

Abstract
Background
The human genome contains “dark” gene regions that cannot be adequately assembled or aligned using standard short-read sequencing technologies, preventing researchers from identifying mutations within these gene regions that may be relevant to human disease. Here, we identify regions with few mappable reads that we call dark by depth, and others that have ambiguous alignment, called camouflaged. We assess how well long-read or linked-read technologies resolve these regions.
Results
Based on standard whole-genome Illumina sequencing data, we identify 36,794 dark regions in 6054 gene bodies from pathways important to human health, development, and reproduction. Of these gene bodies, 8.7% are completely dark and 35.2% are ≥ 5% dark. We identify dark regions that are present in protein-coding exons across 748 genes. Linked-read or long-read sequencing technologies from 10x Genomics, PacBio, and Oxford Nanopore Technologies reduce dark protein-coding regions to approximately 50.5%, 35.6%, and 9.6%, respectively. We present an algorithm to resolve most camouflaged regions and apply it to the Alzheimer’s Disease Sequencing Project. We rescue a rare ten-nucleotide frameshift deletion in CR1, a top Alzheimer’s disease gene, found in disease cases but not in controls.
Conclusions
While we could not formally assess the association of the CR1 frameshift mutation with Alzheimer’s disease due to insufficient sample-size, we believe it merits investigating in a larger cohort. There remain thousands of potentially important genomic regions overlooked by short-read sequencing that are largely resolved by long-read technologies.
Electronic supplementary material
The online version of this article (10.1186/s13059-019-1707-2) contains supplementary material, which is available to authorized users.
Keywords: Camouflaged genes, Dark genes, Long-read sequencing, Pacific Biosciences (PacBio), Oxford Nanopore Technologies (ONT), 10x Genomics, Alzheimer’s Disease Sequencing Project (ADSP), CR1, APOE
Background
Researchers have known for years that large, complex genomes, including the human genome, contain “dark” regions—regions where standard high-throughput short-read sequencing technologies cannot be adequately assembled or aligned—thus preventing our ability to identify mutations within these regions that may be relevant to human health and disease. Some dark regions are what we term “dark by depth” (few or no mappable reads), while others are what we term “dark by mapping quality” (reads aligned to the region, but with a low mapping quality). Regions that are dark by depth may arise because the region is inherently difficult to sequence at the chemistry level (e.g., high GC content [1, 2]), essentially eliminating sequencing reads from that region altogether. Other dark regions arise, not because the sequencing is inherently problematic, but because of bioinformatic challenges. Specifically, many dark regions arise from duplicated genomic regions, where confidently aligning short reads to a unique location is not possible; we term these regions as “camouflaged”. These camouflaged regions are generally either large contiguous tandem repeats (e.g., centromeres, telomeres, and other short tandem repeats), or a larger specific DNA region that has been duplicated (e.g., a gene duplication) either in tandem or in a more distal genome region. In fact, many genes in the human genome were duplicated over evolutionary time and are still transcriptionally and translationally active (e.g., heat-shock proteins) [3–9], while others have been duplicated, but are considered inactive (i.e., pseudogenes). Regardless of whether the duplication is active, however, any genomic region that has been nearly identically duplicated and is large enough to prevent sequencing reads from aligning unambiguously will be “dark”, because the aligner cannot determine which genomic region the read originated from.
When confronted with a read that aligns equally well to two or more camouflaged regions (commonly known as multi-mapping reads [2, 10]), standard next-generation sequence aligners, such as the Burrows-Wheeler Aligner (BWA) [11–13], randomly map the read to one of the regions and assign a low mapping quality. For BWA, specifically, reads that cannot be uniquely mapped are generally assigned a mapping quality (MAPQ) of 0; though, in certain paired-end sequencing scenarios, BWA will assign a high mapping quality if the read mate is confidently mapped nearby (i.e., within the estimated insert-size length).
Recent work has characterized camouflaged regions, in part, including a study that demonstrates how this issue affects all standard RNA-Seq analyses [10] and another that quantifies the number of nucleotides in human reference GRCh38 that are dark from mapping quality of 0 (camouflaged regions), based on 1000 Genome Project data [2]. Robert and Watson demonstrated that expression for 958 genes were either over- or under-represented because of multi-mapping reads across 12 different RNA-Seq processing methods, and no method was immune to the problem [10]. They also demonstrated that many of these genes are directly implicated in human disease. Zheng-Bradley et al. recently re-aligned genomes from the 1000 Genomes Project to GRCh38, and, among other findings, generally demonstrated the breadth of multi-mapping reads across the genome [2]. These data characterize the general problem and report specific genes affected by this issue.
Here, we systematically analyze dark and camouflaged genes to more fully characterize the problem, and we highlight many disease-relevant genes that are directly implicated in neurological diseases and conditions such as Alzheimer’s disease, autism spectrum disorder, amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and others. We also show that linked-read and long-read sequencing technologies substantially reduce the number of dark and camouflaged regions, and we present a method to address camouflaged regions, even in standard short-read sequencing data. As a proof of concept, we applied our method to the Alzheimer’s Disease Sequencing Project (ADSP) data and identified a rare, ten-nucleotide frameshift deletion in the C3b and C4b binding domain of CR1, a top Alzheimer’s disease gene [14–22], that is present in five ADSP cases but zero controls. The ADSP is not large enough to statistically assess association between the CR1 frameshift mutation and Alzheimer’s disease, but this warrants further investigation.
Results
To quantify the number of dark and camouflaged regions in standard short-read whole-genome sequencing data, we obtained whole-genome sequencing data for ten unrelated males from the Alzheimer’s Disease Sequencing Project (ADSP) and scanned each sample for dark and camouflaged regions, averaging across all ten samples; we only used data from males in this study so we could also assess dark and camouflaged regions on the Y chromosome because large portions of the Y chromosome are dark. We ignored incomplete genomic regions (e.g., centromeres). For most of our analyses, we then limited the dark and camouflaged regions to known gene bodies, based on annotations from build 93 of the GRCh38 human reference genome, excluding alternate contig assemblies. For comparison, we performed the same analyses on GRCh38 including alternate contig assemblies, and on build 87 of the Ensembl GRCh37 human reference genome [23]. All ten samples were sequenced using standard Illumina whole-genome sequencing with 100-nucleotide read lengths, where median genome-wide read depths ranged from 33.0x to 45.0x coverage, with an overall median of 37.5x. We performed the same analyses on ten unrelated males from the 1000 Genomes Project [24] that were sequenced using Illumina whole-genome sequencing with 250-nucleotide read lengths, where median genome-wide read depths ranged from 30.0x to 61.0x coverage, with an overall median of 58.5x. Similarly, we assessed how well linked-read or long-read sequencing technologies from 10x Genomics (52x median coverage), PacBio (50x median coverage), and ONT (down-sampled to ~ 52x median coverage) resolve dark and camouflaged regions. Although we were only able to obtain a single high-depth male genome for each long-read technology, we believe our results are a reasonable estimate for how well each technology addresses dark and camouflaged regions. Larger sequencing studies will further clarify our results.
We consider a region “dark” for one of two reasons: (1) insufficient number of reads aligned to the genomic region (dark by depth) and (2) reads aligned to the region, but with insufficient mapping quality for a variant caller to identify mutations in the region (dark by mapping quality). Specifically, we define regions that are dark by depth as those with ≤ 5 aligned reads (Fig. 1a) and regions that are dark by mapping quality as those where ≥ 90% of aligned reads have a mapping quality (MAPQ) < 10 (Fig. 1b). Defining dark-by-depth regions as those with ≤ 5 reads is a relatively strict cutoff and likely underestimates the number of dark regions because 20 to 30 reads is often considered a reasonable minimum to confidently identify heterozygous mutations; overall median read depth is an important factor, however, and we believe a strict cutoff provides a more conservative estimate. We used a mapping quality threshold < 10 to define regions that are dark by mapping quality because that is the standard cutoff used in the Genome Analysis ToolKit (GATK) [25]. Camouflaged regions are those that are dark by mapping quality because the region has been duplicated in the genome (Fig. 1c). We identified sets of camouflaged regions (regions camouflaged by each other) using BLAT [26], where we required at least 98% sequence identity for two regions to be included in the same set.
Fig. 1.

Genomic regions may be “dark” by depth or mapping quality, many of which are “camouflaged”. Large, complex genomes are known to contain “dark” regions where standard high-throughput short-read sequencing technologies cannot be adequately assembled or aligned. We split these dark regions into two types: (1) dark because of low depth and (2) dark because of low mapping quality (MAPQ), which are mostly “camouflaged”. a HLA-DRB5 encodes a Major Histocompatibility Complex protein that plays an important role in immune response and has been associated with several diseases, including Alzheimer’s disease. It is well known to be dark (low depth); specifically, when performing whole-genome sequencing using standard short-read sequencing technologies, an insufficient number of reads align, preventing variant callers from assessing mutations. We calculated sequencing depth across HLA-DRB5 for ten male samples from the Alzheimer’s Disease Sequencing Project (ADSP) that were sequenced using standard Illumina whole-genome sequencing with 100-nucleotide read lengths. Approximately 63.5% (49.6% of coding sequence) of HLA-DRB5 is dark by depth (≤ 5 aligned reads; indicated by red lines). b HSPA1A is a heat-shock protein from the 70-kilodalton (kDa) heat-shock protein family and plays an important role in stabilizing proteins against aggregation. HSPA1A is dark because of low mapping quality (MAPQ < 10 for ≥ 90% of reads at a given position). Approximately 41.1% (53.0% coding sequence) of HSPA1A is dark by mapping quality (indicated by red line). Dark gray bars indicate sequencing reads with a relatively high mapping quality, whereas white bars indicate reads with a low mapping quality (MAPQ = 0). c Many genomic regions that are dark because of mapping quality arise because they have been duplicated in the genome, which we term “camouflaged” (or “camo genes”). When confronted with a read that aligns equally well to more than one location, standard sequence aligners randomly assign the read to one location and give it a low mapping quality. Thus, it is unclear from which gene any of the reads indicated by white bars originated from. HSPA1A and HSPA1B are clear examples of camouflaged genes arising from a tandem duplication. The two genes are approximately 14 kb apart and approximately 50% of the genes are identical
Standard short-read sequencing leaves 36,794 dark regions across 6054 gene bodies, including protein-coding exons from 748 genes
Using whole-genome Illumina sequencing data (100-nucleotide read lengths) from ten unrelated males, we identified 36,794 dark regions (> 15 million nucleotides) in 6054 gene bodies (based on Ensembl GRCh38 build 93 gene annotations) that were either dark by depth or dark by mapping quality (Table 1; Additional file 1: Figure S1a; Additional file 2: Table S1; Additional file 3: Table S2). Stratifying the gene bodies by GENCODE biotype [27], 3804 gene bodies were protein coding, 1232 were pseudogenes, and 753 were long intergenic non-coding RNAs (lincRNA; Fig. 2a). Of all 36,794 dark gene-body regions, 27,982 were intronic, 4351 were in non-coding RNA exons (e.g., lincRNAs and pseudogenes), 2855 were in protein-coding exons (CDS), 908 were in 5′UTR regions, and 698 were in 3′UTR regions (Fig. 2b; Additional file 2: Table S1). Any dark region that spanned a gene element boundary (e.g., intron to exon) was split into separate dark regions. Of the 6054 gene bodies, 527 (8.7%) were 100% dark, 1608 (26.6%) were at least 25% dark, and 2128 (35.2%) were at least 5% dark (Additional file 1: Figure S1b; Additional file 2: Table S1). In intragenic regions, there were a total of 68.7 million nucleotides that were dark in 84,174 regions, totaling 83.8 million dark nucleotides and 90,228 regions, genome wide (Table 1). We also found that aligning GRCh38+alt increased the number of dark nucleotides > 3 times compared to GRCh37.
Table 1.
Dark and camouflaged regions vary by genome build. We identified dark and camouflaged regions throughout the genome for three different builds, including GRCh37, GRCh38, and GRCh38+alt, across five different sequencing technologies (or read lengths for Illumina). Specifically, we measured dark regions for Illumina based on 100-nucleotide read lengths, Illumina based on 250-nucleotide read lengths, 10x Genomics, PacBio, and Oxford Nanopore Technologies (ONT). Here, the counts for dark and camouflaged regions are combined. We found that the number of dark regions and nucleotides, both within gene bodies (represented as GB in the table) and outside gene bodies, varies dramatically by build and technology. Overall, each technology has its respective strengths. GRCh38 including alternate contigs has > 3x more dark nucleotides than GRCh37, and more than 2x more dark regions. Results presented throughout the manuscript are based on GRCh38 (in gray)
| Dark regions | GRCh37 | GRCh38 | GRCh38+alt | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| il100 | il250 | 10x | PacBio | ONT | il100 | il250 | 10x | PacBio | ONT | il100 | PacBio | ONT | |
| Non-GB nucs. | 22.4M | 15.7M | 5.4M | 11.1M | 6.7M | 68.7M | 42.5M | 57.0M | 56.8M | 52.1M | 88.4M | 69.5M | 59.1M |
| Non-GB regs. | 38,931 | 16,247 | 17,481 | 10,615 | 13,441 | 84,174 | 54,418 | 20,650 | 20,276 | 23,613 | 91,263 | 35,136 | 25,682 |
| GB nucs. | 16.3M | 11.4M | 4.2M | 6.7M | 3.7M | 15.1M | 12.2M | 4.3M | 6.4M | 3.3M | 41.6M | 26.9M | 16.2M |
| GBs | 5857 | 4424 | 3828 | 2095 | 4454 | 6054 | 4227 | 3993 | 2170 | 4465 | 7396 | 3332 | 4465 |
| Protein-coding | 3792 | 2814 | 2845 | 1251 | 3464 | 3804 | 2437 | 2875 | 1275 | 3406 | 4291 | 1741 | 3041 |
| Pseudogenes | 1134 | 955 | 454 | 483 | 417 | 1232 | 1080 | 518 | 474 | 425 | 1701 | 876 | 668 |
| lincRNAs | 732 | 492 | 398 | 254 | 476 | 753 | 513 | 459 | 284 | 546 | 920 | 417 | 529 |
| Others | 199 | 163 | 131 | 107 | 97 | 265 | 197 | 141 | 137 | 88 | 484 | 298 | 227 |
| GB regions | 37,874 | 20,030 | 15,076 | 9729 | 9757 | 36,794 | 21,052 | 14,878 | 8999 | 8701 | 59,703 | 29,302 | 20,657 |
| Intronic | 28,751 | 13,971 | 11,700 | 6632 | 8000 | 27,982 | 14,405 | 11,322 | 6126 | 7371 | 41,219 | 18,842 | 14,029 |
| ncRNA exons | 4188 | 2799 | 1052 | 1734 | 959 | 4351 | 3396 | 1216 | 1738 | 878 | 6589 | 3573 | 2117 |
| CDS | 2657 | 1836 | 1313 | 731 | 416 | 2855 | 2221 | 1452 | 766 | 222 | 7885 | 4754 | 2952 |
| 5′UTR | 1106 | 613 | 617 | 258 | 132 | 908 | 518 | 580 | 191 | 90 | 2238 | 1221 | 861 |
| 3′UTR | 1135 | 785 | 381 | 369 | 233 | 698 | 512 | 307 | 178 | 140 | 1769 | 910 | 695 |
| Other UTR | 37 | 26 | 13 | 5 | 6 | 0 | 0 | 1 | 0 | 0 | 3 | 2 | 3 |
| Total nucs. | 38.7M | 27.1M | 9.6M | 17.8M | 10.4M | 83.8M | 54.7M | 61.3M | 63.2M | 55.4M | 130.0M | 96.4M | 75.3M |
| Total regs. | 76,805 | 36,277 | 32,557 | 20,344 | 23,198 | 120,968 | 75,470 | 35,528 | 29,275 | 32,314 | 150,966 | 64,438 | 46,339 |
Fig. 2.

