

Abstract
Chronic wounds are a major complication of diabetes associated with high morbidity and health care expenditures. To investigate the role of colonizing microbiota in diabetic wound healing, clinical outcomes, and response to interventions, we conducted a longitudinal, prospective study of patients with neuropathic diabetic foot ulcers (DFU). Metagenomic shotgun sequencing revealed that strain-level variation of Staphylococcus aureus and genetic signatures of biofilm formation were associated with poor outcomes. Cultured wound isolates of S. aureus elicited differential phenotypes in mouse models that corresponded with patient outcomes, while wound “bystanders” such as Corynebacterium striatum and Alcaligenes faecalis typically considered commensals or contaminants also significantly impacted wound severity and healing. Antibiotic resistance genes were widespread, and debridement, rather than antibiotic treatment, significantly shifted the DFU microbiota in patients with more favorable outcomes. These findings suggest that the DFU microbiota may be a marker for clinical outcomes and response to therapeutic interventions.
Keywords: diabetes, wound healing, microbiome, antibiotic resistance, metagenomics, chronic wounds
Graphical Abstract
eTOC:
Kalan et al. investigate the role of colonizing microbiota in wound healing, clinical outcomes, and response to therapy in patients with chronic diabetic wounds. Strains of the wound pathogen S. aureus were associated with poor outcomes, and sharp debridement therapy depleted anaerobic bacteria in wounds with favorable outcomes.
INTRODUCTION
Chronic, non-healing wounds are common and costly complications of diabetes. Up to one in four persons with diabetes will develop a diabetic foot ulcer (DFU) (Martins-Mendes et al., 2014), and approximately 25% of hospital stays for patients with diabetes are due to infected or ischemic DFU (Ramsey et al., 1999). Complications from DFUs account for two-thirds of all non-traumatic lower extremity amputations performed in the United States (Hoffstad et al., 2015; Martins-Mendes et al., 2014) and 5-year mortality rates surpass those of prostate and breast cancer, among others (Armstrong et al., 2007; Moulik et al., 2003). Improved therapeutic approaches are desperately needed, as morbidity, mortality, and health care expenditures only continue to increase as the prevalence of diabetes escalates worldwide.
Microbial colonization, biofilm formation, and infection are hypothesized to impair healing of DFUs and contribute to severe complications such as osteomyelitis and amputation. Wound infection is believed to underlie up to 90% of amputations (Boulton et al., 2005); yet quantitative cultures of uninfected DFUs were not predictive of outcomes (Gardner et al., 2014). Systemic and topical antimicrobials are often used to treat DFUs, despite their limited efficacy and even though it is often unclear which microorganisms are pathogenic and if some microorganisms may confer a beneficial effect. Culture-based methods, which are biased toward those microorganisms that thrive under laboratory conditions, insufficiently represent fungal and bacterial communities that colonize DFUs and other chronic wounds (Gardner et al., 2013). The role of microbial bioburden in DFU outcomes and complications remains ambiguous, including the significance of microbial load and diversity and the role of specific microorganisms, including known wound pathogens and microorganisms considered as skin commensals or environmental contaminants.
Culture-independent, amplicon-based sequencing methods (i.e. bacterial and fungal ribosomal RNA gene sequencing) have highlighted the polymicrobial and temporally dynamic nature of the bacterial and fungal microbiota colonizing DFU. However, only limited insight has been gained with these methods regarding the role of wound microbiota in patient outcomes, complications, and healing (Kalan et al., 2016; Loesche et al., 2017). A major limitation of such approaches is the poor taxonomic resolution that precludes accurate identification to the species or strain level (Meisel et al., 2016). Mounting evidence suggests that genetically distinct strains within a single species have important functional differences that influence interactions with their host (Byrd et al., 2017). Shotgun metagenomics, the untargeted sequencing of bulk microbial genomes in a specimen, could address this limitation while providing insight into the functions and virulence of the DFU microbiota. While technically and computationally challenging when applied to clinical wound specimens that contain abundant “contaminating” human tissues and cells, shotgun metagenomics has the potential for unprecedented insight into the microbial basis of impaired wound healing while revealing clinically important biomarkers of healing and complication. These biomarkers can then be combined with other individual and contextual factors to identify and target subgroups of patients for prevention and treatment, consistent with the evolving view and potential of precision health (Whitson et al., 2016).
For these reasons, we performed shotgun metagenomic sequencing of DFU samples to identify strain-level diversity and to profile the genomic content of the DFU microbiota. The aims of this study were to: 1) Identify strain-level taxa and functional genetic pathways that are associated with clinical DFU outcomes; 2) Recover corresponding cultured clinical isolates to test their influence on host inflammatory and tissue repair processes; and 3) Determine how therapeutic intervention changes the DFU microbiome.
RESULTS
Overview of study cohort and design
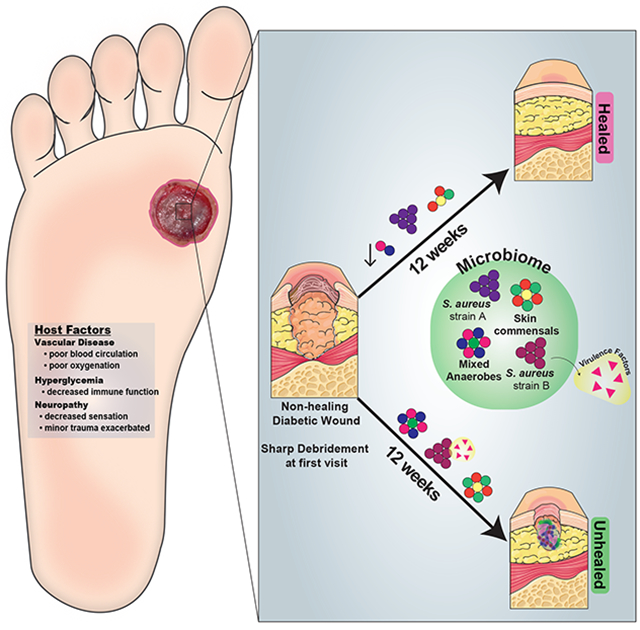
We enrolled 100 subjects with neuropathic, plantar DFU to examine the relationship between wound bioburden and clinical outcome. All enrolled subjects were free of clinical signs of infection at presentation and free from antibiotic exposure for >2 weeks. Specimens were obtained from DFUs by Levine’s swab (Fig. 1A), which samples the deep tissue fluid. Clinical factors were concurrently measured and recorded, including: blood glucose control (total blood glucose; hemoglobin A1c, HgbA1c), inflammation (white blood cell count, WBC; C reactive protein, CRP), ischemia (ankle-brachial index, ABI; toe-brachial index, TBPI), and wound oxygen levels (transcutaneous oxygen pressure at the wound edge). All patients underwent aggressive surgical debridement immediately following the first wound specimen collection at t=0. Specimens were obtained every two weeks, following conservative sharp debridement and non-bacteriostatic saline cleansing, until the wound healed, resulted in an amputation, or remained unhealed at the end of the 26-week follow-up period.
Figure 1: Shotgun metagenomic sequencing of the diabetic foot ulcer microbiome.
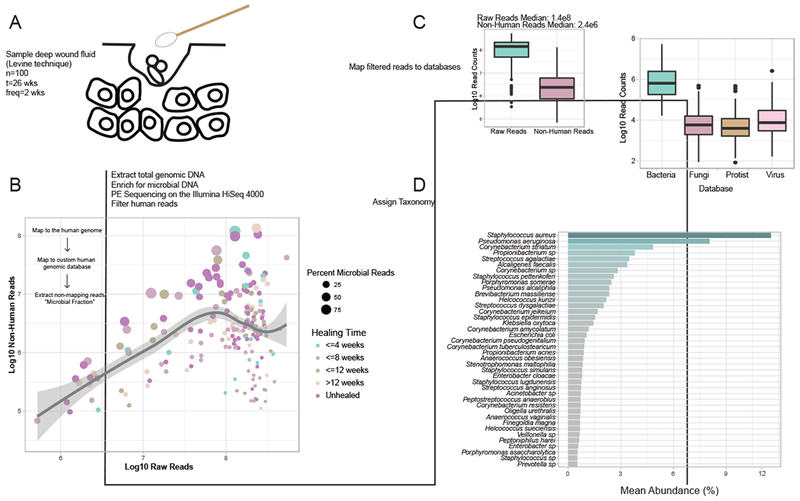
A) The Levine technique (Levine et al., 1976) was used to sample deep wound fluid from ulcers every two weeks over a period of 26 weeks (n=46 subjects). Microbial DNA was enriched from samples by bead based eukaryotic DNA depletion prior to whole shotgun metagenome sequencing (n=195 samples). B) Reads mapping to the human genome and a custom database of human sequences were filtered prior to analysis. Increasing sequencing depth results in a linear increase in the fraction of total microbial reads. C) Non-human reads are mapped to phylogeny-based bacterial, fungal, protist, and viral databases for classification. D) Mean abundance of bacterial species detected in >0.5% abundance of all samples and at least 1% abundance in individual samples.
To achieve the aims of this study, we selected a subset of subjects for shotgun metagenomic sequencing. Subjects were excluded from shotgun sequencing if outcome data were not available, if dropped from the study for unknown reasons or due to another infection (e.g., respiratory infection), or if achieving wound closure prior to the first study visit. This resulted in 195 reconstructed metagenomes from 46 patient timelines. A detailed description of the clinical co-variates is provided in Supplemental Table 1. Complications were experienced by 17 (37%) of the 46 subjects defined as: 1) wound deterioration, 2) development of osteomyelitis, and/or 3) amputations.
Diversity and composition of the DFU metagenome and concordance with 16S rRNA gene amplicon data
We obtained a median of 144,416,914 reads per sample, and microbial reads comprised 0.04% to 92.55% of raw sequence reads (median = 2.52%). Increasing sequence depth increased the number of microbial reads linearly until saturation occurred at approximately 1×108 reads (Fig. 1B). After filtering reads mapping to human genome references, the median number of microbial reads was 2,381,624 reads per sample (Fig. 1C). After mapping reads to multi-kingdom reference databases, bacterial reads comprised the largest proportion of microbial reads detected (96%), with Staphylococcus aureus, Pseudomonas aeruginosa, Corynebacterium striatum, and Alcaligenes faecalis, respectively, comprising the most abundant species of bacteria detected in all samples (Fig. 1C–D).
We assessed the concordance between shotgun metagenomic sequencing and 16S rRNA gene amplicon sequencing for this same cohort (Loesche et al., 2017) using two alpha diversity metrics (Fig. S1). Shannon diversity, a measure of species richness and evenness within a sample, was concordant between the two datasets (ρ=0.36; P≤0.0001). Species richness, measured by the number of genus-level operational taxonomic units (OTU richness; 16S rRNA amplicon sequencing) or genera (shotgun metagenomics) detected per sample, was also concordant (ρ=0.22; P≤0.01) (Fig. S1).
