

Abstract
Hepatoblastoma (HB) is the most common type of pediatric liver cancer. Activation of yes-associated protein (YAP) has been implicated in HB molecular pathogenesis. The transcriptional co-activator Yap regulates downstream gene expression through interaction with the TEA domain (TEAD) proteins. Nonetheless, YAP also displays functions that are independent of its transcriptional activity. The underlying molecular mechanisms by which Yap promotes HB development remain elusive. In the current study, we demonstrated that blocking TEAD function via the dominant-negative form of TEAD2 abolishes Yap-driven HB formation in mice and restrains human HB growth in vitro. When TEAD2 DNA-binding domain was fused with virus protein 16 transcriptional activation domain, it synergized with activated β-catenin to promote HB formation in vivo. Among TEAD genes, silencing of TEAD4 consistently inhibited tumor growth and Yap target gene expression in HB cell lines. Furthermore, TEAD4 mRNA expression was significantly higher in human HB lesions when compared with corresponding nontumorous liver tissues. Human HB specimens also exhibited strong nuclear immunoreactivity for TEAD4. Altogether, data demonstrate that TEAD-mediated transcriptional activity is both sufficient and necessary for Yap-driven HB development. TEAD4 is the major TEAD isoform and Yap partner in human HB. Targeting TEAD4 may represent an effective treatment option for human HB.
Hepatoblastoma (HB) is the most common pediatric liver cancer, with the highest incidence in children occurring before the age of 5 years. Approximately 100 cases of HB are diagnosed in the United States each year.1 Several risk factors associated with HB development have been identified, including low birth weight, familial adenomatous polyposis, and Beckwith-Wiedemann syndrome.2, 3 The clinical management of HB has evolved significantly over the past three decades.4, 5 Neoadjuvant chemotherapy and surgical resection are the standard treatment options for HB. However, drug resistance and distal metastases frequently develop in patients with high-risk HB, often precluding the success of treatments.5
HB is histologically divided into epithelial and mixed epithelial/mesenchymal subtypes.3, 6 The epithelial subtype, consisting of embryonic and fetal tumor cells, is the most frequently occurring HB histotype.3 The molecular mechanisms responsible for HB development remain poorly understood. Genome-wide analyses recently provided a global genomic landscape of genetic events occurring in human HBs.7, 8, 9 Among them, mutations in the β-catenin gene (CTNNB1) are identified in >80% of HBs, suggesting that activation of the Wnt/β-catenin signaling cascade is the dominant driver oncogenic event along HB development and progression.10, 11 Specifically, point mutations in exon 3 or large deletions in the N-terminal region of the CTNNB1 gene are found. β-Catenin mutations cause nuclear translocation and enhance the stabilization of β-catenin protein.10, 11 Subsequently, β-catenin interacts with nuclear cofactors of the T-cell factor/lymphoid enhancer-binding factor family to induce target gene expression. Intriguingly, mouse studies revealed that overexpression of activated forms of β-catenin in the mouse liver is insufficient to induce HB formation in vivo,12 strongly suggesting that additional molecular events cooperating with activated β-catenin are necessary to promote HB tumorigenesis.
The Hippo/yes-associated protein (YAP) cascade is a critical pathway in HB development.13 The Hippo signaling is an evolutionarily conserved cascade playing a vital role in tissue growth, organ size control, and tumorigenesis.14, 15 YAP and its paralog transcriptional co-activator with PDZ-binding motif (TAZ) are the two major effectors downstream of Hippo kinases and act as transcriptional co-activators.14, 15 In the canonical Hippo cascade, large tumor suppressor kinase 1/2, the mammalian Hippo kinases, phosphorylate and inactivate YAP/TAZ. When Hippo kinases are inhibited, YAP/TAZ become dephosphorylated and stabilized. Consequently, YAP/TAZ can translocate into the nucleus, interacting with DNA-binding transcription factors, particularly of the TEA domain (TEAD) family, to activate downstream gene expression and cell growth and survival.14, 15 There are four TEAD genes in mammals (namely, TEAD1, TEAD2, TEAD3, and TEAD4). TEAD transcription factors share similar DNA-binding motifs and may function to regulate a similar set of genes.16, 17 However, each TEAD gene has its own tissue distinctive expression, leading to the tissue-specific role for each TEAD protein.16 Although TEAD-mediated transcriptional regulation has been solidly proved to be a major oncogenic mechanism downstream of YAP/TAZ, mounting evidence indicates that YAP/TAZ possess additional functions independent of TEAD.18 For instance, YAP/TAZ can be incorporated in the β-catenin destruction complex and modulate the Wnt/β-catenin signaling.19 Furthermore, cytoplasmic pools of YAP/TAZ could sequester SMAD proteins and subsequently regulate the transforming growth factor-β signaling.20 Also, YAP/TAZ bind to Snail/Slug and regulate skeletal stem cell self-renewal and differentiation.21
In HB, YAP is activated (as indicated by its nuclear localization) in approximately 80% of human HB samples.13 More important, coordinated and almost ubiquitous Yap and β-catenin activation was only detected in human HB, but not in other malignant liver lesions, such as hepatocellular carcinoma or cholangiocarcinoma, implying the critical role of the two pathways in combination only in pediatric liver cancer.13 In accordance with this hypothesis, using hydrodynamic tail vein injection, it was demonstrated that co-expression of activated forms of Yap (YapS127A) and β-catenin (ΔN90-β-catenin) leads to the exclusive development of pure HB, but not other tumor types, in the mouse liver.13 On the basis of studies in vitro and in vivo, we investigated herein whether YAP oncogenic activity depends on its interactions with TEAD factors in HB. The data obtained indicate that TEAD-mediated transcriptional regulation is required for Yap-driven HB development, and TEAD4 may be the major TEAD isoform in human HB, thus representing a potential therapeutic target in this pediatric malignancy.
Materials and Methods
Plasmids and Reagents
The plasmids used in the experiments, including pT3-EF1α-ΔN90-β-catenin, pT3-EF1α-YapS127A, and pCMV–sleeping beauty transposase, were described in detail previously.13, 22, 23, 24 The dominant-negative form of TEAD2 (dnTEAD2) construct25 was kindly provided by Dr. Duojia Pan (Johns Hopkins University, Baltimore, MD). The TEAD2 DNA-binding domain fused with the virus protein 16 (VP16) transcriptional activation domain (TEAD2VP16) was purchased from Riken BioResource Center (Ibaraki, Japan). The dnTEAD2 and TEAD2VP16 plasmids were cloned into the pT3-EF1α vector via the Gateway cloning technology (Invitrogen, Carlsbad, CA). All plasmids for in vivo experiments were purified with the GenElute HP Endotoxin-Free Maxiprep kit (Sigma-Aldrich, St. Louis, MO). In addition, dnTEAD2 was inserted into a pLenti_CMV_Puro_DEST vector (Addgene, Cambridge, MA; number 17452) via the Gateway PCR cloning strategy. pLenti–enhanced green fluorescent protein plasmid was purchased from Addgene (number 17448) and used as a control. Short hairpin TEAD4 was purchased from Dharmacon (San Francisco, CA; number RHS4533-EG7004). The pLKO.1 plasmid (empty vector control) was obtained from Addgene (number 10879).
Hydrodynamic Tail Vein Gene Delivery
All FVB/N mice were purchased from the Jackson Laboratory (Bar Harbor, ME). The hydrodynamic tail vein gene delivery procedure was performed, as described in our previous studies.24, 26 Briefly, to assess the oncogenic potential of TEAD2VP16, 20 μg pT3-EF1α-TEAD2VP16, either alone or in combination with 20 μg pT3-EF1α-ΔN90-β-catenin, was mixed together with pCMV/sleeping beauty transposase at a ratio of 25:1 and delivered into 6- to 8-week–old FVB/N mice via injection into the lateral tail vein. To determine whether overexpression of dnTEAD2 affects Yap/β-catenin–induced HB formation in mice, 20 μg pT3-EF1α-YapS127A and 20 μg pT3-EF1α-ΔN90-β-catenin were injected together with 60 μg pT3-EF1α-dnTEAD2 or 60 μg pT3-EF1α (empty vector control) and 4 μg pCMV/sleeping beauty transposase into the mice. Mice were monitored continually and euthanized at specific time points, as indicated in the main text or when they became moribund, as previously described.27 Mice were maintained and monitored in accordance with protocols approved by the Committee for Animal Research at the University of California, San Francisco (San Francisco, CA).
Histology, Immunohistochemistry, and Assessment of Mouse Proliferation Rate
Liver specimens were fixed in 10% buffered formalin overnight at 4°C and embedded in paraffin. Hematoxylin and eosin (Thermo Fisher Scientific, Waltham, MA) staining was performed using a standard protocol on human and mouse liver sections (5 μm thick) to characterize the hepatocellular lesions histopathologically. For immunohistochemistry, epitope unmasking in deparaffinized sections was used by boiling the slides for 10 minutes in 10 mmol/L sodium citrate buffer (pH 6.0). Then, sections were blocked with 5% goat serum and the Avidin-Biotin blocking kit (Vector Laboratories, Burlingame, CA), and they were incubated with primary antibodies overnight at 4°C. The antibodies are listed in Table 1. Endogenous peroxidase activity was blocked by 20 minutes of incubation of the slides in 3% H2O2 (dissolved in methanol). Subsequently, the slides were incubated with secondary antibodies for 1 hour at room temperature. Finally, the reaction signal was visualized using the Vectastain ABC Elite Kit (Vector Laboratories) and developed with 3,3′-diaminobenzidine. Sections were counterstained with hematoxylin. As negative control, slides were incubated with secondary antibodies, omitting the primary antibodies. Images were taken using a bright-field microscope connected to a digital color camera (Leica, San Francisco, CA; model DFC295). The proliferation rate of the hepatocellular lesions was determined by calculating the Ki-67 labeling index in mouse livers. At least 2000 cells were counted for each sample.
Table 1.
Primary Antibodies Used for IHC and WB
| Antibody | Company | Catalog no. | Dilution | Method |
|---|---|---|---|---|
| VP16-tag | Santa Cruz Biotechnology (Dallas, TX) | sc-7545 | 1:200 | IHC |
| Myc-tag | Cell Signaling Technology Inc. (Danvers, MA) | 2978 | 1:1000 | WB |
| AFP | Proteintech (Rosemont, IL) | 14550-I-AP | 1:2000 | WB |
| DLK1 | Proteintech | 10636-I-AP | 1:1000 | WB |
| GAPDH | EMD Millipore (Billerica, MA) | 5174P | 1:10,000 | WB |
| Yap | Cell Signaling Technology Inc. | 4912S | 1:1000 | WB |
| Phosphorylated Yap | Cell Signaling Technology Inc. | 13619P | 1:1000 | WB |
| TEAD4 | Sigma-Aldrich | HPA056896 | 1:100 | IHC |
| Ki-67 | Thermo Fisher Scientific | MA5-14520 | 1:150 | IHC |
| β-Catenin | BD Biosciences (San Jose, CA) | 610153 | 1:200 | IHC |
AFP, α-fetoprotein; DLK1, Δ-like noncanonical Notch ligand 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IHC, immunohistochemistry; TEAD4, TEA domain transcription factor 4; VP16, virus protein 16; WB, Western blot analysis.
Protein Extraction and Western Blot Analysis
