

Abstract
Premise
Third‐generation sequencing methods generate significantly longer reads than those produced using alternative sequencing methods. This provides increased possibilities for the study of biodiversity, phylogeography, and population genetics. We developed a protocol for in‐solution enrichment hybridization capture of long DNA fragments applicable to complete plastid genomes.
Methods and Results
The protocol uses cost‐effective in‐house probes developed via long‐range PCR and was used in six non‐model monocot species (Poaceae: African rice, pearl millet, fonio; and three palm species). DNA was extracted from fresh and silica gel–dried leaves. Our protocol successfully captured long‐read plastome fragments (3151 bp median on average), with an enrichment rate ranging from 15% to 98%. DNA extracted from silica gel–dried leaves led to low‐quality plastome assemblies when compared to DNA extracted from fresh tissue.
Conclusions
Our protocol could also be generalized to capture long sequences from specific nuclear fragments.
Keywords: de novo assembly, DNA probes, long‐range PCR, MinION, whole plastome sequencing
High‐throughput sequencing is revolutionizing research in plant evolutionary biology. The development of second‐generation sequencing, also known as next‐generation sequencing (NGS), led to the cost‐effective generation of a massive amount of sequence data (Straub et al., 2012). Although NGS offers many advantages, one shortcoming is that this sequencing method generates short reads (between 100–400 bp). This is problematic for de novo assemblies of plant genomes that prove difficult to resolve due to repetitive sequences resulting from transposable elements, polyploidy, and large genome sizes.
In contrast to NGS, third‐generation sequencing (TGS) directly targets single DNA molecules without prior PCR, enabling “real‐time sequencing” (Bleidorn, 2016). The main improvement of TGS is the significant increase in read length from several to tens of thousands of bases per single read (termed “long reads”). This provides important advantages to improve de novo assemblies (Jiao and Schneeberger, 2017), gap filling (Eckert et al., 2016), or phasing (Laver et al., 2016). Technologies such as MinION, a portable real‐time sequencing device developed by Oxford Nanopore Technologies (ONT; Oxford, United Kingdom), are able to generate mean read lengths ranging from 5 to 20 kbp in standard analyses (and peak up to 2 Mbp) depending on the quality of the DNA (Lee et al., 2016). One drawback is that most TGS technologies have high error rates when compared to NGS (~10% for ONT MinION vs. 0.1% for Illumina; Goodwin et al., 2016). However, new base‐calling algorithms, associated with a posteriori corrections, allow for a significant decrease of sequence errors. With sufficient coverage and proper algorithms, TGS can lead to assemblies with consensus nucleotide accuracy of 99.90% (Lee et al., 2016).
The application of TGS using MinION to large genomes such as plants is problematic mainly because of the generally low output of data currently available from MinION (10–20 Gbp vs. 1500 Gbp for a HiSeq 4000 [Illumina, San Diego, California, USA]). Thus, efficiently sequencing specific regions will depend on genome reduction approaches, such as targeted sequencing (Cronn et al., 2012; Jones and Good, 2016). Genome reduction via sequence capture refers to DNA fragments (nuclear, ribosomal, or plastid) that are directly captured from a total genomic library using probes binding to the complementary DNA sequences. This approach has the advantage of being cost effective, optimizing read depth on the targeted region, and allowing the analysis of more samples per run. However, our ability to capture and sequence long DNA fragments has yet to be properly applied in plants. Indeed, sequence capture is only routinely undertaken on short DNA fragments (Mamanova et al., 2010; Cronn et al., 2012), limiting its usefulness for long read–based TGS.
In this study, we focused on complete plastid genome or plastome sequencing for two main reasons. First, in practical terms, plastome DNA provides an ideal model to test capture protocols because it is generally easy to sequence and good a priori data on structure and composition are available (reference plastomes are available for numerous species). Thus, plastome DNA is a good starting point to validate capture protocols that can then be extended to the nuclear genome. Second, the plastome has been shown to be a cost‐effective marker for the study of plant evolution (Mariac et al., 2014; Twyford and Ness, 2017), and improved sequencing is highly desirable. Indeed, de novo assembly of genomes based on short reads can be problematic because it is linked to the presence of repeated regions, which lead to low‐quality assemblies (Sohn and Nam, 2018). In plastomes, the presence of two large inverted repeat (IR) regions (~20 kbp long) present in most plant species is problematic for de novo assembly (Mariac et al., 2014); long reads would be particularly useful here. This is especially true for non‐model taxa for which high‐quality reference genomes are not available. Given the low output of data from MinION, this technology cannot be easily used to sequence plastomes directly from genomic DNA (e.g., genome skimming). In addition, long‐read sequencing will provide new insights into the structural variation of plastomes (Mower and Vickrey, 2018). The main challenge in efficiently applying TGS to the study of plant evolution will be based on our ability to capture long DNA fragments. To date, long‐read targeted capture has mainly been undertaken on organisms with small genomes such as bacteria or viruses (e.g., Eckert et al., 2016); it has rarely been attempted in organisms with large genomes such as plants. Protocols for DNA enrichment for segments in excess of 20 kbp in length have also been developed (Dapprich et al., 2016). In plants, few studies have undertaken long‐read targeted capture (Giolai et al., 2016, 2017). These protocols prove that capturing long DNA fragments is possible but has yet to be routinely developed for non‐model plants.
