

Abstract
Darunavir (DRV) is a nonpeptidic protease inhibitor (PI) approved for the treatment of human immunodeficiency virus (HIV) infection. DRV displays potent activity against HIV strains resistant to other available PIs. Coadministration with ritonavir (RTV) improves the oral bioavailability of DRV. Inhibition of cytochrome P450 by RTV has been proposed as a mechanism for enhanced DRV bioavailability. However, interaction of these drugs with intestinal transporters has not been elucidated. This study was performed to explore the involvement of P-glycoprotein in transcellular DRV transport in monolayers of human intestinal Caco-2 and in ABCB1 multidrug resistance 1, (MDR1) gene-transfected renal LLC-PK1 (L-MDR1) cell lines. Transepithelial transport of DRV in Caco-2 cell monolayers was 2-fold greater in the basal-to-apical direction compared to that in the opposite direction. RTV had a significant inhibitory effect on the efflux transport of DRV in Caco-2 cells. The apical- to-basal DRV transport was enhanced by P-glycoprotein inhibitors, cyclosporin A and verapamil, as well as multidrug resistance-related protein (MRP/ABCC) inhibitors, probenecid and MK571. Using the L-MDR1 cell line, basal-to-apical DRV transport was much greater than in the opposite direction. Furthermore, cyclosporin A markedly inhibited the basal-to-apical DRV transport. RTV significantly increased the apical-to-basal transport of DRV in L-MDR1 cells, but reduced transport in the opposite direction. DRV inhibited P-glycoprotein-mediated efflux of calcein-acetoxymethyl ester in L-MDR1 cells with the inhibitory potency of 121 µM. These findings suggest that DRV is a substrate of P-glycoprotein and MRP, most likely MRP2. RTV appeared to inhibit P-glycoprotein, thereby enhancing the absorptive transport of DRV.
Keywords: protease inhibitor, transcellular transport, P-glycoprotein, Caco-2 cell
Treatment regimens for human immunodeficiency virus (HIV) infection have been greatly improved by the development of novel classes of anti-HIV drugs, including nucleoside analogues (NRTI), non-nucleoside analogue reverse transcriptase inhibitors (NNRTI) and protease inhibitors (PI). The term highly active antiretroviral therapy (HAART) is used to describe a combination of three or more of these anti-HIV drugs.1,2) During HAART, plasma HIV-1 levels rapidly decline to below the detection limit of standard clinical assays. However, reactivation of the remaining latently infected memory CD4+-T cells appears to be a source of continued virus reproduction.3) Virologic response to HAART treatment depends on viral sensitivity to antiretroviral drugs as well as patient compliance and medication adherence.1) However, long-term HAART treatment has raised issues concerning the development of drug-resistant HIV-1 variants4,5) and chronic side effects caused by the drugs.6,7) With the initiation of HAART, patients generally receive a combination of two NRTIs and either NNRTI or PI. The Department of Health and Human Services recommends an initial treatment regimen with the combination of tenofovir/emtricitabine or zidovudine/lamivudine as the NRTIs and either efavirenz as an NNRTI or atazanavir with ritonavir, fosamprenavir with ritonavir (RTV), or lopinavir/RTV as the PIs.8) However, virological failure continues to occur in a substantial proportion of HIV-infected patients who have received HAART.9)
Darunavir (DRV), formerly TMC-114, is a novel approved PI for the treatment of HIV infection.10,11) It was originally designed to be active against HIV strains resistant to other currently available PIs.12) POWER trials have evaluated the safety and efficacy of DRV in treatment-experienced HIV patients previously given other PIs.13) In these studies, DRV has indicated a significantly greater reduction in plasma HIV-RNA and an increase in CD4 counts compared with the active controls for patients with extensive PI resistance. Currently, there is a paucity of information on DRV resistance. Indeed, such data is mainly derived from clinical trials conducted during the registration of DRV. It is known that DRV is rapidly absorbed from the intestine after oral administration, reaching peak plasma concentrations after 2.5—4.0 h.14) Absorption of DRV is followed by a fast distribution/elimination phase and a subsequent slower elimination phase with a terminal elimination half-life of 15 h in the presence of low-dose RTV as a boosting drug.14,15) DRV is extensively metabolized by intestinal and hepatic cytochrome P450 (CYP) 3A4. Coadministration with small doses of RTV (100 mg) is thought to inhibit hepatic CYP3A4 activity, resulting in an increase in oral DRV bioavailability from 37 to 82%. DRV and its metabolites are mainly excreted in feces (80%), and, to a lesser extent, in urine (14%).15) Most PIs are reported to suppress intestinal ABCB1/P-glycoprotein mediated drug efflux into the lumen.16,17) However, there are no reports whether DRV per se is a transport substrate of P-glycoprotein.
The purpose of this study was to investigate the involvement of P-glycoprotein in transcellular transport of DRV across monolayers of the human intestinal epithelial Caco-2 cell line and the renal epithelial LLC-PK1 cell line stably transfected with the ABCB1 gene.
