

Abstract
Resistance to inhibitors of cholinesterase-8A (Ric-8A) and Ric-8B are essential biosynthetic chaperones for hetero-trimeric G protein α subunits. We provide evidence for the direct regulation of Ric-8A cellular activity by dual phosphorylation. Using proteomics, Western blotting, and mutational analyses, we determined that Ric-8A was constitutively phosphorylated at five serines and threonines by the protein kinase CK2. Phosphorylation of Ser435 and Thr440 in rat Ric-8A (corresponding to Ser436 and Thr441 in human Ric-8A) was required for high-affinity binding to Gα subunits, efficient stimulation of Gα subunit guanine nucleotide exchange, and mediation of Gα subunit folding. The CK2 consensus sites that contain Ser435 and Thr440 are conserved in Ric-8 homologs from worms to mammals. We found that the homologous residues in mouse Ric-8B, Ser468 and Ser473, were also phosphorylated. Mutation of the genomic copy of ric-8 in Caenorhabditis elegans to encode alanine in the homologous sites resulted in characteristic ric-8 reduction-of-function phenotypes that are associated with defective Gq and Gs signaling, including reduced locomotion and defective egg laying. The C. elegans ric-8 phosphorylation site mutant phenotypes were partially rescued by chemical stimulation of Gq signaling. These results indicate that dual phosphorylation represents a critical form of conserved Ric-8 regulation and demonstrate that Ric-8 proteins are needed for effective Gα signaling. The position of the CK2-phosphorylated sites within a structural model of Ric-8A reveals that these sites contribute to a key acidic and negatively charged surface that may be important for its interactions with Gα subunits.
INTRODUCTION
Heterotrimeric guanine nucleotide–binding proteins (G proteins) regulate cellular signaling circuits broadly across physiology, including regulation of nervous, endocrine, sensory, and cardiovascular systems (1–3) G protein heterotrimers consist of α, β, and γ subunits (4). Upon activation of G protein-coupled receptors (GPCRs) by extracellular stimuli, the activated receptors stimulate G protein a subunit guanosine diphosphate (GDP) release and subsequent guanosine triphosphate (GTP) binding. Dissociated Gα-GTP and the Gbγ heterodimer each regulate sets of downstream effector proteins (1, 2, 4). Efficient GPCR signaling requires appropriate G protein subunit biosynthesis, G protein heterotrimer assembly, and trafficking to the plasma membrane (5–7). G protein subunit biosynthesis requires multiple chaperones, which are specific to each subunit (5, 8–13). G protein β subunits are folded within the chaperonin–containing tailless complex polypeptide-1 (CCT) and transferred to the chaperone phosducin-like protein-1 (PhLP-1) (9,14). G protein γ subunit biosynthetic folding may be assisted by dopamine receptor–interacting protein 78 (DRiP78) before Gγ subunit isoprenylation occurs (8, 15). The precise order of events of Gβγ heterodimer assembly involving PhLP-1 is not completely known, but PhLP-1 must be phosphorylated by protein kinase CK2 (casein kinase 2) to release folded Gβγ heterodimers before their insertion into the outer leaflet of an endomembrane, such as the endoplasmic reticulum (ER) or Golgi (14). The specific intracellular site(s) of the subsequent assembly of the G protein heterotrimer is also unknown but might be the ER or Golgi membrane (5, 8, 12,13).
We demonstrated that G protein a subunit biosynthetic folding requires the activity of resistance to inhibitors of cholinesterase-8 (Ric-8) proteins (10, 11). Mammalian Ric-8A is a folding chaperone for the Gαi, Gαq, and Gα13 subunit classes (collectively referred to as Gαi/q/13), and Ric-8B participates in the folding of Gαs and Gαolf subunits in the cytosol. Gα subunit folding occurs before its binding to newly produced Gβγ subunits and the association of the G protein heterotrimer with the membrane. Ric-8A and Ric-8B were originally found to act as GPCR-independent guanine nucleotide exchange factors (GEFs) for Gα subunits (16,17). In biochemical assays, Ric-8 proteins bind to folded Gα-GDP, enhance GDP release, and stabilize nucleotide-free Gα. The binding of Gα to GTP dissociates the Ric-8-Gα complex (16–18). We proposed a model that attempted to unify the in vitro GEF and in vivo molecular chaperone activities of Ric-8 (6). Newly translated Gα subunits that have yet to bind to guanine nucleotide are thought to require Ric-8 to chaperone the highly dynamic Gα subunit in its nucleotide-free state(s) to properly position the Gα Ras GTPase-like and α-helical domains to enable the first GTP-binding event (6, 11, 19,20). If Ric-8 is deleted or inhibited, the G protein may fold in a nonproductive manner without guanine nucleotide, thus generating a misfolded species that is rapidly degraded. In Ric-8A–/– or Ric-8B–/– mouse embryonic stem (mES) cells, the steady-state abundances of Gαi/q/13 or Gαs, respectively, are low, because the Gα subunits are misfolded, fail to become membrane-bound, and are rapidly degraded (10, 11). The substrate for in vitro GEF assays is recombinant Gα subunit that has already been successfully folded during biosynthesis and is bound to GDP. In GEF assays, Ric-8 may catalyze the reverse of the biosynthetic folding reaction and cause the G protein to release GDP. GTP binding to the partially unfolded Ric-8/nucleotide-free G protein intermediate may represent the authentic forward protein folding reaction.
A single copy of the Ric-8 gene was discovered in a Caenorhabditis elegans genetic screen designed to find worm mutants with defective neurotransmission (21, 22). Through epistasis analyses, ric-8 mutant neurotransmission and locomotion defects were attributed to defective Gq signaling, which is necessary for the production of diacylglycerol (DAG), a second messenger required for efficient synaptic vesicle priming (23–25). Activation of signaling pathways downstream of Gs partially rescued ric-8 mutant paralysis defects, demonstrating that Ric-8 was also epistatic to Gs in worms (26). The worm G proteins might not be properly folded in ric-8 mutants, thereby explaining the deficiencies in both Gq and Gs signaling.
Despite the importance of Ric-8 for G protein folding and signaling, understanding of the regulation of Ric-8 activities is limited. On the basis of shotgun proteomic studies of the phosphoproteomes of multiple cell and tissue sources, Ric-8 proteins are known to be phosphorylated (27–32). The two most frequently identified sites of human Ric-8A phosphorylation are Ser436 and Thr441 (corresponding to Ser435 and Thr440 in rat and mouse Ric-8A), which often appear on the same multiphosphorylated proteolytic peptide in mass spectrometry (MS) experiments (28–35). Of two studies that focused specifically on Ric-8, data from one suggest that phosphorylation of Ric-8A Ser501 contributes to the cell cycle–dependent degradation of Ric-8A, whereas findings from the other suggest that phosphorylation reduces the nuclear localization of Ric-8A (36, 37). Boularan et al. (36) observed an increase in Ric-8A ubiquitylation and degradation during mitosis, which followed increased Ric-8A Ser501 phosphorylation during the G2-M phases of the cell cycle. Xing et al. (37) correlated cell treatment with platelet-derived growth factor (PDGF) to increased Ric-8A Ser501 phosphorylation and decreased Ric-8A nuclear localization. Yan et al. (33) suggested that Gα13 promotes the tyrosine phosphorylation of Ric-8A and its translocation to the plasma membrane. Thus, multiple phosphorylation events may influence Ric-8A protein abundance or subcellular localization. However, no studies have demonstrated that phosphorylation status alters the enzymatic activity of Ric-8A as a Gα subunit GEF or molecular chaperone.
Here, we identified the sites of Ric-8A that are phosphorylated by protein kinase CK2 and contribute to stimulation of two activities of Ric-8A: Gα subunit guanine nucleotide exchange and Gα subunit molecular chaperoning. Phosphorylation of recombinant and endogenous Ric-8A was observed by gel mobility shift assays, MS analyses, and Western blotting analyses with newly generated Ric-8A phosphorylation site-specific antibodies. Efficient Gα subunit folding was monitored using a newly developed assay of kinetic folding of a fusion between Gα and green fluorescent protein (Gα-GFP), which showed that folding depended on Ric-8A phosphorylation at Thr440 and, to a lesser degree, at Ser435 (corresponding to Thr441 and Ser436 in human Ric-8A). The requirements of Thr440 and Ser435 phosphorylation for Ric-8A–mediated stimulation of Gαq guanine nucleotide exchange matched the results from the folding assay. The two CK2 consensus regulatory sites are invariant among Ric-8 homologs, and our analysis provided experimental verification of the phosphorylation of these sites in mammalian Ric-8B. Mutation of either site to alanine within the genomic copy of C. elegans ric-8 resulted in strong ric-8 reduction-of-function (rof) mutant phenotypes similar to those of previously attained ric-8 rof alleles, including egg-laying defects and severe locomotor defects that are attributable to defective G protein signaling (22, 23, 38, 39). Our work has identified two constitutively phosphorylated sites that affect Ric-8A activity and suggests a potential for dynamic Ric-8A regulation through dephosphorylation that could alter G protein abundance and signaling in cells.
RESULTS
Ric-8A is a constitutively phosphorylated protein
To determine whether human Ric-8A was phosphorylated, we immunoprecipitated endogenous Ric-8A from human embryonic kidney (HEK) 293 cells and analyzed the immunoprecipitated proteins by standard SDS–polyacrylamide gel electrophoresis (PAGE) or Phos-tag reagent-supplemented PAGE and detection with the Ric-8A monoclonal antibody 3E1 (11). The presence of the Phos-tag reagent can slow the electrophoretic migration of phosphorylated proteins (40), increasing their separation from unphosphorylated forms. We compared human Ric-8A immunoprecipitated from cytosolic and detergent-solubilized membrane fractions prepared from HEK293 cells with recombinant rat Ric-8A purified from Escherichia coli or High Five insect cells. Enzymatic dephosphorylation with alkaline phosphatase of endogenous and recombinant Ric-8A from the eukaryotic cells (HEK293 and insect) resulted in increased electrophoretic mobility, indicating that Ric-8A was phosphorylated (Fig. 1A). E. coli–purified recombinant Ric-8A migrated the same as, or slightly faster than, dephosphorylated Ric-8A from the eukaryotic sources. Ric-8A phosphorylation appeared to be constitutive, and the phosphorylation site(s) appeared to be highly occupied, because none of the lowest-molecular weight Ric-8A species was observed from the eukaryotic sources unless the samples were treated with phosphatase.
fig. 1. Ric-8A is constitutively phosphorylated in cells.
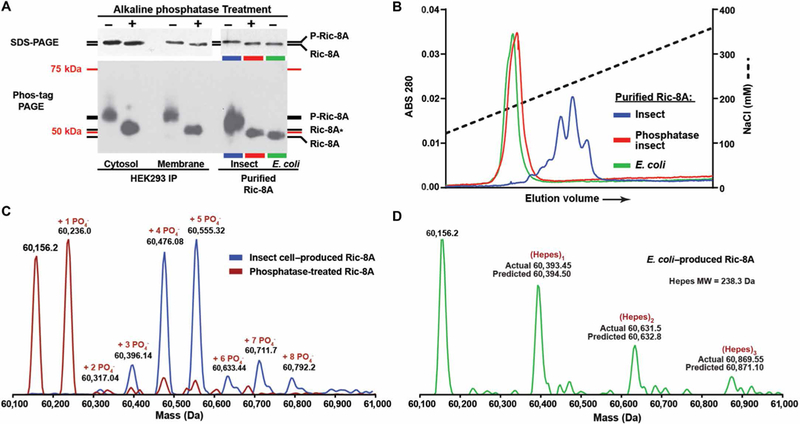
(A) Gel mobility shift assays of endogenous Ric-8A immunoprecipitated (IP) from cytosolic and detergent-extracted membrane fractions (5× material loaded) of HEK293 cells, and recombinant Ric-8A purified from E. coli or insect cells before and after alkaline phosphatase treatment. Top: Standard SDS-PAGE. Bottom: Phos-tag PAGE. Ric-8A* may have phosphosite(s) partially resistant to alkaline phosphatase. The molecular mass markers (75 and 50 kDa) do not accurately reflect the true masses of Ric-8A proteins on the Phos-tag PAGE. Data are representative of more than three independent experiments. (B) Anion exchange chromatography resolution of recombinant Ric-8A purified from E. coli or from insect cells before and after alkaline phosphatase treatment. Data are representative of more than three independent experiments. (C) Mass spectra of Ric-8A proteins were obtained through whole-protein ESI/MS analysis. Spectra are of insect cell–purified recombinant rat Ric-8A (blue trace) and alkaline phosphatase–treated Ric-8A (red trace). (D) The ESI-MS/MS spectrum of E. coli–purified Ric-8A revealed a completely unmodified protein (mass, 60156.2 Da) with a series of Hepes buffer adducts. MW, molecular weight.
We subjected the purified recombinant Ric-8A proteins to highresolution anion exchange chromatography, which can resolve phosphorylated species (Fig. 1B). Ric-8A purified from E. coli eluted as a single peak at ~181 mM NaCl. Ric-8A purified from insect cells eluted as multiple species at higher ionic strengths spanning ~220 to 248 mM NaCl, which is indicative of highly negatively charged states contributed by differentially phosphorylated species. Alkaline phosphatase treatment of the insect cell–produced Ric-8A resulted in elution of the protein as a single peak at ~184 mM NaCl, coinciding with the elution profile of unphosphorylated, E. coli–produced Ric-8A (Fig. 1B). Electrospray ionization (ESI)–MS/MS analysis showed that intact recombinant Ric-8A purified from insect cells was quantitatively phosphorylated at three (6.9% of total Ric-8A), four (34.4% of total Ric-8A), five (38.6% of total Ric-8A), or more independent sites (Fig. 1C and table S1). Alkaline phosphatase treatment produced two major species, unmodified Ric-8A (40.6% of total Ric-8A) and alkaline phosphatase–resistant, singly phosphorylated Ric-8A (46% of total Ric-8A). E. coli–produced Ric-8A was not phosphorylated or modified but formed a series of Hepes (buffer) adducts in the experiment (Fig. 1D).