Here, we present a protocol to capture long reads for plastome sequencing and reassembly using ONT MinION technology. We first developed our protocol for the model plant species Oryza sativa L. (Asian rice). We then applied the protocol to sequence plastomes in several wild species and non‐model but economically important crops. Finally, we tested the ability to capture and assemble plastomes from DNA extracted from silica gel–dried leaves.
MATERIAL AND METHODS
Sampling strategy and DNA extraction
For this study, we focused on seven economically important plant species from Asia, Africa, and South America. First, we developed and validated our long‐read capture protocol using the model plant species O. sativa (Asian rice). We then applied our protocol to several other plant species from the same genus (Oryza L.), family (Poaceae), and finally superorder (Lilianae or monocotyledons): African rice (O. glaberrima Steud.), pearl millet (Cenchrus americanus (L.) Morrone [previously known as Pennisetum glaucum (L.) Leeke]), fonio (Digitaria exilis Stapf), and three species of palms (Podococcus acaulis Hua, Raphia textilis Welw., and Phytelephas aequatorialis Spruce) (Table 1, Appendix 1).
Table 1.
Output data obtained from MinION plastome‐enriched library sequencing
| Species | DNA | Probe origin | Total no. of reads (bp) | Median read length (bp) | Longest read (bp) | % of plastome readsa | X‐fold | Longest plastome read (bp) | Median plastome read length (bp) |
|---|---|---|---|---|---|---|---|---|---|
| Oryza sativa | Fresh | Oryza sativa | 17,129 | 4627 | 26,128 | 70.8 | 5.3 | 25,828 | 4264 |
| Oryza glaberrima | Fresh | Oryza sativa | 81,361 | 3695 | 24,804 | 98.2 | 12.4 | 24,504 | 3398 |
| Cenchrus americanus | Fresh | Oryza sativa | 105,760 | 4914 | 25,468 | 97.0 | 156.3 | 25,167 | 4623 |
| Digitaria exilis | Fresh | Oryza sativa | 141,250 | 3783 | 19,378 | 94.4 | 13.8 | 19,078 | 3489 |
| Podococcus acaulis | Silica gel | Podococcus barteri | 202,924 | 2486 | 13,103 | 15.7 | 25.0 | 12,805 | 2129 |
| Raphia textilis | Silica gel | Podococcus barteri | 83,833 | 2322 | 10,705 | 87.5 | 94.8 | 10,405 | 1997 |
| Phytelephas aequatorialis | Silica gel | Podococcus barteri | 202,925 | 2437 | 15,132 | 79.0 | 21.6 | 14,832 | 2158 |
The percentage of plastome mapped reads was calculated using Burrows–Wheeler alignment to indicated reference plastomes.
DNA was extracted from fresh leaves for O. sativa, O. glaberrima, C. americanus, and D. exilis; while silica gel–dried leaves were used for DNA extraction for P. acaulis, R. textilis, and P. aequatorialis. In both cases, DNA extraction was performed using a MATAB lysis buffer (Sigma‐Aldrich, St. Louis, Missouri, USA) and chloroform isoamyl alcohol (24 : 1) purification method following Mariac et al. (2006).
General probe design
Long‐fragment plastome sequences were captured from the total genomic DNA extracts using two different sets of biotinylated probes: one based on O. sativa and used on related Poaceae species (O. glaberrima, C. americanus, D. exilis) and one based on Podococcus barteri G. Mann & H. Wendl. and used for P. acaulis, R. textilis, and P. aequatorialis. Podococcus barteri is the sister species to P. acaulis, whereas R. textilis and P. aequatorialis are distantly related to P. barteri, being in two different subfamilies (Calamioideae and Ceroxyloideae, respectively). Probe production (Fig. 1; see Appendix 2 for a detailed protocol) was undertaken following the protocol described elsewhere (Cronn et al., 2012; Mariac et al., 2014) and led to an average probe size of 300 bp. First, an initial full‐length plastome was amplified by long‐range PCR (LR‐PCR) using 11 primer pairs taken from Scarcelli et al. (2011) for O. sativa (Appendix S1), and another set of 11 primer pairs taken from Faye et al. (2016) for P. barteri (Appendix S1). LR‐PCR was carried out using the LongAmp Taq PCR kit (#E5200S; New England BioLabs, Ipswich, Massachusetts, USA) following the manufacturer's instructions in a final volume of 50 μL and using 300 ng of DNA. For each probe set, LR‐PCR amplicons were pooled at an equimolar ratio and sheared to reach a mean size fragment of 300 bp, then ligated to adapters for PCR amplification with biotinylated primers.
Figure 1.