MATERIALS AND METHODS
Chemicals
Saquinavir (SQV) was obtained form Nippon Roche Co. (Tokyo Japan). Nelfinavir (NFV) and ritonavir (RTV) were a gift from JT Co. (Tokyo Japan) and Abbott Laboratories Co. (Illinois, U.S.A.), respectively. Darunavir (DRV) was synthesized in a convergent manner as previously described by Ghosh et al.10) Cylcosporin A was obtained from Novartis Pharma (Basel, Switzerland). Verapamil, probenecid, novobiocin, rifamycin SV, bromosulfophthalien, glycylleucine and glyclylsarcosine were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). MK571 was obtained from BIOMOL International, L.P. (Plymouth Meeting, PA, U.S.A.). Calcein-acetoxymethyl (AM) ester was purchased from Molecular Probe Co. (Eugene, OR, U.S.A.). All other chemicals used were of the highest purity available.
Cell Cultures
Caco-2 cells at passage 18, obtained from the American Type Culture Collection (ATCC HTB37), were maintained by serial passage in plastic culture dishes as described previously.18) For the transport studies, Caco-2 cells were seeded on polycarbonate membrane filters (3-µm pores, 4.71-cm2 growth area) inside Transwell cell culture chambers (Costar; Cambridge, MA, U.S.A.) at a cell density of 3×105 cells/well. The Transwell chambers were placed in six-well tissue culture plates with 2.6 ml of medium outside (basal chamber) and 1.5 ml of medium inside (apical chamber). The medium consisted of Dulbecco’s modified Eagle’s medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum (BioReliance; Rockville, MD, U.S.A.) and 1% nonessential amino acids (Invitrogen; Carlsbad, CA, U.S.A.) without antibiotics. The cells were grown in an atmosphere of 5% CO2, 95% air at 37 °C, and given fresh medium every 2 or 3 d. Cells were cultured for a total of 14d. In this study, cells between the 37th and 50th passage were used. Porcine kidney epithelial LLC-PK1 and L-MDR1 cells transfected with human ABCB1 cDNA (generous gifts from Dr. Erin G. Schuetz, St. Jude Children’s Research Hospital, Memphis, TN, U.S.A.) were cultured as described previously.19) In brief, LLC-PK1 and L-MDR1 cells were maintained in complete medium consisting of Medium 199 supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin, and L-MDR1 cells were maintained at 640 nM vincristine. For the transport studies, L-MDR1 and LLC-PK1 cells were seeded on polycarbonate membrane filters (3.0-µm pores, 4.71-cm2 growth area) inside Transwell cell culture chambers at a cell density of 2×106 cells/filter. Cells in each chamber were cultured as described above for 3 d. The medium was replaced by fresh medium after 2 d, and the cells were used in the transport studies 3 d after inoculation.
Transcellular Transport and Intracellular Accumulation of DRV
Transcellular transport of DRV was determined using cell monolayers grown in Transwell chambers. Culture medium on either the basal or apical side of the monolayers was replaced with 2 ml of incubation medium [145 mM NaCl, 3 mM KCl, 1 mM CaCl2, 0.5 mM MgCl2, 5 mM D-glucose, 8 mM Na2HPO4, 1.5 K2HPO4 and 5 mM N-(2-hydroxyethyl)piperazine-N′−2-ethanesulfonic acid (HEPES) (pH 7.4) or MES (pH 6.0)] containing DRV (10 µM) with or without PI, and medium on the opposite side was replaced with 2 ml of fresh incubation medium. In inhibition study, an inhibitor was added to the incubation medium on both sides of the monolayer. The monolayers were incubated in 5% CO2–95% air at 37 °C for up to 6 h, and 130 µl aliquots of medium from the relevant side were taken at the indicated time points. For accumulation studies, the medium was aspirated off at the end of the incubation period, and the monolayers were washed twice with 2 ml of ice-cold incubation medium. The filters with cell monolayers were immersed in 1 ml of extraction solution composed of the mobile phase of HPLC assay/methanol (1 : 1) for 1 h. Supernatants obtained after centrifugation at 200 g for 15 min were used for the HPLC assay. The remaining cells were lysed with 1 ml of 1 N NaOH, and used for the protein assay using a Bio-Rad protein assay kit (Bio-Rad Laboratories; Richmond, CA, U.S.A.) with bovine γ -globulin as a standard.
Calcein-AM Efflux Assay
Efflux assays were performed as described previously.19) A kinetic fluorometric assay was used to study the interaction of DRV with P-glycoprotein. For the calcein-AM efflux assay, L-MDR1 and LLC-PK1 cells were seeded in 96-well tissue culture plates at a cell density of 1×105 cells/well. Cells were cultured in 200 µl of Medium 199 supplemented with 10% fetal bovine serum in each well in an atmosphere of 5% CO2–95% air at 37 °C for 1 d. Cells were plated in 96-well tissue culture plates in Medium 199 containing PI. After a 30-min incubation period, calcein-AM was added to a final concentration of 2 µM, and the plates were placed into a Fluoroscan Ascent Thermo Labsystems, Franklin, MA, U.S.A.). Fluorescence was measured from 0 to 30 min using an excitation of 485 nm and an emission of 530 nm. The rate of calcein accumulation in the presence and absence of DRV was calculated by linear regression analysis using the Ascent software (Thermo Labsystems). Inhibitory potency of DRV for P-glycoprotein was evaluated by fluorescence at 30 min as a percentage of LLCmax, ((L-MDR1[I])-(L-MDR1[0]))×100/((LLC-PK1[0])–(L-MDR1[0])), where [I] represents a concentration of DRV, and [0] means absence of DRV.20)
HPLC Detrermination of DRV
The concentration of DRV was determined using HPLC (model LC-6A; Shimadzu, Kyoto, Japan). One hundred ml of sample were injected into the HPLC column. Separation was performed on a reversed-phase column (Zorbax SB-C18, 5-µm particle size, 150 mm×4 mm i.d.) at 40 °C. The mobile phase was a mixture of solution containing 25 mM sodium acetate and 25 mM hexane-1-sulfuric acid (pH 6.0) and acetonitrile (57 : 43). The flow rate was 1.0 ml/min and DRV was detected by UV absorption at 268 nm. The recovery of known small amount of DRV applied on cell monolayers for 60 min in the extraction method was >93%.
