient siRNA mediated knockdown of STAT3 is shown in Fig. 7A. In contrast, knockdown of CEBPδ had no effect in Huh7 cells alone, but significantly reduced the amount of hepcidin mRNA in Huh7 cells exposed to MCHM. STAT3 knockdown completely abolished hepatocellular hepcidin induction mediated by MCHM (Fig. 7A). Most interestingly, STAT3 and CEBPδ knockdown showed the same effect on CEBPδ mRNA levels in Huh7 cells alone, implicating that a functional STAT3 axis is essential for CEBPδ promoter activity/transcription (Fig. 7B). Exposure of Huh7 cells to MCHM significantly increased CEBPδ mRNA expression, whereas siRNA against STAT3 or CEBPδ significantly reduced CEBPδ mRNA. The combined knockdown of STAT3 and CEBPδ led to basal CEBPδ levels as found in the untreated, single knockdown controls (Fig. 7B). In addition, silencing of CEBPδ significantly decreased STAT3 phosphorylation in Huh7 cells after exposure to MCHM (Fig. 7C). The role of CEBPδ in regulating hepcidin was also analyzed at the promoter level. As demonstrated in Fig. 7D, siRNA mediated knockdown of CEBPδ completely blunted the effect of MCHM on hepcidin promoter activity. However, silencing of CEBPδ does not further suppress the effect of the lacking STAT3 binding site, but abolished the damped effect of MCHM. All together, the results point towards an essential role of STAT3 in mediating the induction of hepatocyte hepcidin by macrophage secreted IL-1β and an involvement of CEBPδ. Furthermore, the concerted effect of both proteins seems to enhance the transcription of hepcidin in our experimental setting mimicking physiological cell ratios and oxygen levels. Furthermore, both proteins might be mutually dependent or closely interlinked.
Fig. 7.

STAT3 is an essential link between macrophage-induced IL-1β signaling and hepatocellular hepcidin upregulation. A) siRNA-mediated knockdown of STAT3 or CEBPδ significantly inhibits basal MCHM-induced hepcidin expression in Huh7 cells. B) CEBPδ upregulation by MCHM is significantly blocked by siRNA against CEBPδ or STAT3. C) siRNA-mediated silencing of CEBPδ decreases the induction of pSTAT3 protein expression after treatment with MCHM. D) Induction of hepcidin promoter activity by MCHM is inhibited by transfection with siRNA against CEBPδ. Huh7 cells were co-transfected with hepcidin wild type promoter (WT) or WT promoter with specific deletion of STAT3-binding site (STAT3bs del) and CEBPδ siRNA or control siRNA for 48 h and then cultivated with MCHM for 24 h. Renilla plasmid was used as control for expression and transfection. The results are expressed as fold induction ± SD of firefly/Renilla luciferase activity relatively to the normoxic control of each construct. Representative data of at least three independent experiments are shown. Hepcidin and CEBPδ mRNA levels were quantified by RT-PCR and presented as mean mRNA normalized to β2-microglobulin ± SD. Significant differences in relation to the respective controls are marked by asterisks (**, P < 0.01; ***, P < 0.001) and significant differences to the macrophage-conditioned medium control by hash tags (##, P < 0.01; ###, P < 0.001).
4. Discussion
Normal liver oxygen tension ranges from around 60–70 mmHg (ca. 10–12% O2) in the periportal area and drops to about 25–35 mmHg (3–5% O2) in the perivenular area [27]. A prominent feature of inflamed or diseased tissues is the presence of even lower oxygen tensions in the range of 1–2% O2. Effector cells of the innate immune system such as tissue resident macrophages in the liver (Kupffer cells) as well as blood circulating monocytes are recruited to inflammatory sites. There they differentiate into macrophages and must maintain their viability and physiologic functions, e.g. iron uptake and recycling from damaged or aged erythrocytes as well as dying hepatocytes even in a low oxygen microenvironment.
This study shows that NOX2, as well as hepcidin mRNA levels are significantly upregulated in THP-1 cells during macrophage differentiation. This could be recapitulated in isolated primary monocytes differentiated into macrophages. However, the expression of macrophage hepcidin is about 8-fold less than in Huh7 cells suggesting a minor but not negligible role of macrophage hepcidin during differentiation and maybe during subsequent inflammatory responses. In addition, we could demonstrate that hepcidin induction during macrophage differentiation is accompanied by a reduction of FPN mRNA as well as a slight increase in TfR1 and ferritin (FTL and FTH) mRNA, which decreased during 48 h to levels found in naïve THP-1 cells. Our results in differentiating macrophages support previous findings by Recalcati et al., who reported that activation of M1 macrophages is associated with an induction of the iron storage protein H ferritin and a reduction of the iron exporter ferroportin leading to iron retention [28]. These findings could suggest that hepcidin plays an important autocrine role in the differentiating macrophage.
