

Summary
The mechanisms underlying the chronic, progressive airways inflammation, remodelling and alveolar structural damage characteristic of human chronic obstructive pulmonary disease (COPD) remain unclear. In the present study, we address the hypothesis that these changes are at least in part mediated by respiratory epithelial alarmin (IL‐33)‐induced production of autoantibodies against airways epithelial cells. Mice immunized with homologous, syngeneic lung tissue lysate along with IL‐33 administered directly to the respiratory tract or systemically produced IgG autoantibodies binding predominantly to their own alveolar type II epithelial cells, along with increased percentages of Tfh cells and B2 B‐cells in their local, mediastinal lymph nodes. Consistent with its specificity for respiratory epithelial cells, this autoimmune inflammation was confined principally to the lung and not other organs such as the liver and kidney. Furthermore, the serum autoantibodies produced by the mice bound not only to murine, but also to human alveolar type II epithelial cells, suggesting specificity for common, cross‐species determinants. Finally, concentrations of antibodies against both human and murine alveolar epithelial cells were significantly elevated in the serum of patients with COPD compared with those of control subjects. These data are consistent with the hypothesis that IL‐33 contributes to the chronic, progressive airways obstruction, inflammation and alveolar destruction characteristic of phenotypes of COPD/emphysema through induction of autoantibodies against lung tissue, and particularly alveolar type II epithelial cells.
Keywords: alveolar type II epithelial cell, autoimmune, chronic obstructive pulmonary disease, emphysema, IL‐33
Abbreviations
- B2 B‐cells
most common type of B‐cell and mainly located in lymphoid follicles of secondary lymphoid organs
- BAL fluid
bronchoalveolar lavage fluid
- COPD
chronic obstructive pulmonary disease
- PBS
phosphate‐buffered saline
- Tfh cells
follicular B helper T‐cells
Introduction
Chronic obstructive pulmonary disease (COPD) now ranks as the third leading cause of death worldwide. It is a potentially devastating lung disease characterized by progressive, irreversible airways obstruction reflecting inflammation and remodelling and, in some patients, impaired gas transfer secondary to pulmonary alveolar destruction.1, 2, 3, 4, 5
Inhalation of environmental smoke, typically from cigarette smoking or open fires, has been recognized for some decades as a key risk factor for the development of COPD, although only approximately 25% of smokers by the age of 80 years develop the disease,6 while only approximately half of all patients diagnosed with COPD have a clear history of smoking or smoke exposure, suggesting alternative environmental triggers of the disease.7 Another phenomenon that remains largely unexplained is the fact that pulmonary inflammation and impairment of lung function may persist and progress even after cessation of smoke exposure,8 suggesting environmental initiation of a more persistent process independent of the original trigger.
Recently, there has been increasing appreciation and acceptance of a possible role for autoimmune disease in the development and progression of COPD.9, 10 Several relatively recent studies have shown that patients with COPD manifest serum antibodies directed against structural components of the airways, including structural proteins and airways structural cells such as epithelial, endothelial and smooth muscle cells, along with evidence of B cellular proliferation in local lymphoid follicles,9, 10, 11, 12, 13, 14, 15, 16, 17, 18 although few have been clearly implicated in the progression of COPD, many are not detectable in every patient diagnosed with the disease and not all are clearly disease‐specific.9, 19 In parallel, other researchers have shown that immune responses to structural airways components can be induced in animals and may be associated with lung function changes reminiscent of human COPD, although not all of these responses are ‘autoimmune’.15, 20, 21, 22
It has also been shown that COPD is characterized by elevated production of the alarmin cytokine IL‐33, particularly with continued cigarette smoke exposure, to a degree that correlates with the rate of progression of the disease.5, 23 IL‐33, mainly produced by airways epithelial cells, fibroblasts and endothelial cells as well as activated immune cells,24 plays important roles in a range of autoimmune diseases.25, 26, 27, 28
In an attempt to link these findings, and in particular to provide a possible explanation for why lung function impairment and particularly alveolar damage may persist and progress despite apparent removal of the provoking stimulus, we hypothesized that the respiratory epithelial alarmin IL‐33, known to be produced in response to inhaled environmental insults such as cigarette smoke, triggers a humoral autoimmune response directed against alveolar epithelial cells. Our aim was to explore whether antibodies can be induced in mice immunized with autologous (syngeneic) normal lung tissue while exposed to exogenous IL‐33 delivered topically to the airways or systemically, and to examine their specificity in mice as well as their cross‐specificity for human alveolar epithelial cells. Finally, we sought evidence of spontaneous production of these antibodies in human patients diagnosed with COPD.
Materials and methods
Mice
Eight‐week‐old C57BL/6 mice were purchased from Vital River Laboratory (Beijing, China) and kept in a specific pathogen‐free facility in the Department of Laboratory Animal Sciences (Capital Medical University, Beijing, China). Experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of Capital Medical University, Beijing, China.
Preparation of lung tissue lysate
C57BL/6 mice were exsanguinated by extirpating/removing the eyeballs following death by overdosage of chloral hydrate anaesthetic. Mice were then intubated by tracheotomy, and the airways washed twice with 0·8‐ml aliquots of sterile phosphate‐buffered saline (PBS) to remove microbiota and cells. After opening the thoracic cavities, the lungs were exposed and washed using sterile PBS introduced through the right ventricle to remove the blood from the pulmonary circulation. The entire lungs were then excised, weighed then homogenized in PBS containing 1% Triton X‐100 and protease inhibitors (a protease inhibitor cocktail tablet; Roche Diagnostics GmbH, Mannheim, Germany). A Pierce BCA protein assay kit (Thermo Scientific, Waltham, MA, USA) was used to quantify protein in the supernatants after centrifugation to remove debris. The lung tissue lysate was finally stored at −80° at documented concentrations pending its use to immunize autologous mice.
Experimental autoimmune‐mediated lung inflammation
To establish animal models employing local exposure, the murine lung tissue lysate (prepared as above, 20 μg total protein in 50 μl PBS) with or without recombinant murine IL‐33 (rmIL‐33, 25 ng in 50 μl saline; R&D Systems, Minneapolis, MN, USA) was administered intranasally to autologous C57BL/6 mice daily from days 1 to 6, then once every other day from days 8 to 25 (Fig. 1a). On day 26, the mice were killed and samples collected for analysis as described below. For experiments employing systemic exposure, C57BL/6 mice were administered rmIL‐33 (1 μg in 100 μl saline; R&D Systems) intraperitoneally for 10 days (from days 1 to 5 and then from days 19 to 23). Concurrently, the mice were injected intradermally at the base of the tail with lung tissue lysate prepared as above (1 mg total protein in 100 μl PBS) twice on days 1 and 19 (Fig. 6a). Again, on day 26 the mice were killed and samples collected for analysis.