The most abundant genera in our data set were, in descending order, Staphylococcus (18.95%), Corynebacterium (14.64%), Pseudomonas (9.37%), and Streptococcus (7.32%) (Fig. 2A). These genera are consistent with cultured isolates and 16S rRNA gene sequencing data from the same cohort (Fig. S1C,D). Shotgun metagenomics identified S. aureus as the major Staphylococcus species, and was dominated by a single strain, S. aureus 7372 (Fig. 2B,C). Staphylococcal species present in lesser abundance included the coagulase negative species S. pettenkoferi, S. epidermidis, S. simulans, and S. lugdunensis. Corynebacterium striatum, a bacterium associated with infection and multi-drug resistance (Hahn et al., 2016), was the most prevalent Corynebacterium spp. classified in DFU and showed a positive correlation with ulcer duration (Fig. S2), while C. jeikeium, C. amycolatum, C. pseudogenitalium, C. tuberculostearicum, and C. resistens were present in lesser abundances (Fig. 2B). Pseudomonas spp. were the third most abundant genera detected with the most abundant species identified as P. aeruginosa followed by P. alcaliphila. P. aeruginosa is regarded as a common pathogen associated with DFU as it is frequently isolated by culture-based methods (Fig. S1D). Streptococcus was the fourth most abundant genera, with S. agalactiae, S. dysgalactiae, and S. anginosus present in descending abundance. Correlation analysis of strains identified in >0.5% abundance to clinical co-variates is described in Fig. S2.
Figure 2: Strain-level resolution of DFU microbiota.
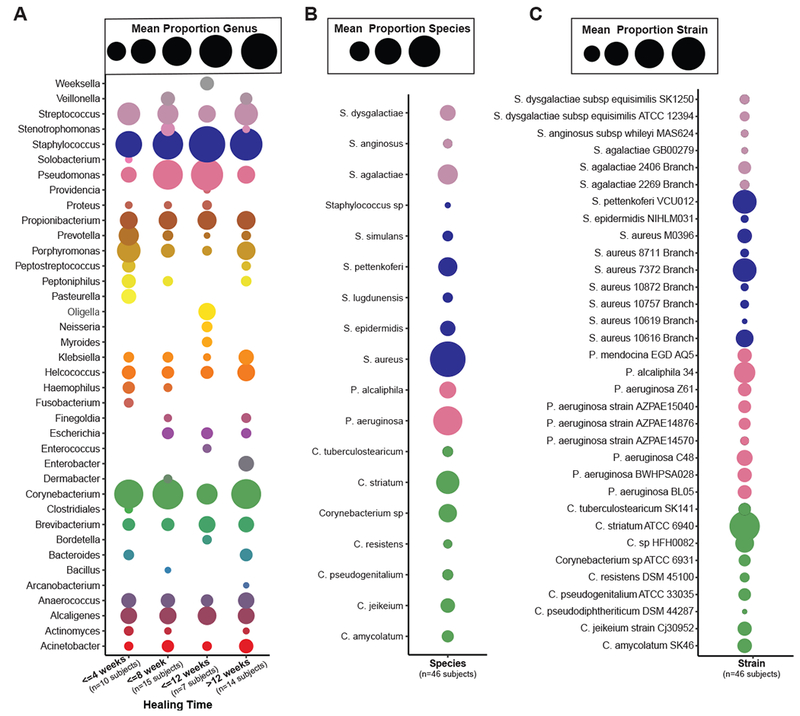
A) Mean relative abundance of genera detected in >0.5% of samples from wounds with different healing rates. B) Most abundant bacterial species detected in >0.5% mean relative abundance from all samples of top genera. C) Most abundant bacterial strains detected in >0.5% mean relative abundance in all samples of top genera. Circle color indicates the taxonomic assignment; Circle size represents mean relative abundance.
Staphylococcus aureus strain diversity is associated with clinical outcomes.
Ninety-four percent of DFU specimens tested were positive for S. aureus in >0.1% abundance. Using a generalized linear model, we observed a significant association between S. aureus community abundance and healing time (Fig. 3A). Since shotgun metagenomic sequencing enables strain-level detection not possible with cultures or amplicon-based sequencing approaches, we further explored the association between S. aureus strain and clinical outcomes using a phylogenetic approach. In our cohort, some strains of S. aureus were broadly distributed across all healing categories. For example, S. aureus 7372 (SA7372) was detected in 28.7% (56/195) of DFU specimens across disparate outcomes (Fig. 3B and D). We also identified several strains of S. aureus that are exclusively associated with unhealed wounds, such as S. aureus 10757 (SA10757), detected in 6.2% (12/195) of all specimens corresponding to 18.2% of non-healing wound specimens (Fig. 3B and E). These two representative strains of ‘generalist’ or ‘specialist’ S. aureus, respectively, were recovered from mixed Gram-positive cocci and anaerobic communities (Fig. 3C).
Figure 3: Staphylococcus aureus strain heterogeneity is associated with clinical outcomes.
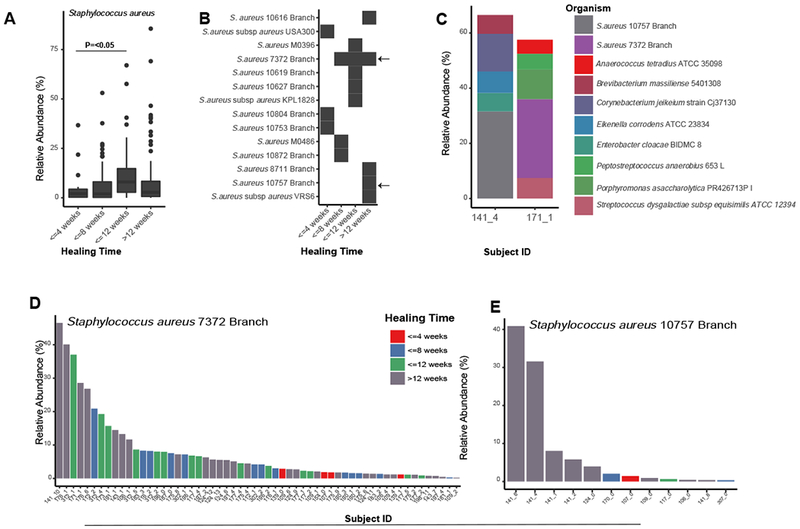
A) Mean relative abundance of S. aureus increases with healing time (P<0.05). B) Distribution of S. aureus strains and healing time. Each row corresponds to a different strain of S. aureus and the black box indicates detection in samples corresponding to each healing time (x-axis). Arrows indicate strains found in many samples (SA7372) and strains found only in non-healing wounds (SA10757). C) Microbiome community composition and taxa identified in >5 % relative abundance in patient specimens used to obtain representative isolates of SA7372 and SA10757. D) Mean relative abundance and distribution of SA7372 and E) SA10757 per sample across the cohort. Color indicates healing time.
We performed whole genome sequencing and comparative analysis of SA10757 and SA7372 to identify unique genomic loci and potential virulence mechanisms that could explain their association with clinical outcome. Both SA10757 and SA7372 are spa type 127, while multilocus sequence type (MLST) analysis determined SA10757 as ST474 and SA7372 as ST1. Both strains contained the agr type III agrABCD operon, encoding genes for the AGR quorum sensing system to produce autoinducing peptide (AIP) that functions to regulate biofilm development and virulence factors including toxins and degradative exoenzymes (Le and Otto, 2015; Novick and Geisinger, 2008). Together, the MLST, spa, and agr type suggest both strains are in the USA400 lineage of S. aureus (File S1).
To further explore their phylogenetic relationship to known lineages of S. aureus, we conducted phylogenomic analyses of our strains with previously published genomes, including the reference strains of S. aureus (ATCC 6538, NCTC 8325, Newman, Mu50, N315), LAMRSA (ST398), USA100 (strain 209), USA200 (MRSA252), USA300 (FPR3757), USA400 (MW2, 0051, MSSA476), and S. epidermidis (see Supplemental Data S2 for accession numbers). We determined both SA7372 and SA10757 form a monophyletic group with the USA400 strains S. aureus MW2, USA400-0051 (a Brazilian isolate), and MSSA476 (Fig. 4A). Further, this USA400 group form a monophyletic clade with the USA100, USA200, and USA300 lineages, separate from S. epidermidis.
Figure 4: Comparative genome analysis of S. aureus DFU isolates.
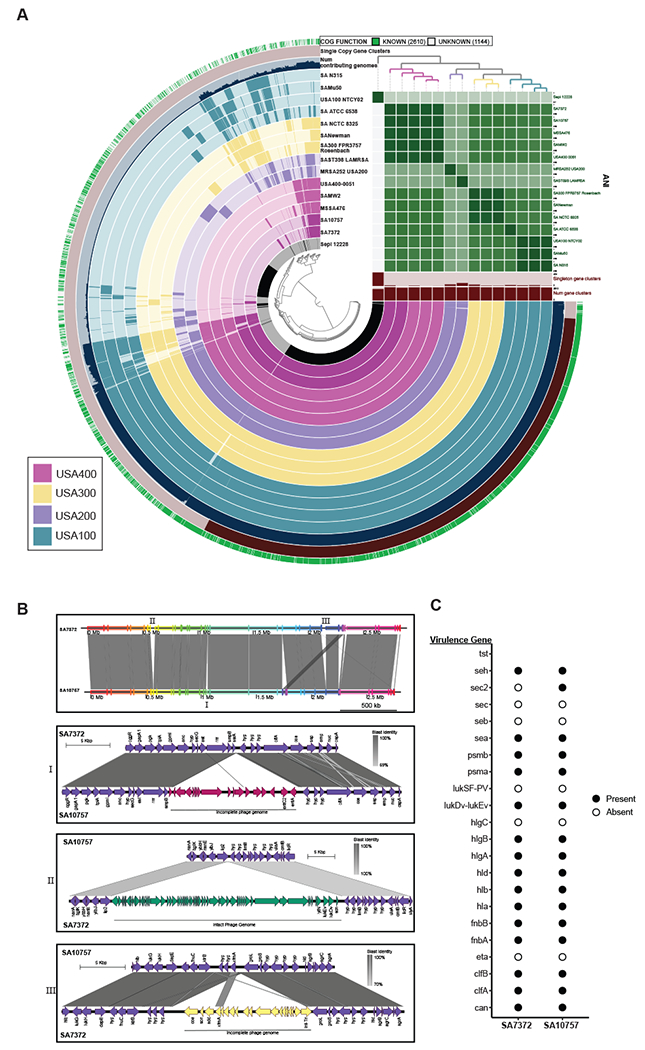
A) Pangenome analysis generated with Anvi’o for 15 S. aureus genomes ordered by gene cluster frequency (opaque=present, transparent=absent). Genomes are colored by monophyletic group. ANI scale 0.95-1 except for S. epidermidis (0.7-1) B) Whole genome and sub-region alignments of SA7372 and SA10757. Homologous blocks are shaded in gray. Phage genomes predicted by PHASTER are denoted with annotation of virulence genes. C) Gene presence (solid) or absence (open) of virulence factors in S. aureus.