Many dark regions involve protein-coding gene regions. We identified 36,794 dark regions (> 15 million nucleotides) in 6054 gene bodies that were either dark by depth or dark by mapping quality. a Stratifying the gene bodies by GENCODE biotype, 3804 gene bodies were protein coding, 1232 were pseudogenes, and 753 were long intergenic non-coding RNAs (lincRNA). b Of all 36,794 dark regions, 27,982 were intronic, 4351 were in ncRNA exons, 2855 were in protein-coding exons (CDS), 908 were in 5′UTR regions, and 698 were in 3′UTR regions. Any dark region that spanned a gene element boundary (e.g., intron to exon) was split into separate dark regions
Focusing only on CDS regions, we identified 2855 dark CDS regions (> 460,000 nucleotides) across 748 protein-coding genes that were dark by either depth or mapping quality (Fig. 3a; Additional file 2: Table S1; Additional file 3: Table S2). We identified 117 (15.6%) of the 748 protein-coding genes that were 100% dark in CDS regions, 402 (53.7%) were at least 25% dark in CDS regions, and 592 (79.1%) were at least 5% dark in CDS regions (Fig. 3b; Additional file 2: Table S1).
Fig. 3.
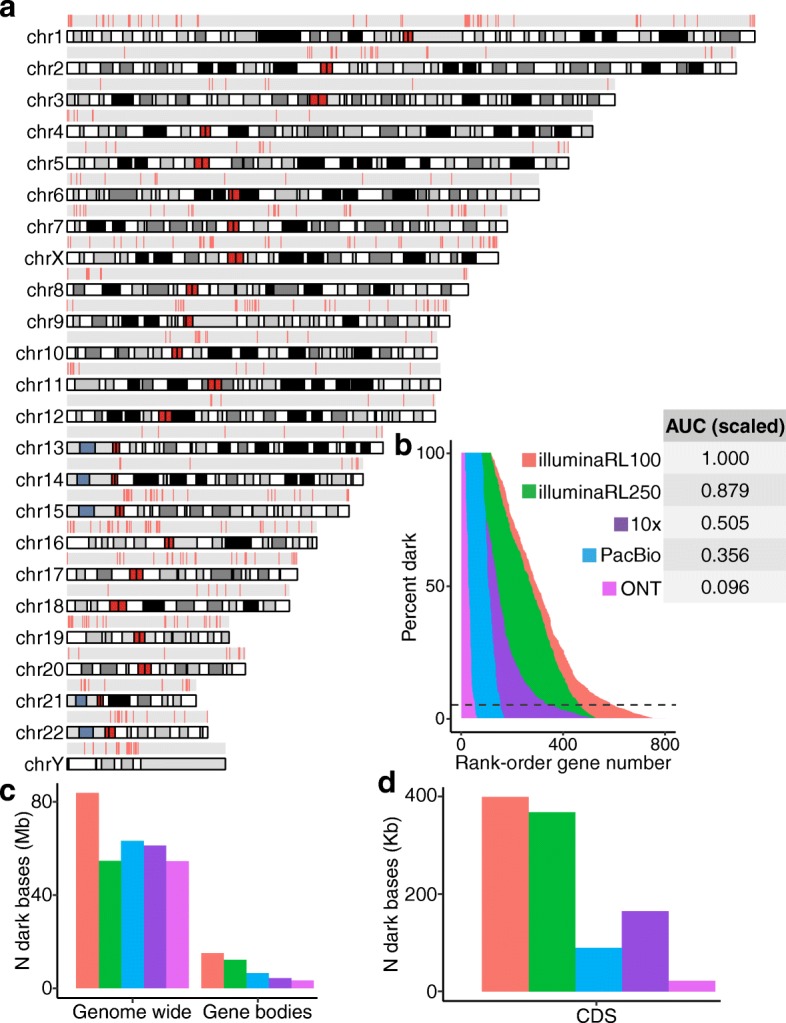
Dark coding regions occur throughout the genome and are largely resolved with long-read sequencing technologies. We identified 2855 dark coding (CDS) regions in 748 protein-coding genes that were dark by either depth or mapping quality (Additional file 2: Table S1; Additional file 3: Table S2). We identified 117 (15.6%) of the 748 protein-coding genes were 100% dark in CDS regions, 402 (53.7%) were at least 25% dark in CDS regions, and 592 (79.1%) were at least 5% dark in CDS regions (Additional file 2: Table S1). a We mapped all protein-coding gene bodies with a dark coding exon to the genome to visualize their genomic location and are generally spread throughout. There are several tight clusters of dark CDS regions on chromosomes 1, 9, 10, and Y, however. b We assessed how well increasing read lengths would resolve dark regions by assessing samples sequenced with Illumina whole-genome sequencing using 250-nucleotide read lengths, as well as long-read technologies 10x Genomics, Oxford Nanopore Technologies (ONT), and Pacific Biosciences (PacBio). Data from the samples sequenced using 250-nucleotide Illumina read lengths reduced the area under the curve (AUC) by 12.1% in CDS regions. Comparing long-read sequencing technologies to the standard Illumina 100-nucleotide read lengths, 10x Genomics, PacBio, and ONT reduced the area under the curve for CDS regions by approximately 49.5%, 64.4%, and 90.4%, respectively. The AUC for each technology is scaled in reference to Illumina sequencing based on 100-nucleotide read lengths (i.e., AUC for Illumina 100-nucleotide read lengths = 1). In contrast to overall results, PacBio outperformed 10x Genomics when looking only at CDS regions (see text). Most analyses focused on genes where at least 5% of the CDS nucleotides are dark, indicated by the dashed line. c, d We also calculated the raw number of dark nucleotides for each technology in GRCh38, genome wide, in full gene bodies, and in CDS regions
Most dark protein-coding regions are specifically camouflaged
Regions may be dark because of either low depth or low mapping quality, but the majority of regions are dark because of mapping quality, and many specifically because they are camouflaged (low mapping quality because of a duplication). We found that 3782 (62.5%) of the 6054 dark gene bodies are dark because of mapping quality, where 2716 (44.9%) were, in fact, camouflaged. Likewise, 436 (73.6%) of the 592 genes that were ≥ 5% dark in CDS regions were dark because they were camouflaged. We also measured the number of times each gene region was duplicated and found that 71.1% of gene regions were replicated three or fewer times in the genome, but 42 regions were duplicated ≥ 100 times (Additional file 1: Figure S2a), with the most repeated regions (six separate intronic regions totaling 833 nucleotides from FGF12 intron six) being replicated 530 times in aggregate. Limiting to only CDS regions, we estimate that 76.2% are replicated three or fewer times, with 45 replicated ≥ 10 times (Additional file 1: Figure S2b), and the most repeated region was from NBPF20, in which 109 nucleotides were replicated 32 times.
Linked- and long-read sequencing technologies resolve substantial portions of the dark regions
Data from the samples sequenced using 250-nucleotide Illumina read lengths reduced the percentage of dark nucleotides by 34.7%, 19.2%, and 8.1% genome-wide, for all gene bodies, and for only CDS regions, respectively, leaving 65.3%, 80.8%, and 91.9% of the original dark nucleotides, respectively (Fig. 3c, d; Additional file 4: Table S3; Additional file 5: Table S4). Comparing linked- and long-read sequencing technologies to the standard Illumina 100-nucleotide read lengths, the ONT platform performed best, both when assessing entire gene bodies, and when considering only CDS regions. Specifically, approximately 42.8%, 28.7%, and 22.1% of the nucleotides remained dark for all gene bodies for PacBio, 10x Genomics, and ONT, respectively (Fig. 3c; Additional file 6: Table S5; Additional file 7: Table S6; Additional file 8: Table S7; Additional file 9: Table S8; Additional file 10: Table S9; Additional file 11: Table S10). Similarly, approximately 22.3%, 41.2%, and 5.4% of CDS nucleotides remained dark for PacBio, 10x Genomics, and ONT, respectively (Fig. 3d; Additional file 6: Table S5; Additional file 7: Table S6; Additional file 8: Table S7; Additional file 9: Table S8; Additional file 10: Table S9; Additional file 11: Table S10). We also calculated the area under the curve (AUC) for each technology, where the AUC is based on the percentage of each gene that is dark. Compared to the AUC for 100-nucleotide Illumina read lengths, Illumina-250 read lengths, PacBio, 10x Genomics, and ONT resolved 12.1%, 64.4%, 49.5%, and 90.4% of CDS gene regions, respectively (Fig. 3b). Only 15 of 117 genes that were originally 100% dark remained 100% dark in the ONT data. In contrast to overall gene-body results, PacBio outperformed 10x Genomics when looking only at CDS regions (Fig. 3c, d). The long-read technologies improved over Illumina mostly by reducing the percentage of nucleotides that are dark by mapping quality (Additional file 1: Figure S1c). Surprisingly, the percentage of gene-body regions that are dark because of low depth is higher for long-read technologies than it is for Illumina (Additional file 1: Figure S1c).
Important pathways and gene families are affected by dark and camouflaged regions
Because such a large number of genes are dark, we characterized the pathways for genes that are not fully represented in standard Illumina short-read sequencing (100-nucleotide reads) datasets. We included all genes where at least 5% of the CDS regions were dark (565 unique gene symbols) and identified several pathways that are important in human health, development, and reproductive function (Fig. 4a; Additional file 12: Table S11). Specific pathway groups included Ub-specific processing proteases (R-HSA-5689880; logP = − 10.70), defensins (R-HSA-1461973; logP = − 9.43), ncRNA 3′-end processing (GO:0043628; logP = − 8.87), gonadal mesoderm development (GO:0007506; logP = − 8.76), spermatogenesis (GO:0007283; logP = − 8.29), spindle assembly (GO:0051225; logP = − 7.56), NLS-bearing protein import into nucleus (GO:0006607; logP = − 6.63), methylation-dependent chromatin silencing (GO:0006346; logP = − 4.98), activation of GTPase activity (GO:0090630; logP = − 4.67), and others. Some specific gene families involved in these pathways include 21 ubiquitin-specific 17-like family members (e.g., USP17L3), 12 defensin genes (e.g., DEFA1 and DEFB4A), 6 testis-specific proteins (e.g., TSPY2), and 13 golgin genes (e.g., GOLGA6B; Additional file 12: Table S11).
Fig. 4.
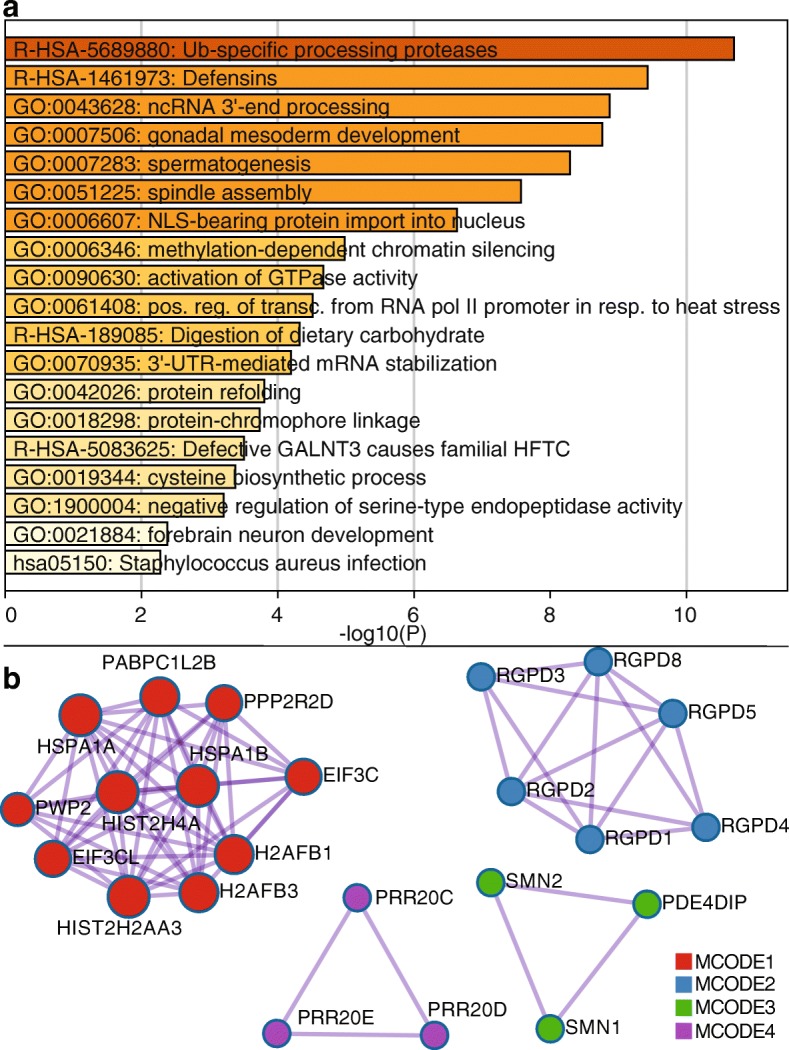
Pathways relevant to human health, development, and reproductive function are affected by dark and camouflaged genes. We characterized the pathways for dark and camouflaged genes using Metascape.org, including only genes where at least 5% of the CDS regions were dark (565 unique gene symbols; based on standard Illumina 100 nucleotide read lengths). a Specific pathway groups included Ub-specific processing proteases (R-HSA-5689880; logP = − 10.70), defensins (R-HSA-1461973; logP = − 9.43), ncRNA 3′-end processing (GO:0043628; logP = − 8.87), gonadal mesoderm development (GO:0007506; logP = − 8.76), spermatogenesis (GO:0007283; logP = − 8.29), spindle assembly (GO:0051225; logP = − 7.56), NLS-bearing protein import into nucleus (GO:0006607; logP = − 6.63), methylation-dependent chromatin silencing (GO:0006346; logP = − 4.98), activation of GTPase activity (GO:0090630; logP = − 4.67), and others. b Looking specifically at known protein-protein interactions, we found 103 proteins with 172 known interactions (Additional file 1: Figure S3) and, within those, identified four groups enriched for protein-protein interactions using the MCODE algorithm [28] (Fig. 4b). All four MCODE groups combined are primarily associated with RNA transport (hsa030313; logP = − 18.59; Additional file 1: Figure S4; accessed March 2019). Individually, the first group (MCODE1) is enriched for proteins involved in systemic lupus erythematosus (hsa05322; logP = − 6.55), cellular response to stress (R-HSA-2262752; logP = − 6.13), and RNA transport (hsa03013; logP = − 4.26; Additional file 1: Figure S5). The second group (MCODE2) is enriched with proteins involved in NLS-bearing protein import into nucleus (GO:0006607; logP = − 18.44; Additional file 1: Figure S6). The third and fourth groups do not have significant enrichment associations, likely because little is known about them; five of the six genes (PRR20C, PRR20D, PRR20E, SMN1, and SMN2) are completely or nearly 100% camouflaged, and several do not even have known expression measurements in GTEx [29] (Additional file 1: Figures S7-S9)
Looking specifically at known protein-protein interactions, we found 103 proteins with 172 known interactions (Additional file 1: Figure S3) and, within those, identified four groups enriched for protein-protein interactions using the MCODE algorithm [28] (Fig. 4b). All four MCODE groups combined are primarily associated with RNA transport (hsa030313; logP = − 18.59; Additional file 1: Figure S4; accessed March 2019). Individually, the first group (MCODE1) is enriched for proteins involved in systemic lupus erythematosus (hsa05322; logP = − 6.55), cellular response to stress (R-HSA-2262752; logP = − 6.13), and RNA transport (hsa03013; logP = − 4.26; Additional file 1: Figure S5). The second group (MCODE2) is enriched with proteins involved in NLS-bearing protein import into nucleus (GO:0006607; logP = − 18.44; Additional file 1: Figure S6). The third and fourth groups do not have significant enrichment associations, likely because little is known about them; five of the six genes (PRR20C, PRR20D, PRR20E, SMN1, and SMN2) are completely or nearly 100% camouflaged, and several do not even have known expression measurements in GTEx [29] (Additional file 1: Figures S7-S9).