We used the Group-based Prediction System (GPS 3.0) to predict rat Ric-8A residues that might be phosphorylated by specific kinases (41). Among the highest-scoring residues were those predicted to be phosphorylated by the acidophilic serine and threonine kinase, protein kinase CK2, including rat Ric-8A residues Ser435, Thr440, Ser523, and Ser527 (Fig. 2A). Multiple whole-proteome phosphopeptide analyses and a few directed studies have reported phosphorylated Ric-8A peptides that contain these four residues and Ser522 (28–35). Using liquid chromatography (LC)–MS/MS, we performed phosphorylation site identification of insect cell–produced rat Ric-8A or E. coli–produced rat Ric-8A that was treated with protein kinase CK2 holoenzyme and repurified. We obtained peptide coverage for 88.7% of insect cell–produced Ric-8A and 97% of protein kinase CK2-treated E. coli–produced Ric-8A (fig. S1). Mascot peptide mass fingerprinting software and collision-induced dissociation (CID) peptide analyses revealed specific sites of insect cell–produced Ric-8A phosphorylation (Ser30, Ser32, Ser298, Ser435, and Thr440) and CK2-treated, E. coli–produced Ric-8A phosphorylation (Ser435, Thr440, Ser522, Ser523, and Ser527) (Fig. 2B and figs. S1 and S2) (42). We did not obtain coverage of peptides of insect cell–produced Ric-8A consisting of residues Ser522, Ser523, or Ser527. Of the five identified phosphorylated residues from insect cell–purified Ric-8A, peptides containing Ser435 and Thr440 were phosphorylated most frequently with 46% (16 of 35) and 37% (13 of 35) of the peptides having confirmed site-specific phosphorylation, respectively. Site-specific phosphorylated peptides for the three additional sites were obtained with far less frequency: Ser30 (4 of 65), Ser32 (3 of 65), and Ser298 (1 of 19) (table S2).
Fig. 2. Identification of the Ric-8A sites phosphorylated by CK2.
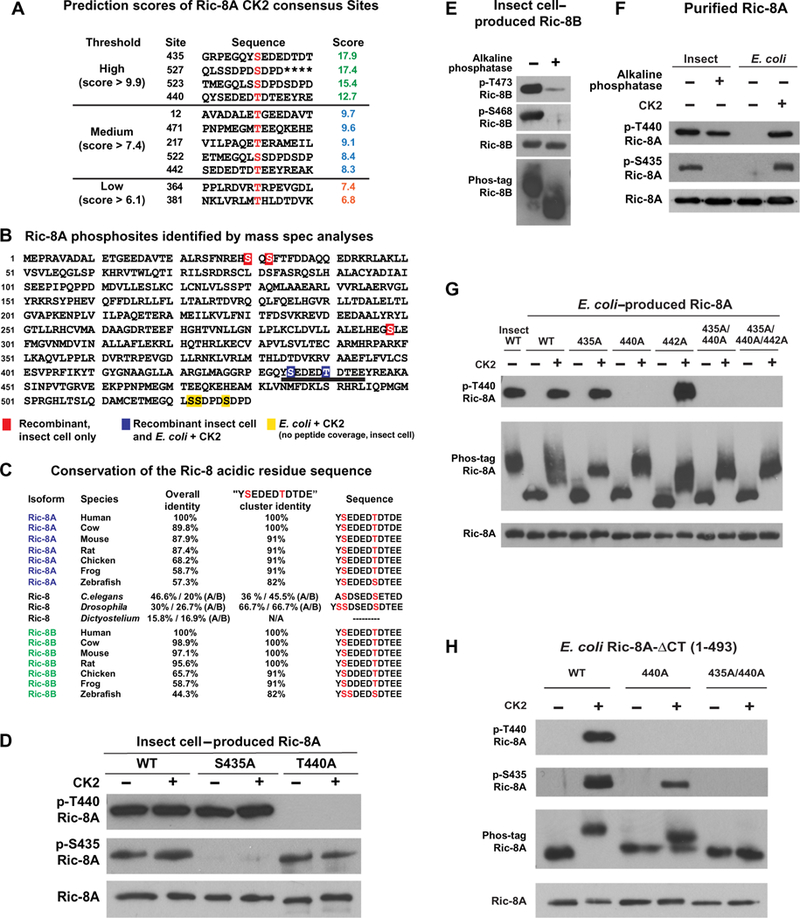
(A) GPS 3.0 was used to evaluate sites within Ric-8A with potential for CK2-mediated phosphorylation (47). The prediction was run at the high-stringency setting using a maximal false-positive rate threshold cutoff of 2%. Higher relative scores represent greater potential for phosphorylation. (B) Phosphosite identification in rat Ric-8A purified from insect cells and from E. coli-produced Ric-8A treated with CK2 was made by MS analysis of peptides from tryptic and chymotryptic digests. Red boxed residues were phosphorylated sites in insect cell–produced Ric-8A, yellow boxed residues were phosphorylated sites in E. coli–produced Ric-8A treated with CK2, and blue boxed residues were phosphorylated sites obtained for both proteins. All phosphosites were identified by Mascot software and confirmed by analysis of CID spectra. Peptide coverage of the three C-terminal phosphosites (Ser522, Ser523, and Ser527) was not attained for insect cell–produced Ric-8A. The 10-residue acidic sequence that contains the two consecutive CK2 sites of Ser435 and Thr440 is underlined. (C) The 10-residue acidic sequence is highly conserved across vertebrate Ric-8 proteins. The phosphosites corresponding to rat Ric-8A Ser435 and Thr440 are invariantly serine or threonine residues in all homologs examined. Dictyostelium Ric-8 is a shortened protein that lacks the acidic sequence stretch altogether. (D) Insect cell–produced rat wild-type (WT), S435A, and T440A Ric-8A proteins were treated with or without CK2 and then analyzed by Western blotting with an antibody specific for the CK2 consensus site (pS/pT)-D-X-E, which recognizes the phosphorylated Thr440 site (p-Thr440), the 6383 Ric-8A antiserum that recognizes the phosphorylated Ser435 site (p-Ser435), and the 1184 antiserum that recognizes Ric-8A. (E) Insect cell–produced mouse Ric-8B was treated with and without alkaline phosphatase and analyzed by Western blotting with the Ric-8A phosphosite-specific antibodies that also detect mouse Ric-8B p-Ser468 and p-Ser472, respectively. Ric-8B antiserum 2413 was used to detect the Ric-8B alkaline phosphatase–dependent gel shift after protein resolution by Phos-tag PAGE. (F) Purified Ric-8A from insect cells was treated with or without alkaline phosphatase, whereas E. coli–produced Ric-8A was treated with or without CK2. The proteins were analyzed by Western blotting with the p-Thr440, p-Ser435, and 1184 antibodies. (G) Insect–produced WT Ric-8A and the indicated Ric-8A alanine substitution mutant proteins produced in E. coli were treated with or without CK2 and analyzed by Western blotting with the p-Thr440 and 1184 Ric-8A antibodies. The proteins were also resolved by Phos-tag PAGE and analyzed by Western blotting with the 1184 Ric-8A antibody, as indicated. (H) WT, T440A, and S435A/T440A Ric-8A-ΔCT purified from E. coli were treated with or without CK2, resolved by SDS-PAGE and Phos-tag PAGE, and analyzed by Western blotting with the p-Thr440, p-Ser435, and 1184 Ric-8A antibodies, as indicated. Data in (D) to (H) are representative of more than three independent experiments.
We examined the tryptic peptide that spanned rat Ric-8A residues 430 to 445 in detail by CID analysis, because previous results from stand-alone Mascot Server peptide mass fingerprint searches suggested the phosphorylation of Tyr434 (Tyr435 in human Ric-8A) and Thr442 (37, 43–45). We found no evidence that either of those residues was phosphorylated. Our peptide fragmentation analysis showed that Ser435 and Thr440 were phosphorylated, most frequently on the same peptide (table S2). The region surrounding the Ser435 and Thr440 phosphosites is a well-conserved, acidic, 10–amino acid stretch, and the residues equivalent to rat Ric-8A Ser435 and Thr440 are invariant (that is, they are either Ser or Thr) across all vertebrate Ric-8A and Ric-8B homologs, as well as the single copies of Ric-8 in C. elegans and Drosophila melanogaster (Fig. 2C) (33). An ancestral Ric-8 homolog found in Dictyostelium (DDB_G0292036) encodes a shorter protein with marginal homology to the first ~430 amino acids of mammalian Ric-8A and therefore lacks the acidic cluster and sites equivalent to Ser435 or Thr440.
We generated or validated two antibodies to specifically detect Ric-8A proteins phosphorylated at positions Ser435 or Thr440. A rabbit (6383) was immunized with a keyhole limpet hemocyanin (KLH)-conjugated synthetic 12–amino acid peptide containing phosphorylated residues corresponding to the Ser435 and Thr440 sites: EGQY(pS) EDED(pT)DT-NH2. Despite immunization with the dually phosphorylated peptide antigen, the resultant immunoglobulin Gs (IgGs) that were enriched from the 6383 rabbit antiserum detected insect cell–produced wild-type rat Ric-8A, rat Ric-8A T440A, but not rat Ric-8A S435A, regardless of whether these proteins were treated in vitro with CK2 holoenzyme (Fig. 2D and fig. S2). Therefore, the 6383 antiserum contains a phosphosite-specific antibody for p-Ser435–Ric-8A. A commercially available antibody raised against the CK2 consensus site (pS/ pT)-D-X-E detected insect cell–produced wild-type rat Ric-8A, rat Ric-8A-S435A, but not rat Ric-8A-T440A, irrespective of in vitro phosphorylation with protein kinase CK2 holoenzyme (Fig. 2D). The only (pS/pT)-D-X-E CK2 consensus site in rat Ric-8A is p-Thr440, which explains why this antibody is specific for rat Ric-8A p-Thr440. This antibody also does not detect phosphorylated human Ric-8A because the Thr441 CK2 consensus site is (pT)-D-X-D (Fig. 2C). Given the sequence identity of the acidic residue regions of Ric-8A and Ric-8B, we used both phosphosite-specific antibodies to assess phosphorylation of the corresponding prospective phosphosites of insect cell–produced mouse Ric-8B (Ser468 and Thr473). Western blotting analyses showed that purified Ric-8B exhibited an alkaline phosphatase-sensitive Phos-tag gel mobility shift and detection with the Ric-8A p-Ser435 and p-Thr440 site-specific antibodies (Fig. 2E). These results showed that constitutive, dual phosphorylation within the conserved acidic residue cluster [YpSEDEDpTDT(D/E)E] is a conserved feature of Ric-8 proteins.
The MS analysis showed that treatment of E. coli–produced Ric-8A in vitro with protein kinase CK2 resulted in Ser435 and Thr440 phosphorylation (Fig. 2B and fig. S2), a result that was corroborated by Western blotting analysis using the phosphosite-specific antibodies (Fig. 2F). Insect cell–produced Ric-8A was detected with the p-Thr440 antibody, and this immunoreactive signal was still detectable after alkaline phosphatase treatment (Fig. 2F), suggesting that p-Thr440 is partially resistant to alkaline phosphatase–mediated dephosphorylation. This resistant form likely represents the population of singly phosphorylated Ric-8A (~46% of total) observed by ESI-MS/MS after alkaline phosphatase treatment (Fig. 1C).
CK2-treated, full-length Ric-8A appeared as a broad band on Phos-tag PAGE Western blots with a mobility shift (Fig. 2G). To determine the contributions of specific sites, we generated the single mutants S435A, T440A, and T442A, the double mutant S435A/T440A, and the triple mutant S435A/T440A/T442A and purified them from E. coli. Thr442 served as a control, because none of our analyses indicated that this site was phosphorylated. All of the CK2-treated mutants exhibited gel mobility shifts (Fig. 2G), indicating that Ser435 and Thr440 are not the only sites phosphorylated by CK2. On the basis of the MS results (Fig. 2B), we hypothesized that the additional CK2-phosphorylated sites Ser522, Ser523, and Ser527 contributed to the broad gel mobility shifts.
A C-terminally truncated Ric-8A protein lacking the last 37 amino acids (Ric-8A-ΔCT or 1–493) is an effective G protein α subunit GEF (18). This protein lacks the Ser522, Ser523, and Ser527 phosphorylation sites. E. coli–produced Ric-8A-ΔCT exhibited CK2-dependent phosphorylation at Ser435 and Thr440 (Fig. 2H). Phos-tag analysis revealed a mobility-shifted band for CK2-treated Ric-8A-ΔCT, whereas Ric-8A-ΔCT-T440A exhibited a lower gel mobility shift, and Ric-8A-ΔCT-S435A/T440A was detected as a single band. These results, combined with the MS data, indicate that Ser435 and Thr440 are likely the only sites efficiently phosphorylated by CK2 within the Ric-8A-ΔCT protein.
Ric-8A phosphorylation enhances G protein binding affinity
To determine the influence of Ric-8A phosphorylation on Gα subunit binding affinity, we adapted a flow cytometry protein interaction assay (FCPIA) (46). Purified glutathione S-transferase (GST) and E. coli– or insect cell–produced GST–Ric-8A, pretreated with or without CK2, were incubated with increasing concentrations of purified Gαi1 fused to cyan fluorescent protein (CFP), complexes were adsorbed to glutathione-coated beads, and the bead-associated fluorescent signals were measured. Phosphorylated Ric-8A had a 20-fold higher affinity for Gαi1 (Kd = 16.3 ± 1.9 nM) than unphosphorylated Ric-8A (Kd = 595.6 ± 205.2 nM) (Fig. 3).
fig. 3. Gαi1 binds phosphorylated Ric-8A with a higher affinity than it has for unphosphorylated Ric-8A.