We then determined in detail the shared and unique gene content between the two DFU strains (Fig. 4B,C). The shared genome consists of 2468 predicted genes comprising 90% of predicted open reading frames. Both strains harbor a ~10,500 bp plasmid identical to pMW2 carrying a beta-lactamase resistance gene cassette (blaRIZ) (Fig. S3). The genome of SA7372 contained 183 unique genes while the genome of SA10757 contained 64 unique genes (Fig. 4B). The majority of unique genes within each genome were of unknown function and predicted to encode hypothetical proteins. Staphylokinase (sak) is present in the SA7372 genome but not SA10757. In addition, SA7372 has an extra copy number of the genes encoding the neutrophil targeting leukotoxin (lukDV, lukEv) (Yoong and Torres, 2013), two extra cell wall hydrolases (lytN) that aide in protection from opsonophagocytic clearance (Becker et al., 2014), and two extra copies of the scn, encoding the Staphylococcal compliment inhibitor protein SCIN. Chromosomal genes conferring resistance to aminoglycoside (ant1), tetracycline (tetA), and macrolide (ermA) antibiotics were unique to the SA10757 genome, in addition to the staphylococcal enterotoxin C-2 (sec2) and enterotoxin A (sea) (Fig. 4B). Known virulence factor genes common to both strains included genes for enterotoxins seh and sea; phenol-soluble modulins psma, psmb; leukotoxin lukEv-lukDv; gamma hemolysins hlgA, hlgB, hlgC; alpha, beta, and delta-hemolysin hla, hlb, hld; fibronectin binding proteins fnbA, fnbB; collagen adhesion cna; and clumping factor clfA, clfB (Fig. 4C).
To identify if unique virulence-related genes are present on mobile genetic elements, we used PHASTER to identify prophage elements within each genome. Staphylococcal enterotoxins sec2 and sea were both present on a phage element predicted to be incomplete but closely related to phage PT1028, suggesting possible defective lytic capabilities resulting in stable integration into the SA10757 genome (Fig. 4B). An intact phage genome closely related to staphylococcus phage 96 containing the leukotoxin, hydrolase, and compliment inhibitor genes was detected within the SA7372 genome (Fig. 4B). Together, these findings provide evidence that closely related S. aureus strains differ at the genomic level in a phage-dependent manner to govern virulence-associated loci. This type of genomic diversification in turn may serve to influence host response and clinical outcome.
Poor outcomes are associated with biofilm- and virulence-related genetic pathways in the DFU metagenome
In addition to enhanced strain-level resolution, shotgun metagenomics also enables the estimation of community-level metabolic and virulence-related activities. We used the SEED database (Overbeek et al., 2005) to first annotate microbial genes and pathways in the DFU shotgun metagenomic dataset. At the SEED subsystem level 1, the most abundant features included expected metabolic activities such as carbohydrate utilization, amino acid and protein metabolism. Of note, genes related to virulence, disease and defense, phages and transposable elements were also within the top functional pathways identified (Fig. 5A). After sub-setting the most abundant features in the dataset, we assessed correlations with clinical co-variates. Hierarchal clustering analysis of the resulting Spearman rank coefficients for SEED subsystem level 3 annotations (detected at >0.1% abundance) revealed that wound depth, surface area, and tissue oxygenation are correlated with differing microbial functional profiles (Fig. 5B). Deep and poorly oxygenated wounds are strongly associated with virulent metabolism, including capsular and extracellular polysaccharide production, saccharide biosynthesis, and non-glycolytic energy production (Fig. 5B). Associations for all SEED annotations can be found in Fig. S4A.
Figure 5: Metagenome annotation reveals functional subsystems associated with clinical factors and outcomes.
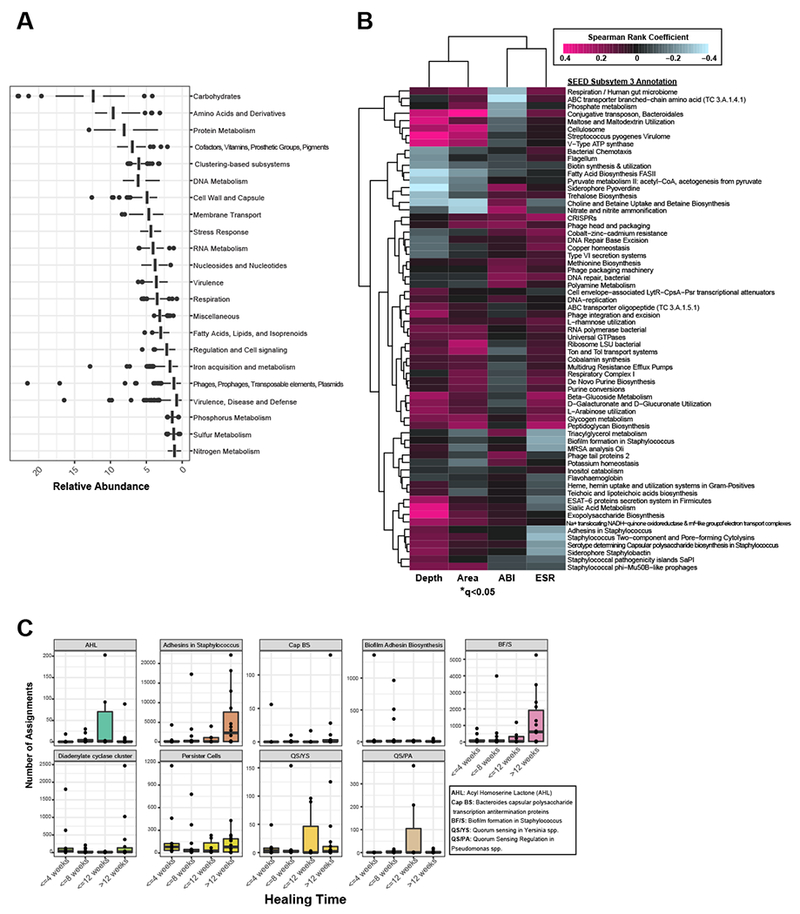
A) Mean relative abundance of the top SEED subsystem level 1 annotations detected in DFU metagenomes. B) Correlation heatmap and hierarchical clustering of SEED subsystem level 3 annotations with clinical co-variates. Color corresponds to Spearman rank coefficient (pink and blue indicating positive and negative correlation, respectively). Wound depth and area cluster separately from ankle brachial index (ABI) and eosinophil sedimentation rate (ESR), markers of inflammation. Asterisk indicates significant associations (q<0.05). C) Number of read assignments to SEED subsystem level 3 annotations, normalized by total read depth per sample, with biofilm-specific terms (y-axis). Samples are stratified by healing time (x-axis). Each plot represents a single term.
Taken together, our data are suggestive of genetic pathways encompassing depressed metabolic activity and heterogeneity, traits of an established biofilm, and further offers support to our hypothesis that in stable, non-healing wounds the microbiome exists in a biofilm state. In situ detection of biofilm is not clinically feasible without tissue biopsy and specialized microscopy techniques. We therefore applied an indirect measurement of biofilm by extracting SEED annotations with terms related to biofilm formation such as ‘adhesins’, ‘biofilm’, and ‘persister cells’. We assessed the number of reads mapping to these functions by healing category, normalized by total read depth per sample at the baseline visit. Slow or non-healing wounds were enriched in biofilm-related functional categories in Staphylococcus, compared to wounds that achieve closure by 12 weeks (Fig. 5C). Subsystem functional roles related to quorum sensing in Pseudomonas spp. (QS/PA and acyl-homoserine lactone production (AHL)) were the most abundant in the wounds that healed between 8-12 weeks, corresponding to the distribution of P. aeruginosa across healing categories (Fig. S4B). The abundance of reads mapping to these categories did not significantly differ between from the initial baseline (pre-debridement) visit and the next post-debridement visit (Fig. S4B).
The differential influence of primary wound isolates on host response and wound healing
To investigate the functional significance of our findings associating the DFU metagenome with clinical outcomes, we utilized patient wound isolates that were cultured in parallel with our culture-independent analysis. We hypothesized that S. aureus strains associated with clinical outcome wound elicit distinct biological responses. We further hypothesized that S. aureus, a conventional wound pathogen, would elicit more severe tissue destruction and inflammation than isolates recovered from wounds that are generally regarded as wound “bystanders”. To this end, we recovered SA7372, SA10757, and isolates of Corynebacterium striatum and Alcaligenes faecalis, to represent strains that are typically considered opportunistic pathogens but are not regularly identified in the clinical laboratory (C. striatum) and non-pathogenic environmental contaminants (A. faecalis). C. striatum and A. faecalis were also the third and fourth most abundant species detected in our cohort, respectively (Fig. 1D, Fig. S5A). To better mimic the wound environment, we grew the bacteria on sterile cotton gauze, using keratinocyte culture media as the primary nutrient source. Each clinical isolate developed biofilm-like structures exhibiting strong attachment to individual cotton fibers of the gauze, which were observed after vigorous washing to remove planktonic cells (Fig. S5B).
To determine if wound isolates differentially influenced cytokine production by keratinocytes, we grew wounded primary keratinocytes in 1% cell-free spent media (CFSM) from mid-log phase planktonic bacterial cultures, or mature 72-hour biofilm cultures. After 8 hours of exposure, we quantitated the secretion of twenty inflammatory cytokines and applied hierarchal clustering to the resulting cytokine profiles (Fig. S5). We did not observe detectable levels of EGF, IFNR, IFN-α2, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-7, or IL-9. Keratinocytes treated with A. faecalis biofilm and planktonic CFSM (Fig. S5) exhibited a strong IL-8 response (1873 ± 202 AF biofilm vs. 36±12 pg/mL control; P=<0.0001). Further, A. faecalis biofilm and to a lesser extent planktonic CFSM resulted in a significant and specific increase in the production of G-CSF, GM-CSF, IL-6, TGF-α, TNF-α, and IP-10 compared to the control and other treatment groups (Fig. 6A). A. faecalis biofilm and C. striatum planktonic CFSM enhanced the production of platelet derived growth factor (PDGF-AB:BB,) while C. striatum planktonic CFSM was associated with an increase in IL-1α and IL-1RA. Exposure to S. aureus CFSM from biofilm or planktonic cultures did not significantly shift cytokine production levels compared to the untreated control group, with the exception of SA7372 biofilm CFSM which resulted in decreased production of TGF-α (Fig. 6A; Fig. S5). This trend was also observed for C. striatum biofilm and planktonic CFSM, whereas A. faecalis CFSM resulted in an increased production of TGF-α.
Figure 6: Primary wound isolates result in differential host responses and wound healing.