There are 76 dark genes with known mutations associated with 326 human diseases
To assess the potential impact missing mutations in dark genes may have on human disease genetics, we measured the number of dark genes with at least 5% dark CDS that have mutations known to be involved in human disease; we calculated the number of genes that are ≥ 5% dark CDS with a mutation in the Human Gene Mutation Database (HGMD) [30]. We found 76 genes associated with 326 unique human diseases (Fig. 5a). Some of the diseases with the most known associated genes include autism spectrum disorder, schizophrenia, hearing loss, spinal muscular atrophy, and inflammatory bowel disease. Some of the diseases most represented in our data are not surprising, given the number of genes involved in the disease, but these data demonstrate the number of diseases impacted by genes that are at least 5% dark CDS. We also performed an enrichment analysis, where the diseases most enriched for dark genes included color blindness (protan color vision defect), X-linked cone-rod dystrophy, and spinal muscular atrophy (Additional file 1: Figure S10).
Fig. 5.

Seventy-six dark genes (≥ 5% CDS) are associated with 326 human diseases, including autism, inflammatory bowel disease, and others. We found 76 genes ≥ 5% dark CDS that harbor mutations associated with 326 unique human diseases, according to the Human Gene Mutation Database (HGMD). a Some of the diseases with the most known associated genes include autism spectrum disorder, schizophrenia, hearing loss, spinal muscular atrophy, and inflammatory bowel disease. Word size represents the number of genes associated with each disease. These data demonstrate the number of diseases impacted by genes that are at least 5% dark CDS, and how important it is to completely resolve dark regions. We also performed an enrichment analysis, where the diseases most enriched for dark genes included color blindness (protan color vision defect), X-linked cone-rod dystrophy, and spinal muscular atrophy (Additional file 1: Figure S10). b Similarly, we quantified the number of diseases each gene was associated with and identified many disease-relevant genes with large portions of dark CDS regions that may harbor critical disease-modifying mutations that currently go undetected. Some of the genes with the most known disease associations include ARX (12.8% dark CDS), NEB (9.5% dark CDS), TBX1 (10.6% dark CDS), RPGR (8.6% dark CDS), HBA2 (9.5% dark CDS), and CR1 (26.0% dark CDS). CR1 is particularly notable for neuroscientists and Alzheimer’s disease geneticists, patients, and their caregivers, given that CR1 is a top-ten Alzheimer’s disease gene. Other notable genes include SMN1 (94.6% dark CDS) and SMN2 (88.0% dark CDS), which are known to harbor mutations (in camouflaged regions) that are involved in spinal muscular atrophy (SMA) and are implicated in ALS. HSPA1A (53.0% dark CDS) and HSPA1B (51.5% dark CDS) also encode two primary 70-kilodalton (kDa) heat-shock proteins. Heat-shock proteins have been implicated in ALS [31, 32]
Similarly, we quantified the number of diseases each gene was associated with (Fig. 5b). We identified many disease-relevant genes with large portions of dark CDS regions that may harbor critical disease-modifying mutations that currently go undetected. Some of the genes with the most known disease associations include ARX (12.8% dark CDS), NEB (9.5% dark CDS), TBX1 (10.6% dark CDS), RPGR (8.6% dark CDS), HBA2 (9.5% dark CDS), and CR1 (26.0% dark CDS). The CR1 gene is particularly notable given that CR1 is a top-ten Alzheimer’s disease gene. Other notable genes include SMN1 (94.6% dark CDS) and SMN2 (88.0% dark CDS), which are known to be involved in spinal muscular atrophy (SMA) and ALS [33–35]. HSPA1A (53.0% dark CDS) and HSPA1B (51.5% dark CDS) also encode two primary 70-kilodalton (kDa) heat-shock proteins, a family of proteins that have been implicated in ALS [31, 32].
Camouflaged genes are consistently dark in gnomAD, but dark-by-depth genes may be sample or dataset specific
Although many dark genes are specifically camouflaged (Additional file 13: Table S12; Additional file 14: Table S13), many are dark by depth in the ADSP data; upon manual comparison between whole-genome sequencing data from the ten ADSP males and coverage plots from the gnomAD consortium dataset (http://gnomad.broadinstitute.org/) [36], we found that camouflaged regions in the ADSP males are consistently dark in the gnomAD data, demonstrating that these camouflaged regions are consistent across datasets. The ADSP data are also included in gnomAD, but they only make up approximately 15% of the data. The dark-by-depth regions are more variable between samples and datasets, however, suggesting these regions may be sensitive to specific aspects of whole-genome sequencing (e.g., library preparation) or downstream analyses. Specific camouflaged genes include SMN1 and SMN2 (Fig. 6a), HSPA1A and HSPA1B (Fig. 6b), NEB (9. Fig. 6c), and CR1 (Fig. 6d). Specific dark-by-depth genes include HLA-DRB5 (Fig. 6e), RPGR (Fig. 6f), ARX (Fig. 6g), and TBX1 (Fig. 6h). All four camouflaged genes are also dark in the gnomAD data. A manual inspection of our dark-by-depth gene list, however, suggests most are not completely dark in gnomAD, but vary by sample or dataset. Specifically, HLA-DRB5 and RPGR in gnomAD appear to be consistent with the ADSP data; ARX and TBX1, however, only appear to be dark in a portion of the gnomAD samples, where about 30% of samples have ≤ 5 reads in their respectively defined dark regions. Dark regions are either similar or more pronounced in the gnomAD whole-exome data than what we observed in the whole-genome data (Figs. 6a–h), highlighting that dark and camouflaged regions are generally magnified in whole-exome data; this is likely because of differences in library preparation and shorter read lengths in exome data. For interest, we also found that APOE—the top genetic risk for Alzheimer’s disease [44–46]—is approximately 6% dark CDS (by depth) for certain ADSP samples with whole-genome sequencing, and the same region is dark in gnomAD whole-exome data (Additional file 1: Figure S11). It is possible some of the dark regions we identified in standard short-read whole-genome data are specific to the ADSP samples, but additional work can clarify this issue. In either case, dark-by-depth regions (Additional file 15: Table S14; Additional file 16: Table S15) should be interrogated within individual datasets, and perhaps for individual samples as a quality control measure.
Fig. 6.

Camouflaged genes are consistently dark in gnomAD, but dark-by-depth genes may be sample or dataset specific. Many dark genes are specifically camouflaged (Additional file 13: Table S12; Additional file 14: Table S13), but many are dark by depth; we found that camouflaged regions in the ADSP are consistently dark in the gnomAD consortium data (http://gnomad.broadinstitute.org/) [36]. Dark-by-depth regions may be more variable between samples and datasets, however, suggesting these regions may be sensitive to specific aspects of whole-genome sequencing (e.g., library preparation) or downstream analyses. a SMN1 and SMN2 are camouflaged by each other (only SMN1 shown). Both genes contribute to spinal muscular atrophy and have been implicated in ALS. b HSPA1A and HSPA1B are also camouflaged by each other (only HSPA1A shown). The heat-shock protein family has been implicated in ALS. c NEB (9.5% dark CDS) is a special case that is camouflaged by itself. NEB is associated with 24 diseases in the HGMD, including nemaline myopathy, a hereditary neuromuscular disorder. NEB is a large gene; thus, 9.5% dark CDS translates to 2424 protein-coding bases. d CR1 is a top Alzheimer’s disease gene that plays a critical role in the complement cascade as a receptor for the C3b and C4b complement components, and potentially helps clear amyloid-beta (Aβ) [37–39]. CR1 is also camouflaged by itself, where the repeated region includes the extracellular C3b and C4b binding domain. The number of repeats and density of certain isoforms have been associated with Alzheimer’s disease [21, 40–43]. e HLA-DRB5 is dark by depth in the ADSP and gnomAD data. HLA-DRB5 has been implicated in several diseases, including Alzheimer’s disease. f RPGR is likewise dark in ADSP and gnomAD and is associated with several eye diseases, including retinitis pigmentosa and cone-rod dystrophy. g ARX is dark-by-depth, but varies by sample or cohort, as approximately 70% of gnomAD samples are not strictly dark by depth. ARX is associated with diseases including early infantile epileptic encephalopathy 1 (EIEE1) and Partington syndrome. h Similarly, TBX1 is not strictly dark by depth in approximately 70% of gnomAD samples. The Y axes for figures a–f indicate median coverage in gnomAD (blue = exomes; green = genomes), whereas the Y axes in g, h represent the proportion of gnomAD samples that have > 5x coverage. Dark and camouflaged regions, as well as the percentage of each gene’s CDS region that is dark, are indicated by red lines. Dark regions in exome data are either similar or more pronounced than what we observed in whole-genome data, highlighting that dark and camouflaged regions are generally magnified in whole-exome data. For interest, we also discovered that APOE—the top genetic risk for Alzheimer’s disease [44–46]—is approximately 6% dark CDS (by depth) for certain ADSP samples with whole-genome sequencing, and the same region is dark in gnomAD whole-exome data (Additional file 1: Figure S11)
SMN1 and SMN2 are camouflaged by each other, where both genes are known to contribute to spinal muscular atrophy, and have been implicated in ALS. HSPA1A and HSPA1B are also camouflaged by each other, and the heat-shock protein family has been implicated in ALS [33, 35]. NEB is a special case that is camouflaged by itself (rather than another gene), and is associated with 24 diseases in the HGMD, including nemaline myopathy, a hereditary neuromuscular disorder. NEB is a large gene (249,151 nucleotides; 25,577 CDS nucleotides); thus, ~ 9.5% dark CDS translates to 2424 dark protein-coding bases. CR1 is a top Alzheimer’s disease gene that plays a critical role in the complement cascade as a receptor for the C3b and C4b complement components, and potentially helps clear amyloid-beta (Aβ) [37–39]. Like NEB, CR1 is also camouflaged by itself, where the repeated region actually includes the extracellular C3b and C4b binding domain. The number of repeats and density of certain isoforms have been associated with Alzheimer’s disease [21, 40–43].
We found HLA-DRB5 is dark by depth in the ADSP and gnomAD data and has been implicated in several diseases, including Alzheimer’s disease. RPGR is likewise dark in ADSP and gnomAD and is associated with several eye diseases, including retinitis pigmentosa and cone-rod dystrophy. We identified ARX as a dark-by-depth gene, but this gene appears to vary by sample or cohort, as only approximately 30% of gnomAD samples are strictly dark by depth, using our cutoff of ≤ 5 reads. ARX is associated with diseases including early infantile epileptic encephalopathy 1 (EIEE1) [47] and Partington syndrome [48]. Similarly, TBX1, which harbors mutations that cause the same phenotype as 22q11.2 deletion syndrome [49], is dark by depth in only approximately 30% of gnomAD samples.
Linked- and long-read technologies resolve many camouflaged regions, with variable success
We selected three camouflaged gene regions to highlight common strengths and differences for how well each linked- or long-read sequencing technology addresses the camouflaged region, including SMN1 and SMN2 (Fig. 7a), HSPA1A and HSPA1B (Fig. 7b), and CR1 (Fig. 7c). The SMN1 and SMN2 genes are camouflaged by each other (gene duplication), as are HSPA1A and HSPA1B. CR1, however, is a special case, where it is camouflaged by a repeated region within itself. Only ONT appeared to be capable of fully addressing the camouflaged region for all three genes. 10x Genomics also performed well under certain circumstances, such as SMN1 and SMN2 (regions where the duplication is > 50 kb away), but did not perform well for HSPA1A and HSPA1B. PacBio performed well for HSPA1A/HSPA1B, but did not perform as well as ONT in CR1 and the SMN1/SMN2 region.
Fig. 7.