Statistical Analysis
Data were analyzed statistically by analysis of variance (ANOVA) followed by Scheffe’s multiple comparison test. A p-value of less than 0.05 was considered statistically significant.
RESULTS
Effect of RTV on the Transepithelial Transport of DRV in Caco-2 Cell Monolayers
Transepithelial transport of DRV (10 µM) in the apical-to-basal direction was time-dependent. Approximately 20% of the applied DRV in the apical chamber permeated into the basal compartment after 6 h (Fig. 1). The basal-to-apical transport of DRV was also time-dependent, with 43% of the applied DRV dose found in the opposite compartment after 6 h. These findings suggest that transepithelial transport of DRV is two-fold greater in the basal-to-apical direction compared to the opposite direction, corresponding to intestinal secretory transport. The apical-to-basal transport of DRV in the presence of RTV (20 µM) was significantly elevated compared to that in the absence of RTV. In contrast, the basal-to-apical transport of DRV was unaffected by the presence of RTV. The results indicate that the presence of RTV in the apical compartment had an inhibitory effect on efflux transport of DRV in the apical membranes of Caco-2 cells. The apical-to-basal transport of DRV in the presence of either cyclosporin A or verapamil, representative inhibitors of P-glycoprotein, was significantly increased compared to that in the absence of inhibitors (data not shown).
Fig. 1.
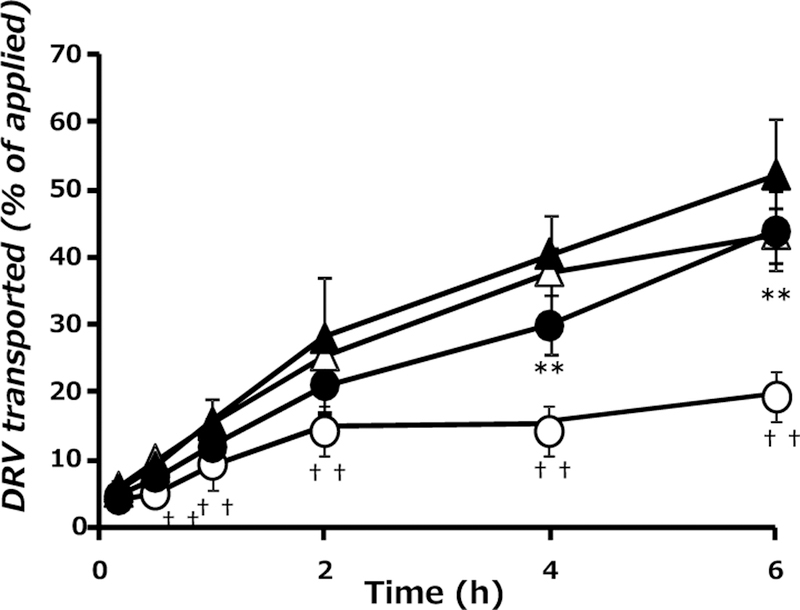
Effect of RTV on the Transepithelial Transport of DRV in Monolayers of Caco-2 Cells
Transport of DRV (10 µM) in the apical-to-basal direction (○, ●) and in the basal-to-apical direction (△, ▲) in the absence (control, open symbol) or presence (closed symbol) of RTV (20 µM). Each point represents the mean ± S.D. of three independent measurements. **p<0.01, significantly different from apical-to-basal transport without RTV. †† p<0.01, significantly different from basal-to-apical transport without RTV.
Effects of Various Inhibitors on Transcellular Transport of DRV in Caco-2 Cell Monolayers
The apical-to-basal transport of DRV was significantly increased in the presence of cyclosporin A, verapamil, probenecid, MK571, rifamycin SV and bromosulfophthalein (BSP) (Fig. 2). Probenecid and MK571 have been reported to inhibit multidrug resistant-associated protein (MRP/ABCC) 2.22) Rifamycin SV and BSP are known to inhibit organic anion transporting protein (OATP/SLCO) family members OATP1B1 and OATP1B3, respectively.23) In contrast, novobiocin, an inhibitor for breast cancer resistance protein (BCRP/ABCG2),24) significantly enhances the basal-to-apical transport of DRV. Glycylleucine and glycylsarcosine, a substrate and inhibitor of H+/oligopeptide cotransporter PEPT1 (SLC15A1), respectively,18) had no effect on the DRV transport. The ratio of basal-to-apical transport to apical-to-basal transport indicates that cyclosporin A, verapamil and RTV strongly suppressed the net secretory transepithelial transport of DRV. Probenecid, MK571 rifamycin SV and BSP had weak but significant inhibitory effects on the secretory transport of DRV. However, novobiocin markedly stimulated the secretory transport of DRV.