Figure 1.
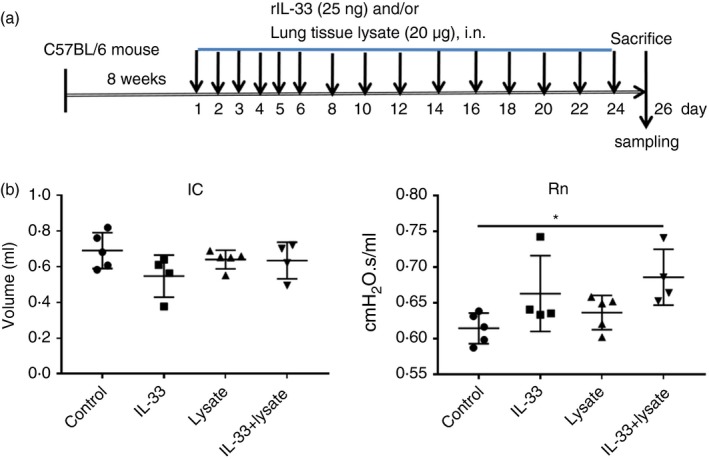
Establishment of the murine model and associated changes in pulmonary mechanics. (a) Protocol of murine exposure. (b) Changes of the volume of inspiratory capacity (IC) and Newtonian resistance (Rn) measured by the FlexiVent invasive airway mechanics system in mice treated with topical IL‐33 or lung lysate alone or together, or diluent control. The data are expressed as the mean ± SEM (n = 4–5/group). *P < 0·05.
Lung function measurement
Lung function was measured using the FlexiVent system (Scireq, Montreal, Canada) as described previously.29 Briefly, mice were anaesthetized with pentobarbital sodium, tracheostomized and ventilated, and connected to the FlexiVent system. The pulmonary mechanics measured at a frequency of at 150 breaths/min with a tidal volume of 8 ml/kg. Lung mechanical properties including inspiratory capacity (IC) and Newtonian resistance (Rn) were recorded.
Analysis of bronchoalveolar lavage fluid
After lung function measurement, bronchoalveolar lavage (BAL) fluid was obtained as previously described.29 The resulting cell pellets were resuspended in PBS for total cell counting using Cell Counter (Bio‐Rad Laboratories, Singapore). Additionally, cytospins were prepared, fixed and stained with Wright‐Giemsa stain for differential cellular counts.
The preparation of whole protein of A549 cells and MLE12 cells
Human alveolar epithelial cells (A549 cells, ATCC‐CCL‐185) and murine lung epithelial cells (MLE12 cells, ATCC‐CRL‐2110) were obtained and maintained in Dulbecco's modified Eagle's minimum essential medium (HyClone Laboratories, logan, UT, USA) supplemented with 10% fetal bovine serum (Gibco Laboratories, Grand Island, NY, USA) and 1% penicillin‐streptomycin solution (100 U/ml penicillin and 100 mg/ml streptomycin) at 37° in 5% CO2 until 80% confluence. The cells were then collected and lysed with ultrasonic. The supernatants were collected and total proteins were quantitated using the Pierce BCA protein assay kit after centrifugation at 13 200 g for 20 min at 4°.
Enzyme‐linked immunosorbent assay
Total concentrations of IgG in serum and cytokines in lung tissue homogenates of mice were measured using commercial enzyme‐linked immunosorbent assay (ELISA) according to the manufacturers’ protocols (eBioscience, San Diego, CA, USA). To measure lung tissue‐specific IgG in the serum of mice, tissue culture plates (Corning Incorporated, Kennebunk, ME, USA) were coated with 100‐μl aliquots of the supernatants of whole lung homogenate (10 μg total protein/ml) overnight at 4°. After washing three times with PBS containing 0·05% Tween‐20 in PBS, the plates were incubated with the blocking buffer at room temperature for 2 hr and then washed as above. After preliminary experimental tests, samples in suitable concentrations were added and incubated for 2 hr at room temperature. After washing, donkey anti‐mouse IgG (H+L) conjugated with peroxidise (1 : 4000; Jackson ImmunoResearch, West Grove, PA, USA) was added to each well, and then the wells were incubated for 1 hr at room temperature and then washed three times. After adding substrate (TMB, 3,3′,5,5′‐tetramethylbenzidine) and incubating for 10 min, the reaction was stopped by adding 100 μl of 2·0 N H2SO4 solution. The optical density of the wells at 450/570 nm was finally measured using an EnSpire Multimode Plate Reader (Perkin Elmer, Waltham, MA, USA).
To measure the presence of ‘autoantibodies’ against alveolar epithelial cells, plates were pre‐coated with whole protein of alveolar epithelial cells A549 cells and MLE12 cells (5 μg total protein/ml) as antigens, and ELISA was performed in human sera (see below) using the same method as described above.
Histology
The lung (left lobe), liver and kidney tissues were collected from the challenged animals, fixed and paraffin‐embedded for histological analysis. Sections (4‐μm thickness) were cut and stained with H&E (haematoxylin and eosin). Images were acquired with a Nikon microscope (Nikon, Tokyo, Japan) and analysed using image‐pro plus 6.0 software (Media Cybernetics, Silver Spring, MD, USA).