Liver tissues and cells were lysed in protein lysis buffer, which contains 30 mmol/L tris (pH 7.5), 150 mmol/L NaCl, 0.5% Na deoxycholate, 1% Nonidet-P40, 10% glycerol, 0.1% SDS, 2 mmol/L EDTA (Thermo Fisher Scientific), and protease and phosphatase inhibitor. The concentration of proteins was determined with the Bio-Rad Protein Assay Kit (Bio-Rad, Hercules, CA) using bovine serum albumin as a standard. Total protein lysates were denatured by gently mixing with Tris-Glycine SDS Sample Buffer (Bio-Rad) in a heat block for 5 minutes. Equal amounts of proteins were separated by SDS-PAGE gels and transferred to nitrocellulose membranes (Bio-Rad) by electroblotting. Nitrocellulose membranes were blocked for 60 minutes and then incubated with specific antibodies (Table 1) at 4°C overnight. After washing, membranes were incubated with a horseradish peroxidase secondary antibody (Jackson Immuno Research Laboratories Inc., West Grove, PA), diluted 1:10,000 for 60 minutes at room temperature and visualized using the Super Signal West Femto (Pierce Chemical Co, New York, NY). ImageJ software version 6.02 (NIH, Bethesda, MD; http://imagej.nih.gov/ij, accessed February 1, 2018) was used for Western blot densitometric analysis.
RT-qPCR
Total RNA was extracted from liver tissues or cells using the Quick-RNA MiniPrep Kit (Genesee Scientific, El Cajon, CA). cDNA was generated using the 5× iScriptTM RT Supermix (Bio-Rad), according to the instructions of the manufacturer. The real-time quantitative RT-PCR (RT-qPCR) was performed using the TaqMan Universal PCR Master Mix (Thermo Fisher Scientific) on an ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA). Analysis of mRNA levels was conducted with the QuantStudio Real-Time PCR software version 1.1 (Thermo Fisher Scientific). The RT-qPCR was conducted as follows: template denaturation at 95°C for 10 minutes, primer annealing at 95°C for 15 seconds, and extension step at 60°C for 1 minute. Forty cycles of amplification were used. All the primers used in the present study are listed in Table 2.
Table 2.
Real-Time Quantitative RT-PCR Primer Sequence Information
| Gene | Forward sequence | Reverse sequence |
|---|---|---|
| rRNA | 5′-CGGCTACCACATCCAAGGAA-3′ | 5′-GCTGGAATTACCGCGGCT-3′ |
| Human | ||
| CTGF (CCN2) | 5′-CCAATGACAACGCCTCCTG-3′ | 5′-GAGCTTTCTGGCTGCACCA-3′ |
| CYR61 (CCN1) | 5′-AGCCTCGCATCCTATACAACC-3′ | 5′-GAGTGCCGCCTTGTGAAAGAA-3′ |
| TEAD1 | 5′-ATGCCAACCATTCTTACAGTGAC-3′ | 5′-ACAGTTCCTTTAAGCCACCTTTC-3′ |
| TEAD2 | 5′-GCCTCCGAGAGCTATATGATCG-3′ | 5′-TCACTCCGTAGAAGCCACCA-3′ |
| TEAD3 | 5′-TGGACCCTCTCAGGACATCAA-3′ | 5′-CCAGGGGCTCATAACTGCTG-3′ |
| TEAD4 | 5′-GGACACTACTCTTACCGCATCC-3′ | 5′-TCAAAGACATAGGCAATGCACA-3′ |
| YAP (YY1AP1) | 5′-TAGCCCTGCGTAGCCAGTTA-3′ | 5′-TCATGCTTAGTCCACTGTCTGT-3′ |
| SURVIVIN (BIRC5) | 5′-AGGACCACCGCATCTCTACAT-3′ | 5′-AAGTCTGGCTCGTTCTCAGTG-3′ |
| AXL | 5′-TGTAAAACGACGGCCAGT-3′ | 5′-CAGGAAACAGCTATGACC-3′ |
| Mouse | ||
| Ctgf (Ccn2) | 5′-GGGCCTCTTCTGCGATTTC-3′ | 5′-ATCCAGGCAAGTGCATTGGTA-3′ |
| Cyr61 (Ccn1) | 5′-CTGCGCTAAACAACTCAACGA-3′ | 5′-GCAGATCCCTTTCAGAGCGG-3′ |
| Survivin (Birc5) | 5′-GCCACGCATCCCAGCTT-3′ | 5′-TTTCAAAATACCACTGTCTCCTTCTC-3′ |
| Axin2 | 5′-CAGATGAACCTGAAGGATACCAAA-3′ | 5′-AGACAACGCTGTTGTTCTCAATG-3′ |
| Tbx3 | 5′-CAGGCAGCCTTCAACTGCTT-3′ | 5′-GGACACAGATCTTTGAGGTTGGA-3′ |
| Cyclind1 (Ccnd1) | 5′-CGTGGCCTCTAAGATGAAGGA-3′ | 5′-CCTCGGGCCGGATAGAGTAG-3′ |
| Yap | 5′-TACTGATGCAGGTACTGCGG-3′ | 5′-TCAGGGATCTCAAAGGAGGAC-3′ |
| Tead1 | 5′-AAGCTGAAGGTAACAAGCATGG-3′ | 5′-GCTGACGTAGGCTCAAACCC-3′ |
| Tead2 | 5′-GAAGACGAGAACGCGAAAGC-3′ | 5′-GATGAGCTGTGCCGAAGACA-3′ |
| Tead3 | 5′-CAACCAGCACAATAGCGTCCA-3′ | 5′-CTGAAAGCTCTGCTCGATGTC-3′ |
| Tead4 | 5′-CAACCTGGAACATCCCACGAT-3′ | 5′-GAAAGCCGAGAACTCCAACAT-3′ |
CTGF, connective tissue growth factor; Cyr61, cysteine rich angiogenic inducer 61.
In Vitro Studies
The Hep293TT cell line was a kind gift of Dr. Gail Tomlinson (University of Texas Southwestern Medical Center, Dallas, TX). Cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Sigma-Aldrich) supplemented with 25 mmol/L HEPES, 10% fetal bovine serum (Gibco, Grand Island, NY), 100 U/mL penicillin, and 100 μg/mL streptomycin (Sigma-Aldrich).28 HepG2 cells, purchased from ATCC (Manassas, VA), were instead cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were grown in a 5% CO2 atmosphere, at 37°C. For the colony formation assay, cells were seeded in 100 × 20-mm culture dishes and, when they reached 50% to 60% confluence, dnTEAD2, enhanced green fluorescent protein, pLKO, or short hairpin TEAD4 lentivirus fluid was added into the culture medium. Seventy-two hours later, the cells were screened, and stable transfected cells were generated by replacing the old medium with fresh medium supplemented with puromycin at the concentration of 2 μg/mL for HepG2 and 1 μg/mL for Hep293TT. After 72 hours of selection, cells were plated at 1000 cells/well for colony formation assays. Two weeks later, colonies were stained with crystal violet and manually counted. To knockdown TEAD genes, cells were seeded in 6-well plates and transfected with 15 pmol siRNA targeting TEAD1-4 (Thermo Fisher Scientific), separately, complexed to Lipofectamine RNAiMAX and Opti-MEMI Reduced Serum Medium (Thermo Fisher Scientific). As control, a scrambled siRNA (number 4390844; Thermo Fisher Scientific) was designed to have no known targets in the cells. Seventy-two hours after transfection, cells were collected for RNA analysis. For cell growth assays, cells were treated with crystal violet staining solution. Subsequently, cells were washed, incubated with lysis solution, and placed on a shaker at room temperature for 20 to 30 minutes. The dissolving solution was added to 96-well plates, and the OD at 590 nm was measured using a BioTek ELx808 Absorbance Microplate Reader (BioTek Instruments, Inc., Winooski, VT). Transient transfection experiments with TEAD2VP16 and TEAD4 (in pCMV6-Neo plasmid; Origene Technologies Inc., Rockville, MD; number SC111037) constructs were conducted in HepG2 and Hep293TT cell lines using the Lipofectamine 2000 Reagent (Thermo Fisher Scientific). Cell proliferation was determined in human HB cell lines, after 24-hour serum starvation (corresponding to the 0 time point) and 24 and 48 hours later using the bromodeoxyuridine/5-bromo-2′-deoxyuridinex Cell Proliferation Assay Kit (Cell Signaling Technology Inc., Danvers, MA). As concerns apoptosis, two distinct stimuli were applied to HB cells (namely, 24-hour serum starvation and staurosporine administration), and apoptotic cell death was assessed using the Cell Death Detection Elisa Plus Kit (Roche Molecular Biochemicals, Indianapolis, IN), following the manufacturer's instructions. Staurosporine (Cell Signaling Technology Inc.) was administered at 1 μmol/L concentration, and apoptosis was evaluated at 4- and 8-hour time points. For both cell proliferation and apoptosis assays, the untreated cells in 0% serum for 24 hours were used as the baseline, and all values were normalized to the baseline reading. All experiments were repeated at least three times in triplicate.
Human Tissue Samples
A collection of 28 paraffin-embedded and formalin-fixed human HB specimens, which were previously collected, was used for the present investigation. Liver cancer patients' clinicopathological features are summarized in Table 3. All HB samples were obtained from the archives of the pathology departments of Semmelweis University (Budapest, Hungary) and the Medical University of Greifswald (Greifswald, Germany). The study was performed under the Institutional Review Board approval of the local Ethical Committee of the Medical University of Greifswald and the Regional Ethical Committee of Semmelweis University. Written informed consent was acquired from all the participants.
Table 3.
Clinicopathological Features of Human HB Patients
| Variables | Value∗ |
|---|---|
| No. of patients | 28 |
| Male | 16 |
| Female | 12 |
| Age (years)† | 4.4 ± 2.8 |
| Tumor morphology | |
| Fetal | 14 |
| Embryonal | 8 |
| Mixed | 6 |
| Recurrence | |
| Yes | 10 |
| No | 18 |
| Lung metastases | |
| Yes | 9 |
| No | 19 |
HB, hepatoblastoma.
Data are given as number of patients, unless otherwise indicated.
Data are given as means ± SD.
Statistical Analysis
All data are presented as means ± SD for each group. Statistical differences between two groups were determined using the U-tests embedded in the Prism 6 software version 6.0 (Graph Pad Software Inc., La Jolla, CA). P < 0.05 was considered statistically significant.
Results
dnTEAD2 Inhibits HB Growth in Vitro and in Vivo
As a first step to investigate whether TEAD-mediated transcriptional activity is indispensable for Yap-driven HB development, the effect of dnTEAD2 was tested in in vitro and in vivo models of HB. Specifically, dnTEAD2 lacks the DNA-binding domain, which results in nuclear sequestration of Yap and inhibition of its transcriptional activity.25
For in vitro experiments, two human HB cell lines, HepG2 and Hep293TT, were used. The HepG2 cell line was isolated from a 15-year–old male29 and has been widely used as a hepatocellular carcinoma cell line. However, molecular studies have recently shown that it contains a large deletion of exon 3 of the β-catenin gene and other molecular features that are consistent with its HB nature.30 Hep293TT cells were isolated from an HB developed in a 5-year–old male. Similar to HepG2, Hep293TT cells contain a N-terminal deletion of the β-catenin exon 3.28 More important, we have recently shown that YAP is activated in both HB cell lines.31 In light of this body of information, dnTEAD2 was overexpressed in HepG2 and Hep293TT cells. As expected, dnTEAD2 effectively inhibited the expression of Yap downstream targets, such as cysteine-rich angiogenic inducer 61 (CRY61), connective tissue growth factor (CTGF), AXL receptor tyrosine kinase (AXL), and baculoviral IAP repeat containing 5 (BIRC5, alias SURVIVIN) in HepG2 and Hep293TT cells (Supplemental Figure S1A). Forced expression of dnTEAD2 strongly suppressed HepG2 and Hep293TT cell growth in the colony formation assay (Supplemental Figure S1B). Consistently, transient overexpression of dnTEAD2 led to decreased proliferation and increased apoptosis in these cells (Supplemental Figure S2). Overexpression of dnTEAD2 neither influenced the expression levels of β-catenin and its target genes (glutamine synthetase or GS and T-box 3 or TBX3) (Supplemental Figure S3) nor affected β-catenin nuclear localization (Supplemental Figure S4) in HepG2 and Hep293TT cells. The results suggest that dnTEAD2 does not regulate the activity of the Wnt/β-catenin pathway in HB cells.
Next, dnTEAD2 was co-expressed with Yap and β-catenin into the mouse liver (Yap/β-catenin/dnTEAD2, n = 5) by hydrodynamic tail vein injection. pT3-EF1α (empty vector) was also co-injected with Yap and β-catenin as control (Yap/β-catenin/pT3, n = 5) (Figure 1A). Similar to that reported for the Yap/β-catenin model,13 all mice injected with Yap/β-catenin/pT3 developed lethal burden of liver tumor and were euthanized between 10 and 11 weeks after injection (Figure 1B). In striking contrast, all Yap/β-catenin/dnTEAD2–injected mice appeared to be healthy, showing no abdominal palpable mass at this time point. Yap/β-catenin/dnTEAD2 mice were euthanized between 12 and 18 weeks after injection. On gross inspection, liver tissues from the five Yap/β-catenin/dnTEAD2 mice appeared to be completely normal (Figure 1C). Histologically, all liver tumor lesions found in Yap/β-catenin/pT3 mice were equivalent to those detected in Yap/β-catenin–injected mice in previous experiments.13 They were classified as mouse HB and were highly proliferating, as demonstrated by Ki-67 immunohistochemical staining (Figure 1C). On the other hand, liver tissues from Yap/β-catenin/dnTEAD2 mice appeared to be normal, indistinguishable from normal livers of wild-type mice (Figure 1C). In accordance with morphologic data, the proliferation rate (as assessed by Ki-67 index) of Yap/β-catenin/dnTEAD2 livers (n = 5) was equivalent to that of normal livers (injected with the empty vector; n = 5) at the same time point (1.7 ± 0.7 versus 2.0 ± 0.8, respectively; not significant), whereas it was significantly higher in Yap/β-catenin/pT3 livers (n = 5; 31.8 ± 9; P < 0.001 versus both Yap/β-catenin/dnTEAD2 and control livers).
Figure 1.