Notably, differentiation-induced macrophage hepcidin transcription is impaired by both NOX2 inhibition and H2O2 degradation by catalase. Likewise, IL-1β mRNA transcription could also be efficiently blocked by NOX inhibition using VAS2870 as well as H2O2 scavenging by 20 mM NAC [29]. These results demonstrate a novel role of H2O2 as a second messenger in macrophage iron homeostasis, as well as in regulating cytokine expression. We hypothesize that enhanced IL-1β transcription during macrophage differentiation results from NOX2-derived H2O2. H2O2 has been proven to act as a second messenger in the activation of nuclear factor-κB (NFκB) signaling [30], thereby inducing the transcription of its target genes including IL-1β [31]. However, further investigation is still needed to understand the detailed underlying mechanisms.
In addition, macrophages are shown to tolerate long periods of chronic hypoxia and acquire the ability to exert their specific functions even in a hypoxic and inflammatory environment by rendering their energy metabolism and changing their phenotype including the secretion of specific cytokines [32]. Interestingly and in contrast to previous studies on hepatocytes [17], macrophage hepcidin expression is not further enhanced under low oxygen. Since the hypoxia-mediated induction of hepcidin in hepatocytes was shown to depend on constitutive active NOX4, which is mainly expressed in the liver, the lack of NOX4 in macrophages could explain the absence of hypoxia response.
However, detailed co-culture experiments using physiological and pathophysiological hepatocyte/macrophage cell ratios, as well as exposure of hepatocytes to macrophage conditioned hypoxic medium significantly increased hepcidin transcription in hepatocytes. This data is in agreement with previous studies using peritoneal macrophages, PMA-differentiated THP-1 cells or macrophage-conditioned medium [[33], [34], [35], [36]]. The stimulatory effect on hepcidin transcription is even more pronounced under physiologically low liver oxygen levels (hypoxia) and drastically enhanced in comparison to the previously discovered hypoxia-mediated hepcidin induction in hepatocyte monocultures [17]. However, our findings are in contrast to Chaston et al. who reported a significant decrease of hepcidin mRNA in Huh7/THP-1 co-cultures by hypoxia and oxidative stress (H2O2) [25]. We believe that this discrepancy is mainly explained by the non-physiological hepatocyte/macrophage cell ratios (1:2) and very high H2O2 bolus treatments (100 μM) that lead to unspecific inhibition of the mRNA transcription machinery thereby suppressing hepcidin [18,37]. Here we study hepcidin regulation at mRNA level, since hepcidin is transcriptionally regulated by STAT3 or BMP/SMAD pathway [38,39]. In addition, it is widely accepted, that mRNA level correspond to the amount of protein and secreted peptide levels are only used in clinical studies if liver biopsies are not available [10]. We are also aware that a direct in vivo proof is missing. However, studies in total or myeloid specific NOX2 knockout mice revealed severe dysregulation in immune responses and persistent inflammation highly affecting the cytokine milieu. In addition, these animals presented with splenomegaly, which might point towards an abnormal erythrocyte turnover impairing systemic iron homeostasis [40,41].
Various studies have described that activated macrophages secrete a number of cytokines e.g. IL-6, IL-1β or TGF-β [42], as well as release H2O2 from superoxide anion mainly produced by NOX2, which are all factors known to stimulate hepcidin expression. Therefore, we hypothesized that under physiologically low oxygen levels the production and secretion of these factors may be increased and responsible for enhanced hepatocyte hepcidin transcription. Interestingly, a microarray study analyzing gene expression profiles of THP-1 cells following addition of PMA, revealed a 16-fold increase in IL-1β suggesting a predominant role of this cytokine during macrophage differentiation [42]. In support of this hypothesis, Lee et al. suggested that in addition to IL-6 also IL-1α and β stimulated hepcidin expression in murine hepatocytes and that this stimulation by both IL-1 forms was even greater than stimulation by IL-6 [33]. Therefore we next performed a cytokine array using PMA-differentiated THP-1 cells under 21% as well 1% O2. Many cytokines such as IL-1β, IL-8, Oncostatin M (OSM) or leukemia inhibitory factor (LIF), which are all known hepcidin regulators are expressed, but only IL-1β expression increased under low oxygen levels.