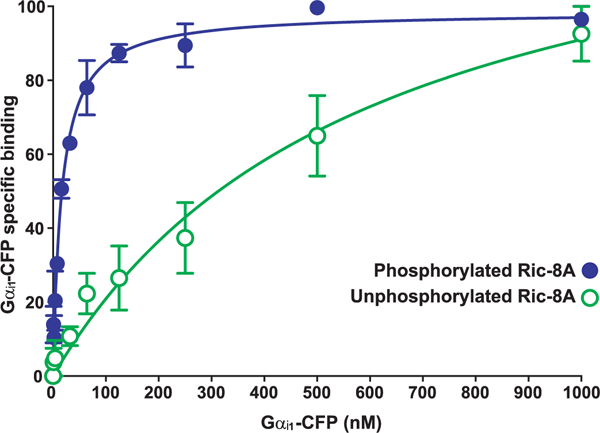
A concentration series of purified Gαi1CFP (1 to 1000 nM) was incubated with insect cell– or E. coli–produced GST-tagged Ric-8A or GST to determine the nonspecific component. Glutathione-coated microspheres were used to adsorb the GST-Ric-8A-Gαi1-CFP complexes before flow cytometric measurement of bead-associated fluorescence. Data were gated to include only singlet beads and then were analyzed for median fluorescence intensity (MFI). Nonspecific binding (GST only) was subtracted from total to yield the percentage of Gαi1-CFP bound specifically to Ric-8A, with 100% indicating the maximum fluorescence of saturated binding. The plotted data are the result of three independent experiments, and error bars indicate the SEM. The data were fitted to one-site specific binding curves with GraphPad Prism software.
Ric-8A GEF activity depends on Ser435 and Thr440 phosphorylation
Ric-8 proteins enhance the guanine nucleotide exchange rate of purified Gα subunits (16, 17,47). With the marked difference in Gα subunit binding affinity between phosphorylated and unphosphorylated Ric-8A, we predicted that phosphorylation also influenced Ric-8A GEF activity for Gα proteins. Therefore, we measured the rates of Gαq steady-state GTP hydrolysis [steady-state guanosine triphosphatase (GTPase) activity] in the presence of phosphorylated and unphosphorylated Ric-8A proteins with and without site-specific mutations. Insect cell–produced Ric-8A (phosphorylated) was a markedly more potent and efficacious GEF [steady-state GTPase EC50 (median effective concentration), ~155.2 ± 21.9 nM; maximal rate, 0.097 ± 0.002 min–1] than the equivalent Ric-8A protein produced in E. coli, which is not phosphorylated (Fig. 4A). The presence of CK2 in the assay affected the GTPase assay, likely by consuming GTP. Therefore, we repurified any recombinant protein that was phosphorylated with CK2 in vitro before using the CK2-phosphorylated proteins in these assays. E. coli–produced Ric-8A that was phosphorylated by CK2 was more potent (steady-state GTPase EC50, ~67.1 ± 17.6 nM) than insect cell–produced Ric-8A, although the maximal rate of stimulated Gαq GTPase activity was reduced only slightly (0.082 ± 0.002 min–1), indicating that phosphorylation of the Ric-8A CK2 consensus site(s) activated the unphosphorylated protein (Fig. 4A). Furthermore, enzymatic dephosphorylation of insect cell–produced Ric-8A with alkaline phosphatase resulted in a fourfold decrease in potency (EC50, ~657.2 ± 53 nM) compared with the phosphorylated form, but only a slightly reduced maximal rate (0.085 ± 0.003 min–1). This intermediate activity was likely due to partial occupancy of the phosphatase-resistant Thr440 site as was observed earlier (Figs. 1 and 2F). GEF assays measuring the kinetics of the Ric-8A–stimulated GTPγS binding of Gαq, Gα13, and Gαi1 (fig. S3) were consistent with the results of the steady-state GTPase assays (Fig. 4A).
Fig. 4. Requirements of the Thr440 and Ser435 phosphosites for Ric-8A-stimulated Gαq steady-state GTP hydrolysis activity.
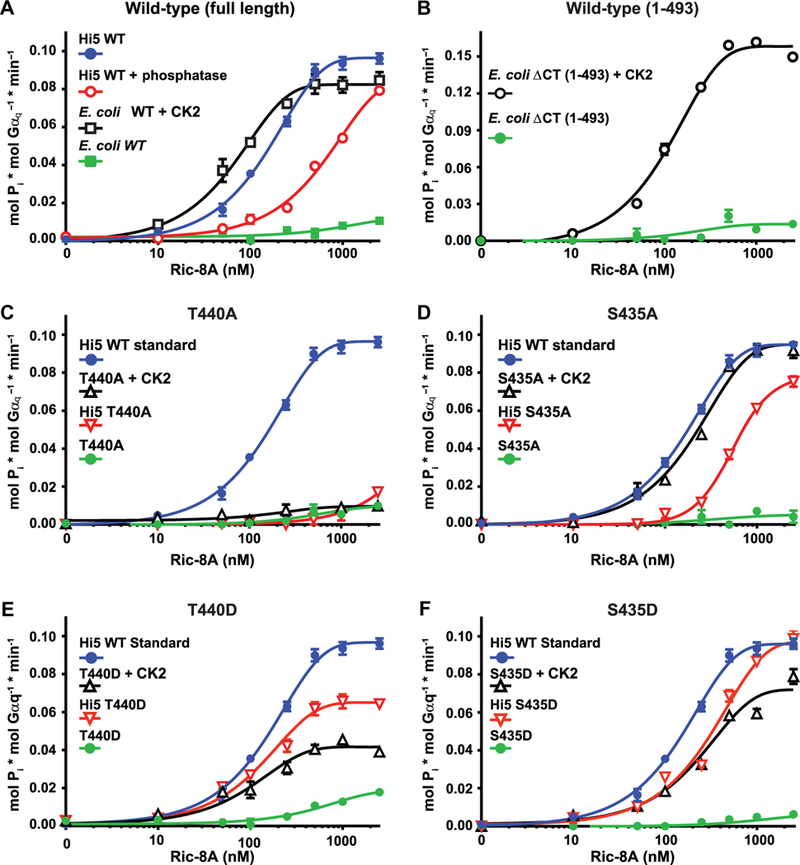
(A to F) Purified Gαq (50 nM) was incubated with the indicated concentrations of purified Ric-8A proteins and [ γ−32P]GTP. The linear rate of GTP hydrolysis was determined by measuring the production of free 32Pi. (A) Full-length WT Ric-8A proteins purified from insect cells (Hi5) and treated with or without alkaline phosphatase or purified from E. coli and treated with or without CK2 were tested for their ability to stimulate the steady-state GTPase activity of Gαq. (B) WT Ric-8A-ACT protein purified from E. coli and treated with or without CK2 was tested for its ability to stimulate the steady-state GTPase activity of Gαq. (C to F) WT Ric-8A purified from insect cells (Hi5) was used as a positive standard of phosphorylated Ric-8A activity for comparison to the activities of (C) Ric-8A–T440A purified from insect cells and Ric-8A–T440A purified from E. coli and treated with or without CK2, (D) Ric-8A–S435A purified from insect cells and Ric-8A-S435A purified from E. coli and treated with or without CK2, (E) Ric-8A-T440D purified from insect cells and Ric-8A-T440D purified from E coli and treated with or without CK2, and (F) Ric-8A-S435D purified from insect cells and Ric-8A-S435D purified from E. coli and treated with or without CK2. Data were plotted on semilog graphs and fitted to one-phase exponential association functions using GraphPad Prism. Experiments were performed in triplicate. Error bars indicate the SEM and were sometimes smaller than the size of the plotted symbols.
E. coli–produced Ric-8A-ΔCT (1–493) was phosphorylated with CK2 and used in the Gαq steady-state GTPase assay to discriminate the contributions of phosphorylated Ser435 and Thr440 from the other experimentally verified phosphorylation sites found near the C terminus of rat Ric-8A, which are not present in the truncated protein (Ser501, Ser522, Ser523, and Ser527) (28, 31, 37, 48). CK2 phosphorylation enhanced the Ric-8A-ΔCT-dependent stimulation of the steady-state GTPase activity of Gαq (Fig. 4B), demonstrating the contributions of phosphorylated Ser435 and Thr440 to the function of Ric-8A as a GEF for Gα (Fig. 4B). The maximal rate stimulated by Ric-8A-ΔCT (0.158 ± 0.003 min–1) exceeded that of full-length, phosphorylated Ric-8A (maximal rate, 0.097 ± 0.002 min–1), suggesting that the C terminus of Ric-8A may have an inhibitory function.
To discriminate between the individual contributions of p-Ser435 and p-Thr440 to GEF activity, we performed experiments with purified phosphorylation-mutant Ric-8A proteins. Ric-8A–T440A had a very low ability to activate Gαq steady-state GTPase activity, and pretreatment with CK2 had no effect (Fig. 4C). Because we determined that Ric-8A–T440A was phosphorylated by CK2 at the other CK2 consensus sites, including Ser435 (Fig. 2, G and H), the lack of activity of CK2-phosphorylated Ric-8A–T440A in the GTPase assay indicated that Ric-8A Thr440 phosphorylation was essential for efficient Ric-8A GEF activity. Insect cell–purified Ric-8A–S435A had markedly decreased potency in activating the steady-state GTPase activity of Gaq (EC50, ~558 ± 135.8 nM) and reduced efficacy (maximal rate, 0.081 ± 0.004 min–1) (Fig. 4C). E. coli–purified Ric-8A–S435A had negligible activity, which was almost fully restored by phosphorylation by CK2 (EC50, 211.0 ± 36.2 nM; maximal rate, 0.095 ± 0.002 min–1).
The quantitative MS analyses of Ric-8A proteins purified from insect cells showed that 34.4 and 38.6% of wild-type Ric-8A were modified with four and five phosphates, respectively (table S1). Ric-8A-S435A lacks one CK2 phosphorylation site; however, only 34.2 and 18.9% of this mutant were modified with three and four phosphates, respectively. This reduction in the frequency of multisite phosphorylation suggests that multisite Ric-8A phosphorylation in cells may be cooperative. Thus, we proposed that the diminished activity of Ric-8A-S435A could be contributed to by direct loss of the Ser435 phosphorylation site and reduced occupancy of the other CK2 phosphorylation sites, especially Thr440. The ability to restore near full potency and efficacy of Ric-8A-S435A by in vitro CK2 phosphorylation suggests that the Ser435 phosphorylation site may promote efficient phosphorylation of Thr440 in cells (Fig. 4D).
We generated mutant forms of Ric-8A with aspartic acid substitutions in positions Thr440 or Ser435 as attempts to produce phosphor-mimetic Ric-8A proteins. Ric-8A–T440D purified from insect cells had less efficacy in the Gαq steady-state GTPase assay (submaximal rate of GTPase activity, 0.065 ± 0.002 min–1) than did wild-type Ric-8A (Fig. 4E). Ric-8A–T440D purified from E. coli had negligible activity, and CK2 treatment of the protein only partially restored stimulatory activity to a submaximal Gαq steady-state GTPase rate (0.042 ± 0.002 min–1). E. coli–purified Ric-8A–T440D exhibited an apparent full SDS-PAGE mobility shift after CK2 treatment, suggesting that the protein was efficiently phosphorylated at the other CK2 consensus sites, including specific confirmation of Ser435 phosphorylation (fig. S4). Accounting for mutational loss of one phosphosite in the Ric-8A-Thr440 site mutants, insect cell-produced Ric-8A–T440D had substantially higher overall occupancy of its other phosphorylated sites in comparison to wild-type Ric-8A or Ric-8A–T440A (table S1). Overall, these results indicate that Ric-8A–T440D has characteristics of a partial phosphomimetic. Although T440D appeared to support cooperativity of Ric-8A multisite phosphorylation, Ric-8A–T440D did not activate Gαq with full efficacy in the steady-state GTPase assay. Insect cell–produced Ric-8A–S435D was not as potent as wild-type Ric-8A but achieved equal efficacy of Gαq steady-state GTPase activation (Fig. 4F). E. coli–produced Ric-8A–S435D had negligible activity. Phosphorylation by CK2 resulted in substantial activation; however, the CK2-treated protein was less potent (EC50, ~287.4 ± 95.9 nM) and efficacious (maximal rate, 0.072 ± 0.01 min–1) when compared to insect cell–produced, wild-type Ric-8A.
Gα subunit folding activity of Ric-8A depends on Ser435 and Thr440 phosphorylation
Plants lack an apparent Ric-8 homolog, which might explain why they have a self-folding or self-activating G protein α subunit homolog (49). The wheat germ extract (WGE) translation and protein folding system produces functionally folded mammalian Gα subunits if reconstituted with purified mammalian Ric-8A (11). We developed a real-time WGE/Gα subunit translation and folding assay that uses Gαi1 with an internal GFP tag (Fig. 5A), which is located at the junction of the αB and αC helices. Insertion of GFP at this site does not perturb Gα function (16,50,51). We added Gαi1-GFP mRNA to WGE, and we detected both parts of the fusion protein (Gαi1 and GFP) by Western blotting even in the absence of Ric-8A (Fig. 5B), which indicated that Gαi1 translation was Ric-8A–independent. Similar amounts of the fusion protein accumulated regardless of the presence of Ric-8A (Fig. 5B). However, the evolution and maturation of GFP fluorescence depended on Ric-8A (Fig. 5A), indicating that folding of GFP required folding of the Gαi1 portion of the fusion protein, which depended on Ric-8A.
fig. 5. Efficient Gα subunit folding in a WGE/Ric-8A reconstitution assay is dependent on Ric-8A Ser435 and Thr440 phosphorylation.