Fig. 2.
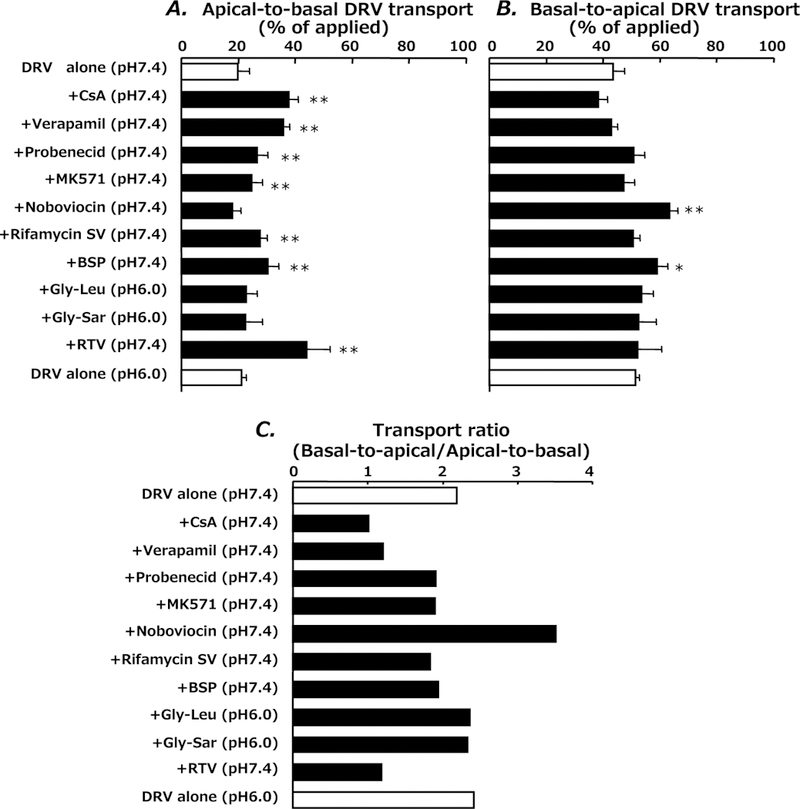
Effect of Various Inhibitors on Transepithelial Transport of DRV in Monolayers of Caco-2 Cells 6 h after Incubation with DRV
Apical-to-basal transport (A), basal-to-apical transport (B) and the transport ratio of basal-to-apical transport divided by apical-to-basal transport (C). Each bar represents the mean±S.D. of three independent measurements. *p<0.05, **p<0.01; significantly different from control (DRV alone).
Effect of Cyclosporin A and Verapamil on Transepithelial Transport of DRV in LLC-PK1 and L-MDR1 Cells
P-glycoprotein is an efflux pump responsible for limiting the oral bioavailability and tissue penetration of saquinavir.25) In order to confirm the involvement of P-glycoprotein in DRV transport, we compared transepithelial transport of DRV in untransfected renal LLC-PK1 cells and ABCB1/MDR1 gene-transfected LLC-PK1 (L-MDR1) cells overexpressing P-glycoprotein in the apical membrane domain. As shown in Fig. 3A, there was no difference in the transport of DRV between the apical-to-basal and basal-to-apical directions in the LLC-PK1 cell monolayers. However, the basal-to-apical transport of DRV was much greater than the apical-to-basal transport in L-MDR1 cell monolayers. Furthermore, cyclosporin A markedly decreased the basal-to-apical transport and increased apical-to-basal transport of DRV in L-MDR1 cells (Fig. 3B). Figures 3C and D show the intracellular accumulation of DRV after incubation with the drug on either the apical or basal side of the monolayer in the presence and absence of cyclosporin A. In all cases, the accumulation of DRV was much lower in L-MDR1 cells in comparison to LLC-PK1 cells. Cyclosporin A significantly increased the accumulation of DRV from both sides of the monolayer. These findings suggest that transepithelial transport of DRV is stimulated via P-glycoprotein in L-MDR1 cells.
Fig. 3.