Previous studies showed that IL-1β upregulates CCAAT-enhancer binding protein (CEBP) δ through IL-6, which then stimulated hepcidin transcription via C/EBP-BS and CRE site B as well as STAT3-BS on the hepcidin promoter [43,44]. A novel regulation of hepcidin by IL-1β is revealed under low oxygen levels in hepatocytes. We could demonstrate that macrophage-released IL-1β at physiological concentrations induces hepcidin over IL-1 receptor activation and STAT3 signaling, but independent of IL-6 in hepatocytes and may suggest an early hepcidin response towards IL-1β released by differentiated macrophages, which might be further regulated when macrophages undergo pro-inflammatory M1 activation. In addition, stimulation was completely blocked by using IL-1 receptor antagonist anakinra or selective deletion of the STAT3-binding site in the hepcidin promoter. This is in agreement with Lee et al. showing that mice with targeted disruption of the gene encoding IL-6 (IL-6−/−) retain their ability to respond to endotoxin injections by increasing hepcidin transcription in the liver or that the addition of IL-6 antibody did not ablate the IL-1-mediated effect [33]. In addition, IL-1β induces the expression of CEBPδ protein, which binds to putative CEBP-BS in the hepcidin promoter to activate transcription. We also confirm the involvement of CEBPδ in transcriptional activation of hepcidin expression by hepatocytes induced by macrophage-conditioned hypoxic medium. CEBPδ is known as inflammatory response gene and its expression is induced by low oxygen in mammary tumors in vivo and in breast cancer cells in vitro [45]. Furthermore, it has been shown that CEBPδ is activated by STAT3 in mammary gland involution [46]. Most interestingly, it is shown that the murine CEBPδ promoter contains a STAT3-BS [47]. To our knowledge, we are the first to demonstrate an induction of CEBPδ by macrophage-conditioned hypoxic medium in hepatocytes depending on the STAT3 signaling pathway. CEBPδ might integrate and amplify low oxygen and IL-1-induced signaling by contributing to a more efficient binding to the CEBP-BS in the hepcidin promoter. In addition, Zhang et al. has shown that inflammation mediated CEBPδ induction is completely blocked in STAT3 KO mice [48]. Likewise, we show that siRNA mediated STAT3 inhibition decrease CEBPδ expression abolishing hepcidin transcription induced by macrophage-conditioned hypoxic medium. Interestingly, deletion of the STAT3 binding site in the HAMP promoter in combination with siRNA-mediated silencing of CEBPδ expression completely blocked luciferase activity in the presence of MCHM. This suggests that STAT3 plays a major role in controlling hepatocyte hepcidin regulation in macrophage co-cultures under physiologically low oxygen levels.
In summary, differentiated macrophages induce hepatocyte hepcidin transcription in a direct or indirect co-culture under (patho)physiological cell ratios. This effect is strongly enhanced under physiologically low oxygen levels in liver (hypoxia). We are the first to reveal the induction of macrophage hepcidin during their differentiation. Taken together, present and previous data demonstrate that oxidases such as NOX2 and NOX4 are powerful inducers of hepcidin via synchronic modulation of both oxygen and peroxide levels in different cell populations present in the liver involving the STAT3 signaling pathway. Thus, hepatocyte-macrophage crosstalk is important for hepatocyte hepcidin regulation and points towards a concerted action of both cell types during inflammatory processes.
Contribution
IS and VR designed the experiments, analyzed the data and wrote the manuscript. SM critically discussed and revised the manuscript.
Conflict of interest disclosure
The authors have nothing to disclose.
Acknowledgements
This study funded by a grant of the DFG to VR and SM (RA 2677/1-1). We would further like to thank Dr. M. Muckenthaler and Dr. U Merle (University of Heidelberg, Germany) for providing us the hepcidin promoter constructs and the IL-1 receptor antagonist (Anakinra).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101209.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Krause A., Neitz S., Magert H.J., Schulz A., Forssmann W.G., Schulz-Knappe P., Adermann K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480(2–3):147–150. doi: 10.1016/s0014-5793(00)01920-7. [DOI] [PubMed] [Google Scholar]; A. Krause, S. Neitz, H.J. Magert, A. Schulz, W.G. Forssmann, P. Schulz-Knappe, K. Adermann, LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity, FEBS Lett 480(2-3) (2000) 147-150. [DOI] [PubMed]