Blockade of Yap-mediated transcriptional regulation via dnTEAD2 suppresses hepatoblastoma growth in mice. A: Study design. B: Survival curve of Yap/β-catenin/pT3 and Yap/β-catenin/dnTEAD2 mice. C: Gross images, hematoxylin and eosin (H&E) staining, and Ki-67 immunohistochemistry of Yap/β-catenin/pT3 and Yap/β-catenin/dnTEAD2 mouse livers at 10.7 and 17 weeks after hydrodynamic injection, respectively. Arrows indicate the proliferation activity of hepatocytes. n = 5 Yap/β-catenin/pT3 and Yap/β-catenin/dnTEAD2 mice (B). Scale bar = 100 μm (C, middle and right columns). Original magnification, ×100 (C). SL, surrounding liver tissues; T, tumor tissues.
Overall, our study indicates that blockade of Yap/TEAD transcriptional activity via dnTEAD2 suppresses HB cell growth in vitro and tumor development in vivo.
Activated TEAD (TEAD2VP16) Synergizes with ΔN90-β-Catenin to Promote HB Development in Mice
Subsequently, it was determined whether Yap/TEAD-mediated transcriptional activation is sufficient to promote HB development in vivo. To induce TEAD activity in vivo, the pT3-EF1α-TEAD2VP16 construct was generated. This construct consists of the fusion between the N-terminal region of TEAD2, containing the TEA domain, and the activation domain of herpes simplex virus VP16, thus allowing TEAD transcriptional activity independent of Yap.32 TEAD2VP16 strictly functions via promoting TEAD-dependent gene modulation while lacking any other TEAD-independent function of Yap, such as regulating the transforming growth factor-β or Wnt/β-catenin signaling pathways.32
Thus, TEAD2VP16 (n = 4) was hydrodynamically transfected either alone or in combination with ΔN90-β-catenin, into the mouse liver (TEAD2VP16/β-catenin, n = 9) (Figure 2). Similar to the results observed when activated Yap was overexpressed alone in the mouse liver,13 overexpression of TEAD2VP16 alone did not trigger liver tumor development (Figure 2). The overexpression of ΔN90-β-catenin alone is unable to promote liver tumor formation in mice.33 In vivid contrast, co-expression of TEAD2VP16/β-catenin resulted in massive liver tumor growth, and mice needed to be euthanized approximately 10 weeks after injection because of high tumor burden (Figure 2). The proliferation index was similar between TEAD2VP16-injected (n = 4) and control (n = 5) mice (1.5 ± 0.8 versus 1.9 ± 1.0, respectively; not significant), whereas a much stronger proliferation was detected in TEAD2VP16/β-catenin at the same time point (n = 5; 24.2 ± 4.6; P < 0.001 versus both TEAD2VP16 and control livers).
Figure 2.