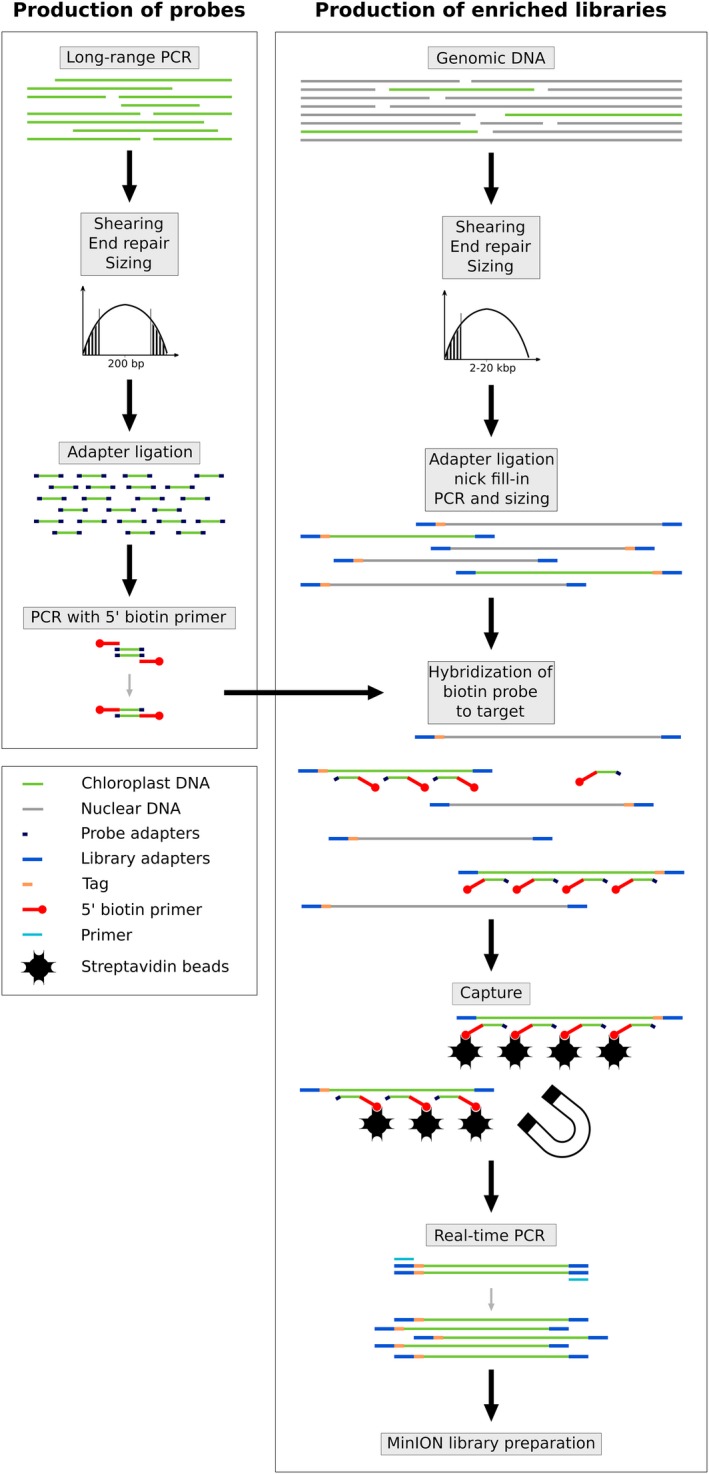
Schematic representation of the protocol used for long‐fragment capture of plastomes (adapted from Mariac et al., 2014).
Library preparation, in‐solution hybridization, multiplexing, and sequencing
Illumina libraries were constructed following the protocol of Rohland and Reich (2012) using 6‐bp barcodes and Illumina indices, with extra steps added to allow for amplification and in‐solution hybridization (Fig. 1, Appendix S2). Briefly, each high‐molecular‐weight DNA was sheared using a g‐TUBE (Covaris, Woburn, Massachusetts, USA) to a mean target size of 10 kbp. DNA fragments less than 2000 bp were removed by a sizing step performed with 0.4× AMPure magnetic beads (Beckman Coulter, Beverly, Massachusetts, USA). DNA was then end‐repaired, ligated with adapters (allowing PCR amplifications), and then nick filled‐in before performing a pre‐hybridization PCR. Optimal cycle number (ranging from five to 12) was defined by real‐time amplification (KK2700; KAPA Biosystems, Roche Sequencing and Life Science, Bâle, Switzerland). After clean‐up and quantification using the NanoQuant Plate (Tecan Group Ltd., Männedorf, Switzerland) and QIAxcel (QIAGEN, Valencia, California, USA), library preparations were mixed with biotin‐labeled probes for hybridization of the targeted regions. DNA probe hybridization complexes were then immobilized with 100 μg of streptavidin‐coated magnetic beads. This step was performed using the Dynabeads M‐280 kilobaseBINDER Kit (#60101; Invitrogen, ThermoFisher Scientific, Waltham, Massachusetts, USA), which is designed for immobilizing double‐stranded DNA molecules longer than 2 kbp.