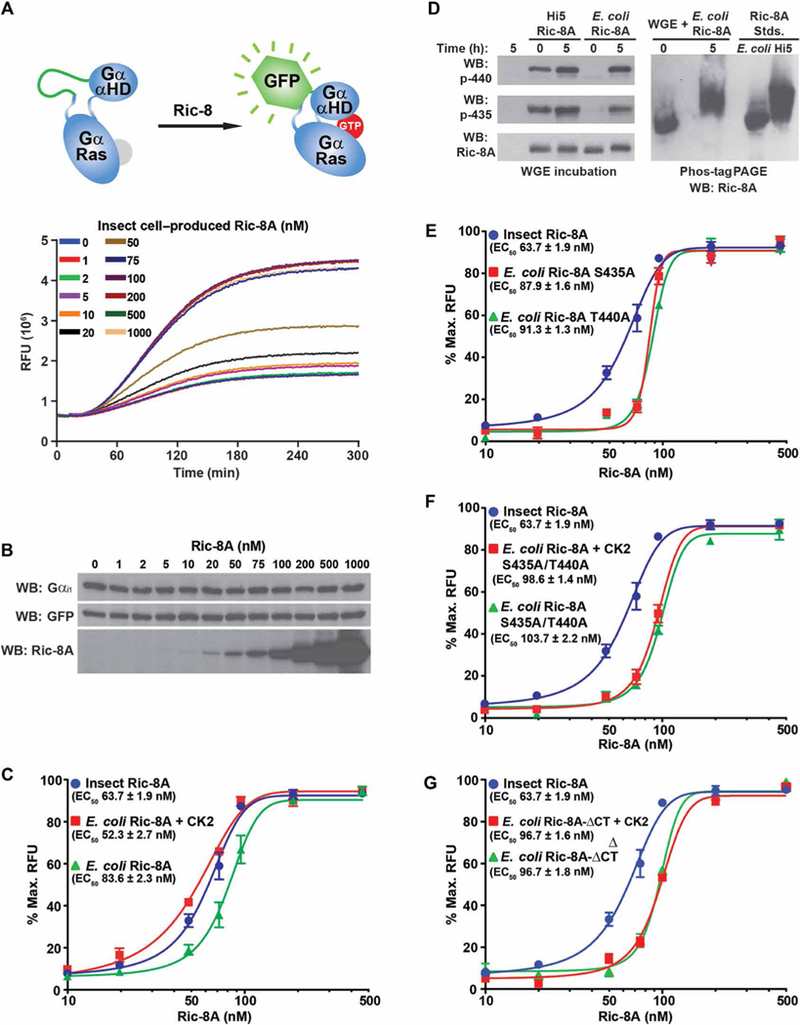
(A) Top: The mRNA encoding a Gαi1 fusion protein with an internal GFP tag was translated in WGE that had been reconstituted with purified rat Ric-8A. Bottom: Folding of the fusion protein was monitored by evolution of GFP fluorescence. (B) Samples for SDS-PAGE were attained at the conclusion of the kinetic Gαi1-GFP translation/folding reactions and analyzed by Western blotting (WB) to detect Gαi1, GFP, and Ric-8A. Data are representative of more than three experiments. (C) Gαi1-GFP mRNA was introduced into WGE translation/ folding reactions reconstituted with the indicated concentrations of insect cell-produced WT Ric-8A or E. coli–produced WT Ric-8A with or without CK2 pretreatment. Maximal Gαi1-GFP relative fluorescence units (RFUs) at 535 nm were plotted versus Ric-8A concentration on semilog plots. The data were fitted to variable Hill slope, four-parameter concentration response functions using the following equation in GraphPad Prism: Y = Ymin + (Ymax – Ymin)/(1 + 10((LogEC50-X)*Hillslope)). EC50 values were estimated from the fitted line functions. Experiments were performed in triplicate, and data are means ± SEM. (D) E. coli–and insect cell–purified Ric-8A proteins were incubated for 5 hours in WGE, resolved by SDS-PAGE and Phos-tag PAGE, and then analyzed by Western blotting. Data are representative of more than three experiments. (E to G) Insect cell–produced WT Ric-8A was used as phosphorylated Ric-8A standard in the WGE/Gαi1-GFP folding assay to compare to the actions of (E) E. coli–produced Ric-8A S435A or T440A, (F) E. coli–produced Ric-8A S435A/T440A treated with or without CK2, or (G) E. coli–produced Ric-8A-ΔCT treated with or without CK2. Data were processed as described in (C). Experiments were performed in triplicate, and data are means ± SEM.
Insect cell–produced Ric-8A promoted Gai1-GFP folding with an EC50 of 63.7 ± 1.9 nM (Fig. 5C). E. coli–purified Ric-8A also promoted Gαi1-GFP folding but was less active, exhibiting a rightward shifted EC50 of 83.6 ± 2.3 nM. E. coli–purified Ric-8A that was phosphorylated with CK2 was the most potent Gαi1-GFP folding protein with an EC50 of 52.3 ± 2.7 nM. Having expected a greater difference between the activity imparted by phosphorylated and unphosphorylated E. coli–purified Ric-8A in this assay, we hypothesized that E. coli–produced Ric-8A became phosphorylated by endogenous WGE kinases over the time course of the experiment (52,53). Western blotting analyses confirmed the phosphorylation of the Ser435 and Thr440 sites of E. coli–produced Ric-8A after incubation in WGE (Fig. 5D). We attempted to test the effects of chemical inhibitors of WGE kinases, but Gαi1-GFP translation was inhibited, preventing measurement of Gαi1-GFP folding. To examine the contributions of specific Ric-8A phosphorylation sites to Gαi1-GFP folding, we used E. coli–produced Ric-8A S435A or T440A proteins or double mutant proteins (Fig. 5, E and F). Ric-8A with individual mutations in Ser435 or Thr440 had reduced ability to promote Gαi1-GFP folding (EC50, 87.9 ± 1.6 nM and 91.3 ± 1.3 nM, respectively). The Ric-8A S435A/T440A double mutant was the least potent of all the Ric-8 proteins tested and promoted Gαi1-GFP folding with an EC50 of 103.7 ± 2.2 nM. The activity of Ric-8A S435A/T440A was not affected by CK2 or, presumably, by kinases in WGE (Fig. 5F).
Finally, the activity of E. coli–produced Ric-8-ΔCT was examined in the folding assay. Ric-8-ΔCT had low potency of Gαi1-GFP folding (EC50, 96.7 ± 1.6 nM) and was not enhanced upon phosphorylation by CK2 (Fig. 5G). This result contrasts with our earlier finding (Fig. 4B), which showed that treatment of Ric-8-ΔCT with CK2 restored efficacious GEF activity, but is consistent with our previous finding that Ric-8-ΔCT does not genetically complement G protein abundance defects in Ric-8A–/–mES cells (54). Together, these data indicate that phosphorylation of the Ric-8A residues Ser435 and Thr440 is critical for Gα subunit GEF and folding activities, but additional elements in the C-terminal region of Ric-8A are required for Gα subunit folding activity in WGE and cells.
Phosphorylation regulates Ric-8A activity in cells
We generated a clonal HEK293T R1C-8A knockout cell line (termed “G7”) using clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated 9 (Cas9) technology (55). We isolated G7 cells after two rounds of dilution cloning and determined that this line had three distinct indel mutations at the human R1C-8A target site (fig. S5A). This indicates that there may be three genomic copies of R1C-8A in HEK293T cells, which is consistent with the known hypotriploid karyotype of this cell line (56–58). We confirmed that the G7 cells had no detectable Ric-8A, a normal amount of Ric-8B, and reduced abundances of Gαq, Gα13, and Gαi1, but not of Gαs (fig. S5B). The affected G proteins are those folded by Ric-8A (11), and the reduced Ga abundances observed in G7 cells are consistent with previous observations of cell lines derived from Ric-8A knockout mouse tissues (mES cells and melanocytes) (10, 59).
We transfected the parental G7 RIC-8A knockout cells with plasmids that expressed wild-type rat Ric-8A or combinations of Ric-8A–encoded proteins with mutant Ser435 and Thr440 phosphorylation sites. We clonally isolated two to three independent cell lines of each Ric-8A transgene. We measured the steady-state abundance of Gα13 to evaluate the activity of each mutant Ric-8A protein for folding G proteins in cells. Whereas the parental G7 cells had a very low amount of Gα13, cell lines expressing wild-type Ric-8A had abundant Gα13 (Fig. 6, A to D). G7 cells expressing Ric-8A–S435A or Ric-8A–T440A only partially complemented the RIC-8A knockout: These cells only had Gα13 at 84% (S435A) and 29% (T440A) of the amount found in G7 cells expressing wild-type Ric-8A (Fig. 6, A and D). Cell lines expressing Ric-8A–S435D or Ric-8A–T440D also partially complemented the RIC–8A-null defect and restored Gα13 abundance to 91 and 75%, respectively, of the amount found in wild-type Ric-8A-expressing cells (Fig. 6, B and D). This suggests that the Ric-8A–T440D protein has partial phosphomimetic characteristics when stably expressed in G7 cells. However, double mutant Ric-8A–S435A/T440A or Ric-8A–S435D/T440D proteins were less capable of rescuing the RIC-8A–null defect; the abundance of Gα13 was 24 and 31%, respectively, of the amount found in G7 cells expressing wild-type Ric-8A (Fig. 6, C and D).
Fig. 6. Ric-8A-T440 is required for efficient G protein chaperoning activity and signaling in cells.
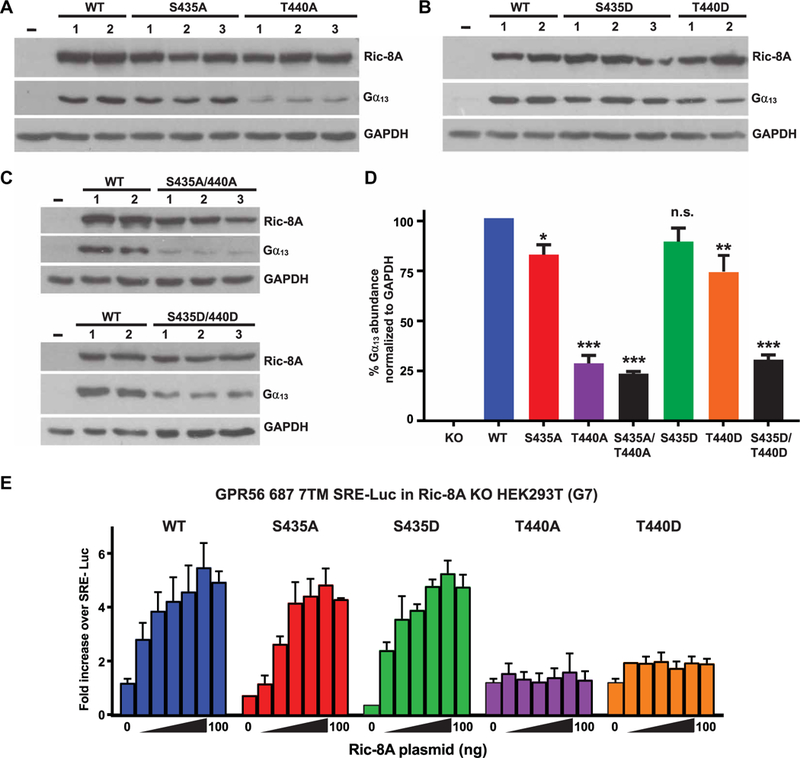
(A to D) A HEK293T cell line (G7) lacking RIC-8A was developed using CRISPR-Cas9 technology. (A to C) Crude membrane preparations from RIC-8A–null cells stably expressing WT or the indicated mutant rat Ric-8A proteins with single alanine or aspartic acid point mutations at the regulatory phosphosites were subjected to quantitative Western blotting for Ric-8A, Gα13, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). (D) Relative Gα13 abundances were quantified by pixel densitometry analysis and normalized to the GAPDH signal. Data are means ± SEM of three independent experiments. Statistical significance was determined by one-way analysis of variance (ANOVA), Dunnett’s multiple comparison to WT: *P < 0.05, **P < 0.005, ***P < 0.0001; n.s., not significant. (E) The effects of Ric-8A on GPCR-mediated G13 signaling activity were measured by dual SRE-Luc assay. RIC-8A–null cells were transiently transfected with plasmids expressing constitutively active GPR56, the SRE-Luc reporter, Renilla luciferase, and the indicated amounts of plasmids encoding WT and phosphosite mutant Ric-8A proteins. The accumulated firefly luciferase signal was measured 24 hours after transfection and normalized to the Renilla luciferase signal. Data were normalized to the signal generated from RIC-8A–null cells expressing the luciferase plasmids alone. Data are means ± SEM of three experiments.
We investigated the ability of the Ric-8A phosphorylation site mutants to support G protein signaling using a serum response elementluciferase (SRE-Luc) assay that we previously developed to measure activity of the G13-coupled GPCR GPR56 (also known as ADGRG1) (60, 61). We transiently transfected G7 cells with a range of amounts of plasmids encoding wild-type or phosphosite mutant Ric-8A proteins and a construct that expresses constitutively active GPR56. The Ric-8A–S435A and Ric-8A–S435D mutants, as well as the wildtype Ric-8A, supported G13 signaling (Fig. 6E). This is consistent with the ability of the stably expressed Ric-8A–Ser435 phosphosite mutants to rescue Gα13 abundance in RIC-8A knockout cells almost as well as did wild-type Ric-8A (Fig. 6D). In contrast, transient expression of the Ric-8A–T440A or Ric-8A–T440D phosphosite mutants did not support G13 signaling (Fig. 6E), which is consistent with earlier observations that the phosphorylation of Ric-8A–Thr440 is critical for Gα subunit folding activity and further supports the conclusion that the T440D mutation is only a partial phosphomimetic. There was apparent discrepancy in the ability of the Ric-8A–T440D mutant to partially complement the Gα13 abundance defect (Fig. 6B) versus its near inability to rescue Gα13 signaling in the SRE-Luc reporter assay (Fig. 6E). This may be due to the inherent differences in the two experiments. The Gα13 abundance assay used G7 cells that stably expressed high amounts of Ric-8A–T440D in 100% of the cell population, whereas the SRE-Luc assay used cells that had reduced amounts of Ric-8A–T440D, both due to transient 24-hour expression and the low concentrations of plasmids used. Furthermore, in the transient transfection–based SRE-Luc assay, transfection efficiencies are typically ~60% efficient (60).