- 2.Park C.H., Valore E.V., Waring A.J., Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001;276(11):7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]; C.H. Park, E.V. Valore, A.J. Waring, T. Ganz, Hepcidin, a urinary antimicrobial peptide synthesized in the liver, J Biol Chem 276(11) (2001) 7806-7810. [DOI] [PubMed]
- 3.Sow F.B., Florence W.C., Satoskar A.R., Schlesinger L.S., Zwilling B.S., Lafuse W.P. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J. Leukoc. Biol. 2007;82(4):934–945. doi: 10.1189/jlb.0407216. [DOI] [PubMed] [Google Scholar]; F.B. Sow, W.C. Florence, A.R. Satoskar, L.S. Schlesinger, B.S. Zwilling, W.P. Lafuse, Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis, J Leukoc Biol 82(4) (2007) 934-945. [DOI] [PubMed]
- 4.Lee P.L., Beutler E. Regulation of hepcidin and iron-overload disease. Annu. Rev. Pathol. 2009;4:489–515. doi: 10.1146/annurev.pathol.4.110807.092205. [DOI] [PubMed] [Google Scholar]; P.L. Lee, E. Beutler, Regulation of hepcidin and iron-overload disease, Annu Rev Pathol 4 (2009) 489-515. [DOI] [PubMed]
- 5.Nemeth E., Ganz T. The role of hepcidin in iron metabolism. Acta Haematol. 2009;122(2–3):78–86. doi: 10.1159/000243791. [DOI] [PMC free article] [PubMed] [Google Scholar]; E. Nemeth, T. Ganz, The role of hepcidin in iron metabolism, Acta Haematol 122(2-3) (2009) 78-86. [DOI] [PMC free article] [PubMed]
- 6.Hentze M.W., Muckenthaler M.U., Galy B., Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell. 2010;142(1):24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]; M.W. Hentze, M.U. Muckenthaler, B. Galy, C. Camaschella, Two to tango: regulation of Mammalian iron metabolism, Cell 142(1) (2010) 24-38. [DOI] [PubMed]
- 7.Ross S.L., Tran L., Winters A., Lee K.J., Plewa C., Foltz I., King C., Miranda L.P., Allen J., Beckman H., Cooke K.S., Moody G., Sasu B.J., Nemeth E., Ganz T., Molineux G., Arvedson T.L. Molecular mechanism of hepcidin-mediated ferroportin internalization requires ferroportin lysines, not tyrosines or JAK-STAT. Cell Metabol. 2012;15(6):905–917. doi: 10.1016/j.cmet.2012.03.017. [DOI] [PubMed] [Google Scholar]; S.L. Ross, L. Tran, A. Winters, K.J. Lee, C. Plewa, I. Foltz, C. King, L.P. Miranda, J. Allen, H. Beckman, K.S. Cooke, G. Moody, B.J. Sasu, E. Nemeth, T. Ganz, G. Molineux, T.L. Arvedson, Molecular mechanism of hepcidin-mediated ferroportin internalization requires ferroportin lysines, not tyrosines or JAK-STAT, Cell Metab 15(6) (2012) 905-917. [DOI] [PubMed]
- 8.Weiss G., Goodnough L.T. Anemia of chronic disease. N. Engl. J. Med. 2005;352(10):1011–1023. doi: 10.1056/NEJMra041809. [DOI] [PubMed] [Google Scholar]; G. Weiss, L.T. Goodnough, Anemia of chronic disease, N Engl J Med 352(10) (2005) 1011-1023. [DOI] [PubMed]
- 9.Konz T., Montes-Bayon M., Vaulont S. Hepcidin quantification: methods and utility in diagnosis. Metallomics. 2014;6(9):1583–1590. doi: 10.1039/c4mt00063c. [DOI] [PubMed] [Google Scholar]; T. Konz, M. Montes-Bayon, S. Vaulont, Hepcidin quantification: methods and utility in diagnosis, Metallomics 6(9) (2014) 1583-1590. [DOI] [PubMed]
- 10.Detivaud L., Nemeth E., Boudjema K., Turlin B., Troadec M.B., Leroyer P., Ropert M., Jacquelinet S., Courselaud B., Ganz T., Brissot P., Loreal O. Hepcidin levels in humans are correlated with hepatic iron stores, hemoglobin levels, and hepatic function. Blood. 2005;106(2):746–748. doi: 10.1182/blood-2004-12-4855. [DOI] [PubMed] [Google Scholar]; L. Detivaud, E. Nemeth, K. Boudjema, B. Turlin, M.B. Troadec, P. Leroyer, M. Ropert, S. Jacquelinet, B. Courselaud, T. Ganz, P. Brissot, O. Loreal, Hepcidin levels in humans are correlated with hepatic iron stores, hemoglobin levels, and hepatic function, Blood 106(2) (2005) 746-748. [DOI] [PubMed]
- 11.Ganz T. Macrophages and systemic iron homeostasis. J. Innate Immun. 2012;4(5–6):446–453. doi: 10.1159/000336423. [DOI] [PMC free article] [PubMed] [Google Scholar]; T. Ganz, Macrophages and systemic iron homeostasis, J Innate Immun 4(5-6) (2012) 446-453. [DOI] [PMC free article] [PubMed]