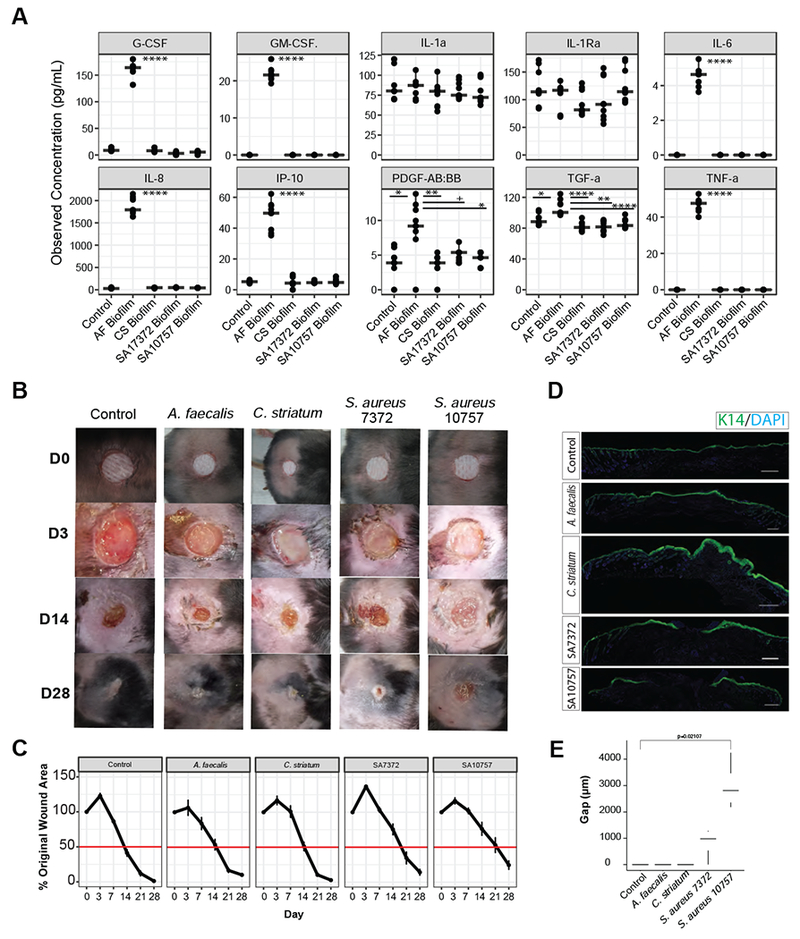
A) Observed concentration of secreted cytokines (pg/mL) from primary keratinocytes exposed for 8 hours to conditioned media from mature biofilms of A. faecalis (Af), C. striatum (CS), S. aureus 7372 (SA7372), or S. aureus 10757 (SA10757). Each condition was repeated with three biological replicates and three technical replicates of each (n=3 replicates from 3 cell cultures per group). Analysis of variance with post hoc multiple comparison testing was performed between each group (****<0.00001, ***<0.0001, **<0.001, *<0.01, +<0.05). B) Biofilms of each strain listed above were allowed to mature over a period of 72 hours on sterile gauze before being placed into full thickness dorsal mouse wounds (n=4 mice per group). Photographs of the wounds were taken at day 0, 3, 7, 14, 21, and 28. Wound measurements were recorded by two independent observers and are plotted in C) over time. Error bars represent standard error of the mean. D) Representative keratin 14 (K14) immunofluorescence staining of each wound at day 28. E) Gap (μM) between wound edges of each sample (n=4 wounds). A two-sided Wilcoxon-rank analysis was performed between each group (*<0.05).
To determine if different strains of primary clinical isolates impact healing rates in vivo, we used a type II diabetic mouse model of impaired wound healing (db/db; Lepr−/−). These experiments were performed with mature biofilms since the most pronounced effects on keratinocyte cytokine expression occurred when treated with biofilm, and because we hypothesize that within wound tissue bacterial isolates grow as biofilms. Full thickness excisional wounds were created on the mouse dorsa using 6 mm punch biopsy and mature biofilms were transferred into the wounds. A non-infected negative control consisted of gauze soaked in PBS. Each wound was photographed and measured on days 0, 3, 7, 14, 21 and 28 (Fig. 6B). Average wound surface area (percent of the original wound size), increased in all groups by day 3, except A. faecalis (control=123%, C. striatum=117%, SA7372=137%, SA10757=116%). Notably the wound margins in the A. faecalis group remained defined while they became irregular, diffuse, and macerated in the other infection groups, including the non-infected control. By day 7, wounds infected with A. faecalis biofilm resumed the same healing-trajectory as the control group (Fig. 6C). The C. striatum group exhibited an early delayed healing phenotype, with a mean wound area of 100.9 % of the original area on day 7, compared to 86.4% mean wound area in the control group. However, by day 14 the C. striatum infected wounds resumed the control healing trajectory. Persistent delayed healing occurred with both clinical S. aureus strains. By day 21 the control group exhibited near complete closure (mean wound area percentage of original=12.1%), while the mean original percent wound area of wounds infected with SA7372 and SA10757 was 34.6% and 53.1% respectively (P=0.01 and P=0.005).
On day 28, the wounds inoculated with the strain of S. aureus detected in unhealed wounds by metagenomics, SA10757, exhibited the slowest healing rate and open wounds remained with a mean percent wound area of 24% of the original size. Wound healing was further assessed by histopathology. Wounds were excised on day 28 to be analyzed by H&E staining (Fig. S5) and immunofluorescent cytokeratin 14 (K14) staining for re-epithelialization (Fig. 6D). We observed that control, A. faecalis and C. striatum treated wounds all showed complete re-epithelialization. In the case of C. striatum treated wounds, we observed a thickening of epithelial layer consistent with delay in wound healing (Pastar et al., 2014). Delayed wound healing was observed in both S. aureus strains as evident by the lack of complete re-epithelialization in the wounds. We measured the gap between each wound edge and confirmed that wounds colonized with SA10757 had the largest gap at day 28 (Fig. 6E). Together, these findings demonstrate the differential influence of S. aureus strain-level variation and other primary wound isolates on healing in vivo and provide functional evidence for the microbial basis of delayed healing in chronic wounds.
Debridement, but not systemic antibiotic therapy, shifts the microbiome in an outcome-dependent manner
Since all DFU patients underwent debridement at the baseline study visit, and a subset of patients were administered systemic antibiotics at some time during the study, we tested how these therapeutic interventions influence DFU microbial communities. We first determined if administration of systemic antibiotics influences the distribution of antimicrobial resistance genes in the DFU microbiota. We first examined the frequency of resistance genes to individual antibiotic chemical classes detected in each sample. At baseline, resistance genes to at least one class of antibiotic were detected in each subject, and genes conferring resistance to up to 10 different classes of antibiotics were detected in some wound specimens (Fig. 7A). Of antibiotic resistance classes detected, the most widespread were genes conferring resistance to beta-lactams, aminoglycosides, and macrolide antibiotics (Fig. 7B). In the thirty patients receiving antibiotic therapy, the resistance genotype did not correlate to the type of antibiotic administered (Fig. S6).
Figure 7: The DFU microbiome’s response to intervention predicts healing time.
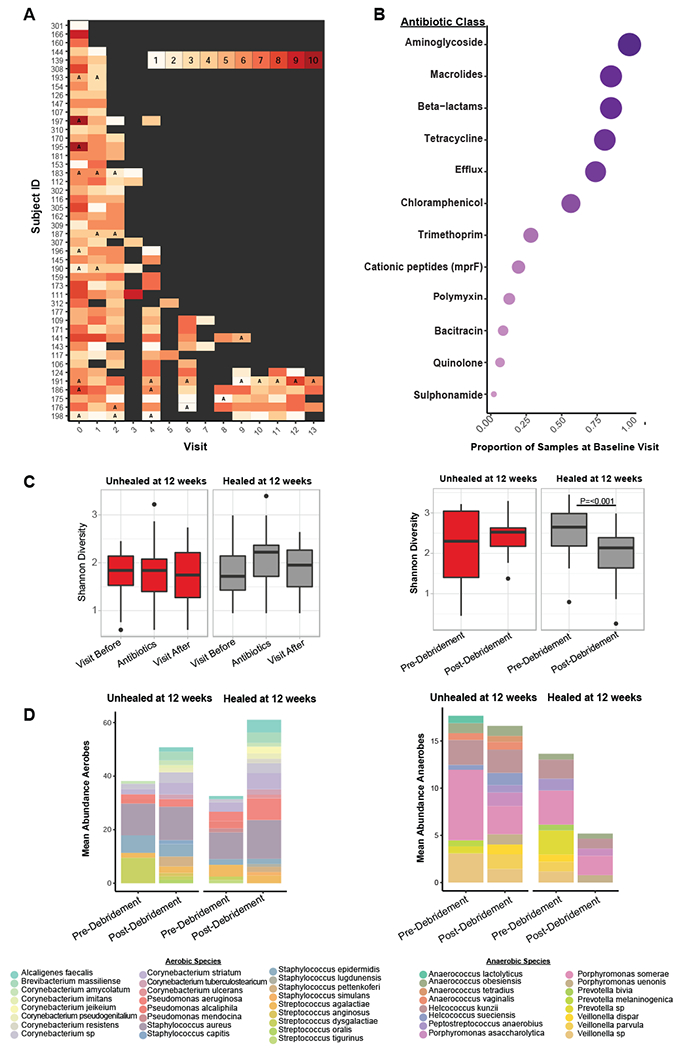
A) Timeline of each subject where the x-axis denotes the visit and the y-axis denotes individual subject IDs. The color of each visit corresponds to the total number of antibiotic resistance classes detected, with increasing darkness in red indicating increasing number of resistance classes detected. Grey boxes indicate a visit where the sample was either not sequenced, the wound was healed, or no resistance genes were detected. Visit 3, 5, and 7 were not sequenced unless it was the last visit a sample was collected before healing was recorded. Types of antibiotics with multiple classes of resistance (e.g., beta-lactamase class A, B, C etc.) were collapsed into a single class (e.g., beta-lactamases). The letter ‘A’ indicates a visit where antibiotics were administered. B) The proportion of samples with resistance genes detected (x-axis) for different classes of antibiotics (y-axis) at the baseline visit. Circle size corresponds to mean proportion. C) Shannon diversity remains unchanged in samples before, during, or after antibiotic administration in healing (n=9 subjects) and unhealed wounds (n=9 subjects) while debridement significantly reduces Shannon diversity in wounds that heal (n=32 subjects) within 12 weeks post-debridement. P<0.001, with non-parametric Wilcoxon rank-sum test. In wounds unhealed at 12 weeks (n=14 subjects) post-debridement a change in Shannon diversity is not observed. D) The mean proportion of common aerobic genera do not shift after debridement. The mean proportion of anaerobic genera are significantly reduced after debridement in wounds that heal within 12 weeks. P=0.002, with non-parametric Wilcoxon rank-sum test. In wounds unhealed at 12 weeks post-debridement the mean proportion of anaerobic genera does not change.