C. elegans with phosphorylation site-deficient Ric-8 exhibit locomotion and egg-laying defects
Mutations in ric-8 that impair function cause severe locomotor defects that are attributable to defective G protein regulation of synaptic vesicle priming and neurotransmitter release in C. elegans (22, 23, 62). The C. elegans ric-8 residues Ser467 and Ser472 are equivalents of the rat Ric-8A residues Ser435 and Thr440, respectively (Fig. 2C). Using direct injection of CRISPR-Cas9 ribonucleoproteins, we individually converted the C. elegans ric-8 Ser467 or Ser472 codons to encode alanine to determine the functional importance of these phosphorylation sites in an organismal system. Homozygous ric-8 (S467A) or ric-8 (S472A) worms were almost completely paralyzed and exhibited rodshaped postures that typify strong locomotor defects (Fig. 7A). Phorbol esters supplant the requirement of ric-8 and rescue Gαq-mediated DAG production for efficient synaptic vesicle priming (63–65). Phorbol ester treatment rescued the rod-shaped postural defects, as shown by the omega-shaped body postures for both ric-8 (S467A) and ric-8 (S472A) homozygous mutants (Fig. 7, A and B). Using real-time video tracking, we quantified locomotor metrics in ric-8 (S467A) and ric-8 (S472A) homozygous worms before and after phorbol ester treatment. Compared to wild-type worms (N2), the number of body bends and the sinusoidal amplitude of the bending movements were reduced in both ric-8 (S467A) and ric-8 (S472A) homozygous mutants (Fig. 7, B and C). After phorbol ester exposure, the defects in body bends and amplitude were completely rescued in ric-8 (S467A) mutants but only partially rescued in ric-8 (S472A) mutants compared to wild-type worms exposed to vehicle (Fig. 7, B and C). By tracking individual worms for 2 min before and after phorbol ester application, we found that phorbol ester treatment not only produced a hyperflexive phenotype but also caused worms to move more rapidly and to change direction with increased frequency (Fig. 7D). The overall distance traveled by each group of worms was increased by phorbol ester; however, ric-8 (S467A) and ric-8 (S472A) mutant worms did not, on average, travel as far as wild-type worms (Fig. 7D, fig. S6, and movies S1 and S2). Note that the defects exhibited by ric-8 (S472A) worms were substantially more severe than those exhibited by the ric-8 (S467A) worms in all assays (Fig. 7, B to D). These data are consistent with our biochemical observations indicating that phosphorylation of mammalian Ric-8A Thr440 is more critical for Gα subunit GEF and folding activities than Ric-8A Ser435 phosphorylation. Corroborating this finding further, we observed that ric-8 (S467A) mutants were defective for egg laying (egl), whereas ric-8 (S472A) mutants were sterile. This finding of egg-laying defects is consistent with past studies in which ric-8 and Gαq rof mutants were discovered as alleles in egl forward genetic screens (22, 23, 38,39). The egg-laying phenotype of the ric-8 (S467A) worms was rescued by phorbol ester application, but ric-8 (S472A) worms did not lay eggs even in the presence of phorbol ester (Fig. 7A).
Fig. 7. Locomotor and postural defects in ric-8 S467A or S472A C. elegans mutants.
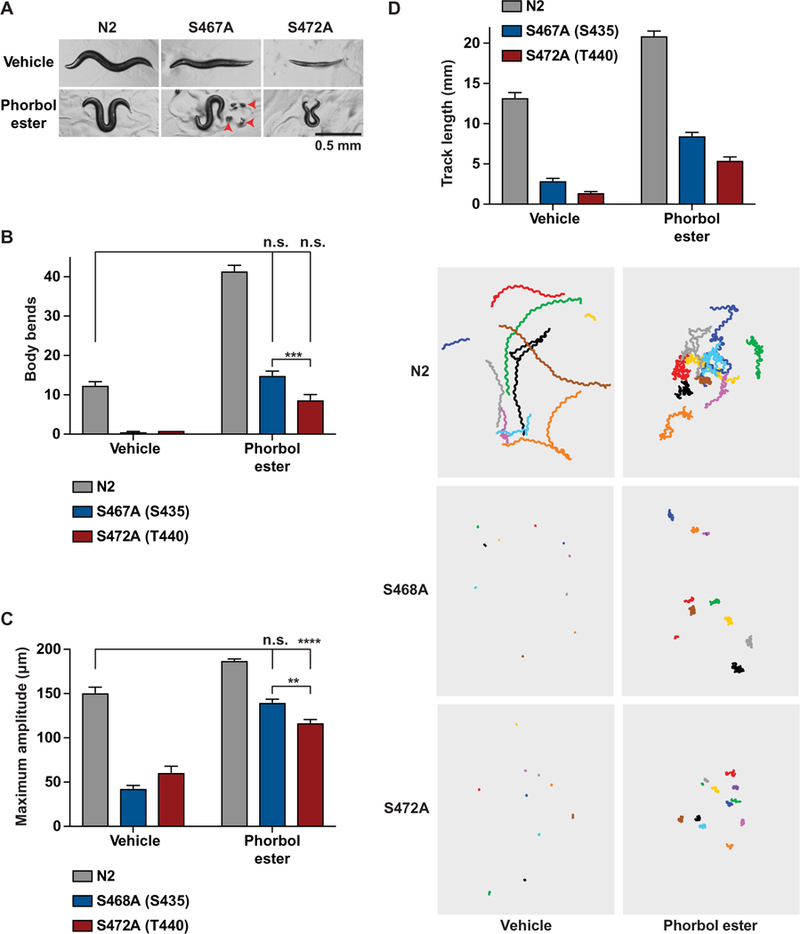
(A to D) WT (N2) and CRISPR-modified C. elegans strains expressing ric-8 with alanine mutations at sites Ser467 or Ser472 were treated with 10 μM phorbol ester or ethanol (vehicle) for 60 min. (A) Representative images show the rod-like body posture phenotype of S467A and S472A mutants on vehicle plates, as compared to the sinusoidal body posture of N2 controls. Phorbol ester–exposed animals had a hyperflexive phenotypes in N2 controls and S467A and S472A mutants, denoted by the omega-shaped body postures. Red arrowheads denote eggs laid by ric-8 S467A animals in response to phorbol ester. Scale bar, 0.5 mm. (B) The numbers of body bends were quantified in freely moving animals over a 2-min period. (C) The maximum bending amplitude was quantified as a measure of hyperflexion. (D) Track length (forward + reverse movement) was quantified in freely moving animals for 2 min before and after 60-min phorbol ester treatment using WormLab acquisition and analysis software. For all experiments, ten 1-day-old adults were assayed per plate with 10 replicated plates per strain for all experiments. Statistical significance was determined by one-way ANOVA with Tukey’s post hoc test. **P < 0.005, ***P < 0.0001.
The structural organization of the five Ric-8A CK2 sites is predicted
Ric-8 protein structural predictions based on homology modeling of proteins that contain similarly repeated Armadillo or Huntington/elongation factor 3/protein phosphatase 2A/TOR1 (HEAT) α-helical elements indicate that Ric-8 adopts a superhelical, crescent-shaped architecture (6, 66). We predicted the structure of Ric-8A with the I-TASSER server (Fig. 8) (67, 68). The experimentally verified CK2-phosphorylated residues were colored as yellow balls, and despite being apart in the linear amino acid sequence (Ser435, Thr440, Ser522, Ser523, Ser527), they are predicted to reside in close proximity to each other on an exterior surface of the concave side of the crescent. We noticed that two similar crescent-shaped Armadillo- or HEAT-repeat proteins, smgGDS and importin-β, bind to Rho/Rac and Ran small GTPases, respectively, and despite having more prevalent cellular functions, they both have weak in vitro GEF activities for their respective small G proteins (6, 69–73). The importin-β/Ran GTPase complex structure (PDB: 1IBR) was compared to four 90° poses of the predicted Ric-8A structure (Fig. 8). An acidic patch of importin-β was described as an important surface for binding to Ran (73). We speculate that Ric-8A may interact with the Ras-like domains of G protein α subunits through the acidic residues and negative charge that is contributed by the experimentally verified CK2-phosphorylated residues.
Fig. 8. Predicted orientation of the CK2 phosphorylation sites in Ric-8A.
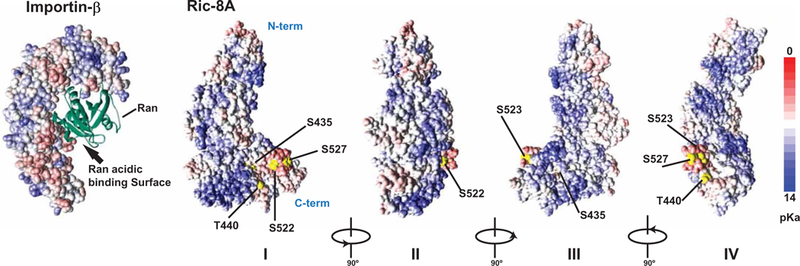
A structural prediction of Ric-8A was made using the I-TASSER server to depict four views of the protein with 90° rotation around a vertical axis (I to IV) (67, 68). The experimentally identified CK2 phosphorylation sites, Ser435, Thr440, Ser522, Ser523, and Ser527, are labeled and colored yellow. The N terminus of Ric-8A is located at the top in each representation. The surface area is colored according to local pKa (logarithmic acid dissociation constant) from 0 (red) to 14 (blue). Rendering was produced using Discovery Studio 4.0 (Accelrys Software Inc.) to visualize structure coordinates. Importin-β bound to Ran small GTPase [left; Protein Data Bank (PDB): 1IBR] is composed of repeated HEAT α-helical elements and has a crescent shape that is similar to the predicted structure of the ~430 N-terminal residues of Ric-8A (73). An acidic surface important for Ran binding in the importin-β crescent is denoted with an arrow. A negatively charged surface predicted to face into the Ric-8A crescent is composed of acidic residues and has a negative charge that is contributed by the experimentally verified CK2-phosphorylated residues.
DISCUSSION
Ric-8A and Ric-8B are essential genes that encode molecular chaperones with in vitro GEF activity and are required for heterotrimeric G protein α subunit biosynthetic protein folding (6). Here, we found that dual phosphorylation of two serine and threonine residues (rat Ric-8A Ser435 and Thr440) within a 10-residue acidic sequence was required for efficient Gα subunit folding and GEF activities, with the phosphorylation of Thr440 having a more prominent contribution to activity. Both residues were constitutively phosphorylated within CK2 consensus sites, and in vitro CK2 treatment was sufficient to phosphorylate the residues to restore full Ric-8A activities. These regulatory phosphosites are conserved among all Ric-8 homologs examined and were demonstrated to be phosphorylated in mammalian Ric-8B (Fig. 2). Mutation of the phosphosites to encode alanine residues in C. elegans resulted in strong ric-8 mutant rof phenotypes that could be partially suppressed through chemical activation of the defective Gq signaling pathway.
Ric-8A Ser435 and Thr440 phosphorylation positively affected both Gαq steady-state GTPase activity (Fig. 4) and G protein folding activity (Fig. 5), findings that advance our view that these two activities may be one and the same and represent the authentic cellular function of Ric-8 proteins as molecular chaperones that fold Gα subunits. Both processes necessarily involve Ric-8-mediated (repositioning of the Gα subunit Ras-like and α-helical domains to affect guanine nucleotide binding (6, 19). In GEF assays, Ric-8 is thought to partially unfold Gα-GDP to facilitate GDP release. Ric-8 then stabilizes the nucleotide-free state and permits GTP binding. In cells, we propose that Ric-8 chaperones nascent Gα subunits in prefolded state(s) that have yet to bind nucleotide. In this role, Ric-8 permits productive, first-time binding of GTP to the G protein; otherwise, the G protein folds in an incorrect manner, perhaps without nucleotide.
The concentration ranges of Ric-8A that exerted effect(s) and the maximal efficacies achieved were different between the G protein GEF and protein folding assays. Ric-8A–stimulated Gαq steady-state GTPase activity spanned ~3-log units of Ric-8A concentration (5 nM to 2.5 μM), whereas Ric-8A–stimulated Gα folding activity had steep Hill slopes with a tighter window of Ric-8A action (~10 to 100 nM). The most potent form of Ric-8A in the folding assay was E. coli–produced protein that was phosphorylated with CK2 (EC50, ~52.3 nM), whereas the least potent was Ric-8A–S435A/T440A, which could not be phosphorylated at the regulatory phosphosites (EC50, ~103.7 nM). At high Ric-8A concentrations, phosphorylation made no difference in Ric-8A promotion of maximal Gα subunit folding efficacy; however, unphosphorylated Ric-8A was a poor stimulator of nucleotide exchange at all concentrations tested. These differences indicate that the folding assay may be a truer measure of Ric-8A activity in the cell. The concentration of endogenous Ric-8A in rabbit reticulocyte lysate was estimated to be ~5 to 10 nM, which is close to the 16.3 nM Kd of phosphorylated Ric-8A binding to Gα (Fig. 3) (11). Accounting for expected inefficiencies of protein translation and folding in an extract system (for example, WGE), the observed dynamic range of reconstituted Ric-8A action (~10 to 100 nM) is reasonably close to that of the endogenous cytosolic concentration. This reflects a potential for dynamism of Ric-8A folding activity in cells and suggests that Ric-8A phosphorylation or dephosphorylation could be subject to regulation to control Gα subunit folding. Because Ric-8A and Ric-8B are constitutively phosphorylated in cells (Figs. 1 and 2), we are actively investigating whether Ric-8 is regulated by cellular phosphatases and, if so, under what conditions.