TEAD2VP16 synergizes with ΔN90-β-catenin to promote hepatoblastoma development in mice. A: Survival curve of TEAD2VP16 and TEAD2VP16/β-catenin mice. B: Gross images and hematoxylin and eosin (H&E) staining of TEAD2VP16 and TEAD2VP16/β-catenin mouse livers both at 10.1 weeks after hydrodynamic injection. n = 4 (A, TEAD2VP16 mice); n = 9 (A, TEAD2VP16/β-catenin mice). Scale bars: 250 μm (B, middle row); 100 μm (B, bottom row). Original magnifications: ×40 (B, middle row); ×100 (B, bottom row); ×400 (B, insets).
Microscopically, altered cells could be appreciated as early as 3 weeks after TEAD2VP16/β-catenin injection (Figure 3A) and were positive in the nucleus and cytoplasm for ectopically injected β-catenin (Figure 3B). At this time point, proliferation was either low or absent (Figure 3C). Tumor nodules started to emerge from the liver parenchyma by 5 weeks (not shown) and occupied most of the hepatic parenchyma by 10 weeks after injection (Figure 3D). Morphologically, the tumor cells were consistent with mouse HB and were characterized by small tumor cell size and small round or oval nuclei (Figure 3E). All tumor cells showed nuclear staining of ectopically injected β-catenin (Figure 3G) and VP16 tag (Figure 3H). Furthermore, the high proliferation rate of TEAD2VP16/β-catenin lesions was underlined by widespread Ki-67 immunoreactivity (Figure 3F).
Figure 3.

Histopathological and biochemical characterization of liver tumors developed in TEAD2VP16/β-catenin and Yap/β-catenin mice. A: Hematoxylin and eosin (H&E) staining of a TEAD2VP16/β-catenin mouse liver 3 weeks after hydrodynamic tail vein injection. Altered cells (arrows) were characterized mainly by nuclei of increased size and bizarre shape. B and C: Corresponding representative immunohistochemical images of β-catenin (B) and Ki-67 (C) in the liver. D–H: Representative H&E staining (D and E) and corresponding images of Ki-67 (F), β-catenin (G), and VP16 tag (H) staining in the liver from a TEAD2VP16/β-catenin mouse 10 weeks after hydrodynamic tail vein injection. Scale bars: 250 μm (D); 100 μm [A, B, G (main image), and H (main image)]; 25 μm [C, E, F, G (inset), and H (inset)]. Original magnifications: ×40 (D); ×100 [A, B, G (main image), and H (main image)]; ×400 [C, E, F, G (inset), and H (inset)].
Next, the biochemical and molecular features of mouse in which HB was induced by Yap/β-catenin and TEAD2VP16/β-catenin constructs were compared. Using Western blot analysis, similar levels of ectopically injected Myc-tagged β-catenin were found in Yap/β-catenin and TEAD2VP16/β-catenin HB tumor specimens (Figure 4A). Endogenous levels of nuclear and activated Yap could be detected in all normal livers and tumor samples. A higher molecular weight band could be detected in nuclear extracts from Yap/β-catenin HB tumors because of the ectopically expressed Flag-tagged Yap (Figure 4B). Overexpression of TEAD2/VP16 did not lead to increased levels of nuclear Yap, suggesting that TEAD2/VP16 promotes HB formation independent of Yap activation. Expression of the hepatocellular tumor marker α-fetoprotein and the HB marker Δ-like noncanonical Notch ligand 1 was equivalent in Yap/β-catenin and TEAD2VP16/β-catenin mouse HBs (Figure 4A). In addition, using RT-qPCR, we found that Yap/TEAD target genes, such as Ctgf, Cyr61, and Survivin, as well as β-catenin target genes, including Axin2, Tbx3, and cyclin D1, were found to be up-regulated in both Yap/β-catenin and TEAD2VP16/β-catenin HB samples (Figure 4C).
Figure 4.