We then prepared the DNA to create the MinION library. A magnetic field was applied to the resulting solution, and the supernatant containing unbounded DNA was discarded. Enriched DNA fragments were then dehybridized from the beads and amplified in 12 to 15 cycles of real‐time PCR in order to obtain the requested quantity for the Nanopore library preparation. The final libraries were then constructed following the ONT MinION library preparation detailed in the 1D Amplicon by ligation (SQK‐LSK108; ONT) protocol for single samples and also in the 1D Native barcoding genomic DNA (with EXP‐NBD103 and SQK‐LSK108) protocol. Briefly, 1 μg of enriched DNA was end‐repaired, extended with a dA‐tailing, ligated with Nanopore barcodes, and then ligated with Nanopore tether‐adapter before loading and sequencing on the MinION flow cell. To benefit from multiplexing and to limit costs and workload, up to four individuals were pooled at an equimolar ratio using ONT barcodes. Prior to each run, flow cells (FLO‐MIN106, R9.4 Version; ONT) were quality tested using MinKNOW software (version 1.2.8; ONT) to ensure the presence of at least 50% (256) active channels. Flow cells were loaded with approximately 275 ± 100 fM capture‐amplified DNA libraries. All costs and product details are provided in Appendix S3.
Non‐enriched MiSeq data
To estimate enrichment rate, we used single‐sample non‐enriched library data sets originating from various Illumina MiSeq sequencing runs for O. sativa, O. glaberrima, C. americanus, and P. aequatorialis. We used MiSeq data here because its higher yield (compared to MinION) allows a better estimation, at a lower cost, of the percentage of plastid reads of a non‐enriched library. For D. exilis, P. acaulis, and R. textilis, we merged 10, two, and 16 samples, respectively, of non‐enriched libraries to provide adequate read counts. Forward sequencing read outputs from each MiSeq run (i.e., R1 files) were first demultiplexed using the demultadapt script (https://github.com/Maillol/demultadapt) to sort reads according to a given barcode list. Adapters at the beginning of each read from the R2 and demultiplexed R1 files were removed using cutadapt‐1.2.1 software (Martin, 2011) with the default parameters. Reads were then filtered by length (size >35 bp) and mean quality values (Q > 30) before being paired using compare_fastq_paired_v5.pl (https://github.com/SouthGreenPlatform/arcad-hts/blob/master/scripts/arcad_hts_3_synchronized_paired_fastq.pl and https://github.com/SouthGreenPlatform/arcad-hts/blob/master/scripts/arcad_hts_2_Filter_Fastq_On_Mean_Quality.pl). A final trimming step using the fastx‐trimmer command from the FASTX‐Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) was undertaken onto the R2 paired files to remove the last six bases of each read to ensure removal of any possible barcode present on short reads.
Bioinformatics
All command lines are available in Appendix S4. Using the MinION Fast5 output format, base‐calling and demultiplexing were undertaken using Albacore v2.5.11 (https://github.com/Albacore/albacore). This generated a FASTQ file from which reads were filtered out. The average quality score was lower than 7. For each barcode, a quality control using the MinionQC R script (https://github.com/roblanf/minion_qc) was performed to check for read mean length and quality scores. Reads were then trimmed using Porechop (https://github.com/rrwick/Porechop) in order to remove the sequencing adapters and barcodes. The only non‐default setting is that splitting reads containing adapters in the middle was disabled, in order to avoid issues during the polishing step using Nanopolish (see below).
For each library, the percentage of plastome‐associated reads was estimated by mapping reads to a reference plastid genome using the Burrows–Wheeler alignment tool (BWA‐MEM, https://github.com/lh3/bwa) with the “‐B 1” option for non‐enriched short‐read data and the “‐x ont2d” option for long‐read data (Li and Durbin, 2009). We then calculated the X‐fold enrichment to evaluate capture efficiency (the ratio of plastome reads obtained with capture relative to plastome reads obtained without capture). Coverage and depth values were calculated using Bedtools (Quinlan and Hall, 2010) genomecov (https://github.com/arq5x/bedtools2). Mismatch percentage values between mapped reads and references were recovered using Tablet version 1.17.08.17 (Milne et al., 2010).
De novo assembly of plastid genomes
We used the Flye assembler version 2.3 (Kolmogorov et al., 2019) for de novo assembly of plastomes based on long MinION reads. For O. sativa, all available reads (17,129) were assembled. For the other species, the number of reads was too high, in excess of 3000× the reference coverage for some data sets, which caused memory usage issues. To alleviate this, the reads were randomly split into sets of approximately equal size. Each set was then assembled individually using the raw Nanopore reads mode. The “min_overlap” parameter (i.e., the minimum overlap between reads) in Flye was adjusted on a species‐by‐species basis ranging from 3000 bp (the default value for our genome size) to 1000 bp, depending on the medium read length for each species. This was done in order to ensure that a sufficient amount of overlaps were detected for the assembly. The draft assemblies were then polished using Nanopolish version 0.9.1 (https://github.com/jts/nanopolish), using minimap2 on the “map‐ont” preset for the overlapping step. Finally, the assemblies were mapped on the reference sequence of each species using the dnadiff tool of MUMmer version 4.0beta2 (Kurtz et al., 2004), which directly provides alignment coordinates and global statistics such as the mean identity percentage of alignments.