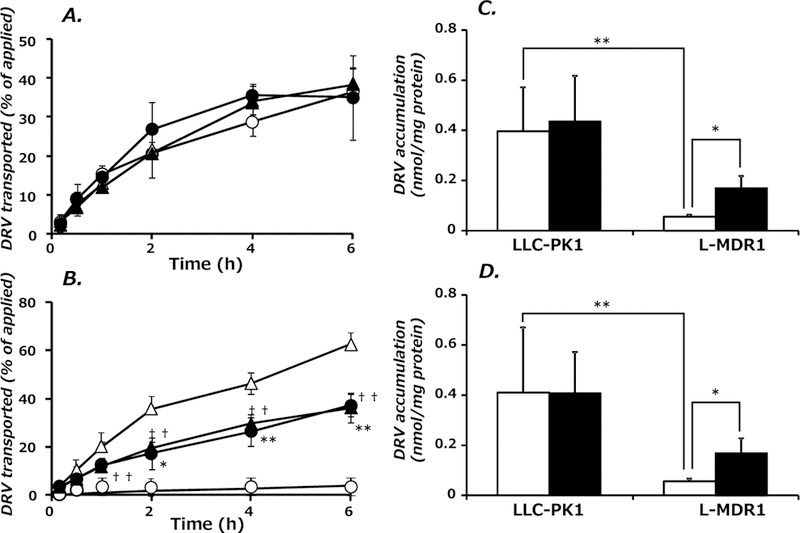
Transepithelial Transport and Intracellular Accumulation of DRV in LLC-PK1 Cells and L-MDR1 Cells
Transport of DRV (10 µM) in the apical-to-basal direction (○, ●) and in the basal-to-apical direction (△, ▲) in the absence (control, open symbol) or presence (closed symbol) of cyclosporin A (10 µM) in LLC-PK1 (A) or L-MDR1 (B) cells. Each point represents the mean±S.D. of three independent measurements. **p<0.01, *p<0.05, significantly different from apical-to-basal transport without cyclosporin A. †† p<0.01, significantly different from basal-to-apical transport without cyclosporin A. Intracellular accumulation of DRV in the absence (control, open column) and presence of cyclosporin A (closed column) 6 h after addition of DRV (10 µM) to the apical side (C) and basal side (D) of the monolayer. Each point represents the mean±S.D. of three independent measurements. **p<0.01, significantly different from DRV accumulation in LLC-PK1 cells without cyclosporine *p<0.05, significantly different from DRV accumulation in L-MDR1 cells without cyclosporin A.
Inhibitory Effects of PIs on Transepithelial Transport of DRV in L-MDR1 Cells
NFV markedly enhanced the apical-to-basal transport of DRV, but suppressed the basal-to-apical transport (Figs. 4A, 4B). RTV or SQV at a concentration of 50 µM significantly increased apical-to-basal DRV transport and decreased DRV transport in the opposite direction. Net secretory transport, evaluated as the ratio of basal-to-apical to apical-to-basal transport, showed NFV to have a potent inhibitory effect on P-glycoprotein-mediated transport of DRV when compared to RTV and SQV (Fig. 4C). RTV and SQV showed inhibitory effects on the net secretory transport of DRV at a concentration of 50 µM.
Fig. 4.
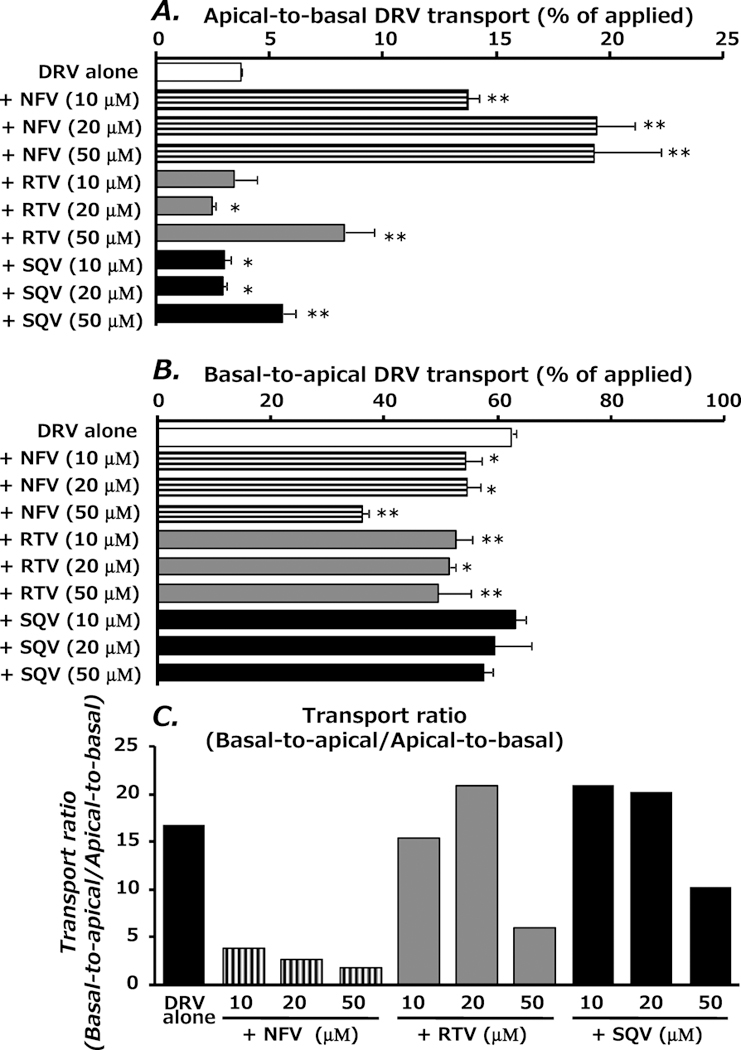
Effect of PIs on the Transepithelial Transport of DRV in L-MDR1 Cells 6 h after Incubation with DRV
Transport of DRV (10 µM) in the apical-to-basal direction (A) or basal-to-apical direction (B). The transport ratio (basal-to-apical/apical-to-basal) is given in (C). Each point represents the mean±S.D. of three independent measurements. **p<0.01, *p<0.05, significantly different from control (DRV alone).