Molecular characterization of liver tumors in TEAD2VP16/β-catenin and Yap/β-catenin mice. A: Western blot analysis of Myc-tag, β-catenin, α-fetoprotein (Afp), and Δ-like noncanonical Notch ligand 1 (Dlk1) proteins in normal liver (NL) from wild-type FVB/N mice as well as in liver tumors from TEAD2VP16/β-catenin and Yap/β-catenin mice. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was used as the loading control. The image is representative of three independent experiments. B: Western blot analysis of nuclear extracts showing nuclear Yap expression in the liver samples. Histone H3 was used as the loading control. C: Real-time quantitative RT-PCR was performed to determine the expression levels of Yap and β-catenin target genes in normal liver and TEAD2VP16/β-catenin and Yap/β-catenin hepatoblastoma (HB) tissues. Data are expressed as means ± SD (C). n ≥ 4 independent replicates (C). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 when compared with normal liver (U-test).
In summary, TEAD2VP16 synergizes with activated β-catenin to promote mouse HB development at equivalent latency and efficacy as YapS127A. Thus, TEAD2VP16 recapitulates, at least grossly, YapS127 oncogenic activity in the mouse liver. Altogether, these data suggest that TEAD-mediated transcriptional regulation is both necessary and sufficient for Yap oncogenic action in HB.
TEAD4 Is the Predominant TEAD Isoform along HB Development
In mammalian cells, there are four TEAD isoforms (namely, TEAD1, TEAD2, TEAD3, and TEAD4).16 The four TEAD proteins bind to similar motifs and, therefore, potentially can all interact with Yap to promote downstream gene expression. Herein, we investigated which TEAD isoform is the most important in HB development. The levels of TEAD1-4 mRNA were assessed by RT-qPCR in a small collection of human HB specimens (n = 14) (Figure 5). All four TEAD genes were expressed in human HBs and corresponding nontumorous livers. Statistical analysis revealed that TEAD2, TEAD3, and TEAD4 mRNA levels were significantly up-regulated in human HBs compared with matching nonneoplastic counterparts, whereas TEAD1 expression was equivalent in tumorous and nontumorous livers. The mRNA findings were in agreement with those that were retrieved from the recent published gene expression array data composed of normal liver tissues (n = 6) and human HBs (n = 51)7 (Supplemental Figure S5). In Yap/β-catenin–induced mouse HB models, the expression level of Tead4 mRNA was strongly induced, Tead1 expression was moderately up-regulated, and the levels of Tead2 and Tead3 were down-regulated (Supplemental Figure S6). Because it was recently demonstrated that nuclear translocation of TEAD is required for Yap-dependent transcriptional activity,34 the cellular expression patterns of TEAD4 were investigated in human HB tumor tissues by immunohistochemistry. Nontumorous liver tissues displayed faint to moderate cytoplasmic staining for TEAD4. In striking contrast, a strong TEAD4 nuclear immunoreactivity was detected in most (25/28; 85.7%) human HB specimens (Figure 6). Similarly, an intense immunolabeling for TEAD4 was detected in the nuclei of HB lesions, but not in surrounding, unaffected tissues, of Yap/β-catenin livers (Supplemental Figure S7).
Figure 5.

TEAD4 is the predominant TEAD isoform in human hepatoblastoma (HB) samples. Real-time quantitative RT-PCR of TEAD1, TEAD2, TEAD3, and TEAD4 mRNA levels in 14 paired human normal liver and corresponding HB samples. Each symbol represents the mean value of an individual sample. ∗∗P < 0.01, ∗∗∗P < 0.001 when compared with normal liver (U-test). NT, nontumorous liver; T, tumor.
Figure 6.

Frequent activation of TEAD4 in human hepatoblastoma (HB) specimens. Immunohistochemical pattern of TEAD4 protein in normal liver (A) and two human HB specimens (B). Normal liver exhibits faint/absent cytoplasmic immunoreactivity for Tead4, whereas HB lesions display strong nuclear accumulation of the protein. The three specimens are depicted in two magnifications. Scale bars: 500 μm (A and B, top row); 100 μm (A and B, bottom row). Original magnifications: ×40 (A and B, top row); ×200 (A and B, bottom row). H&E, hematoxylin and eosin.
Next, the functional relevance of the four TEAD genes for the Yap transcriptional program was investigated in human HB cells. TEAD1, TEAD2, TEAD3, and TEAD4 were silenced in HepG2 and 293TT human HB cell lines using specific siRNAs (Figure 7). As expected, each siRNA was effective in silencing the expression of its specific target in both HepG2 and 293TT cells (Figure 7). The expression of Yap/TEAD target genes, including CTGF, CYR61, AXL, and BIRC5, was analyzed after silencing of each TEAD gene. Depletion of TEAD4 significantly inhibited CTGF, CYR61, AXL, and BIRC5 expression in both HepG2 and Hep293TT cells, whereas silencing of TEAD1, TEAD2, and TEAD3 did not lead to consistent down-regulation of these genes in the two human HB cell lines. Consistently, forced overexpression of TEAD4 led to increased levels of YAP target genes in the two HB cell lines (Supplemental Figure S8). In contrast, no effect on the expression levels of β-catenin targets, such as GS and TBX3, was consistently detected in TEAD4-overexpressed HB cells (Supplemental Figure S8).
Figure 7.

Silencing of TEAD4 inhibits Yap target gene expression in hepatoblastoma (HB) cell lines. HepG2 (A) or Hep293TT (B) human HB cell lines were transfected with scrambled siRNA (siSC), small interfering (si) TEAD1, siTEAD2, siTEAD3, or siTEAD4. The expression of TEAD isoforms as well as that of YAP/TEAD target genes CTGF, CYR61, AXL, and SURVIVIN were analyzed using real-time quantitative RT-PCR. Data are expressed as means ± SD (A and B). n = 3 independent replicates (A and B). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 when compared with siSC (U-test).
Next, it was examined how each TEAD isoform may regulate HB cell growth in vitro. Silencing of TEAD4 led to strong inhibition of proliferation and induction of apoptosis in HepG2 and 293TT cells, whereas intermediate effects on cell growth were detected in TEAD2-silenced cells (Figure 8). Conversely, forced overexpression of TEAD4 by transient transfection resulted in a further increase in proliferation and a decline of apoptosis in the two human HB cell lines (Figure 9). In contrast, no significant differences in cell growth or a paradoxical increase in proliferation were detected in TEAD1- or TEAD3-depleted cells compared with untreated or scrambled siRNA-treated cells (Figure 8).
Figure 8.

Silencing of TEAD4 decreases proliferation and augments apoptosis in hepatoblastoma (HB) cell lines. HepG2 (A) and Hep293TT (B) human HB cell lines were transfected with scrambled siRNA (siSC), small interfering (si) TEAD1, siTEAD2, siTEAD3, or siTEAD4. Cell proliferation and apoptosis were measured. Untreated cells cultured in serum-free medium for 24 hours were used as baseline (ie, 1), and all cell proliferation and apoptosis rates were normalized to the baseline values. Data are expressed as means ± SD (A and B). n = 5 independent replicates (A and B). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 when compared with siSC (U-test).
Figure 9.