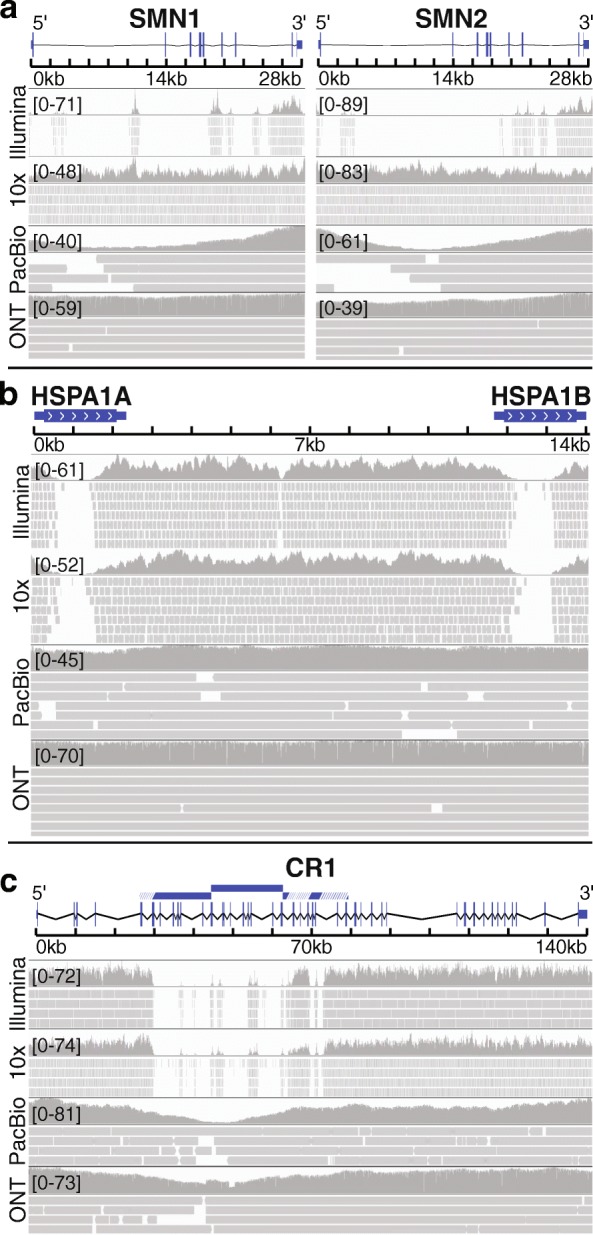
Long-read technologies resolve many camouflaged regions, with variable success. We found that ONT’s long-read technology appeared to resolve all camouflaged regions well with the high sequencing depth. PacBio performed similarly well, and 10x Genomics performs well under certain circumstances. a SMN1 and SMN2 were 94.6% and 88.0% camouflaged CDS, respectively, based on standard Illumina sequencing with 100-nucleotide read lengths (illuminaRL100). Both genes were 0% camouflaged CDS for 10x Genomics, PacBio, and ONT data. 10x Genomics and ONT perform particularly well in these genes, with consistently high mapping coverage. b HSPA1A and HSPA1B were 53.0% and 51.5% camouflaged CDS, respectively, based on illuminaRL100 data. Both genes were 0% camouflaged CDS based on ONT and PacBio data and were 45.8% and 51.8% camouflaged CDS based on 10x Genomics data. In contrast to the results for SMN1 and SMN2, 10x Genomics was unable to resolve the HSPA1A and HSPA1B camouflaged regions. c CR1 was 26.0% camouflaged CDS based on illuminaRL100. 10x Genomics did not improve coverage for CR1; the region remained 26.4% camouflaged CDS. Both ONT and PacBio were 0% camouflaged CDS. While both PacBio and ONT were able to fill the camouflaged region, coverage dropped throughout the region, particularly for PacBio. The duplicated region is indicated by blue bars, where white lines indicate regions that have diverged sufficiently for short-reads to align uniquely. Regions were visualized with IGV. Reads with a MAPQ < 10 were filtered, and insertions, deletions, and mismatches are not shown
Many camouflaged regions can be rescued, including in standard short-read sequencing data
There are many large-scale whole-genome or whole-exome sequencing projects across tens of thousands of individuals that are either completed or underway for a variety of diseases, including cancer (e.g., The Cancer Genome Atlas (TCGA)), autism spectrum disorder (e.g., The Autism Sequencing Consortium (ASC)), Alzheimer’s disease (e.g., The Alzheimer’s Disease Sequencing Project (ADSP)), Parkinson’s disease (e.g., The Parkinson’s Progression Markers Initiative (PPMI)), and ALS (e.g., Target ALS and CReATe). All of these datasets are affected by dark and camouflaged regions that may harbor mutations that either are driving or modify disease in patients. Ideally, all samples would be re-sequenced using the latest technologies over time, but financial resources and biological samples are limited, making it essential to maximize the utility of existing data.
Using a strategy similar to that proposed by Robert and Watson [10], we have developed a method to rescue mutations in most camouflaged regions, including for standard Illumina short-read sequencing data. When confronted with a sequencing read that aligns to two or more regions equally well (with high confidence), most aligners (e.g., BWA [11–13]) will randomly map the read to one of the regions and assign a low mapping quality (MAPQ = 0 for BWA, or MAPQ = 1 for novoalign). Because the reads are already aligned to one of the regions, we can use the following steps to rescue mutations in most camouflaged regions (Fig. 8): (1) extract reads from camouflaged regions; (2) mask all highly similar regions in the reference genome, except one, and re-align the extracted reads; and (3) call mutations using standard methods, while accounting for increased ploidy and potential reference-based artifacts. Reference-based artifacts arise when regions within a given camouflaged set are not 100% identical, causing false positives when reads from both regions are aligned to a single region. Without competing camouflaged regions to confuse the aligner, the aligner will assign a high mapping quality, allowing variant callers to behave normally. This will enable researchers to identify mutations that exist in one of the camouflaged regions, but cannot indicate which specific region the mutation originated from (Fig. 8). After rescuing these mutations, researchers can then perform association studies to determine whether any of the mutations may be implicated in disease, and follow up with targeted sequencing methods to determine the exact camouflage region a mutation lies in.
Fig. 8.

Many camouflaged regions can be rescued, including CR1, even in standard short-read sequencing data. Many large-scale whole-genome or whole-exome sequencing projects exist, covering tens of thousands of individuals. All of these datasets are affected by dark and camouflaged regions that may harbor mutations that either drive or modify disease in patients. Ideally, all samples would be re-sequenced using the latest technologies over time, but financial resources and biological samples are limited, making it essential to maximize the utility of existing data. We developed a method to rescue mutations in most camouflaged regions, including for standard short-read sequencing data. When confronted with a sequencing read that aligns to two or more regions equally well (with high confidence), most aligners (e.g., BWA [11–13]) will randomly assign the read to one of the regions with a low mapping quality (e.g., MAPQ = 0 for BWA). a Because the reads are already aligned to one of the regions, we can use the following steps to rescue mutations in most camouflaged regions: (1) extract reads from camouflaged regions, (2) mask all highly similar regions in the reference genome, except one, and re-align the extracted reads, (3) call mutations using standard methods (adjusting for ploidy), and (4) determine precise location using targeted sequencing (e.g., long-range PCR combined with Sanger, or targeted long-read sequencing [1]). Without competing camouflaged regions to confuse the aligner, the aligner will assign a high mapping quality, allowing variant callers to behave normally. b Exons 10, 18, and 26 in CR1 are identical, according to the reference genome. Standard aligners will randomly scatter reads matching that sequence across these exons and assign a low mapping quality (e.g., MAPQ = 0 for BWA; indicated as hollow reads). Red lines indicate an individual’s mutation that exists in one of these exons, but reads containing this mutation also get scattered and assigned a low mapping quality. c By masking exons 18 and 26, we can align all of these reads to exon 10 with high mapping qualities to determine whether a mutation exists. We cannot determine at this stage which of the three exons the mutation is actually located in, but researchers can test association with a given disease to determine whether the mutation is worth further investigation. d As a proof of principle, we rescued approximately 4214 exonic variants in the ADSP (TiTv = 2.26) using our method, including a frameshift mutation in CR1 (MAF = 0.00019) that is found in five cases and zero controls (three representative samples shown). The frameshift results in a stop codon shortly downstream. The ADSP is not large enough to formally assess association between the CR1 frameshift and Alzheimer’s disease, but we believe the mutation merits follow-up studies given its location (CR1 binding domain) and CR1’s strong association with disease
Re-alignment rescues approximately 4214 exonic variants, including a rare ten-nucleotide frameshift deletion in CR1
As a proof of principle, we applied our method to the Alzheimer’s Disease Sequencing Project (ADSP) case-control data [50] to approximate the number of potential mutations our approach could rescue. The ADSP is a large sequencing project organized, in part, to identify functional mutations that influence Alzheimer’s disease development. Across 10,933 samples from the ADSP, excluding all reference-based artifacts, and variants with a quality by depth (QD) ≤ 2, we were able to rescue approximately 4214 exonic variants with a transition-transversion ration (Ti/Tv) of 2.26 from 137 camouflaged region sets, that are spread across 748 camouflaged genes (Additional file 1: Figure S12; VCF will be provided to the ADSP). Using a more stringent QD (excluding variants with QD ≤ 3), we rescued 3343 variants with a Ti/Tv ratio of 2.35. We only included camouflaged regions from CDS exons for all genes, including those that are < 5% dark CDS.
Because CR1 is a top-10 Alzheimer’s disease gene, we then specifically interrogated it using our method (Fig. 8) for any functional mutations that could be involved in Alzheimer’s disease. Using 13,142 ADSP samples, we identified a rare ten-nucleotide frameshift deletion that is found in five cases but zero controls, all of which are heterozygous (Fig. 8d). Three of the five carriers were of European descent and two were Caribbean Hispanic. All five samples were from the ADSP case-control series; thus, we did not expect them to be related. We verified the three European carriers were unrelated (KING-robust kinship < 0.0442) [51], but the two Caribbean Hispanic carriers appear to be first-degree relatives (KING-robust kinship = 0.3356). For interest, only one of the individuals carried a single APOEε4 allele (ε3/ε4). The other four individuals were homozygous for APOEε3 (ε3/ε3). We were able to determine that the frameshift deletion is in one of exons 10, 18, or 26. We estimate a cohort of approximately 70,000 cases and controls would have approximately 80% statistical power to formally assess this mutation’s involvement in Alzheimer’s disease, assuming a relative risk (RR) of 3.3, at an alpha of 0.0001.
Discussion
While researchers have known for years that dark regions exist in standard short-read sequencing data, little work has been done to characterize the breadth of the issue and to develop possible solutions until more financially feasible linked- or long-read sequencing options are available. Short-read sequencing is unable to adequately address camouflaged regions because the reads cannot fully span camouflaged regions to properly align homologous nucleotides. Linked- and long-read sequencing technologies, such as those from 10x Genomics, Oxford Nanopore Technologies (ONT), and Pacific Biosciences (PacBio), have the potential to address many camouflaged regions because these technologies have median read lengths measured in thousands of nucleotides, rather than only 100–300 nucleotides from standard short-read sequencing technologies (e.g., Illumina). Recent work has even demonstrated that mappable ONT reads can exceed two million nucleotides (e.g, 2,272,580) [52, 53], showing future potential for addressing large camouflaged regions.
In this study, we systematically characterized dark and camouflaged gene regions and proposed a method to address most camouflaged regions in long- or short-read sequencing data. Our solution is specifically applicable to camouflaged regions, not regions that are dark by depth, simply because there are no reads available in those regions. While our solution is conceptually simple, implementing the solution systematically was challenging because of many intricate details, including increased zygosity, and would ideally be integrated into the original alignment and variant-calling process. While the original implementation was challenging, we provide the resulting .bed files for both GRCh37 and GRCh38 that are necessary to rescue mutations from camouflaged regions in any human re-sequencing dataset (https://github.com/mebbert/Dark_and_Camouflaged_genes). We also provide all of our data and source code. The .bed files and source code should make implementing our method relatively straightforward for other groups. As a proof of concept, we were able to rescue approximately 4214 variants in the ADSP dataset from 137 sets of camouflaged gene regions, which are spread across 748 camouflaged genes. Included in these rescued mutations is a ten-nucleotide frameshift deletion in CR1 found in five ADSP cases and zero controls.
The number of genes affected by dark and camouflaged regions was surprisingly high. We identified 36,794 total dark regions across 6054 gene bodies, 3804 of which were protein coding genes. We found that 27,982 of the dark regions were intronic and 2855 were in protein-coding exons (CDS). Others were in pseudogenes (1232) and lincRNAs (753). While most of the dark regions were non-coding (e.g., intronic), these regions may still harbor important mutations that drive or modify human diseases. For example, there are many examples of mutations in non-coding regions driving disease, including repeat expansions [1, 54–61], splice-site mutations (these may be intronic or exonic) [62–76], and regulatory mutations (e.g., UTR regions) [77–86]. There are also many lincRNAs associated with disease [87–96].
There are many patients with diseases known to be genetically inherited, yet remain genetically unexplained because the patients do not have any of the known mutations. Many of the genes we identified as being at least partially dark are known to be involved in numerous diseases, including Alzheimer’s disease, ALS, SMA, autism spectrum disorder, schizophrenia, and others; functional mutations that modify disease likely lie in some of these dark and camouflaged regions. For example, SMN1 and SMN2 are mostly dark (camouflaged) and are known to harbor mutations that cause disease [62, 64–66]. CR1 is another dark gene that is 26.5% dark CDS, being camouflaged to itself, and is strongly implicated in Alzheimer’s disease. In fact, the CR1 camouflaged region includes the C3b and C4b protein binding sites, repeated several times. Interestingly, the C4B gene (encodes the C4b protein) is also 72.8% dark CDS (camouflaged) and may be involved in disease [97, 98]. We are confident that rescuing mutations from camouflaged regions will have a meaningful impact on disease research and may explain some of the missing heritability of Alzheimer’s disease [18, 99–101] and other diseases.
A large number of gene bodies (527) were 100% dark, which means they are entirely overlooked in standard whole-exome, whole-genome, and RNA sequencing studies [10]. Additionally, more than 1600 gene bodies, or nearly 25%, were at least 25% dark and more than 2100 (35.2%) were at least 5% dark; of these, 748 protein-coding genes were at least 5% dark within CDS regions. Understanding what role these genes play in human health and disease will require being able to resolve them in DNA and RNA sequencing experiments.
A critical decision for future large-scale sequencing projects will be regarding which sequencing technology is ideal to maximize the probability of identifying functional mutations driving disease. Unfortunately, the answer is not clear, as each technology has its pros and cons. Based on our results, the ONT platform performed best, overall, resolving 77% of dark gene-body regions (Additional file 1: Figure S1b). Current costs may be prohibitive for large studies, however. The 10x Genomics platform resolved 64.3% of dark gene-body regions, when compared to standard Illumina sequencing. PacBio resolved 49.5% of dark gene-body regions. Even increasing Illumina read lengths from 100 to 250 made a sizeable difference, overall, resolving 12.2% of dark gene-body regions. Both the PacBio and ONT data used in this study had shorter median read lengths than expected, suggesting both technologies can likely perform better than our estimates.
Focusing only on CDS regions, there were 2855 dark CDS regions across 748 protein-coding genes, based on Illumina 100-nucleotide read lengths. ONT outperformed other long-read technologies, resolving 90.4% of dark CDS regions. PacBio and 10x Genomics resolved 64.4% and 49.5%, respectively. We found that 10x Genomics and ONT performed well in the SMN1 and SMN2 genes (Fig. 7), attaining consistently deep, high-quality coverage throughout. PacBio coverage declined in the interior regions of the genes. In other cases, such as CR1 and NEB, 10x Genomics was unable to improve on standard Illumina sequencing, but ONT was able to largely resolve the region. PacBio also performed relatively well, but both ONT and PacBio required higher than normal sequencing depth for those technologies. We believe that 10x Genomics can correct the issues we observed in CR1 and NEB, by implementing a more sophisticated version of our method that also incorporates evidence from their linked-read technology.
Whether each technology is able to reliably resolve dark and camouflaged regions is an important consideration for choosing the best sequencing technology, but we should also consider how reliably each technology is able to resolve structural mutations. In a previous study, we tested how well ONT and PacBio are able to traverse challenging repeat expansions, and whether they are amenable to genetic discovery [1]. We found that both technologies are well-suited, but we have not assessed performance of the 10x Genomics platform across long repeat expansions.