Although read length is an important consideration, the uniformity of reference coverage by the reads can also affect plastome assembly. This is especially problematic for low‐molecular‐weight DNA extractions resulting in shorter read lengths on average (as happened in our study comparing extractions from silica gel–dried leaves vs. from fresh tissue). To test for the impact of the uniformity of reference coverage on the assembly, simulated reads for P. aequatorialis (using DNA extracted from silica gel–dried leaves) were generated using NanoSim v2.1.0 (Yang et al., 2017). A model was first trained on the raw real reads and then 40,000 simulated reads were generated, ensuring they have approximately the same length distribution and error model as the real reads (see Results). The simulated reads were then assembled using the same workflow described above.
RESULTS
Plastome enrichment protocol validation on Oryza sativa
All raw reads for the seven species are available from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (BioProject number PRJNA526996). After read filtering (Q > 7), the median length of the 12,227 mapped plastome reads was 4264 bp (Table 1, Fig. 2B). We recovered the whole plastome with an average coverage depth of 364× for the enriched MinION library, with a standard deviation increasing from 0.25 to 0.37 between enriched and non‐enriched libraries (Fig. 2B, C; Appendix 1). The average mismatch was 11.80% (Appendix 1), and 70.8% of the reads mapped to the reference plastome (Fig. 2D, Appendix 1), representing an approximately fivefold increase in plastome reads when compared to the non‐enriched MiSeq sequenced library (13.32% mapped; Appendix 1). The longest plastome read recovered was 25,828 bp long (Table 1).
Figure 2.
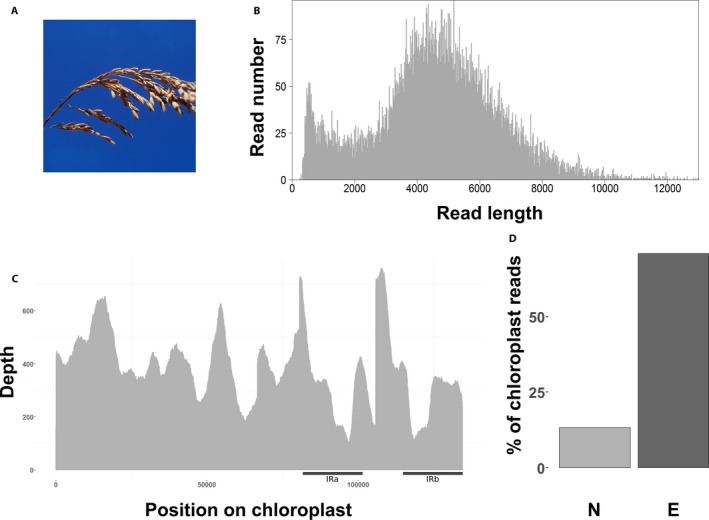
Long‐fragment capture results for Oryza sativa. (A) Panicule of Oryza sativa (© IRD ‐ Jean‐Pierre Montoroi. Reprinted with permission.). (B) Number of reads per read length before mapping. (C) Plastome coverage after mapping. Black bars indicate approximate position of both inverted repeat (IR) regions. (D) Percentage of useful reads mapped to the O. sativa reference plastome (KT289404.1) between non‐enriched (N, light gray) and enriched (E, dark gray) libraries.
Plastome enrichment protocol applied to non‐model species
DNA extraction qualities were variable depending on the source of the leaf material used. DNA extracted from fresh tissue always produced single bands (not degraded) with fragments higher than 20 kbp (Appendix 1). In contrast, DNA extracted from silica gel–preserved material was of lower quality and generally degraded (smear present), with fragments less than 20 kbp long (Appendix 1). For the six non‐model species, sequencing of the non‐enriched libraries resulted in 0.63–7.94% of plastome reads (Appendix 1). In contrast, enriched libraries resulted in 15.7–98.2% plastome reads, corresponding to a 12‐fold to 156‐fold increase in plastome DNA sequences (Fig. 3, Table 1, Appendix 1). The mean average of fragments sequenced from DNA extracted from fresh tissue was 4279 bp versus 2525 bp for DNA extracted from silica gel–dried leaves (Fig. 4A). Sequences mapped to the reference plastomes ranged mainly from 2 kbp to 8 kbp, depending on the species (Fig. 4A). Average coverage depth was 1988× for enriched libraries (Fig. 4B, Appendix 1). The longest read mapped to the plastome ranged from 10,405 to 25,167 bp for R. textilis and C. americanus, respectively (Table 1).
Figure 3.