The differential functionality of the Ric-8A C terminus seems to discriminate in vitro GEF activity and protein folding activity in cells. Unphosphorylated Ric-8-ΔCT (lacking the last 39 amino acids) was a poor GEF, but its activity was enhanced by CK2 phosphorylation of Ser435 and Thr440 to exceed the Gαq steady-state GTPase rate that was stimulated by full-length, phosphorylated Ric-8A (Fig. 1, A and B). However, Ric-8-ΔCT acted with low potency in the WGE folding assay, and no enhancement was observed by CK2 phosphorylation. This is consistent with past findings showing that Ric-8-ΔCT does not rescue the Gα subunit abundance defects of Ric-8A–null mES cells (54). Therefore, the Ric-8A C terminus is important for Gα subunit folding in WGE and cells, but its presence is modestly inhibitory in GEF assays (18). This raises the question of the identity of the Ric-8 surfaces that bind to G protein a subunits and how Ser435 and Thr440 phosphorylation affects binding and Gα folding. The present evidence suggests that Ric-8A interacts with Gα subunit Ras-like domains and induces Ras domain plasticity (18, 19, 74, 75). The comparison of the predicted structure of Ric-8A with the known structure of importin-β and the Ran small GTPase complex may provide important insight. Ran has extensive contacts with the concave face of the importin-β crescent and is engaged by both N-terminal importin-β elements and a highly negatively charged surface near the C terminus that compacts around the GTPase (73, 76–78). We suggest that Ric-8A may bind to the Ras-like domain of Gα subunits in a similar manner (6). Yeast two-hybrid experiments demonstrated an interaction of the Ric-8A N terminus and the last 81 amino acids of Gαi1 (18, 79). A hydrogen-deuterium exchange (HDX) study using Ric-8A-ΔCT and purified Gαi1 showed another probable interaction site within Ric-8A residues 425 to 491, with Ric-8A residues 454 to 470 having high protection from proton exchange when Gαi1 was bound (74). This potential interaction site is proximal to the negatively charged regulatory phosphosite region consisting of residues 435 to 444, although Ser435 and Thr440 were not phosphorylated in the HDX analysis.
The Ric-8A structural prediction suggests that phosphorylated Thr440 and Ser435 reside in a negatively charged exterior surface of the concave side of the Ric-8A crescent. It is tempting to speculate that this is a G protein α subunit–binding surface that acts similarly to the negatively charged surface of importin-β that is required for Ran binding (73, 76, 77). It is also interesting that the experimentally verified CK2 phosphorylation sites (Ser522, Ser523, and Ser527) are predicted to be very close to Ser435/Thr440 in the predicted Ric-8A structure (Fig. 8). We are currently examining the function of these C-terminal, CK2 consensus phosphosites. As indicated, the C terminus of Ric-8A is dispensable for its GEF activity in vitro, but it is required in cells and for WGE-mediated Gα subunit folding.
The role(s) of the additional identified Ric-8A phosphosites also awaits further investigation. Yan et al. (33) purported that Ric-8A was phosphorylated at sites equivalent to rat Ric-8A Tyr434, Ser435, Thr440, and Thr442. Our Mascot software analysis also assigned Tyr434 and Thr442 phosphorylation initially, but we found no evidence for these assignments upon inspection of our peptide CID spectra (Fig. 2B and table S2). Other Ric-8A phosphorylation sites identified here and from multiple studies (for example, S30, S32, S298, and S501) (Fig. 2B and table S2) may serve additional regulatory purposes, including regulation of Ric-8A abundance during mitosis and localization of Ric-8A to the mitotic spindle, plasma membrane, or nucleus (33, 36, 37, 80–83). About 85 to 90% of Ric-8A resides in the cytosol (Fig. 1A), which is the subcellular location of nascent Ga subunit biosynthetic protein folding. The remaining ~10 to 15% of Ric-8A reside on cellular membranes, including the plasma membrane; however, the function of membrane-associated Ric-8A is unknown. We did not notice any overt differences in the phosphorylation states of cytosolic and membrane-associated Ric-8A (Fig. 1A). The existence of membrane-associated Ric-8A suggests additional functionality beyond that of a cytosolic chaperone. Membrane-associated Ric-8A could act as a GEF to generate Gα-GTP for signaling purposes, which is still an unknown, or perhaps there is a chaperone requirement of the nucleotide-free state of Gα during canonical G protein activation mediated by GPCRs. The function of membrane-associated Ric-8A and the role of phosphorylation in mediating Ric-8A subcellular localization are areas of active investigation.
MATERIALS AND METHODS
Antibodies
Rabbit polyclonal antiserum 2414 against Ric-8B, 1184 against Ric-8A, and mouse monoclonal antibody 3E1 were described previously (10, 11, 84). G protein subunit antisera were used to detect Gαq/11 (C19) (Santa Cruz Biotechnology) and Gβ1–4 (B600) (85). Monoclonal rabbit phospho-CK2 motif (pS/pT)-D-X-E antibody (Cell Signaling Technology) was found to specifically recognize phosphorylation at site Thr440 of recombinant rat Ric-8A (pT-D-T-E) but failed to recognize human Ric-8A (pT-D-T-D). Rabbit antiserum (6383) that detected rat Ric-8A phosphorylated at site Ser435 was raised. A synthetic KLH-conjugated synthetic peptide consisting of Ric-8A residues EGQY(pS)EDED(pT)DT as an immunogen [(KLH)-EGQY(pS) EDED(pT)DT] was synthesized by GenScript Inc.; pS and pT denote phosphorylated amino acids that correspond to rat Ric-8A residues Ser435 and Thr440. The peptide was dissolved in phosphate-buffered saline (PBS) and sent to Capralogics Inc. who oversaw the complete rabbit welfare, immunization schedule, and antiserum production. The PBS-solubilized peptide antigen was mixed with Freund’s adjuvant and subcutaneously injected into two white New Zealand rabbits (6382 and 6383) at four time points using a standard immunization schedule. The IgG fraction of the 6383 antiserum was isolated with protein A–Sepharose (PAS) and tested using various phosphorylated Ric-8A proteins to ascertain its specificity for Ric-8A–pS435 (fig. S2A).
Steady-state GTP hydrolysis (GTPase activity) and GTPγS binding assays
[35S]GTPγS and [γ−32P]GTP were purchased from PerkinElmer Life Sciences. Steady-state GTPase assays were conducted as described previously with minor adjustments (86). Reactions were performed in 20 mM Hepes (pH 8.0), 100 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol (DTT), 10 mM MgCl2, 0.05% Genapol C-100, and 2.5 μM [γ−32P]GTP (specific activity ≥ 30,000 cpm/ pmol). To initiate the reactions, 50 nM Gαq was added to the [γ−32P] GTP reaction buffer containing the concentrations of Ric-8A proteins indicated in the figures and incubated for 8 min at 25°C. The reactions were quenched by vortexing into an acidic charcoal slurry, which was pelleted by centrifugation at 3000g (86). The liquid supernatant was subjected to scintillation counting to determine the amount of 32Pi released by GTP hydrolysis. Gα [35S]GTPγS binding assays were performed as described previously (17) using 100 nM Gα and the concentrations of Ric-8A indicated in the figures. Reactions were incubated for 8 min for Gαi1 or Gα13 or for 10 min for Gαq and then were quenched and filtered onto BA85 nitrocellulose filters to capture the [35S]GTPγS-bound Gα. The filters were washed, dried, and subjected to scintillation counting to determine fraction of Gα bound to GTPγS.
WGE Gα-GFP folding assay
A pCI-neo plasmid expressing rat Gαi1 with an internal GFP tag between amino acids 122 and 123 of rat Gαi1 with the amino acid sequence SGGGGS as a linker at the N- and C-terminal ends of GFP was obtained from A. V. Smrcka (University of Michigan). The GFP-tagged Gαi1 pCI-neo plasmid was linearized by digestion with Cla I and purified using a QIAquick Gel Extraction Kit (Qiagen) for use as an in vitro transcription template. Capped GFP-Gαi1 mRNA transcripts were produced using the mMESSAGE/ mMACHINE T7 Transcription Kit (Life Technologies). We purified mRNA with an RNeasy MinElute CleanUp Kit (Qiagen). The mRNA was diluted to 500 μg/ml in water. GFP-Gαi1 mRNA (500 ng) was translated, and protein folding ensued in 25-ml reactions containing 12.5 μl of micrococcal nuclease–treated WGE (Promega), 3.5 μl of complete amino acid mix (Promega), 20 mM potassium acetate, 0.5 μl of Protector RNase Inhibitor (Roche), and purified Ric-8 proteins (10 nM to 2.5 μM) for 5-hour kinetic reactions at 25°C in Nunc 384-well black flat bottom plates. GFP fluorescence (485-nm excitation/ 535-nm emission) was measured using an EnVision Multilabel Plate Reader (PerkinElmer).
Protein expression and purification
Gαq, Gαi1, and Gα13 proteins were purified from insect cell lysates using the Ric-8A association technique as described previously (87). Rat Ric-8A was purified from insect cell lysates as described previously (47) with the following lysis buffer: 20 mM Hepes (pH 8.0), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, phosphatase inhibitor mixture (20 mM imidazole, 1 mM sodium fluoride, 2.5 mM sodium pyrophosphate, and 1 mM β-glycerophosphate), and protease inhibitor mixture [phenylmethylsulfonyl fluoride (23 μg/ml), Na-p-tosyl-L-lysine-chloromethyl ketone (21 μg/ml), L-1-p-tosylamino-2-phenylethyl-chloromethyl ketone (21 μg/ml), leupeptin (3.3 μg/ml), and lima bean trypsin inhibitor (3.3 μg/ml)] (Sigma-Aldrich). GST-tagged recombinant rat Ric-8A was purified from BL21 DE3 pLYSs E. coli. Cells were grown to an OD600 (optical density at 600 nm) of 1.0 by shaking at 225 rpm at 37°C, and then protein production was induced with 30 μM isopropyl-β-D-thiogalactopyranoside, and the cells were shaken at 175 rpm at 18°C for 18 hours. Cells were lysed in 20 mM Hepes (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1 mM DTT, and protease inhibitor mixture by stirring at 4°C for 30 min with 300 mg of lysozyme per liter of culture. Lysates were clarified by centrifugation at 100,000g, and GST–TEV-Ric-8A was isolated from the supernatant with glutathione-Sepharose 4B. GST–Ric-8A was released from the column with buffer containing 20 mM reduced glutathione (pH 8.0). To obtain untagged Ric-8A, the GST was cleaved with tobacco etch virus (TEV) protease and polished by HiTrap Q HP anion exchange chromatography as described previously (47). Purified proteins were stored in 20 mM Hepes (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1 mM DTT, and protease inhibitor mixture.
In vitro kinase reaction
Purified Ric-8A (1 mg) was treated with 15 μg of protein kinase CK2 holoenzyme (P6010L, New England Biolabs Inc.) for 1 hour at 37°C in 1 ml of 20 mM Hepes (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1 mM DTT, 10 mM MgCl2, 250 μM adenosine triphosphate, and 1 mM EGTA. Post-reaction separation of Ric-8A from CK2 was achieved by chromatography over an UnoQ1 anion exchange column (Bio-Rad) using 20 mM Hepes (pH 8.0), 5 mM EDTA buffer, and a linear elution gradient of 100 to 500 mM NaCl. The repurified Ric-8A was concentrated to ~20 μM using an Amicon 30,000 kDa MWCO ultracentrifugal concentrator (Millipore).
Immunoprecipitations
Adherent cells were scraped into cold cell wash buffer containing PBS, protease inhibitor mixture, phosphatase inhibitor mixture, 1 mM EDTA, 1 mM EGTA, Sigma Phosphatase Inhibitor Cocktail 3 (2.5 μM bromotetramisole, 500 nM cantharidin, and 1 nM calyculin), and Kinase Inhibitor mix [100 nM staurosporine (89149–816, Enzo) and 50 μM 4,5,6,7-tetrabromo-2-azabenzimidazole (T0826, Sigma-Aldrich)]. Cells were washed twice in cell wash buffer and then lysed by Dounce homogenization in cell wash buffer containing 1% NP-40. Lysates were centrifuged at 55,000g for 30 min, and the clarified supernatants were precleared by incubating with PAS or protein G–Sepharose (PGS) (Roche) for 60 min. After the PAS or PGS was pelleted by centrifugation at 1000g for 5 min, the supernatant was added to fresh tubes containing Ric-8A antibodies. The tubes were tumbled with PAS or PGS for 12 to 18 hours to capture the antibody-Ric-8A complexes. PAS/PGS beads were then washed in buffer lacking phosphatase inhibitors before treatment with alkaline phosphatase and then were washed four times before being suspended in SDS/DTT sample buffer for SDS-PAGE analysis.
Alkaline phosphatase treatment
Ric-8A proteins were dephosphorylated by incubation with 400 U of Calf Intestine Alkaline Phosphatase (Roche) per 2 mg of Ric-8A at 30°C for 45 to 60 min. Separation of Ric-8A from the alkaline phosphatase was achieved by chromatography over a HiTrap Q HP anion exchange column (GE Healthcare Life Sciences) with 20 mM Hepes (pH 8.0) and 5 mM EDTA with a linear elution gradient of 100 to 500 mM NaCl.
Cloning
QuikChange mutagenesis with PfuTurbo Polymerase (Agilent) was used to produce point mutations in the rat Ric-8A sequence. The following sense-strand primers with paired antisense primers (not shown) were used during mutagenesis: 5’-CGAGGGCCAGTAC-GCAGAGGATGAGGACA-3’ (rat Ric-8A S435A), 5’-AGAGGAT-GAGGACGCCGACACAGAGGAGT-3’ (rat Ric-8A T440A), 5’-CGAGGGCCAGTACGATGAGGATGAGGACA-3’ (rat Ric-8A S435D), and 5’-ACTCCTCTGTGTCGTCGTCCTCATCCTCT-3’ (rat Ric-8A T440D).
Engineering of a RIC-8A–null HEK293T cell line
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco). RIC-8A was deleted from HEK293T cells using CRISPR technology as described previously (55). The RIC-8A guide sequence 5’-AGCTCTGCGGTCATACAACCAGG-3’ was subcloned into the Bbs I site of pSpCas9(BB)-2A-Puro (PX459) (Addgene plasmid #48139) (55). HEK293T cells were transfected with the RIC-8A guide sequence vector and selected with puromycin (2 μg/ml) for 2 days, followed by clonal cell isolation using a 96-well serial dilution cloning format (88). Deletion of RIC-8A in clonal cell line outgrowth cultures was confirmed by Western blotting and sequencing of polymerase chain reaction (PCR)–amplified genomic DNA (fig. S5).