The primary challenge with ONT and PacBio long-read sequencing is, of course, the high error rate, which can be overcome through deeper sequencing because errors in ONT and PacBio sequencing are mostly random [102, 103]. Ultimately, we are confident that, as long-read error rates improve, and costs continue to decline, long-read technologies will be the preferred sequencing choice for large-scale sequencing projects, especially when considering structural mutations.
We identified dark and camouflaged regions in this study by averaging data across ten males with deep Illumina whole-genome sequencing, using 100-nucleotide read lengths. We assessed how well long-read sequencing technologies (PacBio, ONT, and 10X genomics) resolve these regions, but our measurements should only be considered estimates. While long-read sequencing technologies are becoming more common, we were unable to obtain more than one male individual for each long-read technology; we needed male samples to assess all chromosomes, including the Y chromosome. Additionally, the samples we used for each long-read technology were sequenced at a much higher depth than is currently typical for a re-sequencing effort, which is likely over estimating the number of dark regions they resolve for the average use case. Our measurements should be a reasonable estimate of reality, however, and future analyses will be able to refine our estimates.
We used whole-genome sequencing to assess dark and camouflaged regions, but this problem is magnified in whole-exome data, which many large-scale sequencing studies are based on, either completely, or in part. Whole-exome data are typically generated using even shorter read lengths. They are also generally based on capture, which means certain exons are not fully represented. APOE is a prime example, where it is typically well-covered in whole-genome data, but a portion is dark in whole-exome data (Additional file 1: Figure S11). With APOE harboring the largest genetic risk factors for Alzheimer’s disease, it is important to properly characterize the entire gene.
In this study, we characterized dark and camouflaged gene bodies and demonstrated several disease-relevant genes where a significant portion is dark in standard short-read sequencing data, including SMN1 and SMN2, CR1, and sometimes even APOE. We also identified a rare ten-nucleotide frameshift deletion in CR1 that is found in five ADSP cases and zero controls, as a proof of principle (Fig. 8d). Using our method (Fig. 8), we were able to determine that the frameshift deletion is in one of exons 10, 18, or 26. With CR1 being a top Alzheimer’s disease gene without any known functional mutations, we believe it will be important to assess this mutation in a large cohort, to determine whether it plays a role in disease development and progression. We have also proposed a solution to address most camouflaged genes in sequencing data and believe that our approach has the potential to identify functional mutations that are influencing development across a range of diseases, but are currently entirely overlooked by standard short-read sequencing approaches.
Conclusion
There remain thousands of potentially important genomic regions that are overlooked with short-read sequencing, but are largely resolved by linked- or long-read technologies. While these regions represent only a small portion of the entire genome or exome, many of these regions are known to be important in human health and disease. Equally important, however, is that the impact of many other genes is entirely unknown because they are 100% dark. We presented a method that can resolve most camouflaged regions that we believe will help researchers identify mutations that are involved in disease. As a proof of principle, we rescued approximately 4214 variants in the ADSP dataset, including a ten-nucleotide frameshift mutation in CR1. While we cannot formally assess the CR1 frameshift mutation in Alzheimer’s disease (insufficient sample-size), we believe it is worth investigating in a larger cohort. In the long-term, we believe that linked- and long-read sequencing technologies will be the best solution for resolving dark and camouflaged regions.
Methods
Sample selection and preparation
To identify dark and camouflaged regions, and to assess how well other technologies address them, we selected samples from each technology and read length. All samples were aligned to GRCh37, GRCh38, and GRCh38+alt. To assess dark and camouflaged regions in standard Illumina sequencing with 100-nucleotide read lengths, we selected ten unrelated male control samples from the Alzheimer’s Disease Sequencing Project (ADSP) where deep whole-genome sequencing had been performed by randomly selecting one male from ten random families. All ten males were sequenced at the same facility and were from either the “Health/Medical/Biomedical” (HMB-IRB) or “Health/Medical/Biomedical” for non-profit organizations (HMB-IRB-NPU) consent groups, indicated as groups C1 and C2 in the ADSP pedigree files (available through dbGAP). We selected samples from the ADSP because we required samples that met the following criteria: (1) had been sequenced using standard paired-end Illumina sequencing with 100-nucleotide read lengths, (2) had been sequenced with a median depth > 30x, and (3) were publicly available. Median genome-wide read depths ranged from 33.0x to 45.0x coverage, with an overall median of 37.5x. Samples were prepared and sequenced as part of the ADSP [50]. These samples were aligned using BWA (v0.5.9). We could not find samples from the 1000 Genomes Project [24] that met these criteria; either sequencing depths were too shallow or read lengths were too long or short. The ADSP sample IDs we used were as follows: A-CUHS-CU000406, A-CUHS-CU002997, A-CUHS-CU000779, A-CUHS-CU000208, A-CUHS-CU001010, A-CUHS-CU002031, A-CUHS-CU002707, A-CUHS-CU003023, A-CUHS-CU003090, and A-CUHS-CU003128.
To assess dark and camouflaged regions in samples sequenced using Illumina 250-nucleotide read lengths, we selected ten samples from the 1000 Genomes Project that had been sequenced with 250-nucleotide read lengths and had a median depth > 30x. All ten samples were aligned using BWA (v 0.7.5a-r428) [2, 11–13]. Median genome-wide read depths ranged 30.0x to 61.0x coverage, with an overall median of 58.5x. Sample IDs for the Illumina 250-nucleotide read lengths were as follows: NA20845, HG01112, HG01583, HG01051, HG03742, HG00096, HG01565, HG01879, HG01500, and HG03006 (see Availability of Data and Materials section for public links).
We also selected samples generated using the 10x Genomics synthetic long-read sequencing platform and ONT and PacBio long-read sequencing platforms that were publicly available from the respective company. Specifically, we downloaded HG00512 raw FASTQ data from 10x Genomics and aligned it according to 10x Genomics’ standard practices. We used longranger (v2.2.2) and aligned to GRCh38 (longranger wgs --id HG00512 --description=“Han Chinese” --sex=“male” --fastqs=chi/HNKHFCCXX/,chi/HWHFTCCXX/ --reference=“10x-GRCh38-2.1.0/” --jobmode=sge --mempercore=125 –downsample=385). We also aligned to GRCh37. We were unable to align the 10x data to GRCh38+alt because longranger has a limit to the number of contigs it will align to. Median depth for HG00512 was 52x. For ONT, we downloaded the final Cliveome v3 from ONT’s official GitHub page (https://github.com/nanoporetech/ONT-HG1) and aligned it to GRCh37, GRCh38, and GRCh38+alt using minimap2 [104] (ALIGN_OPTS=“x map-pb -a --eqx -L -O 5,56 -E 4,1 -B 5 --secondary=no -z 400,50 -r 2k -Y”; REF=g1kv37/g1kv37.fa; minimap2 -d ${REF}.mmi ${ALIGN_OPTS} ${REF}; minimap2 ${ALIGN_OPTS} -a ${REF}.mmi <reads.fq> | samtools view -T {REF} -F 2308 > output_file). Cliveome v3 was sequenced to a median depth of 52x. We used the same alignment options recommended for PacBio because we found the recommended “map-ont” option in minimap2 performed substantially worse. We used PacBio data generated from HG005 [105], which was sequenced to a median depth of 50x and aligned using minimap2 [104] (pbsv fasta [movie].subreads.bam | minimap2 -t 8 -x map-pb -a --eqx -L -O 5,56 -E 4,1 -B 5 --secondary=no -z 400,50 -r 2 k -Y ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz - | samtools sort > HG005_PacBio_GRCh38.bam). Neither the ONT nor the PacBio alignments included secondary alignments.
Identifying dark and camouflaged gene body regions
To identify dark and camouflaged gene body regions in standard Illumina 100-nucleotide read length data, we first scanned all ten ADSP whole-genome sequence samples for genomic positions that met either of the following criteria: (1) had ≤ 5 reads and (2) had ≥ 90% of reads with a mapping quality (MAPQ) < 10. We then averaged the depth and count of low MAPQ reads across all samples for each position. We used strict cutoffs to identify regions that are clearly dark, but there are many additional regions that fall just beyond our thresholds. This analysis was performed using the Dark Region Finder (DRF; https://github.com/mebbert/DarkRegionFinder; mapq = 9; dark_mass=90; camo_mass=50; dark_depth=5; java -jar -Xmx20g DarkRegionFinder.jar -i <sample>.bam --human-ref genome.fa --min-region-size 1 --camo-mapq-threshold $mapq --min-dark-mapq-mass $dark_mass --min-camo-mapq-mass $camo_mass --dark-depth $dark_depth --camo-bed-output <sample>-camo-dark_depth_${dark_depth}-dark_mass_${dark_mass}-camo_mass_${camo_mass}-mapq_${mapq}.b38.bed --dark-bed-output <sample>-dark-dark_depth_${dark_depth}-dark_mass_${dark_mass}.b38.bed --incomplete-bed-output <sample>-incomplete.b38.bed). Any position that met either criterion was considered dark and categorized as either dark by depth or dark by mapping quality. For gene-body analyses, we then limited the dark regions to gene bodies by intersecting dark regions identified by Dark Region Finder with Ensembl’s GRCh37 build 87 or GRCh38 build 93 gene annotations. We converted the transcript-level annotations to gene-level annotations using bedtools [106] and custom scripts that are available. Any dark region that spanned a gene body element region (e.g., intron-exon boundary) was split into two separate dark regions so we could estimate the number of dark bases in each type of gene body region (e.g., introns, exons, UTRs). For all analyses, we only included dark regions with ≥ 20 contiguous bases. To identify camouflaged regions, specifically, we used BLAT [26] to identify all genomic regions that were highly similar to any given gene body region that was dark by mapping quality. Any region that was ≥ 98% identical (-minIdentity = 98), and that was considered dark (≥ 90% of reads with MAPQ < 10), was considered a match. We generated .bed files for all three genome builds using this method.
Statistics
We quantified the percentage of each gene body that was dark by summing the total number of dark bases in the gene (i.e., between the 5′UTR to the 3′UTR start and end, respectively) and dividing by the total number of bases in the gene. We similarly calculated the percentage of intronic, exonic (including CDS and UTR), and only CDS exons by dividing the total number of dark bases in each category within the gene by the total number of bases within that category. We performed these calculations for data based on Illumina 100-nucleotide reads for all dark regions combined (Additional file 2: Table S1; Additional file 3: Table S2), dark by depth only (Additional file 15: Table S14; Additional file 16: Table S15), dark by mapping quality (Additional file 17: Table S16; Additional file 18: Table S17), and only camouflaged regions (Additional file 13: Table S12; Additional file 14: Table S13). We performed identical calculations for the samples from Illumina 250-nucleotide read length data, 10x Genomics, ONT, and PacBio (Additional file 4: Table S3; Additional file 5: Table S4; Additional file 6: Table S5; Additional file 7: Table S6; Additional file 8: Table S7; Additional file 9: Table S8; Additional file 10: Table S9; Additional file 11: Table S10 and Additional file 19: Table S18; Additional file 20: Table S19; Additional file 21: Table S20; Additional file 22: Table S21; Additional file 23: Table S22; Additional file 24: Table S23; Additional file 25: Table S24; Additional file 26: Table S25; Additional file 27: Table S26; Additional file 28: Table S27; Additional file 29: Table S28; Additional file 30: Table S29; Additional file 31: Table S30; Additional file 32: Table S31; Additional file 33: Table S32; Additional file 34: Table S33; Additional file 35: Table S34; Additional file 36: Table S35; Additional file 37: Table S36; Additional file 38: Table S37; Additional file 39: Table S38; Additional file 40: Table S39; Additional file 41: Table S40; Additional file 42: Table S41). We identified diseases that were known to be associated with genes that are at least 5% dark CDS by searching for mutations in the Human Gene Mutation Database (HGMD) [30]. For the area under the curve (AUC) comparison, we calculated the AUC for the Illumina 100-nucleotide data and normalized that to 1.0. The AUC is the sum of the percentage of dark nucleotides for each gene. The AUC for each other technology is represented as a proportion of the Illumina 100-nucleotide data.
Coverage plots from gnomAD data were obtained from gnomAD-old.broadinstitute.org [36]. We used the old version because the current version of gnomAD (accessed December 2018) does not allow the user to view median read depths, nor the percentage of samples with greater than a given coverage depth. Sequence pileups in representative samples were generated using the Integrative Genomics Viewer (IGV) [107], where reads with a MAPQ < 10 were filtered, and insertions, deletions, and mismatches were not shown. Karyotype plots showing genomic locations for dark and camouflaged regions were generated using KaryotypeR (v1.6.2) [108] in R (v3.5.1). Bar plots were made using ggplot2 (v3.0.0). Pathway analyses and resulting plots were generated using Metascape (accessed December 2018) [109]. Word clouds were generated at wordclouds.com. Gene schematics were generated using the Gene Structure Display Server (GSDS; v2) [110].
We performed an enrichment analysis to assess whether genes that are ≥ 5% dark CDS are enriched for specific diseases. Because we identified 76 genes that have a known mutation associated with disease, and that are ≥ 5% dark CDS, we randomly selected 76 genes from the known HGMD mutations and measured the number of genes with known mutation associated with each disease. We repeated this process 10,000 times and used the following metric as our enrichment score: − 10*log10(empirical_pvalue), rounded to the nearest whole number.
Screening ADSP for functional CR1 mutations in camouflaged region
After discovering that 26% of the CR1 gene’s CDS is camouflaged, we screened all ADSP samples for rare functional mutations that could play a role in Alzheimer’s disease development and progression by applying our proposed method (Fig. 8). To apply our method, we extracted all reads with a mapping quality (MAPQ) < 10 from each camouflaged region within CR1, and from each of the respective camouflage mate regions, using samtools and the GRCh38 .bed file we generated that identifies all camouflaged regions. An example of camouflaged mate regions in CR1 includes exons 10, 18, and 26, which are identical in the reference genome (Fig. 8). As previously mentioned, CR1 is a special case that is camouflaged by regions duplicated within itself, rather than being camouflaged by a different gene; thus, we knew that any mutations we discovered would be from CR1. Our approach works the same regardless of whether a gene is camouflaged by itself or another gene, but we mention that CR1 is camouflaged by itself, for interest. After extracting reads from each camouflaged region, using the .bed file we provide, we then masked all camouflaged regions within CR1 in the reference genome, except for one from each set of camouflaged mates. For example, between exons 10, 18, and 26, we masked exons 18 and 26 in the reference genome, allowing reads from all three exons to align only to exon 10; without competing camouflaged regions to confuse the aligner, all reads from exons 10, 18, and 26 mapped to exon 10 with high quality. Masking regions of the reference genome simply means to change nucleotides to an unmappable character (usually “N”), to prevent any reads from aligning to that region.