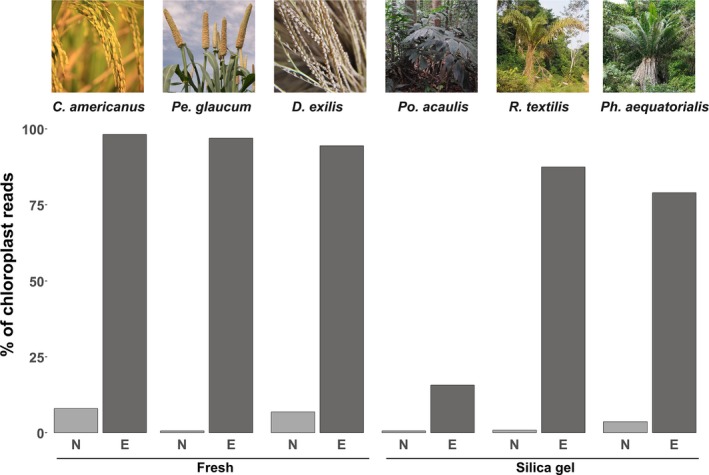
Percentage of useful reads mapped to their respective reference plastome (see Table 1) between Illumina non‐enriched (N, light gray) and MinION enriched (E, dark gray) protocols for the six non‐model species in our study. Photo credits: Cenchrus americanus (CC0 Public Domain; https://pxhere.com/fr/photo/706162); Pennisetum glaucum (© IRD ‐ Cédric Mariac, reprinted with permission); Digitaria exilis (© IRD ‐ A. Barnaud, reprinted with permission); Podococcus acaulis (© IRD ‐ Thomas Couvreur, reprinted with permission); Raphia textilis (© IRD ‐ Thomas Couvreur, reprinted with permission); Phytelephas aequatorialis (© IRD ‐ Thomas Couvreur, reprinted with permission).
Figure 4.
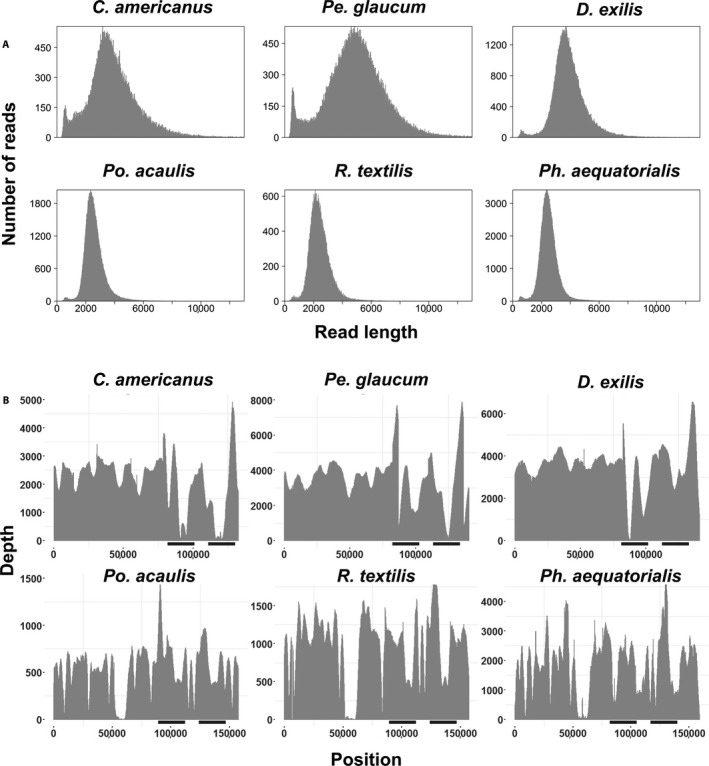
Long‐fragment capture results for six non‐model plant species. (A) Distribution of read lengths before mapping. (B) Plastome coverage results from the enriched long‐read capture protocol. Black bars indicate approximate position of both inverted repeat (IR) regions.
De novo assembly of the plastid genome
When DNA was extracted from fresh leaves, the plastome was assembled in two contigs covering most of the reference (Table 1, Appendix S5 for a visual example in C. americanus). Assembled contig lengths varied from 81,053 to 12,5727 bp long. However, the assembler never managed to fully recover the circular plastid genome throughout a single contig. For DNA extracted from silica gel–dried leaves, where reads were shorter and the coverage more heterogeneous, assembly was suboptimal (Table 1, Appendix S6 for a visual example in P. aequatorialis), with more final contigs (10–17), uncovered regions, and sometimes misassemblies. The longest assembled contigs were also much shorter than those from fresh material (Table 2). In addition, the IR regions were also often not differentiated.
Table 2.
De novo assembly results from real and simulated data in the number of contigs, and coverage and identity percentages to the respective reference plastome genomes (see Table 1)
| Species | Minimum overlap (bp)a | Plastome contigs | Coverage % | Identity % | Longest contig (bp) |
|---|---|---|---|---|---|
| Oryza glaberrima | 3000 | 2 | 92.32 | 99.14 | 109,087 |
| Cenchrus americanus | 3000 | 2 | 99.91 | 98.52 | 81,053 |
| Digitaria exilis | 3000 | 2 | 99.97 | 99.18 | 125,727 |
| Podococcus acaulis | 1000 | 17 | 81.24 | 98.86 | 22,803 |
| Raphia textilis | 1000 | 10 | 83.87 | 98.84 | 21,797 |
| Phytelephas aequatorialis | 1000 | 13 | 87.60 | 98.31 | 20,700 |
| Simulated assemblyb | 1000 | 4 | 99.72 | 99.05 | 107,633 |
Minimum overlap between reads as defined in Flye.
The simulated data were based on the output results of Phytelephas aequatorialis.
Using a simulated data set of reads uniformly distributed across the plastome (Appendix S7) and based on the same quality as P. aequatorialis significantly improved assembly (Table 2). The assembler resulted in four contigs (vs. 13) covering 99.72% (vs. 87.60%) of the reference, and the longest contig was 107,633 bp (vs. 21,797 bp). However, the existence of two distinct repeated regions was still not resolved.