Of the DFUs that were not healed by week 12 (n=14), 50% remained unhealed or resulted in an amputation (n=7). Twelve weeks is also the FDA recommended follow up period to distinguish complete wound closure from transient coverage. Therefore, we used this time point to divide the cohort into ‘healing’ and ‘non-healing’ wound types, and to test the hypothesis that intervention differentially influences the microbiome of healing and non-healing wounds. Antibiotic administration did not have a significant effect on DFU microbiomes as measured by Shannon diversity (a measure of alpha diversity, or within sample diversity) before, during, or after the intervention in both groups (Fig. 7C). We also compared how debridement, an intervention designed to remove necrotic tissue, influences wound microbiota. We determined that a significant reduction in Shannon diversity occurred in the visit immediately after debridement, but only in wounds that healed by 12 weeks (Fig. 7C). When we examined the composition of the community at these time points, we observe that the relative abundances of aerobic bacteria such as S. aureus, Streptococcus agalactiae, and Pseudomonas aeruginosa do not change; however, mixed anaerobic bacteria such as Anaerococcus lactolyticus, Porphyromonas somerae, Prevotella melaninogenica, and Veillonella dispar are reduced after debridement in healed wounds, but not unhealed wounds (Fig. 7D). Tissue oxygenation levels were not significantly different pre- or post-debridement or between healing times. Further, we did not observe a correlation between the abundance of individual anaerobic strains of bacteria within each sample and transcutaneous oxygen pressure (Fig. S6). We also calculated associations between each anaerobic strain detected and each unique strain of S. aureus. SA10757 was positively correlated with three species of Anaerococcus spp.; A. obesiensis, A. vaginalis, and A. lactolyticus (ρ=0.48, 0.48, and 0.64, respectively) while all other strains of S. aureus had weak or negative associations to anaerobic species (Fig. S6).
DISCUSSION
Diabetic foot wounds are complicated by several factors that contribute to impaired tissue regeneration including hyperglycemia, peripheral neuropathy, vascular disease and a complex microbiome. Microbial communities that assemble in wound tissue are difficult to detect and are not necessarily associated with cardinal signs of infection (Lipsky et al., 2012), further complicating prognostics for wound healing outcomes. Chronic wounds have a major societal impact; thus our in-depth investigation of the DFU microbiome, coupled with in vitro and in vivo functional modeling, enhances our understanding of microbial influences on tissue repair pathways, suggests diagnostic/prognostic and therapeutic targets, and has the potential to overcome challenges for improving patient outcomes.
Here we apply shotgun metagenomic sequencing of time-series specimens from patients with DFU to achieve strain-level classification of microbial communities. Despite the growing body of literature dedicated to the study of wound microbiomes, all studies to date have exclusively employed amplicon-based sequencing of phylogenetic marker genes such as the 16S rRNA gene, failing to distinguish individual species. For example, within the Staphylococcus genus are skin commensals and the notorious pathogen S. aureus. Clinically, S. aureus would be regarded differently than the commensal S. epidermidis; thus their classification is critical for determining efficacious treatment strategies. Additionally, shotgun metagenomics allows for strain-level tracking and functional annotation, which both revealed aspects of the DFU microbiome and its association with clinical outcomes in this study.
Strain heterogeneity of S. aureus is associated with disease severity in other dermatological conditions such as atopic dermatitis, where strains from more severe patients elicit stronger immune responses and skin inflammation (Byrd et al., 2017). Diabetic wounds are consistently colonized by S. aureus so we focused our analysis on this skin pathogen to classify strains associated with different wound healing outcomes. We identified S. aureus strains with a wide host range and strains exclusively associated with unhealed wounds. The genome of S. aureus associated with poor wound healing outcomes harbored multiple antibiotic resistance genes and genes encoding staphylococcal enterotoxins. These superantigens result in an exacerbated inflammatory response by non-specific stimulation of large populations of T-cells (Ortega et al., 2010), suggesting colonization by these strains results in persistent inflammation leading to impaired healing progression. Conversely, genome analysis of S. aureus isolates with non-specific associations suggest such strains are experts at immune evasion and warrant additional investigation to link genome diversity with phenotypic differences in pathogenesis.
To investigate the biological consequences of S. aureus strain variation in comparison to wound isolates commonly dismissed as skin flora (C. striatum) or environmental contamination (A. faecalis), we measured inflammatory responses in primary keratinocytes. We found the environmental Gram-negative rod A. faecalis, normally considered nonpathogenic, induces a striking keratinocyte response via production of pro-angiogenic/pro-inflammatory cytokine IL-8 (Arwert et al., 2012; Rennekampff et al., 2000) in addition to cytokines known to stimulate proliferation and enhance wound healing (GM-CSF, G-CSF, PDFG-AB). Diabetic wounds with A. faecalis biofilms healed at an accelerated rate during the early stages of wound healing. These findings suggest a beneficial role for some microbes in tissue repair. Future studies should address how A. faecalis, C. striatum and other “non-pathogenic” wound microbiota function in a polymicrobial setting to influence virulence of wound pathogens, host responses, and healing outcomes. Approaches such as bacterial transcriptomics and metabolomics will further elucidate mechanisms of bacterial interchange with each other and the host tissue.
Given that Corynebacterium was the second most abundant genera classified in our cohort by both 16S rRNA gene sequencing and shotgun metagenomics, consistent with previous chronic wound microbiome studies (Dowd et al., 2008; Gardner et al., 2013; Loesche et al., 2017; Rhoads et al., 2012; Wolcott et al., 2016), we hypothesize that Corynebacterium spp. have a more significant role in wound healing than simple contamination from intact skin. We found that standard clinical microbiology protocols do not routinely classify Corynebacterium spp. without the use of specialized workflows, but instead group aerobic Gram-positive, catalase-positive rods as diphtheroid and consider them to be skin contaminants (Leal et al., 2016). Corynebacterium striatum is the most prevalent and abundant Corynebacterium spp. in DFU, detected in 28% of our specimens. It is also considered an emerging multi-drug resistant microbe (Hahn et al., 2016) and may be an underrecognized cause of diabetic foot osteomyelitis (Patel et al., 2016; Rizvi et al., 2011) suggesting it should not be classified as merely contaminating skin flora in the clinic. Our in vivo findings in a diabetic murine model of impaired healing support this hypothesis, where wounds appeared closed at 28 weeks but histopathology revealed a hyperproliferative epidermal layer consistent with delayed healing.
An aim of this study was to determine the effects of therapeutic intervention, including systemic antibiotic therapy and debridement, on the DFU microbiome. The inherent properties of a diabetic wound environment support microbial colonization, although antibiotic use is not recommended for DFU in the absence of overt clinical infection, such as cellulitis and osteomyelitis (Lipsky et al., 2012). However, some cohorts have reported antibiotic use in 60% of patients (Howell-Jones et al., 2006; Siddiqui and Bernstein, 2010; Tammelin et al., 1998), and it was a DFU from which the first strain of vancomycin-resistant S. aureus was isolated (Tenover et al., 2004). We characterized antibiotic resistance genes across our cohort and over time to determine that resistance genes are widespread in DFU microbiomes, and in some cases harbor genes conferring resistance to ten different classes of antibiotics. Greater than 50% of wound specimens contained resistance genes to the aminogylocoside (e.g., clindamycin), macrolide (e.g., erythromycin), beta-lactam (e.g., amoxicillin), and tetracycline (e.g., minocycline) classes of antibiotics. We further examined the effects of antibiotic administration at the community level and determined that antibiotics do not change the overall diversity in healed or non-healed wounds, suggesting little perturbation to the microbiome within the wound.
In contrast, debridement elicited a decrease in bacterial diversity, driven by a reduction in the abundance of anaerobic bacteria in the overall community, in the subset of wounds that achieved complete re-epithelialization within 12 weeks. It is recommended that all diabetic wounds are surgically sharp debrided, to remove debris, callus, necrotic (senescent, devitalized), and infected tissue (Lipsky et al., 2012). This procedure is thought to ‘re-activate’ stalled healing pathways by inducing an acute wound (Ashrafi et al., 2016). Although correlated with improved healing rates, the association is not significant and less than half of debrided DFUs progress towards healing (Cardinal et al., 2009). Compromised blood flow leads to local tissue ischemia that can promote growth of anaerobic microorganisms. Several studies including our own have concluded that anaerobes are underrepresented in culture-based estimation of DFU isolates (Citron et al., 2007; Gardner et al., 2014; Louie et al., 1976). Our data suggest that several species of anaerobic bacteria are abundant across DFUs in association with mixed aerobes, and successful debridement may disrupt anaerobic networks. While it remains to be determined if this finding is generalizable to other patient cohorts or wound types, it raises the intriguing possibility that the microbiome can serve as a prognostic marker of healing trajectory at the time of debridement.
A limitation is that we did not collect specimens from “normal” wounds or from the adjacent skin to compare our findings. When we compare our findings with traumatic open fractures, a type of acute wound, we found similar colonization patterns, with Corynebacterium and Staphylococcus as the most abundant genera (Bartow-McKenney et al., 2018). We and others have examined the relationship between wound microbiota and adjacent skin microbiota; unsurprisingly, these communities significantly differ, likely owing to the stark contrast in nutrient availability (Gardiner et al., 2017). Future studies should examine the adjacent skin microbiota of DFU and its association with clinical outcome and response to therapy. Further, we did not detect statistical differences in healing rates between male and female subjects. However, only 26% of this cohort was female resulting in lowered statistical power for this type of analysis and limits the conclusions about whether sex influences wound healing rates and the microbiome composition of DFU.
Chronic wounds are a major strain to health care systems and cause significant morbidity and mortality. As the rate of diabetes and obesity increases worldwide, the economic and social burden of chronic wounds such as DFU is projected to snowball (Sen et al., 2009). Therefore, new approaches for their management and treatment are desperately needed. Here, we applied shotgun metagenomic sequencing to clinical DFU specimens to identify microbial taxonomic and genetic markers associated with clinical outcomes. We coupled this analysis with models of host-microbe interaction to demonstrate the strain- and species-specific roles for wound microbiota in host response and wound healing. These insights may lead to targets for improving management and treatment approaches based on microbial-host interactions of the wound.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING:
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Elizabeth Grice (egrice@pennmedicine.upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study Design and Variables
The study design, subject enrollment, and specimen collection are described in previous publications (Kalan et al., 2016; Loesche et al., 2017). Briefly, 100 subjects were enrolled in a prospective-cohort to sample the DFU microbiota and measure outcomes. Samples for microbiota analyses were collected at initial presentation (V0) and every two weeks until the DFU: i) healed; ii) was amputated; or iii) 26 week of follow up elapsed (V1-12). The Institutional Review Boards at the University of Iowa (IRB#200706724) and the University of Pennsylvania approved the study procedures (IRB#815195). Informed consent was obtained from all participants in writing. All metadata describing each sample collected (age, sex, gender) is provided in the supplemental Data S3>metamap.csv file. Subjects were 28-78 years of age (mean age 53 +/− 9 years of age) and 26% were female. All subjects were confirmed diabetic (type I or II) and antibiotic naïve at the beginning of the study, but maintained other medication regimens not related to their DFU. Subjects were not involved in previous procedures.
Wound management was standardized to aggressive sharp debridement of necrotic tissue in the wound bed at baseline and wound edge callus removal every two weeks followed by non-antimicrobial dressing application (i.e., Lyofoam®, Molnlycke Health Care). Ulcers were offloaded with total contact casts (87 subjects) or DH boots (13 subjects). Topical antimicrobial or system antibiotic administration was not included unless an infection-related complication was present occurred within the study period. Data was collected at baseline and longitudinally every two weeks until the wound healed or 26 weeks elapsed for a total of 384 wound specimens. We subset this dataset to maximize read depth and output for shotgun metagenomic sequencing and this is described in the results section.