Flow cytometry
Cells were isolated from the lung, spleen and mediastinal lymph nodes of the challenged animals through triturated and filtered through a 70‐μm nylon cell strainer (BD Falcon, Bedford, MA, USA). After cellular count, the cells were resuspended and incubated in the dark with Fixable Viability Stain 780 for 10 min at room temperature, then washed with PBS. To block Fc receptor‐mediated non‐specific binding, the cells were incubated with anti‐mouse CD16/CD32 (BD Biosciences Pharmingen, San Diego, CA, USA) for 10 min. For identification of follicular B helper T‐cells (Tfh cells), the cells were stained with PerCP‐Cyanine5.5‐conjugated anti‐CD3e (eBioscience), FITC‐conjugated anti‐CD4 (eBioscience) and PE‐CF594‐conjugated anti‐CXCR5 (BD Biosciences Pharmingen). B2 cells were identified with BV605‐conjugated anti‐CD19 (BioLegend, San Diego, CA, USA), FITC‐conjugated anti‐CD43 (BD Biosciences Pharmingen), APC‐conjugated anti‐CD5 (BioLegend) and PerCP‐Cy5.5‐conjugated anti‐B220 (BioLegend). After washing, the stained cells were resuspended in PBS for flow cytometric analysis using a BD LSRFortessa X‐20 instrument (BD Biosciences Pharmingen). Quantification was performed using flowjo software (Tree Star, Ashland, OR, USA).
Lung immunohistochemistry
Lung tissue sections (4 μm) were deparaffinized in xylene then rehydrated in decreasing concentrations of ethanol followed by distilled water.29 The sections were then boiled in citrate buffer for 20 min or digested in 0·25% trypsin for 10 min for antigen retrieval. Subsequently, sections were incubated with 3% H2O2 to inactivate endogenous peroxidase, and with 3% bovine serum albumin and 0·03% Triton X‐100 in PBS for 1 hr to block non‐specific interactions. After washing with PBS containing 0·05% Tween 20, lung sections were incubated overnight at 4° with primary rabbit antibodies against CD3 (T‐cells, 1 : 200; Abcam, Cambridge, UK), CD20 (B‐cells, 1 : 1000; LifeSpan, Seattle, WA, USA), CD138 (plasma cells, 1 : 500; Proteintech, Wuhan, China), F4/80 (macrophages, 1 : 500; Proteintech) and neutrophil elastase (neutrophils, 1 : 2000; Abcam). After washing, the sections were incubated with mouse anti‐rabbit secondary antibody (1 : 50; Jackson ImmunoResearch) for 1 hr. The sections were then washed and incubated with horseradish peroxidase‐conjugated donkey anti‐mouse antibody (1 : 100; Jackson ImmunoResearch) for 1 hr. Positive signals were developed with 3, 3′‐diaminobenzidine (Solarbio, Beijing, China). Immunoreactivity was measured using a microscope (Nikon) connected with a computer imaging system.
Immunofluorescence for lung‐specific IgG
Immunofluorescence was used to detect whether ‘autoantibodies’ in serum of the mice specifically recognize cells and components of murine and human lung tissues. In brief, after dewaxing and antigen‐retrieving, sections derived from lung tissues of normal, untreated C57BL/6 mice were first incubated with 5% normal goat serum in PBS for reducing non‐specific binding. After washing, the sections were incubated with rabbit anti‐mouse lung prosurfactant protein C antibody (for identification of alveolar type II epithelial cells; Abcam) and the sera (1 : 50 diluted) of the C57BL/6 mice immunized with lung tissue lysate (for determining lung‐specific IgG antibodies in the serum) overnight at 4°. Immunoreactivity was detected with a secondary Alexa Fluor‐594 (red)‐labelled goat anti‐rabbit IgG antibody (Zabg‐Bio, Beijing, China) and Alexa Fluor 488 (green)‐labelled goat anti‐mouse IgG antibody (Jackson ImmunoResearch). Then nuclei were stained with 4,6‐diamidino‐2‐phenylindole (DAPI; Zabg‐Bio). Negative control staining was performed in an identical fashion, except that the primary antibodies were omitted. Images of the stained lung tissues were captured using fluorescence microscopy (Nikon).
Clinical specimens
To further identify whether ‘autoantibodies’ produced in the mice also exist in human serum and whether these ‘autoantibodies’ can recognize components of human lungs, human sera were collected from Xuanwu Hospital, Capital Medical University, P. R. China (controls: n = 40; subjects with COPD: n = 43). Furthermore, human lung tissues (n = 3) were obtained from normal donors for lung transplantation. ELISA and immunofluorescence were performed with human sera and lung tissue sections as the above.
Statistical analysis
Statistical analyses were performed using graphpad prism 7.0 software (GraphPad Software, San Diego, CA, USA). In the figures, the data are presented as the mean and standard error of the mean (SEM). One‐way anova was used to compare groups, and Tukey's multiple comparisons test was used to assess for statistical significance. Correlation was evaluated with Pearson's r (two‐tailed) test. P‐values < 0·05 were considered statistically significant.
Results
IL‐33‐mediated autoimmune lung inflammation
To mimic a situation in which excess production of IL‐33 in the airways promotes an autoimmune humoral response to the airway structural cells, we co‐administered rmIL‐33 and the soluble fraction of the whole lung lysates from syngeneic animals, or diluent controls, to C57BL/6 mice repeatedly over a 26‐day period (Fig. 1a). The mean of airways resistance of the mice co‐administered IL‐33 and lung lysate, but not treated with IL‐33 or lysate alone, was significantly elevated compared with those treated with the relevant diluent controls, suggesting possible inflammation and obstruction of the airways (Fig. 1b, right). There were no significant changes in the mean volume of IC between the experimental groups (Fig. 1b, left).
The mice co‐administered IL‐33 and lysate also developed significantly elevated mean numbers of total cells, neutrophils, macrophages, eosinophils and lymphocytes in their BAL fluid compared with those relevant controls. As expected, administration of syngeneic lysate alone exerted no effect, whereas administration of IL‐33 alone significantly increased the mean numbers of total cells and eosinophils, but not the cells of other phenotypes (Fig. 2a; P < 0·01, respectively). Furthermore, co‐administration of IL‐33 and lysate also induced marked infiltration of inflammatory cells into the perivascular and peribronchiolar and, to a lesser extent, the interstitial lung tissue of the targeted animals, but no obvious changes were seen in the mice challenged with IL‐33 or lysate alone, or diluent controls (Fig. 2b).
Figure 2.