Overexpression of TEAD4 increases proliferation and augments apoptosis in hepatoblastoma (HB) cell lines. HepG2 (A) or Hep293TT (B) human HB cell lines were transfected with empty vector or TEAD4. Cell proliferation and apoptosis were measured. Untreated cells cultured in serum-free medium for 24 hours were used as baseline (ie, 1), and all cell proliferation and apoptosis rates were normalized to the baseline values. Data are expressed as means ± SD (A and B). n = 5 independent replicates (A and B). ∗∗∗P < 0.001 when compared with empty vector (U-test).
To further validate the functional role of TEAD4 in HB biology, TEAD4 expression was abolished using shRNA-based gene depletion. HepG2 and 293TT cells were infected with pLKO.1 or short hairpin TEAD4 lentiviral particles. Similar to siRNA experiments, short hairpin TEAD4 strongly suppressed TEAD4 gene expression in HepG2 and 293TT HB cell lines, leading to the decreased expression of CTGF and CYR61 target genes as well as declined cell growth, as measured by colony formation, proliferation, and apoptosis assays (Supplemental Figure S9).
In summary, these data indicate the up-regulation and activation of TEAD4 in human HBs. Modulation of TEAD4 expression strongly influences HB cell growth inhibition in vitro. Together, these data strongly suggest that TEAD4 is the major TEAD family member mediating Yap transcriptional activity in human HB.
Discussion
Deregulation of the Hippo pathway with activation of its downstream effector YAP has been implicated in multiple hepatic malignancies, including hepatocellular carcinoma, cholangiocarcinoma, and HB.35 Besides cancer, Yap is also involved in hepatic stellate cell activation and fibrosis36, 37 as well as in the development of nonalcoholic fatty liver disease.38 Overall, these data imply YAP as a key molecule in various liver diseases. In human HBs, YAP is activated in approximately 70% to 80% of specimens13, 39 and is associated with aggressive or high-risk HBs.7, 40 Furthermore, it has been found that silencing of YAP reduces human HB cell growth in vitro, whereas overexpression of active Yap cooperates with oncogenic β-catenin to induce HB formation in mice.13 Taken together, this body of evidence supports a critical role for YAP along HB development and progression. However, how YAP functions to promote HB formation remains poorly understood. Previously, YAP has been shown to interact with TEAD genes to regulate target gene expression.13 In addition, YAP possesses many functions that are independent of TEAD-mediated transcriptional regulation. For instance, in a study by Azzolin et al,19 it was found that YAP and its paralog TAZ are sequestered in the β-catenin destruction complex, together with AXIN and glycogen synthase kinase–3β, in the absence of Wnt ligands. In this setting, YAP and TAZ are essential to recruit β–transducin repeat containing E3 ubiquitin protein ligase to the complex, leading to β-catenin degradation and inactivation. When Wnt receptors are activated by Wnt ligands, YAP and TAZ are instead released from the destruction complex, ensuring the activation of the β-catenin cascade. Furthermore, it has been demonstrated that YAP/TAZ control the subcellular localization of activated phosphorylated SMAD2/3. Specifically, cytoplasmic pools of Yap/TAZ sequester phosphorylated SMAD2/3 in the cytoplasm and suppress the transforming growth factor-β signaling.20 Herein, we tested the hypothesis that YAP acts via TEAD-mediated transcriptional regulation to promote HB formation in vivo. Blocking TEAD function via dnTEAD2 abolished Yap-driven HB formation. In addition, when TEAD2 was fused with the VP16 transcriptional activator, it functioned in association with activated β-catenin to enhance the growth of HB cells in the absence of activated Yap. Taken together, these findings suggest that TEAD-mediated transcriptional regulation is both required and sufficient for Yap-induced HB development. Additional studies are needed to identify the critical target genes regulated by YAP and their role along HB development.
In mammalian cells, four TEAD family members (namely, TEAD1, TEAD2, TEAD3, and TEAD4) have been discovered. By combining gene expression analysis, immunohistochemistry in human HB samples, together with siRNA-based gene silencing and transient transfection in human HB cell lines, it was found that TEAD4 expression is remarkably up-regulated and localized in the nucleus in human HB samples compared with normal liver tissues. Silencing of TEAD4 consistently led to decreased expression of Yap/TEAD target genes and reduced HB cell growth. A completely opposite trend was detected when TEAD4 was instead forcedly overexpressed in HB cell lines. Furthermore, we discovered that TEAD4 suppression results in down-regulation of Yap, whereas TEAD4 overexpression up-regulates it. This implies the possible existence of a feed-forward mechanism reinforcing YAP activation in HB cells. Thus, TEAD4 might be the major TEAD isoform in human HB. In previous studies, TEAD4 was established as the main TEAD isoform in hepatocellular carcinoma development.41, 42 According to these data, it is tempting to speculate that TEAD4 might represent the key TEAD protein along liver carcinogenesis regardless of the type of tumor. Obviously, this hypothesis requires additional and more thorough validation.
Finally, the present body of evidence suggests that targeting Yap/TEAD-mediated transcriptional regulation is a valid strategy for HB treatment. Consistent with this hypothesis, verteporfin, a small molecule that disrupts YAP/TEAD interaction, robustly hinders HB cell growth.13 However, verteporfin has many other functions independent of Yap/TEAD and is unlikely to be effective clinically as a YAP/TEAD inhibitor.43 Recently, a study demonstrated that Yap and TEAD interact and recruit the Mediator complex (a protein complex that controls various features of transcriptional activation) as well as cyclin-dependent kinase 9 (CDK9) elongating kinases to induce downstream gene expression.43 Therefore, targeting CDK9 might be used as an effective approach to disrupt Yap/TEAD-mediated transcriptional regulation for HB treatment. It would be of high importance to determine whether CDK9 is required for HB cell growth and, consequently, the potency of small-molecule CDK9 inhibitors, such as dinaciclib, flavopiridol, or seliciclib,44 in inhibiting HB cell growth in vitro and in vivo. Nonetheless, it is worth underlining that these small molecules are not specific for CDK9 and they target multiple cellular processes, including cell cycle regulation45 and the unfolded protein response.46 The lack of selectivity against other CDKs might limit their clinical use because of possible toxicity. Thus, more specific drugs directed against CDK9 should be developed. Among them, MC18029547 is a newly developed selective CDK9 inhibitor, and it could be tested both in vitro and in vivo, for the treatment of HB.
In summary, this study demonstrates that TEAD-mediated transcriptional activity is indispensable for Yap-driven HB development, and TEAD4 might be the major TEAD isoform in human HB. Furthermore, the present data support the development and testing of TEAD4 inhibitors as a therapeutic modality for the treatment of human HB.
Acknowledgments
We thank Dr. Gail Tomlinson (University of Texas Southwestern Medical Center, Dallas, TX) for providing the Hep293TT cell line; Dr. Duojia Pan (Johns Hopkins University, Baltimore, MD) for providing the dominant-negative form of the TEA domain 2 construct; and the Medical University of Greifswald (Greifswald, Germany) and Semmelweis University (Budapest, Hungary) for providing human hepatoblastoma specimens.
Footnotes
Supported by NIH grants R01CA204586 (S.P.M. and X.C.) and P30DK026743 (University of California, San Francisco Liver Center); and National Nature Science Foundation of China grant 81802774 (P.L.).
J.Z. and P.L. contributed equally to this work.
Disclosures: None declared.
Supplemental material for this article can be found at https://doi.org/10.1016/j.ajpath.2019.01.016.
Contributor Information
Diego F. Calvisi, Email: diego.calvisi@uni-greifswald.de.
Xin Chen, Email: xin.chen@ucsf.edu.
Supplemental Data
Supplemental Figure S1.

Blockade of Yap transcriptional activity via dnTEAD2 inhibits the growth of hepatoblastoma cell lines. A: mRNA expression levels of CTGF, CYR61, AXL, and SURVIVIN in HepG2 and Hep293TT cell lines overexpressing enhanced green fluorescent protein (EGFP) or dnTEAD2. B: Colony formation assays in HepG2 and Hep293TT cells overexpressing EGFP or dnTEAD2; left panels show representative images, and right panels show the colonies that were counted and the corresponding data. Data are expressed as means ± SD. n ≥ 3 independent experiments (A and B, right panels). ∗∗P < 0.01, ∗∗∗P < 0.001 when compared with EGFP (U-test).
Supplemental Figure S2.

Relative proliferation and apoptosis after overexpression of dnTEAD2 at different time points (24 and 48 hours) in hepatoblastoma cell lines. A and B: HepG2 cells (A) and Hep293TT cells (B); top panels show proliferation, and bottom panels show Apoptosis. Untreated cells cultured in serum-free medium were used as baseline (ie, 1), and all cell proliferation and apoptosis rates were normalized to the baseline values. C: The proapoptotic effect of dnTEAD2 in HepG2 and Hep293TT cells was further augmented with the use of the apoptosis inducer staurosporine control. Data are expressed as means ± SD. n = 9 independent replicates (A–C). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001 when compared with enhanced green fluorescent protein (EGFP; U-test).
Supplemental Figure S3.
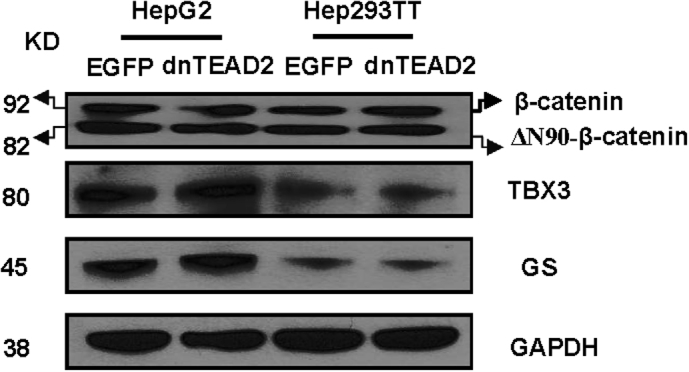
Overexpression of dnTEAD2 does not affect the Wnt/β-catenin pathway. Western blot analysis of β-catenin and its downstream targets, glutamine synthetase (GS) and T-box 3 (TBX3), in HepG2 and Hep293TT cells transfected with enhanced green fluorescent protein (EGFP) or dnTEAD2. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Supplemental Figure S4.