After aligning all reads to a single region within each set of camouflaged regions, we were able to perform standard variant calling using the GATK HaplotypeCaller [25], with two exceptions: (1) instead of treating each camouflaged region as diploid, we increased the ploidy setting in HaplotypeCaller according to the number of copies within a given set of camouflaged regions, and (2) we filtered all reference-based artifacts. Reference-based artifacts arise from aligning reads from two non-identical regions to a single region, causing false-positive mutations. Referring again to our CR1 example, because there are three regions (exons 10, 18, and 26), we set the HaplotypeCaller ploidy to hexaploid. Increasing the ploidy is essential for increased sensitivity, since the number of reads harboring a given variant—which only originate from one of the camouflaged regions—will be overwhelmed by reads from the others, thus preventing the variant caller from identifying the mutation under the assumption that the data are from a diploid region. In other words, if a mutation exists in exon 26, we would expect only approximately 1/6th of reads from exons 10, 18, and 26 to harbor that mutation, rather than approximately 1/2. Because the ADSP is mostly exome data, we limited HaplotypeCaller to CDS exons only. According to the current ADSP phenotype data, one of the samples harboring the CR1 frameshift mutation is a control. The individual has since been officially diagnosed with Alzheimer’s disease, however. We used KING-robust to determine kinship between individuals [51].
To identify reference-based artifacts, all camouflaged CDS regions repeated ≤ 5 times were blatted against the whole genome. DNA sequence from hits with at least 98% sequence identity was locally aligned back to the query sequence. Bio.pairwise2 module in Biopython was used for local alignments using following parameters: match = 1, mismatch = − 3, gapOpen = − 5, gapExtend = − 2. Mismatches or gaps in the resulting aligned sequence were converted into variant positions based on the start position of the query sequence in the genome and the position of the variant within the aligned sequence. Three hundred ninety-one reference-based artifact positions were found using this method. While running our pipeline to rescue variants, any variant called by GATK at one of these positions was filtered out.
Additional files
Supplemental figures. (DOCX 3853 kb)
IlluminaRL100 percent dark genes in hg38. (TXT 401 kb)
IlluminaRL100 dark gene annotations in hg38. (TXT 3152 kb)
IlluminaRL250 percent dark genes in hg38. (TXT 282 kb)
IlluminaRL250 dark gene annotations in hg38. (TXT 1798 kb)
ONT percent dark genes in hg38. (TXT 291 kb)
ONT dark gene annotations in hg38. (TXT 732 kb)
PacBio percent dark genes in hg38. (TXT 145 kb)
PacBio dark gene annotations in hg38. (TXT 759 kb)
10x Genomics percent dark genes in hg38. (TXT 262 kb)
10x Genomics dark gene annotations in hg38. (TXT 1247 kb)
Gene Ontology results for dark genes in IlluminaRL100 for hg38. (CSV 47 kb)
IlluminaRL100 percent camouflaged genes in hg38. (TXT 183 kb)
IlluminaRL100 camouflaged gene annotations in hg38. (TXT 2141 kb)
IlluminaRL100 percent dark-by-depth genes in hg38. (TXT 241 kb)
IlluminaRL100 dark-by-depth gene annotations in hg38. (TXT 647 kb)
IlluminaRL100 percent dark-by-MAPQ genes in hg38. (TXT 253 kb)
IlluminaRL100 dark-by-MAPQ gene annotations in hg38. (TXT 2710 kb)
IlluminaRL250 percent camouflaged genes in hg38. (TXT 142 kb)
IlluminaRL250 camouflaged gene annotations in hg38. (TXT 1279 kb)
IlluminaRL250 percent dark-by-depth genes in hg38. (TXT 147 kb)
IlluminaRL250 dark-by-depth gene annotations in hg38. (TXT 354 kb)
IlluminaRL250 percent dark-by-MAPQ genes in hg38. (TXT 188 kb)
IlluminaRL250 dark-by-MAPQ gene annotations in hg38. (TXT 1574 kb)
ONT percent camouflaged genes in hg38. (TXT 13 kb)
ONT camouflaged gene annotations in hg38. (TXT 122 kb)
ONT percent dark-by-depth genes in hg38. (TXT 278 kb)
ONT dark-by-depth gene annotations in hg38. (TXT 556 kb)
ONT percent dark-by-MAPQ genes in hg38. (TXT 17 kb)
ONT dark-by-MAPQ gene annotations in hg38. (TXT 210 kb)
PacBio percent camouflaged genes in hg38. (TXT 31 kb)
PacBio camouflaged gene annotations in hg38. (TXT 348 kb)
PacBio percent dark-by-depth genes in hg38. (TXT 113 kb)
PacBio dark-by-depth gene annotations in hg38. (TXT 334 kb)
PacBio percent dark-by-MAPQ genes in hg38. (TXT 41 kb)
PacBio dark-by-MAPQ gene annotations in hg38. (TXT 477 kb)
10x Genomics percent camouflaged genes in hg38. (TXT 55 kb)
10x Genomics camouflaged gene annotations in hg38. (TXT 459 kb)
10x Genomics percent dark-by-depth genes in hg38. (TXT 211 kb)
10x Genomics dark-by-depth gene annotations in hg38. (TXT 693 kb)
10x Genomics percent dark-by-MAPQ genes in hg38. (TXT 111 kb)
10x Genomics dark-by-MAPQ gene annotations in hg38. (TXT 730 kb)
Acknowledgements
Biological samples and Associated Phenotypic Data used in primary data analyses were stored at Principal Investigators’ institutions and at the National Cell Repository for Alzheimer’s Disease (NCRAD) at Indiana University funded by NIA. Associated Phenotypic Data used in primary and secondary data analyses were provided by Principal Investigators, the NIA funded Alzheimer’s Disease Centers (ADCs), and the National Alzheimer’s Coordinating Center (NACC) and stored at Principal Investigators’ institutions, NCRAD, and at the National Institute on Aging Alzheimer’s Disease Data Storage Site (NIAGADS) at the University of Pennsylvania, funded by NIA. Contributors to the Genetic Analysis Data included Principal Investigators on projects that were individually funded by NIA, other NIH institutes, private U.S. organizations, or foreign governmental or nongovernmental organizations.
The Alzheimer’s Disease Sequencing Project (ADSP) is comprised of two Alzheimer’s Disease (AD) genetics consortia and three National Human Genome Research Institute (NHGRI) funded Large Scale Sequencing and Analysis Centers (LSAC). The two AD genetics consortia are the Alzheimer’s Disease Genetics Consortium (ADGC) funded by NIA (U01 AG032984), and the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) funded by NIA (R01 AG033193), the National Heart, Lung, and Blood Institute (NHLBI), other National Institute of Health (NIH) institutes and other foreign governmental and non-governmental organizations. The Discovery Phase analysis of sequence data is supported through UF1AG047133 (to Drs. Schellenberg, Farrer, Pericak-Vance, Mayeux, and Haines); U01AG049505 to Dr. Seshadri; U01AG049506 to Dr. Boerwinkle; U01AG049507 to Dr. Wijsman; and U01AG049508 to Dr. Goate, and the Discovery Extension Phase analysis is supported through U01AG052411 to Dr. Goate, U01AG052410 to Dr. Pericak-Vance, and U01 AG052409 to Drs. Seshadri and Fornage. Data generation and harmonization in the Follow-up Phases is supported by U54AG052427 (to Drs. Schellenberg and Wang).
The ADGC cohorts include Adult Changes in Thought (ACT), the Alzheimer’s Disease Centers (ADC), the Chicago Health and Aging Project (CHAP), the Memory and Aging Project (MAP), Mayo Clinic (MAYO), Mayo Parkinson’s Disease controls, University of Miami, the Multi-Institutional Research in Alzheimer’s Genetic Epidemiology Study (MIRAGE), the National Cell Repository for Alzheimer’s Disease (NCRAD), the National Institute on Aging Late Onset Alzheimer’s Disease Family Study (NIA-LOAD), the Religious Orders Study (ROS), the Texas Alzheimer’s Research and Care Consortium (TARC), Vanderbilt University/Case Western Reserve University (VAN/CWRU), the Washington Heights-Inwood Columbia Aging Project (WHICAP) and the Washington University Sequencing Project (WUSP), the Columbia University Hispanic-Estudio Familiar de Influencia Genetica de Alzheimer (EFIGA), the University of Toronto (UT), and Genetic Differences (GD).
The CHARGE cohorts are supported in part by National Heart, Lung, and Blood Institute (NHLBI) infrastructure grant HL105756 (Psaty), RC2HL102419 (Boerwinkle), and the neurology working group is supported by the National Institute on Aging (NIA) R01 grant AG033193. The CHARGE cohorts participating in the ADSP include the following: Austrian Stroke Prevention Study (ASPS), ASPS-Family study, and the Prospective Dementia Registry-Austria (ASPS/PRODEM-Aus), the Atherosclerosis Risk in Communities (ARIC) Study, the Cardiovascular Health Study (CHS), the Erasmus Rucphen Family Study (ERF), the Framingham Heart Study (FHS), and the Rotterdam Study (RS). ASPS is funded by the Austrian Science Fond (FWF) grant number P20545-P05 and P13180 and the Medical University of Graz. The ASPS-Fam is funded by the Austrian Science Fund (FWF) project I904), the EU Joint Programme - Neurodegenerative Disease Research (JPND) in frame of the BRIDGET project (Austria, Ministry of Science) and the Medical University of Graz and the Steiermärkische Krankenanstalten Gesellschaft. PRODEM-Austria is supported by the Austrian Research Promotion agency (FFG) (Project No. 827462) and by the Austrian National Bank (Anniversary Fund, project 15435). ARIC research is carried out as a collaborative study supported by NHLBI contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C). Neurocognitive data in ARIC is collected by U01 2U01HL096812, 2U01HL096814, 2U01HL096899, 2U01HL096902, 2U01HL096917 from the NIH (NHLBI, NINDS, NIA and NIDCD), and with previous brain MRI examinations funded by R01-HL70825 from the NHLBI. CHS research was supported by contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and grants U01HL080295 and U01HL130114 from the NHLBI with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629, R01AG15928, and R01AG20098 from the NIA. FHS research is supported by NHLBI contracts N01-HC-25195 and HHSN268201500001I. This study was also supported by additional grants from the NIA (R01s AG054076, AG049607 and AG033040) and NINDS (R01 NS017950). The ERF study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP grant number 018947 (LSHG-CT-2006-01947) and also received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013)/grant agreement HEALTH-F4-2007-201413 by the European Commission under the program “Quality of Life and Management of the Living Resources” of 5th Framework Programme (no. QLG2-CT-2002-01254). High-throughput analysis of the ERF data was supported by a joint grant from the Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043). The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam; the Netherlands Organization for Health Research and Development (ZonMw); the Research Institute for Diseases in the Elderly (RIDE); the Ministry of Education, Culture and Science; the Ministry for Health, Welfare and Sports; the European Commission (DG XII); and the municipality of Rotterdam. Genetic data sets are also supported by the Netherlands Organization of Scientific Research NWO Investments (175.010.2005.011, 911-03-012); the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC; the Research Institute for Diseases in the Elderly (014-93-015, RIDE2); and the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA), project 050-060-810. All studies are grateful to their participants, faculty, and staff. The content of these manuscripts is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the U.S. Department of Health and Human Services.
The four LSACs are the Human Genome Sequencing Center at the Baylor College of Medicine (U54 HG003273), the Broad Institute Genome Center (U54HG003067), The American Genome Center at the Uniformed Services University of the Health Sciences (U01AG057659), and the Washington University Genome Institute (U54HG003079).
Biological samples and associated phenotypic data used in primary data analyses were stored at Study Investigators institutions and at the National Cell Repository for Alzheimer’s Disease (NCRAD, U24AG021886) at Indiana University funded by NIA. Associated Phenotypic Data used in primary and secondary data analyses were provided by Study Investigators, the NIA funded Alzheimer’s Disease Centers (ADCs), and the National Alzheimer’s Coordinating Center (NACC, U01AG016976) and the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS, U24AG041689) at the University of Pennsylvania, funded by NIA, and at the Database for Genotypes and Phenotypes (dbGaP) funded by NIH. This research was supported in part by the Intramural Research Program of the National Institutes of health, National Library of Medicine. Contributors to the Genetic Analysis Data included Study Investigators on projects that were individually funded by NIA, and other NIH institutes, and by private U.S. organizations, or foreign governmental or nongovernmental organizations.
Funding
This work was supported by the PhRMA Foundation [RSGTMT17 to M.E.]; the Ed and Ethel Moore Alzheimer’s Disease Research Program of Florida Department of Health [8AZ10 and 9AZ08 to M.E., and 6AZ06 to J.F.]; the Muscular Dystrophy Association (M.E.); the National Institutes of Health [NS094137 to J.F., AG047327 to J. F, AG049992 to J.F., NS097261 to R.R., NS097273 to L.P., NS084528 to L.P., NS084974 to L.P., NS099114 to L.P., NS088689 to L.P., NS093865 to L.P.]; Department of Defense [ALSRP AL130125 to L.P.]; Mayo Clinic Foundation (L.P. and J.F.); Mayo Clinic Center for Individualized Medicine (L.P. and J.F.); Amyotrophic Lateral Sclerosis Association (M.E., L.P.); Robert Packard Center for ALS Research at Johns Hopkins (L.P.) Target ALS (L.P.); Association for Frontotemporal Degeneration (L.P.); GHR Foundation (J.F.); and the Mayo Clinic Gerstner Family Career Development Award (J.F.).
Availability of data and materials
The ADSP datasets supporting the conclusions of this article (including the whole-genome and whole-exome data) are available in the National Institute on Aging Genetics of Alzheimer’s Disease Storage (NIAGADS) site and may be requested therein: https://www.niagads.org/adsp/ [50]. Public links to the high-coverage whole-genome data from the 1000 Genomes Project (illumina 250 bp read lengths) are NA20845 (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/NA20845/high_coverage_alignment/), HG01112 (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG01112/high_coverage_alignment/), HG01583
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG01583/high_coverage_alignment/), HG01051
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG01051/high_coverage_alignment/), HG03742
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG03742/high_coverage_alignment/), HG00096
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG00096/high_coverage_alignment/), HG01565
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG01565/high_coverage_alignment/), HG01879
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG01879/high_coverage_alignment/), HG01500
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG01500/high_coverage_alignment/), and HG03006
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG03006/high_coverage_alignment/) [111]. Raw data from the 10x Genomics sample (HG00512) used within this article was downloaded directly from the 10x Genomics website at https://support.10xgenomics.com/de-novo-assembly/datasets/2.1.0/chi [112]. The Cliveome3 data was downloaded from the official Oxford Nanopore Technologies GitHub page: https://github.com/nanoporetech/ONT-HG1/ [113]. The PacBio data used in this publication (HG005) was downloaded from ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/ChineseTrio/HG005_NA24631_son/MtSinai_PacBio/PacBio_minimap2_bam/HG005_PacBio_GRCh37.bam [114].
All scripts are available at https://github.com/mebbert/Dark_and_Camouflaged_genes [115, 116].