Mass spectrometry
Purified Ric-8A was reduced with 12 mM DTT at 60°C, which was followed by alkylation with 14 mM iodoacetamide at room temperature. The protein was then digested overnight with sequencing-grade trypsin or chymotrypsin (Promega) at 37°C. The protease/Ric-8A ratio was 1:20. Digestion was quenched with formic acid, and the sample was desalted by solid-phase extraction with an Empore C18-SD plate (3M). Each digested sample (1 μg) was analyzed by nano LC-MS/MS with a Waters nanoACQUITY HPLC System interfaced to a Thermo Fisher Q Exactive. Peptides were then loaded onto a trapping column and eluted over a 75-μm analytical column at 350 nl/min; both columns were packed with Luna C18 resin (Phenomenex). The mass spectrometer was operated in data-dependent mode, with the Orbitrap operating at 60,000 full width at half maximum (FWHM) and 17,500 FWHM for MS and MS/MS, respectively. The 15 most abundant ions were selected for MS/MS. Data were searched with Mascot to identify phosphorylation sites (Matrix Science). Specific sites of peptide phosphorylation were determined by direct analysis of (CID) fragments.
Flow cytometric analysis of the G protein–Ric-8A interaction
The general methodology of the FCPIA was described previously (46,89). GST-tagged Ric-8A (50 nM) or GST control protein (50 nM) was incubated with purified Gαi1-CFP (1 to 1000 nM) for 1 hour in the dark at room temperature. The Gαi1 fusion contains an internal CFP tag at amino acid 121 (50). Glutathione-coated polystyrene microparticles with a diameter of 2 μm (Spherotech) were vortexed briefly and washed three times with binding buffer [20 mM Hepes (pH 8.0), 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.2 μM MgCl2, 0.1% Lubrol C12E10, 10 μM GDP, and 1% bovine serum albumin]. Beads (2 × 106) were then added to the protein mixtures to make the final volume 150 μl, and the samples were incubated for 1 hour at room temperature before measurements were made with a BD Biosciences LSR II analyzer. Recordings were made for 10,000 events per sample. The data were gated to include only singlet beads (~80% of all events) and subsequently analyzed for MFI. Nonspecific binding (Gαi1-CFP binding to GST) was subtracted from total Gαi1-CFP binding to GST-tagged Ric-8A to yield the percentage of Gαi1-CFP bound specifically to Ric-8A, with 100% indicating the maximum fluorescence of saturated binding. Nonspecific binding was less than 20% of the total signal in all conditions.
Directed dual luciferase assay
HEK293T cells maintained in DMEM and 10% (v/v) FBS were transiently transfected in 24-well format with polyethylenimine with 1 to 100 ng of Ric-8A in pcDNA3.1/Hygro+, 200 ng of constitutively active GPR56-A386M 7TM, 100 ng of the SRE-Luc reporter plasmid pGL4.33 (Promega), and 1 ng of phRLuc (PerkinElmer Life Sciences) (60, 61). Total DNA amounts were normalized with empty pcDNA3.1. At 18 to 24 hours after transfection, cells were serum-starved for 10 hours, harvested in culture medium by trituration, washed in Tyrode’s solution (137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 0.2 mM Na2HPO4, 12 mM NaHCO3, and 5.5 mM D-glucose), and lysed in firefly luciferase reagent (NanoLight Technologies Inc.). Renilla luciferase buffer containing 3 μM coelenterazine H quenched firefly luminescence and induced Renilla luciferase luminescence (90). Luminescence was read using a TriStar2 plate reader (Berthold). All firefly luciferase data were normalized to the Renilla luciferase signal and plotted as fold increase over the signal obtained from cells that were not transfected with the Ric-8A plasmid.
Phos-tag gel electrophoresis and Western blotting
Phos-tag PAGE was performed using methods developed by English et al. (91) with minor changes. Phos-tag PAGE gels were cast as follows. The resolving layer consisted of 8% 29:1 acrylamide/ bisacrylamide (161–0156, Bio-Rad), 350 mM bis-tris (pH 6.8; BP301100, Fisher Scientific), 75 μM Phos-tag (304–93521, Wako Chemical Industries), 150 μM Zn(NO3)2, 0.05% ammonium persulfate (APS), and 0.1% tetramethylethylenediamine (TEMED). The resolving gel was overlaid with isopropanol and allowed to polymerize. The stacking gel was composed of 4% 29:1 acrylamide/bisacrylamide, 350 mM bis-tris (pH 6.8), 0.05% APS, and 0.1% TEMED. The resolving gel was rinsed five times with 350 mM bis-tris (pH 6.8) before the stacking gel was poured. EDTA-free protein samples were heated at 95°C for 5 min and allowed to cool before being loaded onto the gels. The running buffer was 50 mM tris-HCl (pH 7.8), 50 mM Mops, 0.1% SDS, and 5 mM sodium bisulfite (pH 7.2). Sodium bisulfite (5 mM) was added to the running buffer immediately before electrophoresis was performed. Each gel was run at a constant 100 to 150 V for 2.5 to 3.5 hours and then equilibrated in transfer buffer [50 mM bicine, 50 mM bis-tris, 0.1% (v/v) EDTA, 20% (v/v) methanol, 2.5 mM sodium pyrophosphate, 5 mM sodium bisulfite, and 0.1% (v/v) SDS] with shaking at 22°C for at least 30 min to release the protein-bound Phos-tag reagent. Wet transfer to nitrocellulose was then performed at 4°C for 20 hours at 25 V.
C. elegans ric-8 S467A and S472A mutant generation and locomotion assays
The N2 Bristol C. elegans strain was modified by CRISPR-Cas9 gene editing using direct injection of CRISPR-Cas9 ribonucleoprotein (92). In brief, single-stranded oligonucleotides (ssODNs) containing 40– to 50–base pair 5’ and 3’ homology arms flanking the 20-nucleotide target site were designed containing the edit of interest, a unique in-frame restriction site, and conservative nucleotide changes to prevent single-guide RNA (sgRNA)/Cas9 cleavage. The ssODNs were ordered as Ultramers from Integrated DNA Technologies (IDT). Where possible, the NGG PAM motif was conservatively changed to prevent sgRNA/Cas9 cleavage of the ssODN homology-directed repair (HDR) template. Commercially available recombinant Streptococcus pyogenes Cas9 nuclease was purchased from IDT. A synthetic sgRNA (5’-AAAACACGCGUCGGACAGUG-3’) was purchased from Synthego Inc. The ssODN HDR repair template sequences were S467A (5’-GTCACCTCGCAAATTTGGGTCTTCTCGGACAAATCAA TCAACCAAAACACGCagCtGACAGTGAaGATAGTGAAACT-GAGGATTATAATCAGATTAAGGATAGgtatgctatactttttgtgc-3’) and S472A (5’-GTCACCTCGCAAATTTGGGTCTTCTCGGACA-AATCAATCAACCAAAACACGCtagcGACtcTGAaGATgc-cGAAACTGAGGATTATAATCAGATTAAGGATAGgtatgc-tatactttttgtgc-3’). Correctly edited animals were back-crossed six times to N2, and the ric-8 locus was analyzed by PCR amplification of genomic DNA and Sanger sequencing. Both ric-8(S467A/+) and ric-8(S472A/+) heterozygous worms were reproductive and healthy, whereas ric-8(S467A/S467A) and ric-8(S472A/S472A) homozygous worms exhibited severe locomotor and postural defects, with ric-8(S472A/S472A) animals being viable but sterile (23). 12-O-Tetradecanoylphorbol 13-acetate (phorbol 12-myristate 13-acetate) (10 μM) or ethanol was added to 35-mm nematode growth medium plates and spread with 35 μl of OP50 bacteria and allowed to incubate overnight at room temperature. The next day, the plates were loaded with 1-day-old adult worms and assayed 1 hour after loading. Each individual plate carried 10 worms that were video-tracked and analyzed for 2 min using the automated WormLab System (MBF Bioscience)
Supplementary Material
Acknowledgments
We thank A. Smrcka and S. Malik for critical discussion and the gift of the px459 and GFP-Gαi1-pCI-neo plasmids; H. Dohlman and J. English for advice regarding Phos-tag PAGE; M. Dumont, E. Mathew, and K. McGlynn for advice and assistance in flow cytometry assays; and E. Marvin and C. Yu for technical support.
Funding: This work was supported by NIH grants R01-GM088242 (to G.G.T.), R01-NS094678 (to A.A.B.), MDA382300 (to A.A.B.), and T32-GM06841 (to M.M.P.-S.); Howard Hughes Medical Institute “Med-into-Grad” Fellowship (to M.M.P.-S.); and a predoctoral fellowship from the PhRMA Foundation (to M.M.P.-S.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The MS proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) through the PRIDE partner repository with the data set identifier: PXD009816 (93).
REFERENCES AND NOTES
- 1.Hepler JR, Gilman AG, G proteins. Trends Biochem. Sci. 17, 383–387 (1992). [DOI] [PubMed] [Google Scholar]
- 2.Hamm HE, The many faces of G protein signaling. J. Biol. Chem. 273, 669–672 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Lefkowitz RJ, Seven transmembrane receptors: Something old, something new. Acta Physiol. 190, 9–19 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Gilman AG, G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649 (1987). [DOI] [PubMed] [Google Scholar]
- 5.Marrari Y, Crouthamel M, Irannejad R, Wedegaertner PB, Assembly and trafficking of heterotrimeric G proteins. Biochemistry 46, 7665–7677 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papasergi MM, Patel BR, Tall GG, The G protein a chaperone Ric-8 as a potential therapeutic target. Mol. Pharmacol. 87, 52–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willardson BM, Tracy CM, in GPCR Signalling Complexes—Synthesis, Assembly, Trafficking and Specificity, Dupré DJ, Hébert TE, Jockers R, Eds. (Springer; Netherlands, 2012), pp. 131–153. [Google Scholar]
- 8.Dupré DJ, Robitaille M, Richer M, Éthier N, Mamarbachi AM, Hébert TE, Dopamine receptor-interacting protein 78 acts as a molecular chaperone for Gg subunits before assembly with Gp. J. Biol. Chem. 282, 13703–13715 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Lukov GL, Hu T, McLaughlin JN, Hamm HE, Willardson BM, Phosducin-like protein acts as a molecular chaperone for G protein βγ dimer assembly. EMBOJ. 24, 1965–1975 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabay M, Pinter ME, Wright FA, Chan P, Murphy AJ, Valenzuela DM, Yancopoulos GD, Tall GG, Ric-8 proteins are molecular chaperones that direct nascent G protein α subunit membrane association. Sci. Signal. 4, ra79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan P, Thomas CJ, Sprang SR, Tall GG, Molecular chaperoning function of Ric-8 is to fold nascent heterotrimeric G protein α subunits. Proc. Natl. Acad. Sci. U.SA. 110, 3794–3799 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wedegaertner PB, Chu DH, Wilson PT, Levis MJ, Bourne HR, Palmitoylation is required for signaling functions and membrane attachment of Gqα and Gsα. J. Biol. Chem. 268, 25001–25008 (1993). [PubMed] [Google Scholar]
- 13.Evanko DS, Thiyagarajan MM, Wedegaertner PB, Interaction with Gβγ is required for membrane targeting and palmitoylation of Gαs and Gαq. J. Biol. Chem. 275, 1327–1336 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Lukov GL, Baker CM, Ludtke PJ, Hu T, Carter MD, Hackett RA, Thulin CD, Willardson BM, Mechanism of assembly of G protein βγ subunits by protein kinase CK2-phosphorylated phosducin-like protein and the cytosolic chaperonin complex. J. Biol. Chem. 281, 22261–22274 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Takida S, Wedegaertner PB, Heterotrimer formation, together with isoprenylation, is required for plasma membrane targeting of Gβγ. J. Biol. Chem. 278, 17284–17290 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Tall GG, Krumins AM, Gilman AG, Mammalian Ric-8A (synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J. Biol. Chem. 278, 8356–8362 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Chan P, Gabay M, Wright FA, Tall GG, Ric-8B is a GTP-dependent G protein αs guanine nucleotide exchange factor. J. Biol. Chem. 286, 19932–19942 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas CJ, Briknarova K, Hilmer JK, Movahed N, Bothner B, Sumida JP, Tall GG, Sprang SR, The nucleotide exchange factor Ric-8A is a chaperone for the conformationally dynamic nucleotide-free state of Gαi1. PLOS ONE 6, e23197 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Eps N, Thomas CJ, Hubbell WL, Sprang SR, The guanine nucleotide exchange factor Ric-8A induces domain separation and Ras domain plasticity in Gαi1. Proc. Natl. Acad. Sci. U.S.A. 112, 1404–1409 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tall GG, Ric-8 regulation of heterotrimeric G proteins. J. Recept. Signal Transduct. Res. 33, 139–143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen M, Alfonso A, Johnson CD, Rand JB, Caenorhabditis elegans mutants resistant to inhibitors of acetylcholinesterase. Genetics 140, 527–535 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, Rand JB, A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc. Natl. Acad. Sci. U.S.A. 93, 12593–12598 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller KG, Emerson MD, McManus JR, Rand JB, RIC-8 (Synembryn): A novel conserved protein that is required for Gqα signaling in the C. elegans nervous system. Neuron 27, 289–299 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singer WD, Brown HA, Sternweis PC, Regulation of eukaryotic phosphatidylinositol-specific phospholipase C and phospholipase D. Annu. Rev. Biochem. 66, 475–509 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Smrcka AV, Hepler JR, Brown KO, Sternweis PC, Regulation of polyphosphoinositide-specific phospholipase C activity by purified Gq. Science 251, 804–807 (1991). [DOI] [PubMed] [Google Scholar]