Clinical factors:
Demographic variables were collected at the baseline visit including age, sex, diabetes type and duration and duration of the study ulcer using subject self-report and medical records. At each study visit glycemic control was measured by levels of haemoglobin A1c and blood glucose. Inflammatory (Erythrocyte sedimentation rate (ESR), C-reactive protein) and immune (white blood cell counts) markers were determined with standard laboratory tests. Each subject was also assessed for ischemia using the ankle-brachial and toe-brachial index and for neuropathy using the 5.07 Semmes-Weinstein monofilament test. Transcutaneous oxygen pressure was measured at baseline and at each follow-up visit, using a transcutaneous oxygen monitor (Novametrix 840®, Novametrix Medical Systems Inc.). Ulcer location was categorized as forefoot, midfoot, or hindfoot. The level of necrotic tissue was defined as black, grey or yellow tissue in the wound bed measured using a likert scale as the percentage of the total wound area binned according to 0-25%, 25-50%, 50-75% or 75-100% necrotic tissue.
Outcomes:
Healing and infection-related complications were assessed every two weeks. Ulcer closure was determined using the Wound Healing Society’s definition of “an acceptably healed wound,” (Margolis et al., 1996). “Infection-related complications” included wound deterioration defined as development of frank erythema and heat, and an increase in size > 50% over baseline, new osteomyelitis, and/or amputations due to infection. Two members of the research team independently assessed each DFU for erythema and heat. Two members of the research team independently assessed size using the VeVMD® digital software system (Vista Medical, Winnipeg, Manitoba, Canada) and procedures previously described (Gardner et al., 2012). A cotton-tipped swab, placed in the deepest aspect of the DFU, was marked where the swab intersected with the plane of the peri-wound skin. The distance between the tip of the swab and the mark was measured as ulcer depth using a centimetre ruler.
Osteomyelitis was assessed using radiographs and MRI at baseline and during follow-up visits when subjects presented with new tracts to bone, wound deterioration, elevated temperature, elevated white count, elevated erythrocyte sedimentation rate, or elevated C-reactive protein. If these indicators were absent at follow-up, radiographs were not retaken. Subjects experiencing new amputations had their medical records reviewed by the research team to ensure amputations were due to DFU infection, and not some other reason.
Laboratory Animals
Twenty 8-wk-old female BKS.Cg-Dock7m+/+Leprdb/J mice were purchased from Jackson laboratories. All mice were housed and maintained in a BSL II and specific pathogen free conditions, under the care of a veterinarian, at the University of Pennsylvania under and approved IACUC protocol (804065). All animal experiments were performed in full compliance with standards outlined in the “Guide for the Care and Use of Laboratory Animals” by the Laboratory Animal Resources (LAR) as specified by the Animal Welfare Act (AWA) and Office of Laboratory Animal Welfare (OLAW) and approved by the Governing Board of the National Research Council (NRC). Mice used in this study were diabetic due to the Lepr mutation. All mice were naïve for drugs and were not involved in previous procedures.
Cells
Primary human keratinocytes were derived from neonatal male foreskins and obtained from the University of Pennsylvania Skin Biology and Diseases Resource Center (Core B, Tissue and Keratinocyte Procurement Core), under protocols approved by the University of Pennsylvania Institutional Review Board. Keratinocytes were male in origin and of the species homo sapiens. Keratinocytes were cultured in keratinocyte media with 1% antibiotic/antimycotic (15240062, Life Technologies).
METHOD DETAIL
Metagenomic Sequencing of DFU specimens:
After cleansing the ulcer with sterile saline, specimens were collected using the Levine technique by rotating a swab over a 1-cm2 area of viable non-necrotic wound tissue for 5 seconds with sufficient pressure to extract tissue fluid. DNA was isolated from swab specimens as previously described (Loesche et al., 2017). To minimize signal from contaminating eukaryote DNA (human), a microbial DNA enrichment step was performed prior to Illumina library preparation with the NEBNext Microbiome DNA Enrichment kit (New England Biolabs). Purified DNA was quantified using the Qubit dsDNA High-Sensitivity Assay Kit (Invitrogen). Illumina sequence libraries were prepared using the NexteraXT DNA Library Preparation Kit (Illumina) and followed by quality control (Bioanalyzer), quantification (Kapa assay), pooling, and sequencing at either the University of Maryland Institute for Genome Sciences or with CosmosID. 150 bp paired-end sequencing of pooled samples was performed over four runs of two full flow cells (16 lanes) on the HiSeq 4000 to generate 200-400M reads per sample. Samples were randomized throughout the study, but were not blinded with respect to outcomes.
The raw read data were first preprocessed in collaboration with CosmosID to filter and remove human DNA sequences by mapping reads to the human genome and a custom built database of human DNA sequences, followed by additional filtering for repeat regions using the Tandem Repeat Finder algorithm (https://tandem.bu.edu/trf/trf.html). Finally, filtered reads were mapped to custom curated bacterial, fungal, viral, and antibiotic resistance genomic databases. Taxonomic identification was assigned with an in-house K-mer based algorithm refined against a whole genome phylogenetic tree to identify unique species and strains developed at CosmoID and described in Hourigan et al (Hourigan et al., 2018).
Filtered reads were further processed with an in-house pipeline (Grice lab) that included additional read QC and linker cleaning steps using the cutadapt software (Martin, 2011). Microbial ecology metrics including Shannon diversity index and number of observed species (richness) were calculated in the R statistical environment using the vegan package (Oksanen et al., 2018; R Core Development Team, 2017). Functional annotation was assigned with the SUPERFOCUS software (Silva et al., 2016) which classifies each metagenomic sequence into a SEED subsystem (Overbeek et al., 2014) run in the sensitive mode, using the diamond aligner and db_98 with the following command: python SUPERF0CUS_0. 27/superfocus. py –q . –m 1 –a diamond –fast 0 –o all_samples –t 24 –dir SUPER–FOCUS/
Antibiotic resistance gene classification was collapsed into functional categories (e.g., class A, B, and C beta-lactamases were grouped as “beta-lactamase”).
Raw data are deposited in the NCBI Sequence Read Archive (SRA) under the BioProject Accession number PRJNA506988.
Whole Genome Sequencing
DNA was isolated from pure culture following the same procedure as microbiome samples described above. Purified DNA was used as input to the NexteraXT library preparation kit following the manufacturers protocol (Illumina). Libraries were sequenced on the NextSeq500. Raw data was processed with cutadapt (Martin, 2011) to trim adapter sequences and a minimum quality score of 20. Reads were assembled with Unicycler (Wick et al., 2017) and annotated with Prokka (Seemann, 2014). Contigs were then ordered with CAR (Lu et al., 2014) prior to full genome alignment using MAUVE (Darling et al., 2004). The two S. aureus genomes were compared for shared and unique genes using Roary (Page et al., 2015) and default settings. Phage sequences were detected by running genomic contigs through the PHASTER (Arndt et al., 2016) algorithm and classified as complete, questionable or incomplete. Phylogenomic analysis was performed within the Anvi’o software environment (Eren et al., 2015) using MCL (van Dongen and Abreu-Goodger, 2012) to identify clusters of amino acid similarity and muscle (Edgar, 2004) to align whole genomes. The pangenome was built with the following parameters:
anvi–pan–genome –g STAPHREFN0V21–GENOMES, db ––project-name “STAPHREF_PAN_N0V21” –– output–dir STAPHREF_N0V21 ––num–threads 6 ––minbit 0.5 ––mc I–inflation 10 ––use–ncbi–blast ––min–occurrence 1 ––enforce–hierarchical–clustering
Average nucleotide identity was calculated with the anvi-compute-ani function using PyANI. All genomes included in the analysis including NCBI accession numbers are listed in Supplemental Data S2.
Detailed comparative analysis of subregions within the SA7372 and SA10757 genomes was performed with EasyFig (Sullivan et al., 2011) using blastn and annotations were directly read from genbank generated files from prokka.
Bacterial Isolation and Manipulation
Isolation:
Bacterial isolates were grown from wound swabs collected in trypticase soy broth (TSB) and described in Gardner et al. 2013. Identification was achieved with the Vitek Legacy (Biomerieux) or full length 16S rRNA gene Sanger sequencing.
Planktonic and Biofilm growth:
Isolates were grown overnight at 37°C on TSA. Colonies were scraped into Keratinocyte media without antibiotics to an OD600 nm of 0.08-0.1. Keratinocyte media was made by mixing Keratinocyte SFM (kit 17005042, Life Technologies) with Medium 154 (M154500, Life Technologies) supplemented with human keratinocyte growth supplement (S0015, Life Technologies) at a 1:1 ratio. For planktonic cultures the inoculums were then added in a 1/10 dilution to a final volume of 5 mL of Keratinocyte media. Biofilms were inoculated in a 1/10 dilution to a final volume of 3 mL keratinocyte media in 6 well polystyrene plates containing a 1 inch × 1 inch piece of sterile cotton gauze. Cultures were incubated at 37°C for 72 hrs, shaking at 200 rpm to allow adhesion and growth, with the media changed every 24 hrs. After incubation, the media was removed and the biofilms attached to the gauze were washed with 5 × 1 mL sterile phosphate buffered saline to remove non-adherent cells. For imaging, the biofilms were stained with the LIVE BacLight Bacterial Gram Stain Kit (ThermoFisher Scientific, Waltham, MA) with SYTO9 (480nm/500nm) and hexidium iodide (480nm/625nm) as a 2 mL solution in water and according to the kit instructions. The stain was removed and deionized water was added to the biofilms prior to imaging on a Leica SP5-II inverted confocal microscope. Images were post-processed with Volocity software (PerkinElmer, Waltham, MA, USA). The maximum projection for each image was used to generate Fig. S5. Quantitative counts of either planktonic or biofilm cultures (after disruption by vortexing for 3 × 1 min each) was performed by serial dilution in phosphate buffered saline. The dilutions were plated onto non-selective media (TSA) and incubated for 16-18 hrs at 37°C. Colonies were counted and the total CFUs calculated.
Keratinocyte Cytokine Analysis:
Primary human keratinocytes were obtained from the Penn Skin Biology and Disease Resource-based Center (SBDRC), Core B, Tissue and Keratinocyte Procurement Core. Keratinocytes were derived from neonatal (male) foreskins through procedures approved by the University of Pennsylvania Institutional Review Board. Keratinocytes were seeded at 0.3 × 106 cells/well in a 6-well tissue culture plate (2.5 mL total volume per well) and cultured in keratinocyte media (see above) with 1% antibiotic/antimycotic (15240062, Life Technologies). Media was changed the day after seeding, and then every other day until confluent. Confluence was achieved after ~4 days. Planktonic or biofilm cultures were prepared as described above except planktonic cultures were grown to mid-log phase. Based on quantitative counts of each culture, biofilm or planktonic cultures were normalized to a count of 2.8 × 107 cfu/mL, which was the lowest cell density of the four test groups S. aureus 10757, S. aureus 7372, A. faecalis, and C. striatum. Cultures were centrifuged to remove the majority of cells and filtered through a 0.22 uM filter for sterility. Each filtered planktonic culture was then diluted 1/50 and each filtered biofilm culture diluted 1/100 into keratinocyte media before being placed on the keratinocytes. Prior to exposure, a single ‘scratch’ was made across each cell monolayer with a 200μl pipette tip to mimic a wound and then washed three times with sterile saline to remove cell debris. The keratinocytes were exposed to the filtered spent media from biofilm or planktonic cultures for 8 hours. Three biological replicates of each condition were performed. Media was collected from each well into 1.5 mL microcentrifuge tubes, centrifuged at maximum speed for 10-15 min. The supernatant was filtered through a 0.22 uM filter and placed at −20 degrees C until analysis. Secreted cytokine concentrations were determined by Luminex Multiplex Assay Human Cytokine/Chemokine Panel I (MilliporeSigma) and was performed at the University of Pennsylvania Human Immunology Core (P30-CA016520). Two technical replicates of each biological replicate for mid-log phase treatments were performed (n=6 per treatment) and three technical replicates of each biological replicate were performed for the biofilm treated cells and control cells with no treatment (n=9 per treatment).