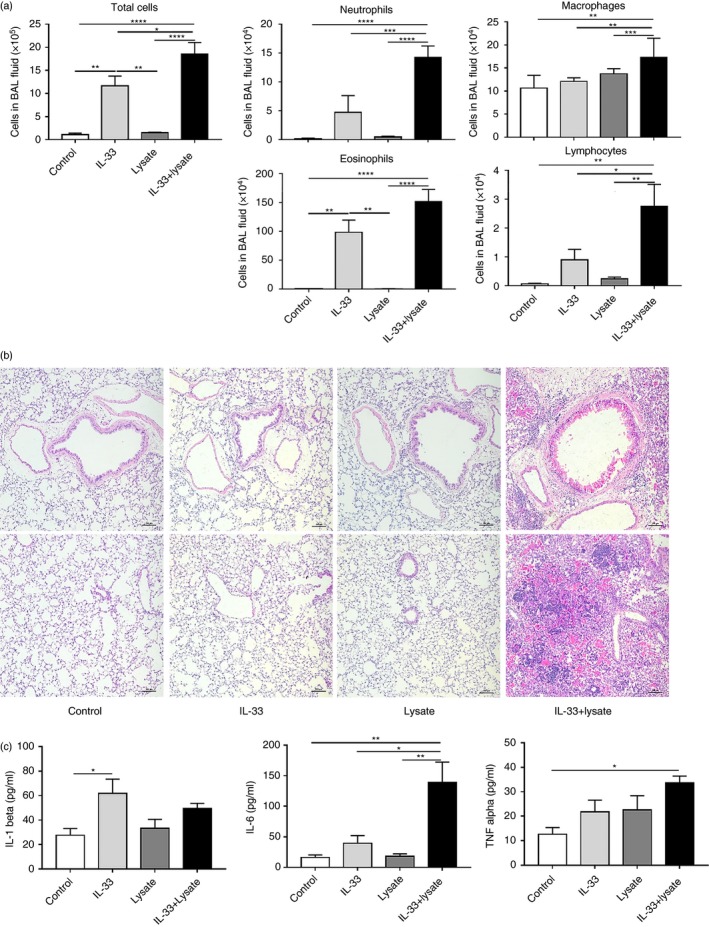
Tissue and airways cellular infiltration and concentrations of pro‐inflammatory cytokines in the lungs of the experimental mice. (a) Total and differential cell counts in the bronchoalveolar lavage (BAL) fluid (n = 5/group). (b) Representative photomicrographs of sections of formalin‐fixed, paraffin‐embedded lung tissue stained with haematoxylin and eosin (H&E) showing peribronchiolar and perivascular (top panel) and parenchymal (bottom panel) inflammation in different fields of the same section (original magnification × 100). (c) Concentrations of the cytokines IL‐1β, IL‐6 and tumour necrosis factor (TNF)‐α in lung tissue homogenates quantified by enzyme‐linked immunosorbent assay (ELISA). The data are expressed as the mean ± SEM (n = 5/group). *P < 0·05, **P < 0·01, ***P < 0·001 and ****P < 0·0001.
In addition, the mean concentrations of the pro‐inflammatory cytokines IL‐6 (P < 0·01) and tumour necrosis factor (TNF)‐α (P < 0·05), but not IL‐1β, were significantly increased in the lung tissue homogenates of the animals co‐administered IL‐33 and lysate (there was a small but significant increase in the mean concentration of IL‐1β in the animals administered IL‐33 alone; Fig. 2c).
Total and lung tissue‐specific IgG production
Enzyme‐linked immunosorbent assay revealed that the mean concentration of total serum IgG was elevated in the mice co‐administered IL‐33 and lysate compared with those administered lysate alone, but not those administered IL‐33 alone or the relevant diluent controls (Fig. 3a). More interestingly, the mean serum concentration of lung tissue‐specific IgG was clearly and significantly elevated in the mice co‐administered IL‐33 and lysate compared with those administered IL‐33 or lysate alone: in these groups, the mean response was statistically equivalent to that seen in the animals administered diluent control (Fig. 3b). Immunofluorescent staining showed that the lung tissue‐specific IgG induced in the serum of the mice co‐administered IL‐33 and lysate bound predominantly, but not exclusively, to alveolar type II epithelial cells and smooth muscle cells (Fig. 3c, middle and bottom panels). In contrast, and as expected, there was no evidence of lung tissue‐specific IgG in the serum of normal control mice (Fig. 3c, top panel).
Figure 3.

Quantification and localization of lung tissue‐specific IgG in the murine model. Concentrations of total serum IgG (a) and lung tissue lysate‐specific IgG (b) quantified by enzyme‐linked immunosorbent assay (ELISA). The data are expressed as the mean ± SEM (n = 5/group). **P < 0·01, ***P < 0·001. (c) Location of serum IgG binding to alveoli and airways of lung tissue sections from a normal mouse as detected by indirect immunofluorescence. Representative photomicrographs show sections stained with normal murine serum (i) and serum from mice administered IL‐33+lung tissue lysate (ii) and (iii). Lung prosurfactant protein C (SPC, red) was used to label alveolar type II epithelial cells; 4,6‐diamidino‐2‐phenylindole (DAPI, blue) probed for cell nuclei and lung tissue‐specific IgG was identified by green immunofluorescence.
Numbers and distribution of T follicular helper cells and B2 B‐cells
To assess whether the production of lung tissue‐specific IgG in our experimental animals was associated with evidence of expansion of Tfh cells and B2 B lymphocytes, we measured the percentages of CD3+ CD4+ CXCR5+ Tfh cells (Fig. 4) and the percentages of CD19+ CD43− B220+ CD5− B2 B‐cells (Fig. 5) in cell suspensions isolated from the spleens, mediastinal lymph nodes and lung tissues of the groups of challenged animals using flow cytometry. The animals co‐administered IL‐33 and lysate had significantly elevated mean percentages of Tfh cells in the spleen compared with those administered IL‐33 or lysate alone or diluent control (Fig. 4a,b), and of B2 B‐cells in the mediastinal lymph nodes compared with the animals administered IL‐33 alone (Fig. 5a,b). Interestingly, there was a significant correlation between the percentages of Tfh cells and B2 B‐cells in the mediastinal lymph nodes, but not in the spleen or lung tissues (Fig. 5c).
Figure 4.

Tissue distribution of follicular B helper T‐cells (Tfh) cells in the different tissues. (a) Tfh cells from whole lung tissue, mediastinal lymph nodes and spleen were identified by flow cytometry. (b) Percentages of CD3+ CD4+ CXCR5+Tfh cells of total lymphocytes in the spleen, mediastinal lymph nodes and lung tissue of mice immunized with IL‐33, lung lysate, both or diluent control. The data are expressed as the mean ± SEM (n = 3/group). *P < 0·05, **P < 0·01, ***P < 0·001.