Overexpression of dnTEAD2 does not affect β-catenin localization. A: Western blot analysis of cytoplasmic and nuclear levels of β-catenin in HepG2 and Hep293TT cells transfected with enhanced green fluorescent protein (EGFP) or dnTEAD2. β-Tubulin and histone H3 were used as loading controls for cytoplasmic and nuclear protein fractions, respectively. B: Immunofluorescence staining of β-catenin protein in HepG2 and Hep293TT cells transfected with EGFP or dnTEAD2. Scale bar = 75 μm (B).
Supplemental Figure S5.

TEAD1, TEAD2, TEAD3, and TEAD4 mRNA expression in human normal liver (NL) and corresponding hepatoblastoma (HB) samples based on expression array data. Dot plot representation of the relative expression of TEAD1-4 genes in human normal liver or corresponding HB samples. Data are expressed as means ± SD of independent replicates. n = 6 (human normal liver); n = 51 (HB samples). ∗∗P < 0.01, ∗∗∗∗P < 0.0001 when compared with human normal liver (U-test).
Supplemental Figure S6.

Molecular characterization of liver tumors in Yap/β-catenin mice. Real-time quantitative RT-PCR was performed to determine the expression levels of Tead1-4 genes in normal liver (NL) and Yap/β-catenin hepatoblastoma (HB) tissues. Data are expressed as means ± SD. n = 4 (NL tissues, HB tissues, and independent replicates). ∗∗P < 0.01, ∗∗∗P < 0.001 when compared with normal liver (U-test).
Supplemental Figure S7.

Representative immunohistochemical patterns for β-catenin, Yap, and Tead4 proteins in a large hepatoblastoma (HB) developed from a Yap/β-catenin mouse 10 weeks after hydrodynamic injection (w.p.i.). Representative hematoxylin and eosin (H&E) staining and corresponding immunohistochemical images of β-catenin, Yap, and Tead4 staining in the HB lesion (better appreciable in insets) from a Yap/β-catenin mouse 10 weeks after hydrodynamic injection. Scale bars: 250 μm (top panels); 100 μm (bottom panels). Original magnifications: ×40 (top panels); ×100 (bottom panels); ×400 (insets).
Supplemental Figure S8.

mRNA expression of YAP and its targets (CTGF, CYR61, AXL, and SURVIVIN) as well as β-catenin targets (GS and TBX3) in HepG2 and Hep293TT cell lines transiently transfected with TEAD4 cDNA. Data are expressed as means ± SD. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 when compared with empty vector (U-test).
Supplemental Figure S9.