- 12.Scott C.L., Guilliams M. The role of Kupffer cells in hepatic iron and lipid metabolism. J. Hepatol. 2018;69(5):1197–1199. doi: 10.1016/j.jhep.2018.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; C.L. Scott, M. Guilliams, The role of Kupffer cells in hepatic iron and lipid metabolism, J Hepatol 69(5) (2018) 1197-1199. [DOI] [PMC free article] [PubMed]
- 13.Krenkel O., Tacke F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017;17(5):306–321. doi: 10.1038/nri.2017.11. [DOI] [PubMed] [Google Scholar]; O. Krenkel, F. Tacke, Liver macrophages in tissue homeostasis and disease, Nat Rev Immunol 17(5) (2017) 306-321. [DOI] [PubMed]
- 14.Mandrekar P., Szabo G. Signalling pathways in alcohol-induced liver inflammation. J. Hepatol. 2009;50(6):1258–1266. doi: 10.1016/j.jhep.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; P. Mandrekar, G. Szabo, Signalling pathways in alcohol-induced liver inflammation, J Hepatol 50(6) (2009) 1258-1266. [DOI] [PMC free article] [PubMed]
- 15.Corradini E., Pietrangelo A. Iron and steatohepatitis. J. Gastroenterol. Hepatol. 2012;27(Suppl 2):42–46. doi: 10.1111/j.1440-1746.2011.07014.x. [DOI] [PubMed] [Google Scholar]; E. Corradini, A. Pietrangelo, Iron and steatohepatitis, J Gastroenterol Hepatol 27 Suppl 2 (2012) 42-46. [DOI] [PubMed]
- 16.Sangkhae V., Nemeth E. Regulation of the iron homeostatic hormone hepcidin. Adv. Nutr. 2017;8(1):126–136. doi: 10.3945/an.116.013961. [DOI] [PMC free article] [PubMed] [Google Scholar]; V. Sangkhae, E. Nemeth, Regulation of the Iron Homeostatic Hormone Hepcidin, Adv Nutr 8(1) (2017) 126-136. [DOI] [PMC free article] [PubMed]
- 17.Silva I., Rausch V., Peccerella T., Millonig G., Seitz H.K., Mueller S. Hypoxia enhances H2O2-mediated upregulation of hepcidin: evidence for NOX4-mediated iron regulation. Redox Biol. 2018;16:1–10. doi: 10.1016/j.redox.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; I. Silva, V. Rausch, T. Peccerella, G. Millonig, H.K. Seitz, S. Mueller, Hypoxia enhances H2O2-mediated upregulation of hepcidin: Evidence for NOX4-mediated iron regulation, Redox Biol 16 (2018) 1-10. [DOI] [PMC free article] [PubMed]
- 18.Millonig G., Ganzleben I., Peccerella T., Casanovas G., Brodziak-Jarosz L., Breitkopf-Heinlein K., Dick T.P., Seitz H.K., Muckenthaler M.U., Mueller S. Sustained submicromolar H2O2 levels induce hepcidin via signal transducer and activator of transcription 3 (STAT3) J. Biol. Chem. 2012;287(44):37472–37482. doi: 10.1074/jbc.M112.358911. [DOI] [PMC free article] [PubMed] [Google Scholar]; G. Millonig, I. Ganzleben, T. Peccerella, G. Casanovas, L. Brodziak-Jarosz, K. Breitkopf-Heinlein, T.P. Dick, H.K. Seitz, M.U. Muckenthaler, S. Mueller, Sustained submicromolar H2O2 levels induce hepcidin via signal transducer and activator of transcription 3 (STAT3), J Biol Chem 287(44) (2012) 37472-37482. [DOI] [PMC free article] [PubMed]
- 19.Millonig G., Hegedusch S., Becker L., Seitz H.K., Schuppan D., Mueller S. Hypoxia-inducible factor 1a under rapid enzymatic hypoxia: cells sense decrements of oxygen but not hypoxia per se. Free Radic. Biol. Med. 2009;46(2):182–191. doi: 10.1016/j.freeradbiomed.2008.09.043. [DOI] [PubMed] [Google Scholar]; G. Millonig, S. Hegedusch, L. Becker, H.K. Seitz, D. Schuppan, S. Mueller, Hypoxia-inducible factor 1a under rapid enzymatic hypoxia: Cells sense decrements of oxygen but not hypoxia per se, Free Radic Biol Med 46(2) (2009) 182-191. [DOI] [PubMed]
- 20.S. Mütze, J. Arnhold, W. Stremmel, S. Mueller, Sensitive detection of hydrogen peroxide in biological systems: a non-enzymic approach, in: S.G. Pandalai (Ed.), Recent Research Developments in Analytical Biochemistry, Transworld Research Network2002, pp. 165-184.
- 21.Mueller S., Millonig G., Seitz H.K., Waite G.N. Chemiluminescence detection of hydrogen peroxide. In: Pantopoulos K., Schipper H., editors. Principles of Free Radical Biomedicine. Nova Publishers; New York: 2012. pp. 283–302. [Google Scholar]; S. Mueller, G. Millonig, H.K. Seitz, G.N. Waite, Chemiluminescence detection of hydrogen peroxide, in: K. Pantopoulos, H. Schipper (Eds.), Principles of Free Radical Biomedicine,, Nova Publishers, New York, 2012, pp. 283-302.
- 22.Bazzi M.D., Nelsestuen G.L. Differences in the effects of phorbol esters and diacylglycerols on protein kinase C. Biochemistry. 1989;28(24):9317–9323. doi: 10.1021/bi00450a011. [DOI] [PubMed] [Google Scholar]; M.D. Bazzi, G.L. Nelsestuen, Differences in the effects of phorbol esters and diacylglycerols on protein kinase C, Biochemistry 28(24) (1989) 9317-9323. [DOI] [PubMed]