Diabetic Mouse Wounding and Infection:
At 12 weeks old, a dorsal patch of skin was shaved and the mice were then housed individually for three days. On day 4, after anesthesia by inhaled isoflurane, an 8-mm full-thickness excisional wound was created with a punch biopsy tool (Miltek). Seventy-two hour biofilms grown on cotton gauze and precisely cut to 8-mm with a clean sterile biopsy tool were then placed into the wounds and covered with a transparent film (Tegaderm, 3M). Each piece of 8-mm size gauze contained between 1.23 – 9.26 × 108 CFU.
Four mice per treatment group were included, the control group was treated with uninoculated sterile gauze treated in the same manner as the biofilm groups (described above). Each wound was photographed and measured at the time of wounding (t=0), day 3, day 7, day 14, and day 28 post-wounding. After day 3 the gauze was removed and the wound was recovered with the transparent film. Wound area was measured in Adobe photoshop by two independent persons that were blinded to treatment group, and the average of each measurement recorded for each individual wound. Four mice were analyzed per group, and results confirmed in at least 2 independent experiments. Samples sizes were calculated based on previous studies of wound closure in this particular mouse model (Grice et al., 2010).
Histopathology and immunofluorescence:
Tissue close to wound was harvested and fixed in 4% paraformaldehyde, and paraffin-embedded. Serial sections (5 μm) were cut and every 4th section was stained with H&E by routine methods. Serial sections were deparaffinized, blocked with 10% goat serum in PBS containing 0.1% Tween-20 and stained with Keratin-14 polyclonal antibody (Biolegend, #905301) at 1:1000 dilution in blocking buffer for 1 hour at room temperature. K14 was detected with goat-anti-Rabbit IgG conjugated with Alexa fluor 488 (Invitrogen, #A-11034) at 1:1000 dilution and counterstained with DAPI. All wide-field H&E and fluorescent images were taken using a 20× objective of Keyence BZ-X710 inverted fluorescence microscope equipped with a motorized stage. The images were stitched using the inbuilt algorithm accompanying the BZ-X-wide image viewer used with Keyence microscopes. The epithelial gap was measured between wound edges using ImageJ over 3 serial sections per wound. Four wounds per treatment group were analyzed.
QUANTIFICATION AND STATISTICAL ANALYSIS
The R Statistical Package (R Core Development Team, 2017) was used to generate all figures that are not photographs and compute statistical analysis. Statistical significance was verified through analysis of variance with post hoc multiple comparison testing between each group or the non-parametric Wilcoxon rank-sum analysis as reported in the text. Correlations between SEED functional categories and clinical features were determined by calculating the Spearman rank coefficient. Multiple hypothesis test correction was done using the false discovery rate (FDR). Alpha diversity metrics were calculated using the ‘vegan’ package in R. The number of participants, cell culture replicates, or animals is denoted by “n” and is described in detail in the results text and figure legends.
DATA AND SOFTWARE AVAILABILITY
Raw data are deposited in the NCBI Sequence Read Archive (SRA) under the BioProject Accession number PRJNA506988. The raw data can be found at the following link: https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA506988
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Keratin-14 polyclonal antibody | Biolegend | Cat#905301; RRID: AB_2565048 |
| Alexa fluor 488 goat-anti-Rabbit IgG | Invitrogen | Cat#A-11034: RRID: AB_2576217 |
| Bacterial and Virus Strains | ||
| Alcaligenes faecalis LK36 | Isolated from DFU | NCBI BioProject: PRJNA506988. See deposited data for accession numbers |
| Corynebacterium striatum LK37 | Isolated from DFU | |
| Staphylococcus aureus LK34 | Isolated from DFU | |
| Staphylococcus aureus LK35 | Isolated from DFU | |
| Biological Samples | ||
| Diabetic Foot Ulcer Specimens | Described in: Kalan et al., 2016; Loesche et al., 2017, and this paper | See metamap.csv in supplemental dataset_scripts for full metadata. |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Keratinocyte SFM media | Life technologies | Cat#17005042 |
| Keratinocyte growth supplement | Life technologies | Cat#S0015 |
| Medium 154 | Life technologies | Cat#M154500 |
| Antibiotic-Antimycotic (100×) | Life technologies | Cat#15240062 |
| Critical Commercial Assays | ||
| NEBNext® Microbiome DNA Enrichment Kit | New England Biolabs | Cat#E2612S |
| Qubit™ dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat#Q32854 |
| Nextera XT DNA Library preparation Kit | Illumina | Cat#FC-131-1096 |
| KAPA Library Quantification Kit | Kappa Biosystems | Cat#KK4824 |
| LIVE BacLight Bacterial Gram Stain Kit | Thermo Fisher Scientific | Cat#L7005 |
| MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel - Immunology Multiplex Assay (Custom designed for EGF,G-CSF, IFN-α-2, IFN-γ, IL-1α, IL-1β, IL-1rα, IL-2 , IL-3, IL-4 , IL-5, IL-6, IL-7 , IL-8/CXCL8, IP-10/CXCL10, TNFα , TGFα, PDGF-AB/BB , IL-9, GM-CSF) | Millipore Sigma | Cat#HCYTO MAG-60K |
| Deposited Data | ||
| Genome sequence: Alcaligenes faecalis LK36 | This paper | NCBI BioProject: PRJNA506988; BioSample: SAMN10134346 |
| Genome sequence: Corynebacterium striatum LK37 | This paper | NCBI BioProject: PRJNA506988 BioSample: SAMN10134347 |
| Genome sequence: Staphylococcus aureus LK34 | This paper | NCBI BioProject: PRJNA506988 BioSample: SAMN10134345 |
| Genome sequence: Staphylococcus aureus LK35 | This paper | NCBI BioProject: PRJNA506988 BioSample: SAMN10134344 |
| Experimental Models: Cell Lines | ||
| Human: Primary foreskin keratinocytes | Penn Skin Biology and Disease Resource-based Center | NA |
| Experimental Models: Organisms/Strains | ||
| Mouse BKS.Cg-Dock7m+/+Leprdb/J | The Jackson Laboratory | Cat#000642 |
| Software and Algorithms | ||
| Volocity software, v6.3 | Quorum Technologies | http://quorumtechnologies.com/index.php/component/content/category/31-volocity-software |
| ImageJ, v1.52a | National Institute of Health | https://imagej.nih.gov/ij/ |
| cutadapt | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| vegan package | Oksanen et al., 2018 | https://cran.r-project.org/web/packages/vegan/index.html |
| SUPERFOCUS | Silva et al., 2016 | https://github.com/metageni/SUPER-FOCUS |
| Unicycler | Wick et al., 2017 | https://github.com/rrwick/Unicycler |
| Prokka | Seemann, 2014 | https://github.com/tseemann/prokka |
| CAR | Lu et al., 2014 | http://genome.cs.nthu.edu.tw/CAR/ |
| MAUVE | Darling et al., 2004 | http://darlinglab.org/mauve/mauve.html |
| Roary | Page et al., 2015 | https://sanger-pathogens.github.io/Roary/ |
| PHASTER | Arndt et al., 2016 | http://phaster.ca/ |
| Anvi’o | Eren et al., 2015 | http://merenlab.org/software/anvio/ |
| MCL | Van Dongen and Abreu-Goodger, 2012 | https://micans.org/mcl/ |
| Muscle | Edgar, 2004 | http://www.drive5.com/muscle/ |
| EasyFig | Sullivan et al | http://mjsull.github.io/Easyfig/ |
| Other | ||
| Millex-GV Syringe Filter Unit, 0.22 μm, PVDF, | Millipore Sigma | Cat#SLGV033RS |
| 8mm biopsy punch tool | Miltex | Cat#21909-146 |
| Sterile gauze pad, 12 ply | Dynarex Corporation, New York | Cat#3352 |
Highlights:
Wound microbiota was profiled longitudinally in patients with diabetic foot ulcers
Staphylococcus aureus strains were associated with poor outcomes
S. aureus and other wound isolates promoted differential wound healing responses
Debridement depleted anaerobic bacteria in wounds with favorable outcomes
ACKNOWLEDGMENTS
Funding: The National Institutes of Health, National Institute of Nursing Research (R01-NR-009448 to S.E.G, R01-NR-015639 to E.A.G, and P20 NR018081 to SEG), National Institute of Arthritis, Musculoskeletal, and Skin Diseases (R01-AR-006663 and R00-AR-060873 to E.A.G.), Burroughs Wellcome Fund PATH Award (E.A.G), Linda Pechenik Montague Investigator Award (E.A.G.). This research was also supported by the Penn Skin Biology and Disease Resource-based Center (P30-AR-069589). M.L. was supported by the Dermatology Research Training Grant, T32-AR-007465. We would also like to acknowledge Gordon Ruthel and the Penn Vet Imaging Center for support with confocal microscopy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL ITEMS
Supplemental File 1: Supplemental Figures 1–6 and Supplemental Table 1
Supplemental Data S1; related to Figure 4: MSLT profiles for S.aureus isolates
Supplemental Data S2; related to Figure 4: Summary of genomes and accession numbers used to generate figure 4A.
Supplemental Data S3: datasets and code for all Figures 1–6.
DECLARATION OF INTERESTS
The authors have no competing interests to declare.