Abbreviations
- ADSP
Alzheimer’s Disease Sequencing Project
- ALS
Amyotrophic lateral sclerosis
- CDS
Coding sequence
- FTD
Frontotemporal dementia
- MAPQ
Mapping quality
- ONT
Oxford Nanopore Technologies
- PacBio
Pacific Biosciences
Authors’ contributions
ME, LP, and JF developed and designed the study and wrote the manuscript. ME and TJ performed all analyses. JR, SY, NT, YA, VB, EL, DK, PC, LP, PR, JK, MC, OA, and RR contributed important intellectual ideas and feedback. SY, YA, NT, OA, and RR helped obtain the data. KW and JS performed the experiments. EL, DK, and PC provided the samples. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The Mayo Clinic Institutional Review Board (IRB) approved all procedures for this study, and we followed all appropriate protocols. All participants included in this study provided written informed consent, and all experimental methods comply with the Helsinki Declaration.
Consent for publication
All participants were properly consented for this study.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mark T. W. Ebbert, Email: ebbert.mark@mayo.edu
Tanner D. Jensen, Email: jensen.tanner@mayo.edu
Karen Jansen-West, Email: jansen.karen@mayo.edu.
Jonathon P. Sens, Email: sens.jonathon@mayo.edu
Joseph S. Reddy, Email: reddy.joseph@mayo.edu
Perry G. Ridge, Email: perry.ridge@byu.edu
John S. K. Kauwe, Email: kauwe@byu.edu
Veronique Belzil, Email: belzil.veronique@mayo.edu.
Luc Pregent, Email: pregent.luc@mayo.edu.
Minerva M. Carrasquillo, Email: carrasquillo.minerva@mayo.edu
Dirk Keene, Email: cdkeene@uw.edu.
Eric Larson, Email: larson.e@ghc.org.
Paul Crane, Email: pcrane@uw.edu.
Yan W. Asmann, Email: asmann.yan@mayo.edu
Nilufer Ertekin-Taner, Email: taner.nilufer@mayo.edu.
Steven G. Younkin, Email: younkin.steven@mayo.edu
Owen A. Ross, Email: ross.owen@mayo.edu
Rosa Rademakers, Email: rademakers.rosa@mayo.edu.
Leonard Petrucelli, Email: petrucelli.leonard@mayo.edu.
John D. Fryer, Email: fryer.john@mayo.edu
References
- 1.Ebbert MTW, Farrugia SL, Sens JP, Jansen-West K, Gendron TF, Prudencio M, et al. Long-read sequencing across the C9orf72 “GGGGCC” repeat expansion: implications for clinical use and genetic discovery efforts in human disease. Mol Neurodegener. 2018;13:46. doi: 10.1186/s13024-018-0274-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng-Bradley X, Streeter I, Fairley S, Richardson D, Clarke L, Flicek P, et al. Alignment of 1000 Genomes Project reads to reference assembly GRCh38. Gigascience. 2017;6:1–8. doi: 10.1093/gigascience/gix038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Callaway E. Human brain shaped by duplicate genes. Nature. 2012. 10.1038/nature.2012.10584.
- 4.Charrier C, Joshi K, Coutinho-Budd J, Kim J-E, Lambert N, de Marchena J, et al. Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell. 2012;149:923–935. doi: 10.1016/j.cell.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennis MY, Nuttle X, Sudmant PH, Antonacci F, Graves TA, Nefedov M, et al. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell. 2012;149:912–922. doi: 10.1016/j.cell.2012.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karlin S, Brocchieri L. Heat shock protein 60 sequence comparisons: duplications, lateral transfer, and mitochondrial evolution. Proc Natl Acad Sci U S A. 2000;97:11348–11353. doi: 10.1073/pnas.97.21.11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin Y, Cheng Y, Jin J, Jin X, Jiang H, Yan H, et al. Genome duplication and gene loss affect the evolution of heat shock transcription factor genes in legumes. PLoS One. 2014;9:e102825. doi: 10.1371/journal.pone.0102825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen AD, Gotelli NJ, Cahan SH. The evolution of heat shock protein sequences, cis-regulatory elements, and expression profiles in the eusocial Hymenoptera. BMC Evol Biol. 2016;16:15. doi: 10.1186/s12862-015-0573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sørensen JG, Kristensen TN, Loeschcke V. The evolutionary and ecological role of heat shock proteins. Ecol Lett. 2003;6:1025–1037. doi: 10.1046/j.1461-0248.2003.00528.x. [DOI] [Google Scholar]
- 10.Robert C, Watson M. Errors in RNA-Seq quantification affect genes of relevance to human disease. Genome Biol. 2015;16:177. doi: 10.1186/s13059-015-0734-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013.
- 14.Lambert J-C, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 15.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert J-C, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kauwe JSK, Cruchaga C, Karch CM, Sadler B, Lee M, Mayo K, et al. Fine mapping of genetic variants in BIN1, CLU, CR1 and PICALM for association with cerebrospinal fluid biomarkers for Alzheimer’s disease. PLoS One. 2011;6:e15918. doi: 10.1371/journal.pone.0015918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ridge PG, Hoyt KB, Boehme K, Mukherjee S, Crane PK, Haines JL, et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging. 2016;41:200.e13–200.e20. doi: 10.1016/j.neurobiolaging.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebbert MTW, Ridge PG, Wilson AR, Sharp AR, Bailey M, Norton MC, et al. Population-based analysis of Alzheimer’s disease risk alleles implicates genetic interactions. Biol Psychiatry. 2014;75:732–737. doi: 10.1016/j.biopsych.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ridge PG, Ebbert MTW, Kauwe JSK. Genetics of Alzheimer’s disease. Biomed Res Int. 2013;2013:254954. doi: 10.1155/2013/254954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahmoudi Rachid, Feldman Sarah, Kisserli Aymric, Duret Valérie, Tabary Thierry, Bertholon Laurie-Anne, Badr Sarah, Nonnonhou Vignon, Cesar Aude, Neuraz Antoine, Novella Jean, Cohen Jacques. Inherited and Acquired Decrease in Complement Receptor 1 (CR1) Density on Red Blood Cells Associated with High Levels of Soluble CR1 in Alzheimer’s Disease. International Journal of Molecular Sciences. 2018;19(8):2175. doi: 10.3390/ijms19082175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naj AC, Jun G, Beecham GW, Wang L-S, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. 2018;46:D754–D761. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.1000 Genomes Project Consortium. Abecasis GR, Auton A, Brooks LD, MA DP, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frankish Adam, Diekhans Mark, Ferreira Anne-Maud, Johnson Rory, Jungreis Irwin, Loveland Jane, Mudge Jonathan M, Sisu Cristina, Wright James, Armstrong Joel, Barnes If, Berry Andrew, Bignell Alexandra, Carbonell Sala Silvia, Chrast Jacqueline, Cunningham Fiona, Di Domenico Tomás, Donaldson Sarah, Fiddes Ian T, García Girón Carlos, Gonzalez Jose Manuel, Grego Tiago, Hardy Matthew, Hourlier Thibaut, Hunt Toby, Izuogu Osagie G, Lagarde Julien, Martin Fergal J, Martínez Laura, Mohanan Shamika, Muir Paul, Navarro Fabio C P, Parker Anne, Pei Baikang, Pozo Fernando, Ruffier Magali, Schmitt Bianca M, Stapleton Eloise, Suner Marie-Marthe, Sycheva Irina, Uszczynska-Ratajczak Barbara, Xu Jinuri, Yates Andrew, Zerbino Daniel, Zhang Yan, Aken Bronwen, Choudhary Jyoti S, Gerstein Mark, Guigó Roderic, Hubbard Tim J P, Kellis Manolis, Paten Benedict, Reymond Alexandre, Tress Michael L, Flicek Paul. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Research. 2018;47(D1):D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bader GD, Hogue CWV. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, et al. A novel approach to high-quality postmortem tissue procurement: the GTEx Project. Biopreserv Biobank. 2015;13:311–319. doi: 10.1089/bio.2015.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 31.Seminary ER, Sison SL, Ebert AD. Modeling protein aggregation and the heat shock response in ALS iPSC-derived motor neurons. Front Neurosci. 2018;12:86. doi: 10.3389/fnins.2018.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalmar B, Lu C-H, Greensmith L. The role of heat shock proteins in amyotrophic lateral sclerosis: the therapeutic potential of Arimoclomol. Pharmacol Ther. 2014;141:40–54. doi: 10.1016/j.pharmthera.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 33.Corcia P, Camu W, Halimi JM, Vourc’h P, Antar C, Vedrine S, et al. SMN1 gene, but not SMN2, is a risk factor for sporadic ALS. Neurology. 2006;67:1147–1150. doi: 10.1212/01.wnl.0000233830.85206.1e. [DOI] [PubMed] [Google Scholar]
- 34.Corcia P, Camu W, Praline J, Gordon PH, Vourch P, Andres C. The importance of the SMN genes in the genetics of sporadic ALS. Amyotroph Lateral Scler. 2009;10:436–440. doi: 10.3109/17482960902759162. [DOI] [PubMed] [Google Scholar]
- 35.Blauw HM, Barnes CP, van Vught PWJ, van Rheenen W, Verheul M, Cuppen E, et al. SMN1 gene duplications are associated with sporadic ALS. Neurology. 2012;78:776–780. doi: 10.1212/WNL.0b013e318249f697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, et al. Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1992;89:10016–10020. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogers J, Li R, Mastroeni D, Grover A, Leonard B, Ahern G, et al. Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes. Neurobiol Aging. 2006;27:1733–1739. doi: 10.1016/j.neurobiolaging.2005.09.043. [DOI] [PubMed] [Google Scholar]
- 39.Kisserli A, Tabary T, Cohen JHM, Duret V, Mahmoudi R. High-resolution melting PCR for complement receptor 1 length polymorphism genotyping: an innovative tool for Alzheimer’s disease gene susceptibility assessment. J Vis Exp. 2017. 10.3791/56012. [DOI] [PMC free article] [PubMed]
- 40.Fonseca MI, Chu S, Pierce AL, Brubaker WD, Hauhart RE, Mastroeni D, et al. Analysis of the putative role of CR1 in alzheimer’s disease: genetic association, expression and function. PLoS One. 2016;11:e0149792. doi: 10.1371/journal.pone.0149792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brouwers N, Van Cauwenberghe C, Engelborghs S, Lambert JC, Bettens K, Le Bastard N, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry. 2012;17:223–233. doi: 10.1038/mp.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kucukkilic Ezgi, Brookes Keeley, Barber Imelda, Guetta-Baranes Tamar, Morgan Kevin, Hollox Edward J. Complement receptor 1 gene (CR1) intragenic duplication and risk of Alzheimer’s disease. Human Genetics. 2018;137(4):305–314. doi: 10.1007/s00439-018-1883-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crane A, Brubaker WD, Johansson JU, Trigunaite A, Ceballos J, Bradt B, et al. Peripheral complement interactions with amyloid β peptide in Alzheimer’s disease: 2. Relationship to amyloid β immunotherapy. Alzheimers Dement. 2018;14:243–252. doi: 10.1016/j.jalz.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu Rev Med. 1996;47:387–400. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- 45.Roses AD, Saunders AM. APOE is a major susceptibility gene for Alzheimer’s disease. Curr Opin Biotechnol. 1994;5:663–667. doi: 10.1016/0958-1669(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 46.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kato M, Saitoh S, Kamei A, Shiraishi H, Ueda Y, Akasaka M, et al. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome) Am J Hum Genet. 2007;81:361–366. doi: 10.1086/518903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Partington MW, Turner G, Boyle J, Gécz J. Three new families with X-linked mental retardation caused by the 428-451dup(24bp) mutation in ARX. Clin Genet. 2004;66:39–45. doi: 10.1111/j.0009-9163.2004.00268.x. [DOI] [PubMed] [Google Scholar]
- 49.Zweier C, Sticht H, Aydin-Yaylagül I, Campbell CE, Rauch A. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am J Hum Genet. 2007;80:510–517. doi: 10.1086/511993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naj AC, Lin H, Vardarajan BN, White S, Lancour D, Ma Y, et al. Quality control and integration of genotypes from two calling pipelines for whole genome sequence data in the Alzheimer’s disease sequencing project. Genomics. 2018. 10.1016/j.ygeno.2018.05.004. [DOI] [PMC free article] [PubMed]
- 51.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W-M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Payne A, Holmes N, Rakyan V, Loose M. Whale watching with BulkVis: a graphical viewer for Oxford Nanopore bulk fast5 files. BioRxiv. 2018. 10.1101/312256. [DOI] [PMC free article] [PubMed]
- 53.Payne A, Holmes N, Rakyan V, Loose M. BulkVis: a graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics. 2018. 10.1093/bioinformatics/bty841. [DOI] [PMC free article] [PubMed]
- 54.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Orr HT, Chung MY, Banfi S, Kwiatkowski TJ, Servadio A, Beaudet AL, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- 56.Lindblad K, Savontaus ML, Stevanin G, Holmberg M, Digre K, Zander C, et al. An expanded CAG repeat sequence in spinocerebellar ataxia type 7. Genome Res. 1996;6:965–971. doi: 10.1101/gr.6.10.965. [DOI] [PubMed] [Google Scholar]
- 57.Squitieri F, Andrew SE, Goldberg YP, Kremer B, Spence N, Zeisler J, et al. DNA haplotype analysis of Huntington disease reveals clues to the origins and mechanisms of CAG expansion and reasons for geographic variations of prevalence. Hum Mol Genet. 1994;3:2103–2114. doi: 10.1093/hmg/3.12.2103. [DOI] [PubMed] [Google Scholar]
- 58.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 61.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 62.Kashima T, Rao N, David CJ, Manley JL. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum Mol Genet. 2007;16:3149–3159. doi: 10.1093/hmg/ddm276. [DOI] [PubMed] [Google Scholar]
- 63.Ward AJ, Cooper TA. The pathobiology of splicing. J Pathol. 2010;220:152–163. doi: 10.1002/path.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cartegni L, Hastings ML, Calarco JA, de Stanchina E, Krainer AR. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet. 2006;78:63–77. doi: 10.1086/498853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 66.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 67.Takahara K, Schwarze U, Imamura Y, Hoffman GG, Toriello H, Smith LT, et al. Order of intron removal influences multiple splice outcomes, including a two-exon skip, in a COL5A1 acceptor-site mutation that results in abnormal pro-alpha1(V) N-propeptides and Ehlers-Danlos syndrome type I. Am J Hum Genet. 2002;71:451–465. doi: 10.1086/342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Habara Y, Takeshima Y, Awano H, Okizuka Y, Zhang Z, Saiki K, et al. In vitro splicing analysis showed that availability of a cryptic splice site is not a determinant for alternative splicing patterns caused by +1G-->A mutations in introns of the dystrophin gene. J Med Genet. 2009;46:542–547. doi: 10.1136/jmg.2008.061259. [DOI] [PubMed] [Google Scholar]