Figure 5.

Tissue distribution of B2 B‐cells in the experimental mice. (a) B2 B‐cells from whole lung tissue, mediastinal lymph nodes and spleen were identified by flow cytometry. (b) Percentages of CD19+ CD43− B220+ CD5− B2 B‐cells of total lymphocytes in the spleen, mediastinal lymph nodes and lung tissue of mice immunized with IL‐33, lung lysate, both or diluent control. The data are expressed as the mean ± SEM (n = 3/group). *P < 0·05. (c) Correlations between percentages of follicular B helper T‐cells (Tfh) cells and B2 B‐cells in the different tissues.
IL‐33‐mediated lung tissue‐specific inflammation in mice systemically immunized with lung tissue lysate
To examine the extent to which administration of IL‐33 along with autologous lung tissue lysate to the experimental animals induced inflammation confined to the lung tissue or additionally elsewhere, we modified our challenge protocol so that the animals were challenged systemically, rather than topically in the airways, with IL‐33 delivered intraperitoneally and lung tissue lysate intradermally (Fig. 6a).
Figure 6.

Systemic immunization with lung tissue lysate with or without IL‐33. (a) Schedule of murine treatments. C57BL/6 mice were administered IL‐33 (1 μg/100 μl) intraperitoneally and/or lung tissue lysate (1 mg/100 μl) intradermally. (b) Total cells and differential cell counts in the bronchoalveolar lavage (BAL) fluid. The data are expressed as the mean ± SEM (n = 5/group). *P < 0·05, **P < 0·01, ***P < 0·001 and ****P < 0·0001. (c) Representative photomicrographs of sections of formalin‐fixed, paraffin‐embedded lung, liver and kidney tissues stained with haematoxylin and eosin (H&E; original magnification × 100).
Cellular analysis revealed that systemic challenge of the mice with IL‐33 and lysate was again associated with significance in the mean numbers of total cells, neutrophils, macrophages and eosinophils in the BAL fluid as compared with challenge with IL‐33 or lysate alone or the relevant diluent controls, with isolated lysate challenge exerting no detectable effect as previously and as expected (Fig. 6b). Correspondingly, there was evidence of inflammatory cellular infiltration into sections of lung tissue of the mice administered IL‐33 and lysate, which was distributed principally around the airways and blood vessels (Fig. 6c, top panel). The extent of this airway inflammation was, however, considerably less than that observed in the mice administered IL‐33 and lysate topically to the airways (compare with Fig. 2b). As with topical challenge, there was evidence of cellular infiltration, albeit less marked, into the lung tissue sections of the mice administered IL‐33 alone compared with those administered syngeneic lung tissue or diluent control alone.
In the same animals, we sought evidence of infiltration of inflammatory cells into sections of liver and kidney tissue. H&E staining revealed no obvious infiltration of inflammatory cells into the kidney (Fig. 6c, middle panel), although there was evidence of borderline infiltration of inflammatory cells into liver tissue, again principally around the blood vessels in the mice administered IL‐33 with lysate or IL‐33 alone (Fig. 6c, bottom panel).
Lymphocytes formed the majority of the inflammatory cells infiltrating the lung tissue
To identify the phenotypes of the inflammatory cells infiltrating the lung tissue of the animals administered IL‐33 along with autologous lung lysate, we examined sections of lung tissue from animals challenged systemically rather than topically, where the infiltrating cells were fewer in number and consequently more certainly identifiable. CD3+ T‐cells and CD20+ B‐cells constituted the majority of the inflammatory cells infiltrating around the blood vessels and airways in sections of the lung tissues of mice treated with IL‐33 with lung tissue lysate, although the numbers of elastase+ neutrophils and CD138+ plasma cells were also slightly elevated (Fig. 7).
Figure 7.
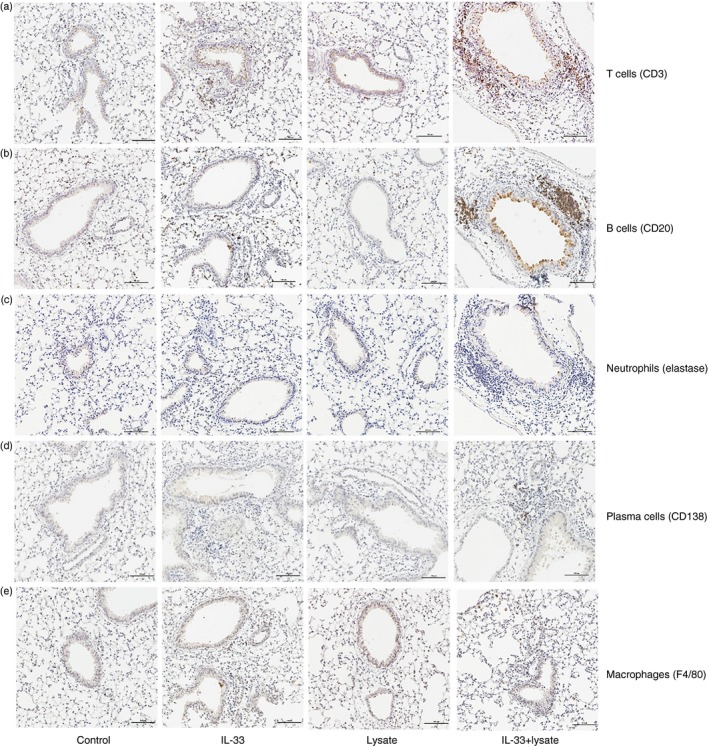
Immunohistochemistry showing phenotypes of inflammatory cells infiltrating the lung tissue of mice immunized with syngeneic lung lysate with or without IL‐33, IL‐33 alone and diluent control. Representative photomicrographs showing (a) T‐cells (CD3+), (b) B‐cells (CD20+), (c) neutrophils (neutrophil elastase+), (d) plasma cells (CD138+) and (e) macrophages (F4/80+) in sections of paraffin‐embedded lung tissues.