DISCUSSION
We show that targeted capture hybridization of long plastome DNA fragments with sufficient coverage (362× to 3318×) is possible in plants (Table 1, Appendix 1). In addition, we show a significant enrichment of our target region (the plastome) when compared to non‐enriched data (Figs. 2D, 3; Appendix 1). The different steps of our protocol (Fig. 1, Appendix 2) are not fundamentally different from previous plastome short‐read capture protocols (e.g., Mariac et al., 2014) based on in‐house probe preparation, shearing, adapter ligation, hybridization, and finally capture (Fig. 1, Appendix 2). Thus, our approach requires minimal adaptation from previous cost‐ and time‐effective protocols and should therefore be of broad interest. The main technical change focused on the beads used to capture long DNA fragments. For that, we used the kilobaseBINDER Kit (Invitrogen), which is said to capture DNA fragments longer than 2 kbp. The sizing step we performed at 0.4× using AMPure removes fragments smaller than 2 kbp and corresponds to the maximum allowed size with the AMPure beads. However, other approaches are possible to achieve sizing with higher molecular weight and could be tested (e.g., gel extraction, automated size selection system). Based on our protocol and costs provided in Appendix S3, we estimate a cost of €33.77 (US$42.00) per individual to sequence a plastome at 30× (costs estimated February 2019). We stress, however, that this value is only an estimate, and the aim of our protocol was not focused on cost effectiveness.
When capturing plastomes across a range of different species, we find a difference in enrichment percentage ranging from 15.7% to 98.2% of useful reads (X‐fold: 12.4–156.3; Table 1, Fig. 4). Differences in genome versus plastome ratios between species might explain the variation of on‐target mapped reads percentage compared to non‐enriched libraries. In general, our results suggest that species with smaller genomes show higher mapped read percentages, although this trend should be confirmed with more data and specific tests. Alternatively, the quality of the material used for DNA extractions (e.g., the cellular type and the degradation state) can also explain such variations. The low enrichment observed for P. acaulis (Table 1, Fig. 3) could potentially be linked to a large genome size, although we do not have an estimate of its genome.
A common coverage gap is observed among the plastid genomes of the three palm species because of a region that was not covered by the probes (see Faye et al., 2016). Lower coverage of other regions can be explained by biases that occur during DNA shearing, PCR amplification, and hybridization capture, considering a CG content effect. Probe bulk normalization from long‐range PCR also has to be taken into account. However, we obtained a decreased standard deviation of the whole target coverage homogeneity (Appendix 1), suggesting that our protocol did not introduce more on‐target coverage heterogeneity. Nevertheless, applying an alternative capture method such as region‐specific extraction (Dapprich et al., 2016) could help maintain overall good coverage by accessing highly complex, variable, repeat‐masked, or unknown regions that prohibit adequate probe binding.
Probes were designed to hybridize across the entire targeted region (Fig. 1), as is generally done using short‐read approaches (Stull et al., 2013; Mariac et al., 2014). However, a recent study showed that probes targeting small regions are also effective in capturing long reads surrounding the targeted region. Indeed, Gasc and Peyret (2017) were able to reconstruct a 21.6‐kbp fragment using probes designed for a small 471‐bp microbial gene target. This shows that long‐read capture will also be very useful for targeted sequence capture of nuclear regions.
We demonstrated the capacity of heterologous plastome probes to capture target DNA in other species or genera in Arecaceae and Poaceae. For example, probes designed on P. barteri hybridized well to other palm genera in different subfamilies. This underlines the good portability of probes for capturing plastomes across a broad evolutionary spectrum (Stull et al., 2013), even for long‐fragment capture.
Limits and challenges
Although we were able to successfully capture long plastome fragments using our enrichment protocol, assembling plastomes from these data remains challenging. Indeed, the best assembly resulted in two mapped contigs, and the worst assembly resulted in 17 (Table 2). Assembly of plastomes is well known to be problematic (Twyford and Ness, 2017), mainly because of the presence of near identical IR regions. Indeed, the similarity of the two IR regions is too high for assemblers to decipher between IR regions when resolving the assembly graph for the entry and exit point of those sequences. Thus, when the sequenced reads are shorter than the IR regions themselves, it becomes difficult to correctly assemble the plastome into a single contig. This can be seen, for example, in C. americanus (Appendix S5), where the resulting two contigs do not completely cross with one of the IR regions, thus failing to reach a single contig. Of course, this problem is amplified when dealing with overall shorter reads sequenced from low‐molecular‐weight DNA (see Appendix S6 for an example). In our case, DNA fragments from silica gel–dried leaves were shorter than those extracted from fresh leaves (Table 1). Moreover, we observed a decrease of the average library fragment size during the preparation steps and after PCR because of preferential amplification of shorter fragments, as observed by Giolai et al. (2016) and Eckert et al. (2016).