Inhibitory Effect of DRV on P-Glycoprotein-Mediated Calcein-AM Efflux
In order to evaluate inhibitory effects of DRV on P-glycoprotein transport activity, calcein-AM extrusion test was performed using LLC-PK1 and L-MDR1 cells. As shown in Fig. 5, cellular accumulation of calcein was time dependent. Calcein accumulated much faster in LLC-PK1 cells than in L-MDR1 cells, confirming that calcein-AM was actively excluded in L-MDR1 cells by P-glycoprotein. RTV increased calcein accumulation in a dose-dependent manner in L-MDR1 cells, suggesting inhibition of P-glycoprotein activity. LLCmax of DRV was 121 µM and this potent inhibitory effect was comparable to that of RTV (LLCmax=111 µM).20)
Fig. 5.
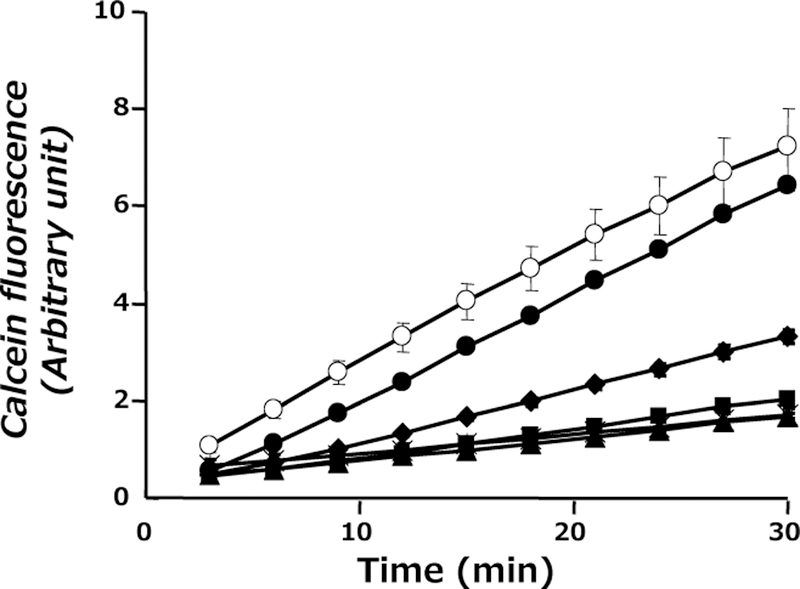
The Effect of DRV on Intracellular Uptake of Calcein-AM in LLC-PK1 and L-MDR1 Cells Monolayers
LLC-PK1 cells without DRV (○), L-MDR1 cells without DRV (×), L-MDR1 cells with DRV at concentrations of 10 µM (▲), 25 µM (■), 50 µM (◆) and 100 µM (●). Each point represents the mean±S.D. of five independent measurements.
DISCUSSION
It has been reported that most of the currently available PIs are transport substrates of P-glycoprotein.25,26) P-glycoprotein expressed in intestinal epithelial cells is thought to decrease absorption of orally administered PIs. Low levels of intestinal absorption together with CYP P450 activity are major factors in the first-pass effect of these drugs.16,26) For example, it was reported that the area under the time-plasma concentration curves (AUC) after oral administration of SQV in mdr1-knockout mice was elevated five-fold compared to that of wild-type mice.27) Moreover, Meaden et al. reported that P-glycoprotein expression levels in lymphocytes of patients coadministered with RTV and SQV were negatively correlated with the cellular accumulation of these PIs.21) It was also suggested that P-glycoprotein expressed in the blood-brain barrier and blood-placenta barrier participate in the restricted distribution of indinavir, SQV, NFV or amprenavir into the brain and placenta, respectively.28,29) Therefore, P-glycoprotein is thought to play a key role in the pharmacokinetics and therapeutic efficacy of most PIs. It was reported that DRV had a weak inhibitory effect (IC50>100 µM) on P-glycoprotein expressed in MDR1-gene transfected Madin–Darby canine kidney (MDCK) cells.30) However, it was not known whether DRV is recognized by P-glycoprotein as a transport substrate.