Lung tissue‐specific IgG antibodies were cross‐reactive between species and also identifiable in the serum of patients with chronic obstructive pulmonary disease
To explore whether the lung tissue‐specific IgG identified in the serum of our experimental mice was capable of binding to human lung tissue, immunofluorescence was used to detect murine IgG bound to sections of normal human lung tissue obtained from lung transplantation donors, which were incubated with sera from the mice administered IL‐33 along with lung tissue lysate. We did indeed find that the sera of mice administered IL‐33 with lysate, but not those normal murine serum, contained IgG antibodies bound to alveolar type II epithelial cells (Fig. 8a). Correspondingly, ELISA showed that the mean serum concentration of IgG antibody binding to a whole protein extract of human alveolar type II epithelial cells (A549 cells) was significantly higher in a group of patients with COPD (n = 43) compared with normal control subjects (n = 40; P < 0·0001; Fig. 8b). In parallel, the mean serum concentration of IgG antibody binding to a whole protein extract of murine alveolar type II epithelial cells (MLE12 cells) was also elevated in these same patients with COPD compared with the controls (P < 0·01; Fig. 8c).
Figure 8.
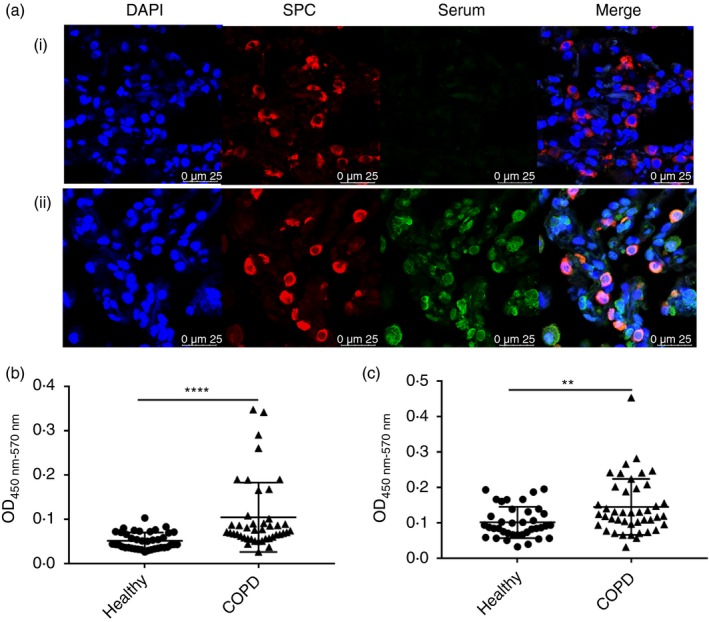
Detection of human lung tissue‐specific IgG. (a) Immunoreactivity for murine serum IgG incubated with human lung sections detected using immunofluorescence. Representative microscopic images show sections stained with normal murine serum (i) and serum from mice immunized with IL‐33+lung tissue lysate (ii). Lung prosurfactant protein C (SPC, red) was used to label alveolar type II epithelial cells; 4,6‐diamidino‐2‐phenylindole (DAPI, blue) probed for cell nuclei and lung tissue‐specific IgG (green) was identified by green immunofluorescence. Concentrations of (b) specific IgG against human II alveolar epithelial cells (A549 cells) and (c) murine II alveolar epithelial cells (MLE12 cells) in sera of healthy control subjects (n = 40) and patients with chronic obstructive pulmonary disease (COPD; n = 43). The data are expressed as the mean ± SEM. **P < 0·01, ****P < 0·0001.
Discussion
The pathogenetic mechanisms of COPD and particularly those by which airways inflammation and alveolar damage persist and progress in some patients even after the removal of perceived, provoking stimuli remain elusive. The development of autoimmune tissue damage in susceptible patients is one plausible explanation for this phenomenon, which has been explored in the recent literature.11 The triggering of an autoimmune response to airways epithelial determinants is perhaps facilitated by local damage of these cells by environmental and inflammatory insults in a high alarmin environment conducive to the priming of T‐cells and the production of tissue‐specific antibodies acting locally or systemically to perpetuate local inflammation and lung parenchymal damage, resulting in the development of COPD.10, 19 Although some ‘autoimmune’ models of COPD in animals have been proposed,20, 22 none of these has addressed the possibility of sensitization to autologous lung tissue. Several studies have confirmed elevated expression of respiratory epithelial alarmins such as IL‐33 both locally in the airways epithelium and systemically in the circulation of patients with COPD.23 In the present study, we demonstrate that local co‐administration of IL‐33 and damaged, autologous lung tissue locally to the airways or systemically results in the production of IgG autoantibodies against this autologous, ‘self’ lung tissue, associated with airways inflammation and tissue damage resembling that observed in patients with COPD. While a direct mechanistic link between the development of the autoantibody and the airways inflammatory changes remains to be elucidated, it seems reasonable to conclude that the two phenomena are related. It is also interesting to note that the development of this phenomenon followed concomitant exposure of the animals to IL‐33 and autologous lung lysate, but not either component in isolation, implying that the juxtaposition of local tissue damage and elevated IL‐33 production are essential for its development. To the best of our knowledge, this compelling finding has never been reported previously.
In addition, this phenomenon was accompanied by elevated, local expression of a range of pro‐inflammatory cytokines, including IL‐6 and TNF‐α, although not IL‐1β, which have also been implicated in causing respiratory epithelial cell damage and promoting autoimmunity in patients with COPD.30, 31, 32
Although some studies have identified autoantibodies specific for components of lung tissue that are detectable not only in the circulation, but also in the lung tissue of patients with COPD,19 we could detect no obvious differences in the concentrations of lung tissue‐specific antibodies in the lung homogenates from the animals administered IL‐33 with lung lysate or either component alone (data not shown). We speculate that this is because detection of such antibodies within the lung homogenates, as distinct from the circulation of the experimental animals is impaired by their binding to their autoimmune targets in the lung parenchyma and/or degradation during preparation of the tissue homogenates. Encouragingly, our data also show that lung tissue‐specific autoantibodies produced under our experimental conditions react predominantly with alveolar type II epithelial cells and airways smooth muscle cells, suggesting that these particular cells are putatively major targets of these autoantibodies, although we do not know yet precisely what components of these cells they recognize. This is the subject of ongoing research in our laboratory.