- 69.Anna A, Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet. 2018;59:253–268. doi: 10.1007/s13353-018-0444-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zeng L, Liu W, Feng W, Wang X, Dang H, Gao L, et al. A novel donor splice-site mutation of major intrinsic protein gene associated with congenital cataract in a Chinese family. Mol Vis. 2013;19:2244–2249. [PMC free article] [PubMed] [Google Scholar]
- 71.Hori T, Fukao T, Murase K, Sakaguchi N, Harding CO, Kondo N. Molecular basis of two-exon skipping (exons 12 and 13) by c.1248+5g>a in OXCT1 gene: study on intermediates of OXCT1 transcripts in fibroblasts. Hum Mutat. 2013;34:473–480. doi: 10.1002/humu.22258. [DOI] [PubMed] [Google Scholar]
- 72.Känsäkoski J, Jääskeläinen J, Jääskeläinen T, Tommiska J, Saarinen L, Lehtonen R, et al. Complete androgen insensitivity syndrome caused by a deep intronic pseudoexon-activating mutation in the androgen receptor gene. Sci Rep. 2016;6:32819. doi: 10.1038/srep32819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fang LJ, Simard MJ, Vidaud D, Assouline B, Lemieux B, Vidaud M, et al. A novel mutation in the neurofibromatosis type 1 (NF1) gene promotes skipping of two exons by preventing exon definition. J Mol Biol. 2001;307:1261–1270. doi: 10.1006/jmbi.2001.4561. [DOI] [PubMed] [Google Scholar]
- 74.Symoens S, Malfait F, Vlummens P, Hermanns-Lê T, Syx D, De Paepe A. A novel splice variant in the N-propeptide of COL5A1 causes an EDS phenotype with severe kyphoscoliosis and eye involvement. PLoS One. 2011;6:e20121. doi: 10.1371/journal.pone.0020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sanz DJ, Hollywood JA, Scallan MF, Harrison PT. Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS One. 2017;12:e0184009. doi: 10.1371/journal.pone.0184009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramalho AS, Beck S, Penque D, Gonska T, Seydewitz HH, Mall M, et al. Transcript analysis of the cystic fibrosis splicing mutation 1525-1G>A shows use of multiple alternative splicing sites and suggests a putative role of exonic splicing enhancers. J Med Genet. 2003;40:e88. doi: 10.1136/jmg.40.7.e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ridge PG, Karch CM, Hsu S, Arano I, Teerlink CC, Ebbert MTW, et al. Linkage, whole genome sequence, and biological data implicate variants in RAB10 in Alzheimer’s disease resilience. Genome Med. 2017;9:100. doi: 10.1186/s13073-017-0486-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lettice LA, Heaney SJH, Purdie LA, Li L, de Beer P, Oostra BA, et al. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. 2003;12:1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- 79.Emison ES, McCallion AS, Kashuk CS, Bush RT, Grice E, Lin S, et al. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. 2005;434:857–863. doi: 10.1038/nature03467. [DOI] [PubMed] [Google Scholar]
- 80.de Vooght KMK, van Wijk R, van Solinge WW. Management of gene promoter mutations in molecular diagnostics. Clin Chem. 2009;55:698–708. doi: 10.1373/clinchem.2008.120931. [DOI] [PubMed] [Google Scholar]
- 81.Short PJ, McRae JF, Gallone G, Sifrim A, Won H, Geschwind DH, et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature. 2018;555:611–616. doi: 10.1038/nature25983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.De Gobbi M, Viprakasit V, Hughes JR, Fisher C, Buckle VJ, Ayyub H, et al. A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science. 2006;312:1215–1217. doi: 10.1126/science.1126431. [DOI] [PubMed] [Google Scholar]
- 83.Grant SF, Reid DM, Blake G, Herd R, Fogelman I, Ralston SH. Reduced bone density and osteoporosis associated with a polymorphic Sp1 binding site in the collagen type I alpha 1 gene. Nat Genet. 1996;14:203–205. doi: 10.1038/ng1096-203. [DOI] [PubMed] [Google Scholar]
- 84.Benko S, Fantes JA, Amiel J, Kleinjan D-J, Thomas S, Ramsay J, et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet. 2009;41:359–364. doi: 10.1038/ng.329. [DOI] [PubMed] [Google Scholar]
- 85.Jeong Y, Leskow FC, El-Jaick K, Roessler E, Muenke M, Yocum A, et al. Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat Genet. 2008;40:1348–1353. doi: 10.1038/ng.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rahimov F, Marazita ML, Visel A, Cooper ME, Hitchler MJ, Rubini M, et al. Disruption of an AP-2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat Genet. 2008;40:1341–1347. doi: 10.1038/ng.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14:723–730. doi: 10.1038/nm1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen W-L, Lin J-W, Huang H-J, Wang S-M, Su M-T, Lee-Chen G-J, et al. SCA8 mRNA expression suggests an antisense regulation of KLHL1 and correlates to SCA8 pathology. Brain Res. 2008;1233:176–184. doi: 10.1016/j.brainres.2008.07.096. [DOI] [PubMed] [Google Scholar]
- 89.Kogo R, Shimamura T, Mimori K, Kawahara K, Imoto S, Sudo T, et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011;71:6320–6326. doi: 10.1158/0008-5472.CAN-11-1021. [DOI] [PubMed] [Google Scholar]
- 90.Ishii N, Ozaki K, Sato H, Mizuno H, Saito S, Takahashi A, et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet. 2006;51:1087–1099. doi: 10.1007/s10038-006-0070-9. [DOI] [PubMed] [Google Scholar]
- 91.Khalil AM, Faghihi MA, Modarresi F, Brothers SP, Wahlestedt C. A novel RNA transcript with antiapoptotic function is silenced in fragile X syndrome. PLoS One. 2008;3:e1486. doi: 10.1371/journal.pone.0001486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chubb JE, Bradshaw NJ, Soares DC, Porteous DJ, Millar JK. The DISC locus in psychiatric illness. Mol Psychiatry. 2008;13:36–64. doi: 10.1038/sj.mp.4002106. [DOI] [PubMed] [Google Scholar]
- 93.Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925–938. doi: 10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Matouk IJ, DeGroot N, Mezan S, Ayesh S, Abu-lail R, Hochberg A, et al. The H19 non-coding RNA is essential for human tumor growth. PLoS One. 2007;2:e845. doi: 10.1371/journal.pone.0000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lin R, Maeda S, Liu C, Karin M, Edgington TS. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene. 2007;26:851–858. doi: 10.1038/sj.onc.1209846. [DOI] [PubMed] [Google Scholar]
- 96.Yang Z, Zhou L, Wu L-M, Lai M-C, Xie H-Y, Zhang F, et al. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann Surg Oncol. 2011;18:1243–1250. doi: 10.1245/s10434-011-1581-y. [DOI] [PubMed] [Google Scholar]
- 97.Zorzetto M, Datturi F, Divizia L, Pistono C, Campo I, De Silvestri A, et al. Complement C4A and C4B gene copy number study in Alzheimer’s disease patients. Curr Alzheimer Res. 2017;14:303–308. doi: 10.2174/1567205013666161013091934. [DOI] [PubMed] [Google Scholar]
- 98.Trouw LA, Nielsen HM, Minthon L, Londos E, Landberg G, Veerhuis R, et al. C4b-binding protein in Alzheimer’s disease: binding to Abeta1-42 and to dead cells. Mol Immunol. 2008;45:3649–3660. doi: 10.1016/j.molimm.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 99.Ridge PG, Mukherjee S, Crane PK, Kauwe JSK. Alzheimer’s disease genetics consortium. Alzheimer’s disease: analyzing the missing heritability. PLoS One. 2013;8:e79771. doi: 10.1371/journal.pone.0079771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ebbert MTW, Ridge PG, Kauwe JSK. Bridging the gap between statistical and biological epistasis in Alzheimer’s disease. Biomed Res Int. 2015;2015:870123. doi: 10.1155/2015/870123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ebbert Mark T.W., Boehme Kevin L., Wadsworth Mark E., Staley Lyndsay A., Mukherjee Shubhabrata, Crane Paul K., Ridge Perry G., Kauwe John S.K. Interaction between variants in CLU and MS4A4E modulates Alzheimer's disease risk. Alzheimer's & Dementia. 2016;12(2):121–129. doi: 10.1016/j.jalz.2015.08.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Weirather JL, de Cesare M, Wang Y, Piazza P, Sebastiano V, Wang X-J, et al. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Res. 2017;6:100. doi: 10.12688/f1000research.10571.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ardui S, Ameur A, Vermeesch JR, Hestand MS. Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res. 2018;46:2159–2168. doi: 10.1093/nar/gky066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zook JM, Catoe D, McDaniel J, Vang L, Spies N, Sidow A, et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci Data. 2016;3:160025. doi: 10.1038/sdata.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinformatics. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gel B, Serra E. karyoploteR: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics. 2017;33:3088–3090. doi: 10.1093/bioinformatics/btx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, et al. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe. 2015;18:723–735. doi: 10.1016/j.chom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hu B, Jin J, Guo A-Y, Zhang H, Luo J, Gao G. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 2015;31:1296–1297. doi: 10.1093/bioinformatics/btu817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.1000 Genomes Project Consortium. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.10x Genomics. chi - Datasets - De Novo Assembly - Official 10x Genomics Support. chi - Datasets - De Novo Assembly - Official 10x Genomics Support. 2018. https://support.10xgenomics.com/de-novo-assembly/datasets/2.1.0/chi. Accessed 25 Apr 2019.
- 113.Oxford Nanopore Technologies. Cliveome ONTHG1 data release. Cliveome ONTHG1 data release. 2019. https://github.com/nanoporetech/ONT-HG1. Accessed 26 Apr 2019.
- 114.Wang Y-C, Olson ND, Deikus G, Shah H, Wenger AM, Trow J, et al. High-coverage, long-read sequencing of Han Chinese trio reference samples. BioRxiv. 2019. 10.1101/562611. [DOI] [PMC free article] [PubMed]
- 115.Ebbert MTW, Jensen TD, Petrucelli L, Fryer JD. Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. sorce code. Github Repository. 2019. https://github.com/mebbert/Dark_and_Camouflaged_genes. Accessed 26 Apr 2019. [DOI] [PMC free article] [PubMed]
- 116.Ebbert MTW, Jensen TD, Petrucelli L, Fryer JD. Dark and camouflaged gene scripts. Zenodo. 2019. 10.5281/zenodo.2652499.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figures. (DOCX 3853 kb)
IlluminaRL100 percent dark genes in hg38. (TXT 401 kb)
IlluminaRL100 dark gene annotations in hg38. (TXT 3152 kb)
IlluminaRL250 percent dark genes in hg38. (TXT 282 kb)
IlluminaRL250 dark gene annotations in hg38. (TXT 1798 kb)
ONT percent dark genes in hg38. (TXT 291 kb)
ONT dark gene annotations in hg38. (TXT 732 kb)
PacBio percent dark genes in hg38. (TXT 145 kb)
PacBio dark gene annotations in hg38. (TXT 759 kb)
10x Genomics percent dark genes in hg38. (TXT 262 kb)
10x Genomics dark gene annotations in hg38. (TXT 1247 kb)
Gene Ontology results for dark genes in IlluminaRL100 for hg38. (CSV 47 kb)
IlluminaRL100 percent camouflaged genes in hg38. (TXT 183 kb)
IlluminaRL100 camouflaged gene annotations in hg38. (TXT 2141 kb)
IlluminaRL100 percent dark-by-depth genes in hg38. (TXT 241 kb)
IlluminaRL100 dark-by-depth gene annotations in hg38. (TXT 647 kb)
IlluminaRL100 percent dark-by-MAPQ genes in hg38. (TXT 253 kb)
IlluminaRL100 dark-by-MAPQ gene annotations in hg38. (TXT 2710 kb)
IlluminaRL250 percent camouflaged genes in hg38. (TXT 142 kb)
IlluminaRL250 camouflaged gene annotations in hg38. (TXT 1279 kb)
IlluminaRL250 percent dark-by-depth genes in hg38. (TXT 147 kb)
IlluminaRL250 dark-by-depth gene annotations in hg38. (TXT 354 kb)
IlluminaRL250 percent dark-by-MAPQ genes in hg38. (TXT 188 kb)
IlluminaRL250 dark-by-MAPQ gene annotations in hg38. (TXT 1574 kb)
ONT percent camouflaged genes in hg38. (TXT 13 kb)
ONT camouflaged gene annotations in hg38. (TXT 122 kb)
ONT percent dark-by-depth genes in hg38. (TXT 278 kb)
ONT dark-by-depth gene annotations in hg38. (TXT 556 kb)
ONT percent dark-by-MAPQ genes in hg38. (TXT 17 kb)
ONT dark-by-MAPQ gene annotations in hg38. (TXT 210 kb)
PacBio percent camouflaged genes in hg38. (TXT 31 kb)
PacBio camouflaged gene annotations in hg38. (TXT 348 kb)
PacBio percent dark-by-depth genes in hg38. (TXT 113 kb)
PacBio dark-by-depth gene annotations in hg38. (TXT 334 kb)
PacBio percent dark-by-MAPQ genes in hg38. (TXT 41 kb)
PacBio dark-by-MAPQ gene annotations in hg38. (TXT 477 kb)
10x Genomics percent camouflaged genes in hg38. (TXT 55 kb)
10x Genomics camouflaged gene annotations in hg38. (TXT 459 kb)
10x Genomics percent dark-by-depth genes in hg38. (TXT 211 kb)
10x Genomics dark-by-depth gene annotations in hg38. (TXT 693 kb)
10x Genomics percent dark-by-MAPQ genes in hg38. (TXT 111 kb)
10x Genomics dark-by-MAPQ gene annotations in hg38. (TXT 730 kb)
Data Availability Statement
The ADSP datasets supporting the conclusions of this article (including the whole-genome and whole-exome data) are available in the National Institute on Aging Genetics of Alzheimer’s Disease Storage (NIAGADS) site and may be requested therein: https://www.niagads.org/adsp/ [50]. Public links to the high-coverage whole-genome data from the 1000 Genomes Project (illumina 250 bp read lengths) are NA20845 (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/NA20845/high_coverage_alignment/), HG01112 (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG01112/high_coverage_alignment/), HG01583
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG01583/high_coverage_alignment/), HG01051
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG01051/high_coverage_alignment/), HG03742
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG03742/high_coverage_alignment/), HG00096
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG00096/high_coverage_alignment/), HG01565
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG01565/high_coverage_alignment/), HG01879
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG01879/high_coverage_alignment/), HG01500
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG01500/high_coverage_alignment/), and HG03006
(ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/HG03006/high_coverage_alignment/) [111]. Raw data from the 10x Genomics sample (HG00512) used within this article was downloaded directly from the 10x Genomics website at https://support.10xgenomics.com/de-novo-assembly/datasets/2.1.0/chi [112]. The Cliveome3 data was downloaded from the official Oxford Nanopore Technologies GitHub page: https://github.com/nanoporetech/ONT-HG1/ [113]. The PacBio data used in this publication (HG005) was downloaded from ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/ChineseTrio/HG005_NA24631_son/MtSinai_PacBio/PacBio_minimap2_bam/HG005_PacBio_GRCh37.bam [114].
All scripts are available at https://github.com/mebbert/Dark_and_Camouflaged_genes [115, 116].