- 26.Schade MA, Reynolds NK, Dollins CM, Miller KG, Mutations that rescue the paralysis of Caenorhabditis elegans ric-8 (synembryn) mutants activate the Gαs pathway and define a third major branch of the synaptic signaling network. Genetics 169, 631–649 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demirkan G, Yu K, Boylan JM, Salomon AR, Gruppuso PA, Phosphoproteomic profiling of in vivo signaling in liver by the mammalian target of rapamycin complex 1 (mTORC1). PLOS ONE 6, e21729 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP, A quantitative atlas of mitotic phosphorylation. Proc. Nat. Acad. Sci. U.S.A. 105, 10762–10767 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA, Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal. 4, rs5 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, Burlingame AL, Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mo/. Ce//. Proteomics 11, 215–229 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humphrey SJ, Yang G, Yang P, Fazakerley DJ, Stockli J, Yang JY, James DE, Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 17, 1009–1020 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, Olsen JV, Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 3, 876 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan M, Ha JH, Dhanasekaran DN, Ga13 stimulates the tyrosine phosphorylation of Ric-8A. J. Mol. Signal. 10, 3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJR, Mohammed S, Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 12, 260–271 (2013) [DOI] [PubMed] [Google Scholar]
- 35.Goswami T, Li X, Smith AM, Luderowski EM, Vincent JJ, Rush J, Ballif BA, Comparative phosphoproteomic analysis of neonatal and adult murine brain. Proteomics 12, 2185–2189 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boularan C, Kamenyeva O, Cho H, Kehrl JH, Resistance to inhibitors of cholinesterase (Ric)-8A and Gαi contribute to cytokinesis abscission by controlling vacuolar protein-sorting (Vps)34 activity. PLOS ONE 9, e86680 (2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xing B, Wang L, Guo D, Huang J, Espenel C, Kreitzer G, Zhang JJ, Guo L, Huang X-Y, Atypical protein kinase Cλ is critical for growth factor receptor-induced dorsal ruffle turnover and cell migration. J. Biol. Chem. 288, 32827–32836 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trent C, Tsung N, Horvitz HR, Egg-laying defective mutants of the nematode Caenorhabditis e/egans. Genetics 104, 619–647 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brundage L, Avery L, Katz A, Kim UJ, Mendel JE, Sternberg PW, Simon MI, Mutations in a C. elegans Gqα gene disrupt movement, egg laying, and viability. Neuron 16, 999–1009 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T, Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Xue Y, Liu Z, Cao J, Ma Q, Gao X, Wang Q, Jin C, Zhou Y, Wen L, Ren J, GPS 2.1: Enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng. Des. Sel. 24, 255–260 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Biemann K, Sequencing of peptides by tandem mass spectrometry and high-energy collision-induced dissociation. Methods Enzymol. 193, 455–479 (1990). [DOI] [PubMed] [Google Scholar]
- 43.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP, A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rigbolt KTG, Prokhorova TA, Akimov V, Henningsen J, Johansen PT, Kratchmarova I, Kassem M, Mann M, Olsen JV, Blagoev B, System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal. 4, rs3 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Han G, Ye M, Liu H, Song C, Sun D, Wu Y, Jiang X, Chen R, Wang C, Wang L, Zou H, Phosphoproteome analysis of human liver tissue by long-gradient nanoflow LC coupled with multiple stage MS analysis. Eectrophoresis 31, 1080–1089 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Roman DL, Ota S, Neubig RR, Polyplexed flow cytometry protein interaction assay: A novel high-throughput screening paradigm for RGS protein inhibitors. J. Biomol. Screen. 14, 610–619 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tall GG, Gilman AG, Purification and functional analysis of Ric-8A: A guanine nucleotide exchange factor for G-protein α subunits. Methods Enzymol. 390, 377–388 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Daub H, Olsen JV, Bairlein M, Gnad F, Oppermann FS, Körner R, Greff Z, Kéri G, Stemmann O, Mann M, Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol. Cell 31, 438–448 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Urano D, Jones JC, Wang H, Matthews M, Bradford W, Bennetzen JL, Jones AM, G protein activation without a GEF in the plant kingdom. PLoS Genet. 8, e1002756 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gibson SK, Gilman AG, Gia and Gp subunits both define selectivity of G protein activation by α2-adrenergic receptors. Proc. Natl. Acad. Sci. U.SA. 103, 212–217 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kozasa T, Gilman AG, Purification of recombinant G proteins from Sf9 cells by hexahistidine tagging of associated subunits. Characterization of α12 and inhibition of adenylyl cyclase by αz. J. Biol. Chem. 270, 1734–1741 (1995). [DOI] [PubMed] [Google Scholar]
- 52.Espunya MC, López-Giraldez T, Hernan I, Carballo M, Martinez MC, Differential expression of genes encoding protein kinase CK2 subunits in the plant cell cycle. J. Exp. Bot. 56, 3183–3192 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Salinas P, Fuentes D, Vidal E, Jordana X, Echeverria M, Holuigue L, An extensive survey of CK2 α and β subunits in Arabidopsis: Multiple isoforms exhibit differential subcellular localization. Plant Cell Physiol. 47, 1295–1308 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Oner SS, Maher EM, Gabay M, Tall GG, Blumer JB, Lanier SM, Regulation of the G-protein regulatory-Gαi signaling complex by nonreceptor guanine nucleotide exchange factors. J. Biol. Chem. 288, 3003–3015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F, Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bylund L, Kytölä S, Lui W-O, Larsson C, Weber G, Analysis of the cytogenetic stability of the human embryonal kidney cell line 293 by cytogenetic and STR profiling approaches. Cytogenet. Genome Res. 106, 28–32 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Lin Y-C, Boone M, Meuris L, Lemmens I, Van Roy N, Soete A, Reumers J, Moisse M, Plaisance S, Drmanac R, Chen J, Speleman F, Lambrechts D, Van de Peer Y, Tavernier J, Callewaert N, Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 5, 4767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stepanenko AA, Dmitrenko VV, HEK293 in cell biology and cancer research: Phenotype, karyotype, tumorigenicity, and stress-induced genome-phenotype evolution. Gene 569, 182–190 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Patel BR, Tall GG, Ric-8A gene deletion or phorbol ester suppresses tumorigenesis in a mouse model of GNAQQ209L-driven melanoma. Oncogenesis 5, e236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stoveken HM, Hajduczok AG, Xu L, Tall GG, Adhesion G protein-coupled receptors are activated by exposure of a cryptic tethered agonist. Proc. Natl. Acad. Sci. U.S.A. 112, 6194–6199 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stoveken HM, Bahr LL, Anders MW, Wojtovich AP, Smrcka AV, Tall GG, Dihydromunduletone is a small-molecule selective adhesion G protein60. coupled receptor antagonist. Mol. Pharmacol. 90, 214–224 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miller KG, Rand JB, A role for RIC-8 (Synembryn) and GOA-1 (Goα) in regulating a subset of centrosome movements during early embryogenesis in Caenorhabditis elegans. Genetics 156, 1649–1660 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maruyama IN, Brenner S, A phorbol ester/diacylglycerol-binding protein encoded by the unc-13 gene of Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 88, 5729–5733 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahmed S, Maruyama IN, Kozma R, Lee J, Brenner S, Lim L, The Caenorhabditis elegans unc-13 gene product is a phospholipid-dependent high-affinity phorbol ester receptor. Biochem. J. 287 (Pt 3), 995–999 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller KG, Emerson MD, Rand JB, Goα and diacylglycerol kinase negatively regulate the Gqα pathway in C. elegans. Neuron 24, 323–333 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Figueroa M, Victoria Hinrichs M, Bunster M, Babbitt P, Martinez-Oyanedel J, Olate J, Biophysical studies support a predicted superhelical structure with armadillo repeats for Ric-8. Protein Sci. 18, 1139–1145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roy A, Kucukural A, Zhang Y, I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y, I-TASSER server for protein 3D structure prediction. BMC Bioinf. 9, 40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hamel B, Monaghan-Benson E, Rojas RJ, Temple BRS, Marston DJ, Burridge K, Sondek J, SmgGDS is a guanine nucleotide exchange factor that specifically activates RhoA and RhoC. J. Biol. Chem. 286, 12141–12148 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schuld NJ, Vervacke JS, Lorimer EL, Simon NC, Hauser AD, Barbieri JT, Distefano MD, Williams CL, The chaperone protein SmgGDS interacts with small GTPases entering the prenylation pathway by recognizing the last amino acid in the CAAX motif. J. Biol. Chem. 289, 6862–6876 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hiraoka K, Kaibuchi K, Ando S, Musha T, Takaishi K, Mizuno T, Asada M, Menard L, Tomhave E, Didsbury JR, Snyderman R, Takai Y, Both stimulatory and inhibitory GDP/GTP exchange proteins, smg GDS and rho GDI, are active on multiple small GTP-binding proteins. Biochem. Biophys. Res. Commun. 182, 921–930 (1992). [DOI] [PubMed] [Google Scholar]
- 72.Lonhienne TG, Forwood JK, Marfori M, Robin G, Kobe B, Carroll BJ, Importin-β is a GDP-to-GTP exchange factor of Ran: Implications for the mechanism of nuclear import. J. Biol. Chem. 284, 22549–22558 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vetter IR, Arndt A, Kutay U, Görlich D, Wittinghofer A, Structural view of the Ran-Importin β interaction at 2.3 Å resolution. Cell 97, 635–646 (1999). [DOI] [PubMed] [Google Scholar]
- 74.Kant R, Zeng B, Thomas CJ, Bothner B, Sprang SR, Ric-8A, a G protein chaperone with nucleotide exchange activity induces long-range secondary structure changes in Gα. eLife 5, e19238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Black LA, Thomas CJ, Nix GN, Terwilliger MC, Sprang SR, Ross JB, Nanosecond dynamics of Gαil bound to nucleotides or Ric-8A, a Gα chaperone with GEF activity. Biophys. J. 111, 722–731 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zachariae U, Grubmüller H, Importin-β: Structural and dynamic determinants of a molecular spring. Structure 16, 906–915 (2008). [DOI] [PubMed] [Google Scholar]
- 77.Fukuhara N, Fernandez E, Ebert J, Conti E, Svergun D, Conformational variability of nucleo-cytoplasmic transport factors. J. Biol. Chem. 279, 2176–2181 (2004). [DOI] [PubMed] [Google Scholar]
- 78.Cansizoglu AE, Chook YM, Conformational heterogeneity of karyopherin beta2 is segmental. Structure 15, 1431–1441 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Afshar K, Willard FS, Colombo K, Johnston CA, McCudden CR, Siderovski DP, Gonczy P, RIC-8 is required for GPR-½-dependent Gα function during asymmetric division of C. elegans embryos. Cell 119, 219–230 (2004). [DOI] [PubMed] [Google Scholar]
- 80.Couwenbergs C, Spilker AC, Gotta M, Control of embryonic spindle positioning and Gα activity by C. elegans RIC-8. Curr. Biol. 14, 1871–1876 (2004). [DOI] [PubMed] [Google Scholar]
- 81.Yang F, Camp II DG, Gritsenko MA, Luo Q, Kelly RT, Clauss TR, Brinkley WR, Smith RD, Stenoien DL, Identification of a novel mitotic phosphorylation motif associated with protein localization to the mitotic apparatus. J. CellSci. 120, 4060–4070 (2007). [DOI] [PubMed] [Google Scholar]
- 82.Hampoelz B, Hoeller O, Bowman SK, Dunican D, Knoblich JA, Drosophila Ric-8 is essential for plasma-membrane localization of heterotrimeric G proteins. Nat. Cell Biol. 7, 1099–1105 (2005). [DOI] [PubMed] [Google Scholar]
- 83.Klattenhoff C, Montecino M, Soto X, Guzmán L, Romo X, Garcia MA, Mellstrom B, Naranjo JR, Hinrichs MV, Olate J, Human brain synembryn interacts with Gsα and Gqα and is translocated to the plasma membrane in response to isoproterenol and carbachol. J. Cell. Physiol. 195, 151–157 (2003). [DOI] [PubMed] [Google Scholar]
- 84.Malik S, Ghosh M, Bonacci TM, Tall GG, Smrcka AV, Ric-8 enhances G protein βγ-dependent signaling in response to βγ-binding peptides in intact cells. Mol. Pharmacol. 68, 129–136 (2005). [DOI] [PubMed] [Google Scholar]
- 85.Linder ME, Middleton P, Hepler JR, Taussig R, Gilman AG, Mumby SM, Lipid modification of G proteins: α subunits are palmitoylated. Proc. Natl. Acad. Sci. U.SA. 90, 3675–3679 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ross EM, Quantitative assays for GTPase-activating proteins. Methods Enzymol. 344,601–617 (2002). [DOI] [PubMed] [Google Scholar]
- 87.Chan P, Gabay M, Wright FA, Kan W, Oner SS, Lanier SM, Smrcka AV, Blumer JB, Tall GG, Purification of heterotrimeric G protein α subunits by GST-Ric-8 association: Primary characterization of purified Gαolf. J. Biol. Chem. 286, 2625–2635 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ryan JA, Cell cloning by serial dilution in 96 well plates protocol (Corning Inc, 2008). [Google Scholar]
- 89.Sarvazyan NA, Remmers AE, Neubig RR, Determinants of Gi1α and βγ binding. Measuring high affinity interactions in a lipid environment using flow cytometry. J. Biol. Chem. 273, 7934–7940 (1998). [DOI] [PubMed] [Google Scholar]
- 90.Dyer BW, Ferrer FA, Klinedinst DK, Rodriguez R, A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Anal. Biochem. 282, 158–161 (2000). [DOI] [PubMed] [Google Scholar]
- 91.English JG, Shellhammer JP, Malahe M, McCarter PC, Elston TC, Dohlman HG, MAPK feedback encodes a switch and timer for tunable stress adaptation in yeast. Sci. Signal. 8, ra5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Prior H, Jawad AK, MacConnachie L, Beg AA, Highly efficient, rapid and co-CRISPR-independent genome editing in Caenorhabditis elegans. G3 7, 3693–3698 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vizcaíno JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Ríos D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolomé S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H, ProteomeXchange provides globally co-ordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.