Our data are consistent with the hypothesis that autoantibodies against alveolar epithelial cells are produced only when epithelial damage occurs in an IL‐33‐enriched environment. This impression is reinforced by our data showing that the percentages of Tfh cells in the spleen and B2 B‐cells in the mediastinal lymph nodes were elevated only in the groups of mice administered IL‐33 along with lung tissue lysate and not either component alone. Tfh cells are known to play a particular role in the activation of B‐cells and their development into memory cells.33 The fact that the percentages of Tfh cells and B2 B‐cells were positively correlated in the mediastinal lymph nodes further suggests the possibility that the B2 B‐cells producing the antibodies are propagated locally at this site. Further, challenge of the animals with IL‐33 along with lysate, but neither alone, resulted in local, elevated expression of IL‐6, which has been implicated in inducing differentiation of primary T‐cells to Tfh cells.4 Recent findings suggest that IL‐33 may also induce the development of Tfh cells.34
Although IL‐33 is well recognized as a permissive influence in the development of autoimmune diseases involving the airways,34 the mechanisms by which it does this remain unclear. IL‐33 is able to promote the activity of immune cells while disabling regulatory T‐cells, activities that might sever immunological tolerance.35 IL‐33 is known to be produced principally by alveolar type II epithelial cells, and is rapidly released following activation or damage of the cells by viruses, allergens and other environmental insults impacting on the airways.23, 36 Concomitantly it is accepted that endogenous fragments from damaged airways epithelium and even lung extracellular matrix proteins, when ‘exposed’ by injury, may become self‐determinants serving as new autoimmune epitopes.37 Our data clearly imply, however, that over‐production of IL‐33 or lung tissue damage alone are insufficient to induce lung inflammation and autoantibody production in the experimental mice, and that it is the coincidence of these that triggers an autoimmune response putatively relevant to the pathogenesis of COPD. Clearly, the precise role of IL‐33 in this process, for example as an adjuvant or to overcome immune tolerance of self‐antigens, remains to be explored.
Our data from the animals administered IL‐33 and autologous lung lysate both nasally and systemically clearly show that the resulting inflammation was clearly sited in the lung, but barely visible in other organs such as the liver and kidney, suggesting that the autoimmune response initiated by these ‘self’ lung tissue components under the influence of IL‐33 is tissue‐specific. Further, our data indicate that the autoantibodies appearing in the serum of our experimental mice bind not only to alveolar type II epithelial cells in lung tissue sections from normal mice but also those in sections from normal human subjects, suggesting that these lung tissue‐specific antibodies have tissue specificity rather than species specificity. In further support of this conclusion, we have further shown that the mean concentration of IgG antibody against both human and murine alveolar type II epithelial cells was significantly elevated in the serum of a group of patients with COPD compared with normal, control subjects. It remains to be seen what components of these cells are recognized by these autoantibodies and how they might be implicated in causing disease.
Finally, as referred to in recent overviews of the possible role of autoimmune immunological reactions in the pathogenesis of COPD, there remains the issue of causality.4, 11 Our present data are consistent with the hypothesis that airways tissue damage in a local IL‐33‐rich environment results in the production of autoantibodies that sustain this damage and are potentially responsible for the development of at least some of the pathological features of COPD, such as progressive alveolar destruction. Litsiou and colleagues have observed ‘lymphoid neogenesis’ in the lungs of smoke‐exposed mice, and so conclude that autoimmunity is likely a cause rather than a consequence of airways damage.38 Other researchers have concluded that an autoimmune response against lung tissue components in COPD is a consequence of the release or ‘exposure’ of these components in damaged lung tissue.19 These concepts are not, of course, mutually exclusive and, whatever the truth, it seems salutary to regard autoimmunity as an important mechanism for sustaining and perhaps advancing persistent airways inflammation and damage. However, how these autoantibodies contribute to the pathogenesis of COPD certainly needs to be further explored. They might exhibit their function through maintaining chronic inflammation and remodelling of small airways, or destroying alveolar type II epithelial cells by antibody‐dependent cytotoxicity, activating complement systems or antibody‐dependent cellular phagocytosis, or might be through all of them. In addition, the autoantibodies detected are polyclonal antibodies. Obviously, making monoclonal antibodies are critical strategies for further understanding the details, which are also ongoing in our lab now.
In conclusion, in the presence of IL‐33, topical or systemic administration of homologous, syngeneic lung tissue lysate to mice induces periluminal and perivascular pulmonary inflammation and production of autoantibodies reacting with both murine and human alveolar epithelial cells. Conversely, elevated concentrations of these cross‐species autoantibodies are detectable in the serum of patients with COPD. Hence, IL‐33 might initiate and potentiate adaptive immune responses to ‘self’ lung components, driving diseases such as COPD. It is possible to envisage that serum concentrations of antibodies against airways structural cells and markers of elevated local IL‐33 production may be developed as diagnostic or prognostic markers when managing COPD, with due diligence to the heterogeneity of the clinical manifestations and natural history of the disease in individual subjects if management is to be ‘personalized’.
Disclosures
The authors have no conflicts of interest to declare.
Acknowledgement
This work was supported by grants from the National Natural Science Foundation of China (81471594 and 81770049).