We explored transcellular transport of DRV using Caco-2 cells, which have been demonstrated to express ATP-binding cassette (ABC) transporter family members including ABCB11/P-glycoprotein, MRPs (ABCC2–6) and BCRP, OATP (SLCO) family members OATP-A and OATP-B, organic cation transporter OCT1 (SLC22A1), and organic anion transporter OAT2 (SLC22A7).22,31–33) In the present study, typical P-glycoprotein inhibitors, cyclosporin A and verapamil, enhanced the apical-to-basal transport of DRV in Caco-2 cells, suggesting that P-glycoprotein mediates efflux transport of DRV in the apical membranes of Caco-2 cells. The ratio of basal-to-apical to apical-to-basal transport was approximately 2.2, indicating that net secretary transport of DRV is preferred in Caco-2 epithelial cells. Probenecid and MK571, nonspecific MRP inhibitors, also had significant but much weaker effects on the apical-to-basal transport of DRV in Caco-2 cells. Huisman et al. reported that indinavir, SQV and RTV were transported by MRP2, but not by MRP1 and MRP3.34) Moreover, Williams et al. reported that SQV could be a substrate for P-glycoprotein, MRP1 and MRP2 with the transportability of P-glycoprotein>MRP2 MRP1.35) Therefore, our results suggest that MRP2 could mediate, at least in part, the apical efflux of DRV in Caco-2 cells. Novobiocin, a typical BCRP inhibitor,24) stimulated the basal-to-apical transport of DRV in Caco-2 cells. This finding suggests that BCRP is not involved in the transcellular transport of DRV, because BCRP is localized in the apical membranes of Caco-2 cells where it mediates efflux of substrates. However, we cannot exclude the possibility that unidentified novobiocin-sensitive transporter(s) expressed in the basolateral membranes of Caco-2 cells may also mediate the efflux transport of DRV. The OATP non-specific inhibitors, rifamycin SV and BSP,32,33) showed weak but significant stimulatory effects on the apical-to-basal transport. Interestingly, this represents the opposite net direction of transport for DRV, suggesting a partial involvement of OATP-A and/or OATP-B in transcellular transport of this drug in Caco-2 cells. In the inhibition studies with glycylleucine and glycylsarcosine, we found no contribution of PEPT1 to apical DRV transport in Caco-2 cells. These findings suggest that P-glycoprotein mediates predominantly efflux transport of DRV in the apical membranes of Caco-2 cells, but other apical membrane-localized efflux transporters MRP2 and/or BCRP, and also OATP might be involved, at least in part, in transcellular transport of DRV.
The present work using L-MDR1 cells provides the first direct evidence that DRV is a transport substrate of P-glycoprotein. The inhibitory potency of DRV was comparable to that of RTV. In HIV therapy, the bioavailability of DRV (100 mg two times a day) is improved by oral coadministration with RTV (600 mg) i.e., from 37% for DRV alone to 82% in combination with RTV.15) It is generally thought that the boosting effect of RTV is due to the inhibition of oxidative metabolism in the intestine and liver by specifically targeting CYP3A4 activity.15) Indeed, the apparent inhibition constant (Ki) of RTV for CYP3A4 and human hepatic microsomes were reported to be 0.10 µM and 0.17 µM, respectively, suggesting RTV is a potent inhibitor of CYP3A4.36) However, our results demonstrate that the mechanisms of action of RTV in terms of improving the bioavailability of DRV involve inhibition of the efflux transport systems of the intestinal lumen in addition to the intestinal/hepatic metabolism. Further investigation is required in order to explore the relative contribution of these two mechanisms towards the improvement of oral bioavailability of DRV in clinical treatment.
In conclusion, the present study has revealed that DRV is a transport substrate of P-glycoprotein in Caco-2 cells and in MDR1 gene-transfected renal LLC-PK1 cells. MRP, most likely MRP2, was found to be partially involved in the apical efflux transport of DRV in Caco-2 cells. Furthermore, our results suggest RTV inhibits P-glycoprotein, thereby enhancing the apical-to-basal transport (i.e., absorptive pathway) of DRV.
Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) for Hideyuki Saito (KAKENHI 17390158) and Akinobu Hamada (KAKENHI 19590149).
REFERENCES
- 1).Bartlett JA, Fath MJ, Demasi R, Hermes A, Quinn J, Mondou E, Rousseau F, AIDS, 20, 2051–2064, (2006). [DOI] [PubMed] [Google Scholar]
- 2).Piacenti FJ, Pharmacotherapy, 26, 1111–1133 (2006). [DOI] [PubMed] [Google Scholar]
- 3).Belmonte L, Baré P, de Bracco MM, Ruibal-Ares BH, Curr. Med. Chem, 10, 303–312 (2003). [DOI] [PubMed] [Google Scholar]