Blockade of Yap transcriptional activity via short hairpin TEAD4 (shTEAD4) inhibits the growth of hepatoblastoma cell lines. A: mRNA expression levels of TEAD4, CTGF, and CYR61 in HepG2 and Hep293TT cell lines overexpressing pLKO or shTEAD4. B: Colony formation assay in HepG2 and Hep293TT cell lines overexpressing pLKO or shTEAD4. C: Cell proliferation and apoptosis are detected in HepG2 and Hep293TT cell lines overexpressing pLKO or shTEAD4. The cells were cultured under serum starvation for 24 hours. D: The proapoptotic effect of shTEAD4 in HepG2 and Hep293TT cells was further augmented with the use of the apoptosis inducer staurosporine control. Data are expressed as means ± SD. n = 3, HepG2 and Hep293TT cell lines (A); n = 12, HepG2 cell line (B); n = 6 Hep293TT cell line (B); n = 9, HepG2 and Hep293TT cell lines (C). ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001 when compared with pLKO or shTEAD4 (U-test).
References
- 1.Pateva I.B., Egler R.A., Stearns D.S. Hepatoblastoma in an 11-year-old: case report and a review of the literature. Medicine. 2017;96:e5858. doi: 10.1097/MD.0000000000005858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spector L.G., Birch J. The epidemiology of hepatoblastoma. Pediatr Blood Cancer. 2012;59:776–779. doi: 10.1002/pbc.24215. [DOI] [PubMed] [Google Scholar]
- 3.Czauderna P., Lopez-Terrada D., Hiyama E., Haberle B., Malogolowkin M.H., Meyers R.L. Hepatoblastoma state of the art: pathology, genetics, risk stratification, and chemotherapy. Curr Opin Pediatr. 2014;26:19–28. doi: 10.1097/MOP.0000000000000046. [DOI] [PubMed] [Google Scholar]
- 4.Aronson D.C., Meyers R.L. Malignant tumors of the liver in children. Semin Pediatr Surg. 2016;25:265–275. doi: 10.1053/j.sempedsurg.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Meyers R.L., Maibach R., Hiyama E., Haberle B., Krailo M., Rangaswami A., Aronson D.C., Malogolowkin M.H., Perilongo G., von Schweinitz D., Ansari M., Lopez-Terrada D., Tanaka Y., Alaggio R., Leuschner I., Hishiki T., Schmid I., Watanabe K., Yoshimura K., Feng Y., Rinaldi E., Saraceno D., Derosa M., Czauderna P. Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration. Lancet Oncol. 2017;18:122–131. doi: 10.1016/S1470-2045(16)30598-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell D., Ranganathan S., Tao J., Monga S.P. Novel advances in understanding of molecular pathogenesis of hepatoblastoma: a Wnt/beta-catenin perspective. Gene Expr. 2017;17:141–154. doi: 10.3727/105221616X693639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sumazin P., Chen Y., Trevino L.R., Sarabia S.F., Hampton O.A., Patel K., Mistretta T.A., Zorman B., Thompson P., Heczey A., Comerford S., Wheeler D.A., Chintagumpala M., Meyers R., Rakheja D., Finegold M.J., Tomlinson G., Parsons D.W., Lopez-Terrada D. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology. 2017;65:104–121. doi: 10.1002/hep.28888. [DOI] [PubMed] [Google Scholar]
- 8.Jia D., Dong R., Jing Y., Xu D., Wang Q., Chen L., Li Q., Huang Y., Zhang Y., Zhang Z., Liu L., Zheng S., Xia Q., Wang H., Dong K., He X. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology. 2014;60:1686–1696. doi: 10.1002/hep.27243. [DOI] [PubMed] [Google Scholar]
- 9.Eichenmuller M., Trippel F., Kreuder M., Beck A., Schwarzmayr T., Haberle B., Cairo S., Leuschner I., von Schweinitz D., Strom T.M., Kappler R. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol. 2014;61:1312–1320. doi: 10.1016/j.jhep.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 10.Cairo S., Armengol C., De Reynies A., Wei Y., Thomas E., Renard C.A. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008;14:471–484. doi: 10.1016/j.ccr.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Udatsu Y., Kusafuka T., Kuroda S., Miao J., Okada A. High frequency of beta-catenin mutations in hepatoblastoma. Pediatr Surg Int. 2001;17:508–512. doi: 10.1007/s003830000576. [DOI] [PubMed] [Google Scholar]
- 12.Lee S.A., Ho C., Roy R., Kosinski C., Patil M.A., Tward A.D., Fridlyand J., Chen X. Integration of genomic analysis and in vivo transfection to identify sprouty 2 as a candidate tumor suppressor in liver cancer. Hepatology. 2008;47:1200–1210. doi: 10.1002/hep.22169. [DOI] [PubMed] [Google Scholar]
- 13.Tao J., Calvisi D.F., Ranganathan S., Cigliano A., Zhou L., Singh S., Jiang L., Fan B., Terracciano L., Armeanu-Ebinger S., Ribback S., Dombrowski F., Evert M., Chen X., Monga S.P.S. Activation of beta-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology. 2014;147:690–701. doi: 10.1053/j.gastro.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu V., Plouffe S.W., Guan K.L. The Hippo pathway in organ development, homeostasis, and regeneration. Curr Opin Cell Biol. 2017;49:99–107. doi: 10.1016/j.ceb.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu F.X., Zhao B., Guan K.L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin K.C., Park H.W., Guan K.L. Regulation of the hippo pathway transcription factor TEAD. Trends Biochem Sci. 2017;42:862–872. doi: 10.1016/j.tibs.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Y., Huang T., Cheng A.S., Yu J., Kang W., To K.F. The TEAD family and its oncogenic role in promoting tumorigenesis. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17010138. pii: E138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang K., Qi H.X., Hu Z.M., Chang Y.N., Shi Z.M., Han X.H., Han Y.W., Zhang R.X., Zhang Z., Chen T., Hong W. YAP and TAZ take center stage in cancer. Biochemistry. 2015;54:6555–6566. doi: 10.1021/acs.biochem.5b01014. [DOI] [PubMed] [Google Scholar]
- 19.Azzolin L., Panciera T., Soligo S., Enzo E., Bicciato S., Dupont S., Bresolin S., Frasson C., Basso G., Guzzardo V., Fassina A., Cordenonsi M., Piccolo S. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell. 2014;158:157–170. doi: 10.1016/j.cell.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 20.Varelas X., Samavarchi-Tehrani P., Narimatsu M., Weiss A., Cockburn K., Larsen B.G., Rossant J., Wrana J.L. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev Cell. 2010;19:831–844. doi: 10.1016/j.devcel.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 21.Tang Y., Feinberg T., Keller E.T., Li X.Y., Weiss S.J. Snail/Slug binding interactions with YAP/TAZ control skeletal stem cell self-renewal and differentiation. Nat Cell Biol. 2016;18:917–929. doi: 10.1038/ncb3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho C., Wang C., Mattu S., Destefanis G., Ladu S., Delogu S., Armbruster J., Fan L., Lee S.A., Jiang L., Dombrowski F., Evert M., Chen X., Calvisi D.F. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology. 2012;55:833–845. doi: 10.1002/hep.24736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marti P., Stein C., Blumer T., Abraham Y., Dill M.T., Pikiolek M., Orsini V., Jurisic G., Megel P., Makowska Z., Agarinis C., Tornillo L., Bouwmeester T., Ruffner H., Bauer A., Parker C.N., Schmelzle T., Terracciano L.M., Heim M.H., Tchorz J.S. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology. 2015;62:1497–1510. doi: 10.1002/hep.27992. [DOI] [PubMed] [Google Scholar]
- 24.Hu J., Che L., Li L., Pilo M.G., Cigliano A., Ribback S., Li X., Latte G., Mela M., Evert M., Dombrowski F., Zheng G., Chen X., Calvisi D.F. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci Rep. 2016;6:20484. doi: 10.1038/srep20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu-Chittenden Y., Huang B., Shim J.S., Chen Q., Lee S.J., Anders R.A., Liu J.O., Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X., Calvisi D.F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol. 2014;184:912–923. doi: 10.1016/j.ajpath.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu P., Ge M., Hu J., Li X., Che L., Sun K., Cheng L., Huang Y., Pilo M.G., Cigliano A., Pes G.M., Pascale R.M., Brozzetti S., Vidili G., Porcu A., Cossu A., Palmieri G., Sini M.C., Ribback S., Dombrowski F., Tao J., Calvisi D.F., Chen L., Chen X. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology. 2017;66:167–181. doi: 10.1002/hep.29183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen T.T., Rakheja D., Hung J.Y., Hornsby P.J., Tabaczewski P., Malogolowkin M., Feusner J., Miskevich F., Schultz R., Tomlinson G.E. Establishment and characterization of a cancer cell line derived from an aggressive childhood liver tumor. Pediatr Blood Cancer. 2009;53:1040–1047. doi: 10.1002/pbc.22187. [DOI] [PubMed] [Google Scholar]
- 29.Aden D.P., Fogel A., Plotkin S., Damjanov I., Knowles B.B. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature. 1979;282:615–616. doi: 10.1038/282615a0. [DOI] [PubMed] [Google Scholar]
- 30.López-Terrada D., Cheung S.W., Finegold M.J., Knowles B.B. Hep G2 is a hepatoblastoma-derived cell line. Hum Pathol. 2009;40:1512–1515. doi: 10.1016/j.humpath.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 31.Liu P., Calvisi D.F., Kiss A., Cigliano A., Schaff Z., Che L., Ribback S., Dombrowski F., Zhao D., Chen X. Central role of mTORC1 downstream of YAP/TAZ in hepatoblastoma development. Oncotarget. 2017;8:73433–73447. doi: 10.18632/oncotarget.20622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ota M., Sasaki H. Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development. 2008;135:4059–4069. doi: 10.1242/dev.027151. [DOI] [PubMed] [Google Scholar]
- 33.Tward A.D., Jones K.D., Yant S., Cheung S.T., Fan S.T., Chen X., Kay M.A., Wang R., Bishop J.M. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc Natl Acad Sci U S A. 2007;104:14771–14776. doi: 10.1073/pnas.0706578104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin K.C., Moroishi T., Meng Z., Jeong H.S., Plouffe S.W., Sekido Y., Han J., Park H.W., Guan K.L. Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat Cell Biol. 2017;19:996–1002. doi: 10.1038/ncb3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yimlamai D., Fowl B.H., Camargo F.D. Emerging evidence on the role of the Hippo/YAP pathway in liver physiology and cancer. J Hepatol. 2015;63:1491–1501. doi: 10.1016/j.jhep.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Machado M.V., Michelotti G.A., Pereira T.A., Xie G., Premont R., Cortez-Pinto H., Diehl A.M. Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J Hepatol. 2015;63:962–970. doi: 10.1016/j.jhep.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mannaerts I., Leite S.B., Verhulst S., Claerhout S., Eysackers N., Thoen L.F., Hoorens A., Reynaert H., Halder G., van Grunsven L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol. 2015;63:679–688. doi: 10.1016/j.jhep.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 38.Chen P., Luo Q., Huang C., Gao Q., Li L., Chen J., Chen B., Liu W., Zeng W., Chen Z. Pathogenesis of non-alcoholic fatty liver disease mediated by YAP. Hepatol Int. 2018;12:26–36. doi: 10.1007/s12072-017-9841-y. [DOI] [PubMed] [Google Scholar]
- 39.Li H., Wolfe A., Septer S., Edwards G., Zhong X., Abdulkarim A.B., Ranganathan S., Apte U. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int. 2012;32:38–47. doi: 10.1111/j.1478-3231.2011.02646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh V., Manalang M., Singh M., Apte U. A brief report of immunohistochemical markers to identify aggressive hepatoblastoma. Appl Immunohistochem Mol Morphol. 2018;26:654–657. doi: 10.1097/PAI.0000000000000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mao B., Hu F., Cheng J., Wang P., Xu M., Yuan F., Meng S., Wang Y., Yuan Z., Bi W. SIRT1 regulates YAP2-mediated cell proliferation and chemoresistance in hepatocellular carcinoma. Oncogene. 2014;33:1468–1474. doi: 10.1038/onc.2013.88. [DOI] [PubMed] [Google Scholar]
- 42.Tschaharganeh D.F., Chen X., Latzko P., Malz M., Gaida M.M., Felix K., Ladu S., Singer S., Pinna F., Gretz N., Sticht C., Tomasi M.L., Delogu S., Evert M., Fan B., Ribback S., Jiang L., Brozzetti S., Bergmann F., Dombrowski F., Schirmacher P., Calvisi D.F., Breuhahn K. Yes-associated protein up-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology. 2013;144:1530–1542.e12. doi: 10.1053/j.gastro.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galli G.G., Carrara M., Yuan W.C., Valdes-Quezada C., Gurung B., Pepe-Mooney B., Zhang T., Geeven G., Gray N.S., de Laat W., Calogero R.A., Camargo F.D. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol Cell. 2015;60:328–337. doi: 10.1016/j.molcel.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morales F., Giordano A. Overview of CDK9 as a target in cancer research. Cell Cycle. 2016;15:519–527. doi: 10.1080/15384101.2016.1138186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Danilov A.V., Hu S., Orr B., Godek K., Mustachio L.M., Sekula D., Liu X., Kawakami M., Johnson F.M., Compton D.A., Freemantle S.J., Dmitrovsky E. Dinaciclib induces anaphase catastrophe in lung cancer cells via inhibition of cyclin-dependent kinases 1 and 2. Mol Cancer Ther. 2016;15:2758–2766. doi: 10.1158/1535-7163.MCT-16-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen T.K., Grant S. Dinaciclib (SCH727965) inhibits the unfolded protein response through a CDK1- and 5-dependent mechanism. Mol Cancer Ther. 2014;13:662–674. doi: 10.1158/1535-7163.MCT-13-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H., Pandey S., Travers M., Sun H., Morton G., Madzo J. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell. 2018;175:1244–1258. doi: 10.1016/j.cell.2018.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]