References
- 1. Global Initiative for Chronic Obstructive Lung Disease . (2019) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. URL www.goldcopd.org [accessed on 14 November 2018]
- 2. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet 2017; 389:1931–40. [DOI] [PubMed] [Google Scholar]
- 3. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 2011; 378:1015–26. [DOI] [PubMed] [Google Scholar]
- 4. Caramori G, Casolari P, Barczyk A, Durham AL, Di Stefano A, Adcock I. COPD immunopathology. Semin Immunopathol 2016; 38:497–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Byers DE, Alexander‐Brett J, Patel AC, Agapov E, Dang‐Vu G, Jin X et al Long‐term IL‐33‐producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest 2013; 123:3967–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hobbs BD, Hersh CP. Integrative genomics of chronic obstructive pulmonary disease. Biochem Biophys Res Commun 2014; 452:276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in non‐smokers. Lancet 2009; 374:733–43. [DOI] [PubMed] [Google Scholar]
- 8. Shapiro SD. End‐stage chronic obstructive pulmonary disease: the cigarette is burned out but inflammation rages on. Am J Respir Crit Care Med 2001; 164:39–340. [DOI] [PubMed] [Google Scholar]
- 9. Packard TA, Li QZ, Cosgrove GP, Bowler RP, Cambier JC. COPD is associated with production of autoantibodies to a broad spectrum of self‐antigens, correlative with disease phenotype. Immunol Res 2013; 55:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bagdonas E, Raudoniute J, Bruzauskaite I, Aldonyte R. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int J Chron Obstruct Pulmon Dis 2015; 10:995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Caramori G, Ruggeri P, Di Stefano A, Mumby S, Girbino G, Adcock IM et al Autoimmunity and COPD: clinical implications. Chest 2018; 153:1424–31. [DOI] [PubMed] [Google Scholar]
- 12. Wen L, Krauss‐Etschmann S, Petersen F, Yu X. Autoantibodies in chronic obstructive pulmonary disease. Front Immunol 2018; 9:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seys LJ, Verhamme FM, Schinwald A, Hammad H, Cunoosamy DM, Bantsimba‐Malanda C et al Role of B cell‐activating factor in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2015; 192:706–18. [DOI] [PubMed] [Google Scholar]
- 14. Brusselle GG, Demoor T, Bracke KR, Brandsma CA, Timens W. Lymphoid follicles in (very) severe COPD: beneficial or harmful? Eur Respir J 2009; 34:219–30. [DOI] [PubMed] [Google Scholar]
- 15. Xiong Y, Gao S, Luo G, Cheng G, Huang W, Jiang R et al Increased circulating autoantibodies levels of IgG, IgA, IgM against cytokeratin 18 and cytokeratin 19 in chronic obstructive pulmonary disease. Arch Med Res 2017; 48:79–87. [DOI] [PubMed] [Google Scholar]
- 16. Feghali‐Bostwick CA, Gadgil AS, Otterbein LE, Pilewski JM, Stoner MW, Csizmadia E et al Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008; 177:156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B et al Oxidative stress‐induced antibodies to carbonyl‐modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2011; 184:796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nunez B, Sauleda J, Anto JM, Julia MR, Orozco M, Monso E et al Anti‐tissue antibodies are related to lung function in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2011; 183:1025–31. [DOI] [PubMed] [Google Scholar]
- 19. Daffa NI, Tighe PJ, Corne JM, Fairclough LC, Todd I. Natural and disease‐specific autoantibodies in chronic obstructive pulmonary disease. Clin Exp Immunol 2015; 180:155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taraseviciene‐Stewart L, Scerbavicius R, Choe KH, Moore M, Sullivan A, Nicolls MR et al An animal model of autoimmune emphysema. Am J Respir Crit Care Med 2005; 171:734–42. [DOI] [PubMed] [Google Scholar]
- 21. Morissette MC, Jobse BN, Thayaparan D, Nikota JK, Shen P, Labiris NR et al Persistence of pulmonary tertiary lymphoid tissues and anti‐nuclear antibodies following cessation of cigarette smoke exposure. Respir Res 2014; 15:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brandsma CA, Timens W, Geerlings M, Jekel H, Postma DS, Hylkema MN et al Induction of autoantibodies against lung matrix proteins and smoke‐induced inflammation in mice. BMC Pulm Med 2010; 10:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kearley J, Silver JS, Sanden C, Liu Z, Berlin AA, White N et al Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin‐33‐dependent response to infection. Immunity 2015; 42:566–79. [DOI] [PubMed] [Google Scholar]
- 24. Cayrol C, Girard JP. IL‐33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol 2014; 31:31–7. [DOI] [PubMed] [Google Scholar]
- 25. Russi AE, Ebel ME, Yang Y, Brown MA. Male‐specific IL‐33 expression regulates sex‐dimorphic EAE susceptibility. Proc Natl Acad Sci USA 2018; 115:E1520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu SL, Wong CK, Tam LS. The alarmin functions of high‐mobility group box‐1 and IL‐33 in the pathogenesis of systemic lupus erythematosus. Expert Rev Clin Immunol 2013; 9:739–49. [DOI] [PubMed] [Google Scholar]
- 27. Bu X, Zhang T, Wang C, Ren T, Wen Z. IL‐33 reflects dynamics of disease activity in patients with autoimmune hemolytic anemia by regulating autoantibody production. J Transl Med 2015; 13:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Macedo RB, Kakehasi AM, Andrade MV. IL33 in rheumatoid arthritis: potential contribution to pathogenesis. Rev Bras Reumatol 2016; 56:451–7. [DOI] [PubMed] [Google Scholar]
- 29. Yao XJ, Huang KW, Li Y, Zhang Q, Wang JJ, Wang W et al Direct comparison of the dynamics of IL‐25‐ and ‘allergen’‐induced airways inflammation, remodelling and hypersensitivity in a murine asthma model. Clin Exp Allergy 2014; 44:765–77. [DOI] [PubMed] [Google Scholar]
- 30. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 2016; 138:16–27. [DOI] [PubMed] [Google Scholar]
- 31. Broekman W, Amatngalim GD, de Mooij‐Eijk Y, Oostendorp J, Roelofs H, Taube C et al TNF‐alpha and IL‐1beta‐activated human mesenchymal stromal cells increase airway epithelial wound healing in vitro via activation of the epidermal growth factor receptor. Respir Res 2016; 17:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Accortt NA, Chung JB, Bonafede M, Limone BL, Mannino DM. Retrospective analysis to describe associations between tumor necrosis factor alpha inhibitors and COPD‐related hospitalizations. Int J Chron Obstruct Pulmon Dis 2017; 12:2085–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. King C. New insights into the differentiation and function of T follicular helper cells. Nat Rev Immunol 2009; 9:757–66. [DOI] [PubMed] [Google Scholar]
- 34. Drake LY, Kita H. IL‐33: biological properties, functions, and roles in airway disease. Immunol Rev 2017; 278:173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen CC, Kobayashi T, Iijima K, Hsu FC, Kita H. IL‐33 dysregulates regulatory T cells and impairs established immunologic tolerance in the lungs. J Allergy Clin Immunol 2017; 140:1351–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Le Goffic R, Arshad MI, Rauch M, L'Helgoualc'h A, Delmas B, Piquet‐Pellorce C et al Infection with influenza virus induces IL‐33 in murine lungs. Am J Respir Cell Mol Biol 2011; 45:1125–32. [DOI] [PubMed] [Google Scholar]
- 37. Gu BH, Madison MC, Corry D, Kheradmand F. Matrix remodeling in chronic lung diseases. Matrix Biol 2018; 73:52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Litsiou E, Semitekolou M, Galani IE, Morianos I, Tsoutsa A, Kara P et al CXCL13 production in B cells via Toll‐like receptor/lymphotoxin receptor signaling is involved in lymphoid neogenesis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 187:1194–202. [DOI] [PubMed] [Google Scholar]