- 4).Kozal MJ, Hullsiek KH, Macarthur RD, Berg-Wolf M, Peng G, Xiang Y, Baxter JD, Uy J, Telzak EE, Novak RM, HIV Clin. Trials, 8, 357–370 (2007). [DOI] [PubMed] [Google Scholar]
- 5).Siliciano JD, Siliciano RF, J. Antimicrob. Chemother, 54, 6–9 (2004). [DOI] [PubMed] [Google Scholar]
- 6).Barbaro G, Am. J. Ther, 13, 248–260 (2006). [DOI] [PubMed] [Google Scholar]
- 7).Haugaard SB, Expert Opin. Drug Metab. Toxicol, 2, 429–445 (2006). [DOI] [PubMed] [Google Scholar]
- 8).Boyle BA, AIDS Read, 14, 71–74 (2004). [PubMed] [Google Scholar]
- 9).Røge BT, Barfod TS, Kirk O, Katzenstein TL, Obel N, Nielsen H, Pedersen C, Mathiesen LR, Lundgren JD, Gerstoft J, HIV Med, 5, 344–351 (2004). [DOI] [PubMed] [Google Scholar]
- 10).Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H, J. Med. Chem, 49, 5252–5261 (2006). [DOI] [PubMed] [Google Scholar]
- 11).Koh Y, Matsumi S, Das D, Amano M, Davis DA, Li J, Leschenko S, Baldridge A, Shioda T, Yarchoan R, Ghosh AK, Mitsuya H, J. Biol. Chem, 282, 28709–28720 (2007). [DOI] [PubMed] [Google Scholar]
- 12).Mitsuya H, Maeda K, Das D, Ghosh AK, Adv. Pharmacol, 56, 169–197 (2008). [DOI] [PubMed] [Google Scholar]
- 13).Clotet B, Bellos N, Molina JM, Cooper D, Goffard JC, Lazzarin A, Wöhrmann A, Katlama C, Wilkin T, Haubrich R, Cohen C, Farthing C, Jayaweera D, Markowitz M, Ruane P, Spinosa-Guzman S, Lefebvre E, Lancet, 369, 1169–1178 (2007). [DOI] [PubMed] [Google Scholar]
- 14).Boffito M, Winston A, Jackson A, Fletcher C, Pozniak A, Nelson M, Moyle G, Tolowinska I, Hoetelmans R, Miralles D, Gazzard B, AIDS, 21, 1449–1455 (2007). [DOI] [PubMed] [Google Scholar]
- 15).Rittweger M, Arastéh K, Clin. Pharmacokinet, 46, 739–756 (2007). [DOI] [PubMed] [Google Scholar]
- 16).Benet LZ, Izumi T, Zhang Y, Silverman JA, Wacher VJ, J. Controlled Release, 62, 25–31 (1999). [DOI] [PubMed] [Google Scholar]
- 17).Lee CG, Gottesman MM, Cardarelli CO, Ramachandra M, Jeang KT, Ambudkar SV, Pastan I, Dey S, Biochemistry, 37, 3594–3601 (1998). [DOI] [PubMed] [Google Scholar]
- 18).Saito H, Inui K, Am. J. Physiol, 265, G289–G294 (1993). [DOI] [PubMed] [Google Scholar]
- 19).Hamada A, Miyano H, Watanabe H, Saito H, J. Pharmacol. Exp. Ther, 307, 824–828 (2003). [DOI] [PubMed] [Google Scholar]
- 20).Shiraki N, Hamada A, Yasuda K, Fujii J, Arimori K, Nakano M, Biol. Pharm. Bull, 23, 1528–1531 (2000). [DOI] [PubMed] [Google Scholar]
- 21).Meaden ER, Hoggard PG, Newton P, Jtjia JF, Aldam D, Cornforth D, Lloyd J, Williams I, Back DJ, Khoo SH, J. Antimicrob. Chemother, 50, 583–588 (2002). [DOI] [PubMed] [Google Scholar]
- 22).Hirohashi T, Suzuki H, Chu XY, Tamai I, Tsuji A, Sugiyama Y, J. Pharmacol. Exp. Ther, 292, 265–270 (2000). [PubMed] [Google Scholar]
- 23).Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K, Hepatology, 36, 164–172 (2002). [DOI] [PubMed] [Google Scholar]
- 24).Shiozawa K, Oka M, Soda H, Yoshikawa M, Ikegami Y, Tsurutani J, Nakatomi K, Nakamura Y, Doi S, Kitazaki T, Mizuta Y, Murase K, Yoshida H, Ross DD, Kohno S, Int. J. Cancer, 108, 146–151 (2004). [DOI] [PubMed] [Google Scholar]
- 25).Jain R, Agarwal S, Majumdar S, Zhu X, Pal D, Mitra AK, Int. J. Pharm, 303, 8–19 (2005). [DOI] [PubMed] [Google Scholar]
- 26).Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, Wilkinson GR, J. Clin. Invest, 101, 289–294 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Park S, Sinko PJ, J. Pharmacol. Exp. Ther, 312, 1249–1256 (2005). [DOI] [PubMed] [Google Scholar]
- 28).Anderson BD, May MJ, Jordan S, Song L, Roberts MJ, Leggas M, Drug Metab. Dispos, 34, 653–659 (2006). [DOI] [PubMed] [Google Scholar]
- 29).Salama NN, Kelly EJ, Bui T, Ho RJ, J. Pharm. Sci, 94, 1216–1225 (2005). [DOI] [PubMed] [Google Scholar]
- 30).Tong L, Phan TK, Robinson KL, Babusis D, Strab R, Bhoopathy S, Hidalgo IJ, Rhodes GR, Ray AS, Antimicrob. Agents Chemother, 51, 3498–3504 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Englund G, Rorsman F, Rönnblom A, Karlbom U, Lazorova L, Gråsjö J, Kindmark A, Artursson P, Eur. J. Pharm. Sci, 29, 269–277 (2006). [DOI] [PubMed] [Google Scholar]
- 32).Sai Y, Kaneko Y, Ito S, Mitsuoka K, Kato Y, Tamai I, Artursson P, Tsuji A, Drug Metab. Dispos, 34, 1423–1431 (2006). [DOI] [PubMed] [Google Scholar]
- 33).Seithel A, Karlsson J, Hilgendorf C, Björquist A, Ungell AL, Eur. J. Pharm. Sci, 28, 291–299 (2006). [DOI] [PubMed] [Google Scholar]
- 34).Huisman MT, Smit JW, Crommentuyn KM, Zelcer N, Wiltshire HR, Beijnen JH, Schinkel AH, AIDS, 16, 2295–2301 (2002). [DOI] [PubMed] [Google Scholar]
- 35).Williams GC, Liu A, Knipp G, Sinko PJ, Antimicrob. Agents Chemother, 46, 3456–3462 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36).Ernest CS, Hall SD, Jones DR, J. Pharmacol. Exp. Ther, 312, 583–591 (2005). [DOI] [PubMed] [Google Scholar]