REFERENCES
- Armstrong DG, Wrobel J, and Robbins JM (2007). Guest Editorial: are diabetes-related wounds and amputations worse than cancer? Int Wound J 4, 286–287. [DOI] [PubMed] [Google Scholar]
- Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, and Wishart DS (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44, W16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arwert EN, Hoste E, and Watt FM (2012). Epithelial stem cells, wound healing and cancer. Nat Rev Cancer 12, 170–180. [DOI] [PubMed] [Google Scholar]
- Ashrafi M, Sebastian A, Shih B, Greaves N, Alonso-Rasgado T, Baguneid M, and Bayat A (2016). Whole genome microarray data of chronic wound debridement prior to application of dermal skin substitutes. Wound Repair Regen 24, 870–875. [DOI] [PubMed] [Google Scholar]
- Bartow-McKenney C, Hannigan GD, Horwinski J, Hesketh P, Horan AD, Mehta S, and Grice EA (2018). The microbiota of traumatic, open fracture wounds is associated with mechanism of injury. Wound Repair Regen.2, 127–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S, Frankel MB, Schneewind O, and Missiakas D (2014). Release of protein A from the cell wall of Staphylococcus aureus. Proc Natl Acad Sci U S A 111, 1574–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton AJ, Vileikyte L, Ragnarson-Tennvall G, and Apelqvist J (2005). The global burden of diabetic foot disease. Lancet 366, 1719–1724. [DOI] [PubMed] [Google Scholar]
- Byrd AL, Deming C, Cassidy SKB, Harrison OJ, Ng WI, Conlan S, Program NCS, Belkaid Y, Segre JA, and Kong HH (2017). Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Science translational medicine 9, (397). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinal M, Eisenbud DE, Armstrong DG, Zelen C, Driver V, Attinger C, Phillips T, and Harding K (2009). Serial surgical debridement: a retrospective study on clinical outcomes in chronic lower extremity wounds. Wound Repair Regen 17, 306–311. [DOI] [PubMed] [Google Scholar]
- Citron DM, Goldstein EJ, Merriam CV, Lipsky BA, and Abramson MA (2007). Bacteriology of moderate-to-severe diabetic foot infections and in vitro activity of antimicrobial agents. J Clin Microbiol 45, 2819–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling ACE, Mau B, Blattner FR, and Perna NT (2004). Mauve: Multiple Alignment of Conserved Genomic Sequence With Rearrangements. Genome Research 14, 1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA, and Wolcott RD (2008). Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol 8, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Esen OC, Quince C, Vineis JH, Morrison HG, Sogin ML, and Delmont TO (2015). Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner M, Vicaretti M, Sparks J, Bansal S, Bush S, Liu M, Darling A, Harry E, and Burke CM (2017). A longitudinal study of the diabetic skin and wound microbiome. PeerJ 5, e3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner SE, Frantz RA, Hillis SL, Blodgett TJ, Femino LM, and Lehman SM (2012). Volume measures using a digital image analysis-system are reliable in diabetic foot ulcers. WOUNDS: A Compendium of Clinical Research and Practice 24, 146–151. [PMC free article] [PubMed] [Google Scholar]
- Gardner SE, Haleem A, Jao YL, Hillis SL, Femino JE, Phisitkul P, Heilmann KP, Lehman SM, and Franciscus CL (2014). Cultures of Diabetic Foot Ulcers Without Clinical Signs of Infection Do Not Predict Outcomes. Diabetes Care. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner SE, Hillis SL, Heilmann K, Segre JA, and Grice EA (2013). The neuropathic diabetic foot ulcer microbiome is associated with clinical factors. Diabetes 62, 923–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EG, Snitkin ES, Yockey LJ, Bermudez DM, NISC Comparative Sequencing Program, Liechty KW, and Segre JA (2010). Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. PNAS 107(33), 14799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WO, Werth BJ, Butler-Wu SM, and Rakita RM (2016). Multidrug-Resistant Corynebacterium striatum Associated with Increased Use of Parenteral Antimicrobial Drugs. Emerg Infect Dis 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffstad O, Mitra N, Walsh J, and Margolis DJ (2015). Diabetes, lower-extremity amputation, and death. Diabetes Care 38, 1852–1857. [DOI] [PubMed] [Google Scholar]
- Hourigan SK, Subramanian P, Hasan NA, Ta A, Klein E, Chettout N, Huddleston K, Deopujari V, Levy S, Baveja R, et al. (2018). Comparison of Infant Gut and Skin Microbiota, Resistome and Virulome Between Neonatal Intensive Care Unit (NICU) Environments. Front Microbiol 9, 1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell-Jones RS, Price PE, Howard AJ, and Thomas DW (2006). Antibiotic prescribing for chronic skin wounds in primary care. Wound Repair Regen 14, 387–393. [DOI] [PubMed] [Google Scholar]
- Kalan L, Loesche M, Hodkinson BP, Heilmann K, Ruthel G, Gardner SE, and Grice EA (2016). Redefining the Chronic-Wound Microbiome: Fungal Communities Are Prevalent, Dynamic, and Associated with Delayed Healing. mBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le KY, and Otto M (2015). Quorum-sensing regulation in staphylococci-an overview. Front Microbiol 6, 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal SM Jr., Jones M, and Gilligan PH (2016). Clinical Significance of Commensal Gram-Positive Rods Routinely Isolated from Patient Samples. J Clin Microbiol 54, 2928–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsky BA, Berendt AR, Cornia PB, Pile JC, Peters EJ, Armstrong DG, Deery HG, Embil JM, Joseph WS, Karchmer AW, et al. (2012). Executive summary: 2012 Infectious Diseases Society of America clinical practice guideline for the diagnosis and treatment of diabetic foot infections. Clin Infect Dis 54, 1679–1684. [DOI] [PubMed] [Google Scholar]
- Loesche M, Gardner SE, Kalan L, Horwinski J, Zheng Q, Hodkinson BP, Tyldsley AS, Franciscus CL, Hillis SL, Mehta S, et al. (2017). Temporal Stability in Chronic Wound Microbiota Is Associated With Poor Healing. J Invest Dermatol 137, 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie TJ, Bartlett JG, Tally FP, and Gorbach SL (1976). Aerobic and anaerobic bacteria in diabetic foot ulcers. Annals of internal medicine 85, 461–463. [DOI] [PubMed] [Google Scholar]
- Lu CL, Chen KT, Huang SY, and Chiu HT (2014). CAR: contig assembly of prokaryotic draft genomes using rearrangements. BMC Bioinformatics 15, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis D, Berlin JA, and Strom BL (1996). Interobserver agreement, sensitivity, and specificity of a “healed” chronic wound. Wound Repair and Regeneration 4, 335–338. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17, 10–12. [Google Scholar]
- Martins-Mendes D, Monteiro-Soares M, Boyko EJ, Ribeiro M, Barata P, Lima J, and Soares R (2014). The independent contribution of diabetic foot ulcer on lower extremity amputation and mortality risk. J Diabetes Complications 28, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel JS, Hannigan GD, Tyldsley AS, SanMiguel AJ, Hodkinson BP, Zheng Q, and Grice EA (2016). Skin Microbiome Surveys Are Strongly Influenced by Experimental Design. J Invest Dermatol 136, 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulik PK, Mtonga R, and Gill GV (2003). Amputation and mortality in new-onset diabetic foot ulcers stratified by etiology. Diabetes Care 26, 491–494. [DOI] [PubMed] [Google Scholar]
- Novick RP, and Geisinger E (2008). Quorum sensing in staphylococci. Annu Rev Genet 42, 541–564. [DOI] [PubMed] [Google Scholar]
- Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, et al. (2018). vegan: Community Ecology Package. R package version 2.4-6. [Google Scholar]
- Ortega E, Abriouel H, Lucas R, and Galvez A (2010). Multiple roles of Staphylococcus aureus enterotoxins: pathogenicity, superantigenic activity, and correlation to antibiotic resistance. Toxins (Basel) 2, 2117–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crecy-Lagard V, Diaz N, Disz T, Edwards R, et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33, 5691–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42, D206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, and Parkhill J (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastar I, Stojadinovic O, Yin NC, Ramirez H, Nusbaum AG, Sawaya A, Patel SB, Khalid L, Isseroff RR, and Tomic-Canic M (2014). Epithelialization in Wound Healing: A Comprehensive Review. Adv Wound Care (New Rochelle) 3, 445–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SA, Iacovella J, and Cornell RS (2016). Corynebacterium Striatum: A Concerning Pathogen of Osteomyelitis in the Diabetic Patient. Journal of the American Podiatric Medical Association 106, 9–9. [Google Scholar]
- R Core Development Team. (2017). R: A language and environment for statistical computing (Vienna, Austria: R Foundation for Statistical Computing; ). [Google Scholar]
- Ramsey SD, Newton K, Blough D, McCulloch DK, Sandhu N, Reiber GE, and Wagner EH (1999). Incidence, outcomes, and cost of foot ulcers in patients with diabetes. Diabetes Care 22, 382–387. [DOI] [PubMed] [Google Scholar]
- Rennekampff HO, Hansbrough JF, Kiessig V, Dore C, Sticherling M, and Schroder JM (2000). Bioactive interleukin-8 is expressed in wounds and enhances wound healing. J Surg Res 93, 41–54. [DOI] [PubMed] [Google Scholar]
- Rhoads DD, Cox SB, Rees EJ, Sun Y, and Wolcott RD (2012). Clinical identification of bacteria in human chronic wound infections: culturing vs. 16S ribosomal DNA sequencing. BMC Infect Dis 12, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi M, Khan F, Raza A, Shukla I, and Sabir AB (2011). Emergence of coryneforms in osteomyelitis and orthopaedic surgical site infections. Australas Med J 4, 412–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069.24642063 [Google Scholar]
- Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, Gottrup F, Gurtner GC, and Longaker MT (2009). Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen 17, 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui AR, and Bernstein JM (2010). Chronic wound infection: facts and controversies. Clin Dermatol 28, 519–526. [DOI] [PubMed] [Google Scholar]
- Silva GG, Green KT, Dutilh BE, and Edwards RA (2016). SUPER-FOCUS: a tool for agile functional analysis of shotgun metagenomic data. Bioinformatics 32, 354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MJ, Petty NK, and Beatson SA (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammelin A, Lindholm C, and Hambraeus A (1998). Chronic ulcers and antibiotic treatment. J Wound Care 7, 435–437. [DOI] [PubMed] [Google Scholar]
- Tenover FC, Weigel LM, Appelbaum PC, McDougal LK, Chaitram J, McAllister S, Clark N, Killgore G, O’Hara CM, Jevitt L, et al. (2004). Vancomycin-resistant Staphylococcus aureus isolate from a patient in Pennsylvania. Antimicrob Agents Chemother 48, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen S, and Abreu-Goodger C (2012). Using MCL to extract clusters from networks. Methods Mol Biol 804, 281–295. [DOI] [PubMed] [Google Scholar]
- Whitson HE, Johnson KS, Sloane R, Cigolle CT, Pieper CF, Landerman L, and Hastings SN (2016). Identifying Patterns of Multimorbidity in Older Americans: Application of Latent Class Analysis. J Am Geriatr Soc 64, 1668–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick RR, Judd LM, Gorrie CL, and Holt KE (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 13, e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolcott RD, Hanson JD, Rees EJ, Koenig LD, Phillips CD, Wolcott RA, Cox SB, and White JS (2016). Analysis of the chronic wound microbiota of 2,963 patients by 16S rDNA pyrosequencing. Wound Repair Regen 24, 163–174. [DOI] [PubMed] [Google Scholar]
- Yoong P, and Torres VJ (2013). The effects of Staphylococcus aureus leukotoxins on the host: cell lysis and beyond. Curr Opin Microbiol 16, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are deposited in the NCBI Sequence Read Archive (SRA) under the BioProject Accession number PRJNA506988. The raw data can be found at the following link: https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA506988