- 23.Schwende H., Fitzke E., Ambs P., Dieter P. Differences in the state of differentiation of THP-1 cells induced by phorbol ester and 1,25-dihydroxyvitamin D3. J. Leukoc. Biol. 1996;59(4):555–561. [PubMed] [Google Scholar]; H. Schwende, E. Fitzke, P. Ambs, P. Dieter, Differences in the state of differentiation of THP-1 cells induced by phorbol ester and 1,25-dihydroxyvitamin D3, J Leukoc Biol 59(4) (1996) 555-561. [PubMed]
- 24.Xu Q., Choksi S., Qu J., Jang J., Choe M., Banfi B., Engelhardt J.F., Liu Z.G. NADPH oxidases are essential for macrophage differentiation. J. Biol. Chem. 2016;291(38):20030–20041. doi: 10.1074/jbc.M116.731216. [DOI] [PMC free article] [PubMed] [Google Scholar]; Q. Xu, S. Choksi, J. Qu, J. Jang, M. Choe, B. Banfi, J.F. Engelhardt, Z.G. Liu, NADPH Oxidases Are Essential for Macrophage Differentiation, J Biol Chem 291(38) (2016) 20030-20041. [DOI] [PMC free article] [PubMed]
- 25.Chaston T.B., Matak P., Pourvali K., Srai S.K., McKie A.T., Sharp P.A. Hypoxia inhibits hepcidin expression in HuH7 hepatoma cells via decreased SMAD4 signaling. Am. J. Physiol. Cell Physiol. 2011;300(4):C888–C895. doi: 10.1152/ajpcell.00121.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]; T.B. Chaston, P. Matak, K. Pourvali, S.K. Srai, A.T. McKie, P.A. Sharp, Hypoxia inhibits hepcidin expression in HuH7 hepatoma cells via decreased SMAD4 signaling, Am J Physiol Cell Physiol 300(4) (2011) C888-C895. [DOI] [PMC free article] [PubMed]
- 26.Liu Z., Simpson R.J., Cheers C. Interaction of interleukin-6, tumour necrosis factor and interleukin-1 during Listeria infection. Immunology. 1995;85(4):562–567. [PMC free article] [PubMed] [Google Scholar]; Z. Liu, R.J. Simpson, C. Cheers, Interaction of interleukin-6, tumour necrosis factor and interleukin-1 during Listeria infection, Immunology 85(4) (1995) 562-567. [PMC free article] [PubMed]
- 27.Lee-Montiel F.T., George S.M., Gough A.H., Sharma A.D., Wu J., DeBiasio R., Vernetti L.A., Taylor D.L. Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp. Biol. Med. 2017;242(16):1617–1632. doi: 10.1177/1535370217703978. [DOI] [PMC free article] [PubMed] [Google Scholar]; F.T. Lee-Montiel, S.M. George, A.H. Gough, A.D. Sharma, J. Wu, R. DeBiasio, L.A. Vernetti, D.L. Taylor, Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems, Exp Biol Med (Maywood) 242(16) (2017) 1617-1632. [DOI] [PMC free article] [PubMed]
- 28.Recalcati S., Locati M., Marini A., Santambrogio P., Zaninotto F., De Pizzol M., Zammataro L., Girelli D., Cairo G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010;40(3):824–835. doi: 10.1002/eji.200939889. [DOI] [PubMed] [Google Scholar]; S. Recalcati, M. Locati, A. Marini, P. Santambrogio, F. Zaninotto, M. De Pizzol, L. Zammataro, D. Girelli, G. Cairo, Differential regulation of iron homeostasis during human macrophage polarized activation, Eur J Immunol 40(3) (2010) 824-835. [DOI] [PubMed]
- 29.Ohnishi T., Bandow K., Kakimoto K., Kusuyama J., Matsuguchi T. Long-time treatment by low-dose N-acetyl-L-cysteine enhances proinflammatory cytokine expressions in LPS-stimulated macrophages. PLoS One. 2014;9(2) doi: 10.1371/journal.pone.0087229. [DOI] [PMC free article] [PubMed] [Google Scholar]; T. Ohnishi, K. Bandow, K. Kakimoto, J. Kusuyama, T. Matsuguchi, Long-time treatment by low-dose N-acetyl-L-cysteine enhances proinflammatory cytokine expressions in LPS-stimulated macrophages, PLoS One 9(2) (2014) e87229. [DOI] [PMC free article] [PubMed]
- 30.Zhang J., Johnston G., Stebler B., Keller E.T. Hydrogen peroxide activates NFkappaB and the interleukin-6 promoter through NFkappaB-inducing kinase. Antioxidants Redox Signal. 2001;3(3):493–504. doi: 10.1089/15230860152409121. [DOI] [PubMed] [Google Scholar]; J. Zhang, G. Johnston, B. Stebler, E.T. Keller, Hydrogen peroxide activates NFkappaB and the interleukin-6 promoter through NFkappaB-inducing kinase, Antioxid Redox Signal 3(3) (2001) 493-504. [DOI] [PubMed]
- 31.Scheibel M., Klein B., Merkle H., Schulz M., Fritsch R., Greten F.R., Arkan M.C., Schneider G., Schmid R.M. IkappaBbeta is an essential co-activator for LPS-induced IL-1beta transcription in vivo. J. Exp. Med. 2010;207(12):2621–2630. doi: 10.1084/jem.20100864. [DOI] [PMC free article] [PubMed] [Google Scholar]; M. Scheibel, B. Klein, H. Merkle, M. Schulz, R. Fritsch, F.R. Greten, M.C. Arkan, G. Schneider, R.M. Schmid, IkappaBbeta is an essential co-activator for LPS-induced IL-1beta transcription in vivo, J Exp Med 207(12) (2010) 2621-2630. [DOI] [PMC free article] [PubMed]
- 32.Riboldi E., Porta C., Morlacchi S., Viola A., Mantovani A., Sica A. Hypoxia-mediated regulation of macrophage functions in pathophysiology. Int. Immunol. 2013;25(2):67–75. doi: 10.1093/intimm/dxs110. [DOI] [PubMed] [Google Scholar]; E. Riboldi, C. Porta, S. Morlacchi, A. Viola, A. Mantovani, A. Sica, Hypoxia-mediated regulation of macrophage functions in pathophysiology, Int Immunol 25(2) (2013) 67-75. [DOI] [PubMed]
- 33.Lee P., Peng H., Gelbart T., Wang L., Beutler E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc. Natl. Acad. Sci. U. S. A. 2005;102(6):1906–1910. doi: 10.1073/pnas.0409808102. [DOI] [PMC free article] [PubMed] [Google Scholar]; P. Lee, H. Peng, T. Gelbart, L. Wang, E. Beutler, Regulation of hepcidin transcription by interleukin-1 and interleukin-6, Proc Natl Acad Sci U S A 102(6) (2005) 1906-1910. [DOI] [PMC free article] [PubMed]
- 34.Matak P., Chaston T.B., Chung B., Srai S.K., McKie A.T., Sharp P.A. Activated macrophages induce hepcidin expression in HuH7 hepatoma cells. Haematologica. 2009;94(6):773–780. doi: 10.3324/haematol.2008.003400. [DOI] [PMC free article] [PubMed] [Google Scholar]; P. Matak, T.B. Chaston, B. Chung, S.K. Srai, A.T. McKie, P.A. Sharp, Activated macrophages induce hepcidin expression in HuH7 hepatoma cells, Haematologica 94(6) (2009) 773-780. [DOI] [PMC free article] [PubMed]
- 35.Jacolot S., Ferec C., Mura C. Iron responses in hepatic, intestinal and macrophage/monocyte cell lines under different culture conditions. Blood Cells Mol. Dis. 2008;41(1):100–108. doi: 10.1016/j.bcmd.2008.01.006. [DOI] [PubMed] [Google Scholar]; S. Jacolot, C. Ferec, C. Mura, Iron responses in hepatic, intestinal and macrophage/monocyte cell lines under different culture conditions, Blood Cells Mol Dis 41(1) (2008) 100-108. [DOI] [PubMed]
- 36.Verga Falzacappa M.V., Vujic Spasic M., Kessler R., Stolte J., Hentze M.W., Muckenthaler M.U. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood. 2007;109(1):353–358. doi: 10.1182/blood-2006-07-033969. [DOI] [PubMed] [Google Scholar]; M.V. Verga Falzacappa, M. Vujic Spasic, R. Kessler, J. Stolte, M.W. Hentze, M.U. Muckenthaler, STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation, Blood 109(1) (2007) 353-358. [DOI] [PubMed]
- 37.Mueller S., Millonig G., Waite G.N. The GOX/CAT system: a novel enzymatic method to independently control hydrogen peroxide and hypoxia in cell culture. Adv. Med. Sci. 2009:1–15. doi: 10.2478/v10039-009-0042-3. [DOI] [PubMed] [Google Scholar]; S. Mueller, G. Millonig, G.N. Waite, The GOX/CAT system: A novel enzymatic method to independently control hydrogen peroxide and hypoxia in cell culture, Adv Med Sci (2009) 1-15. [DOI] [PubMed]
- 38.De Domenico I., Ward D.M., Kaplan J. Hepcidin regulation: ironing out the details. J. Clin. Investig. 2007;117(7):1755–1758. doi: 10.1172/JCI32701. [DOI] [PMC free article] [PubMed] [Google Scholar]; I. De Domenico, D.M. Ward, J. Kaplan, Hepcidin regulation: ironing out the details, J Clin Invest 117(7) (2007) 1755-1758. [DOI] [PMC free article] [PubMed]
- 39.Ganz T. Hepcidin and iron regulation, 10 years later. Blood. 2011;117(17):4425–4433. doi: 10.1182/blood-2011-01-258467. [DOI] [PMC free article] [PubMed] [Google Scholar]; T. Ganz, Hepcidin and iron regulation, 10 years later, Blood 117(17) (2011) 4425-4433. [DOI] [PMC free article] [PubMed]
- 40.Pepping J.K., Vandanmagsar B., Fernandez-Kim S.O., Zhang J., Mynatt R.L., Bruce-Keller A.J. Myeloid-specific deletion of NOX2 prevents the metabolic and neurologic consequences of high fat diet. PLoS One. 2017;12(8) doi: 10.1371/journal.pone.0181500. [DOI] [PMC free article] [PubMed] [Google Scholar]; J.K. Pepping, B. Vandanmagsar, S.O. Fernandez-Kim, J. Zhang, R.L. Mynatt, A.J. Bruce-Keller, Myeloid-specific deletion of NOX2 prevents the metabolic and neurologic consequences of high fat diet, PLoS One 12(8) (2017) e0181500. [DOI] [PMC free article] [PubMed]
- 41.Lee K., Won H.Y., Bae M.A., Hong J.H., Hwang E.S. Spontaneous and aging-dependent development of arthritis in NADPH oxidase 2 deficiency through altered differentiation of CD11b+ and Th/Treg cells. Proc. Natl. Acad. Sci. U. S. A. 2011;108(23):9548–9553. doi: 10.1073/pnas.1012645108. [DOI] [PMC free article] [PubMed] [Google Scholar]; K. Lee, H.Y. Won, M.A. Bae, J.H. Hong, E.S. Hwang, Spontaneous and aging-dependent development of arthritis in NADPH oxidase 2 deficiency through altered differentiation of CD11b+ and Th/Treg cells, Proc Natl Acad Sci U S A 108(23) (2011) 9548-9553. [DOI] [PMC free article] [PubMed]
- 42.Kohro T., Tanaka T., Murakami T., Wada Y., Aburatani H., Hamakubo T., Kodama T. A comparison of differences in the gene expression profiles of phorbol 12-myristate 13-acetate differentiated THP-1 cells and human monocyte-derived macrophage. J. Atheroscler. Thromb. 2004;11(2):88–97. doi: 10.5551/jat.11.88. [DOI] [PubMed] [Google Scholar]; T. Kohro, T. Tanaka, T. Murakami, Y. Wada, H. Aburatani, T. Hamakubo, T. Kodama, A comparison of differences in the gene expression profiles of phorbol 12-myristate 13-acetate differentiated THP-1 cells and human monocyte-derived macrophage, J Atheroscler Thromb 11(2) (2004) 88-97. [DOI] [PubMed]
- 43.Kanamori Y., Murakami M., Sugiyama M., Hashimoto O., Matsui T., Funaba M. Interleukin-1beta (IL-1beta) transcriptionally activates hepcidin by inducing CCAAT enhancer-binding protein delta (C/EBPdelta) expression in hepatocytes. J. Biol. Chem. 2017;292(24):10275–10287. doi: 10.1074/jbc.M116.770974. [DOI] [PMC free article] [PubMed] [Google Scholar]; Y. Kanamori, M. Murakami, M. Sugiyama, O. Hashimoto, T. Matsui, M. Funaba, Interleukin-1beta (IL-1beta) transcriptionally activates hepcidin by inducing CCAAT enhancer-binding protein delta (C/EBPdelta) expression in hepatocytes, J Biol Chem 292(24) (2017) 10275-10287. [DOI] [PMC free article] [PubMed]
- 44.Kanamori Y., Murakami M., Matsui T., Funaba M. JNK facilitates IL-1beta-induced hepcidin transcription via JunB activation. Cytokine. 2018;111:295–302. doi: 10.1016/j.cyto.2018.09.014. [DOI] [PubMed] [Google Scholar]; Y. Kanamori, M. Murakami, T. Matsui, M. Funaba, JNK facilitates IL-1beta-induced hepcidin transcription via JunB activation, Cytokine 111 (2018) 295-302. [DOI] [PubMed]
- 45.Balamurugan K., Wang J.M., Tsai H.H., Sharan S., Anver M., Leighty R., Sterneck E. The tumour suppressor C/EBPdelta inhibits FBXW7 expression and promotes mammary tumour metastasis. EMBO J. 2010;29(24):4106–4117. doi: 10.1038/emboj.2010.280. [DOI] [PMC free article] [PubMed] [Google Scholar]; K. Balamurugan, J.M. Wang, H.H. Tsai, S. Sharan, M. Anver, R. Leighty, E. Sterneck, The tumour suppressor C/EBPdelta inhibits FBXW7 expression and promotes mammary tumour metastasis, EMBO J 29(24) (2010) 4106-4117. [DOI] [PMC free article] [PubMed]
- 46.Thangaraju M., Rudelius M., Bierie B., Raffeld M., Sharan S., Hennighausen L., Huang A.M., Sterneck E. C/EBPdelta is a crucial regulator of pro-apoptotic gene expression during mammary gland involution. Development. 2005;132(21):4675–4685. doi: 10.1242/dev.02050. [DOI] [PubMed] [Google Scholar]; M. Thangaraju, M. Rudelius, B. Bierie, M. Raffeld, S. Sharan, L. Hennighausen, A.M. Huang, E. Sterneck, C/EBPdelta is a crucial regulator of pro-apoptotic gene expression during mammary gland involution, Development 132(21) (2005) 4675-4685. [DOI] [PubMed]
- 47.Zhang Y., Sif S., DeWille J. The mouse C/EBPdelta gene promoter is regulated by STAT3 and Sp1 transcriptional activators, chromatin remodeling and c-Myc repression. J. Cell. Biochem. 2007;102(5):1256–1270. doi: 10.1002/jcb.21356. [DOI] [PubMed] [Google Scholar]; Y. Zhang, S. Sif, J. DeWille, The mouse C/EBPdelta gene promoter is regulated by STAT3 and Sp1 transcriptional activators, chromatin remodeling and c-Myc repression, J Cell Biochem 102(5) (2007) 1256-1270. [DOI] [PubMed]
- 48.Zhang L., Pan J., Dong Y., Tweardy D.J., Dong Y., Garibotto G., Mitch W.E. Stat3 activation links a C/EBPdelta to myostatin pathway to stimulate loss of muscle mass. Cell Metabol. 2013;18(3):368–379. doi: 10.1016/j.cmet.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; L. Zhang, J. Pan, Y. Dong, D.J. Tweardy, Y. Dong, G. Garibotto, W.E. Mitch, Stat3 activation links a C/EBPdelta to myostatin pathway to stimulate loss of muscle mass, Cell Metab 18(3) (2013) 368-379. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.