Optimizing read length in such a way that single reads are longer than the entire IR region should significantly help in the assembly process. In this sense, DNA shearing could be removed in order to increase the average size of the reads. Technical limitations would, however, include (1) the ability of streptavidin beads to immobilize fragments of tens of thousands of base pairs and (2) the long‐range PCR amplification step of the enriched fragments, which is necessary to produce an input of several hundred nanograms for the construction of Nanopore libraries. The latter is probably the most limiting because it is difficult to produce amplicons more than 10 kbp long, and even if this is achieved, representation bias must be considered. Finally, we show, via simulations, that the uniformity of read coverage across the reference is important for assembly (Table 2). Indeed, uniformly distributed reads, even of lower quality, lead to better assemblages than poor coverage of the reference (Table 2). Therefore, uniform coverage of the reference by the captured reads plays a significant role in the correct and improved assembly even for suboptimal DNA extractions.
A final concern, which is not restricted to long‐read sequencing, is the presence of plastid DNA in the nuclear genome (i.e., nuclear plastid DNAs [NUPTs]). The vast majority of NUPTs are of small size (<1000 bp; Yoshida et al., 2014) and thus are normally not captured and sequenced using our protocol (we sized libraries for reads >2 kbp, see Appendix 2). Longer NUPTs up to several kilo base pairs long have also been reported (Yoshida et al., 2014). However, differentiating these NUPTs from true plastid DNA is difficult because of their length and because they are generally associated with low divergence levels (p‐distance <0.01%; Yoshida et al., 2014). It is thus possible that our protocol does capture NUPTs longer than 2 kbp, influencing our enrichment efficiency (the X‐fold value). However, this did not affect our de novo assembly of the Oryza plastome (99.14% identity between the de novo assembly and the reference plastome), even though rice contains many NUPTs, including long ones (Yoshida et al., 2014).
AUTHOR CONTRIBUTIONS
C.M., Y.V., and T.L.P.C. conceived the idea; R.M., T.L.P.C., C.M., and Y.V. provided material; C.M., Y.V., J.F.M., S.S., and M.A. designed the protocol; K.B., C.M., and M.C. undertook the experiments; C.M., K.B., F.S., N.S., and V.K. analyzed the data. K.B. and T.L.P.C. led the writing, and all authors read and commented on the final version.
Supporting information
APPENDIX S1. Both sets of long‐range PCR primers, elongation times, and annealing temperature values used.
APPENDIX S2. Raw output data of the three MinION runs undertaken in our study.
APPENDIX S3. Detailed list of materials and costs to implement the capture protocol.
APPENDIX S4. Bioinformatic codes used for analyses of MinION data.
APPENDIX S5. Mummer visualization of assembled long reads for Cenchrus americanus.
APPENDIX S6. Mummer visualization of assembled long reads for Phytelephas aequatorialis.
APPENDIX S7. Mapping of Phytelephas aequatorialis–like simulated reads (using LAST software [http://last.cbrc.jp/]) to the reference plastome NC_029957.1.
ACKNOWLEDGMENTS
This work was publicly funded through ANR (the French National Research agency) under the “Investissement d'Avenir” program (reference ANR‐10‐LABX‐001‐01 Labex Agro) coordinated by the Agropolis Fondation. The authors thank Oliver Lucas from Oxford Nanopore Technologies and Christian‐Julian Villabona‐Arenas for help and advice. The exportation and use of the Phytelephas aequatorialis sample was authorized by the Ministerio del Ambiente of Ecuador (permit MAE‐DNB‐CM_2018‐0082); the exportation and use of the Podococcus barteri and P. acaulis samples was authorized by CENAREST of Gabon (permits AR0020/16, AR0036/15). We thank Dr. Ya Yang, two anonymous reviewers, and Beth Parada (managing editor, Applications in Plant Sciences) for comments that improved the manuscript.
APPENDIX 1. Details of accessions/voucher information, DNA extraction, reference genomes used, and sequencing results for the seven species used in this study.
| Species | Country of origin | Voucher/accession no. | DNA quality | Type of library preparation | Probes used | No. of passed reads | Reference | % reads mapped reference | Average coverage depth | Standard deviation | % mismatch | % reference uncovered | % 10× | % 50× |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Oryza sativa | Japan | (no voucher) NIP – IRD Montpellier | Band, >20 kbp | Chloroplast enrichment | Oryza sativa | 17,129 | KT289404.1 | 70.80 | 362.69 | 0.36 | 11.80 | 0.000 | 100.000 | 99.996 |
| Shotgun | None | 151,234 | KT289404.1 | 13.32 | 34.15 | 0.25 | 0.20 | 0.000 | 99.898 | 4.146 | ||||
| Oryza glaberrima | Senegal | (no voucher) CG14 – IRD Montpellier | Band, >20 kbp | Chloroplast enrichment | Oryza sativa | 81,361 | KM088021.1 | 98.23 | 2033.68 | 0.43 | 9.60 | 0.019 | 99.934 | 99.295 |
| Shotgun | None | 1,201,104 | KM088021.1 | 7.94 | 99.56 | 0.44 | 1.80 | 0.034 | 99.546 | 88.834 | ||||
| Pennisetum glaucum | Senegal | (no voucher) PE01455 – IRD Montpellier | Band, >20 kbp | Chloroplast enrichment | Oryza sativa | 105,757 | KJ490012.1 | 97.03 | 3318.44 | 0.39 | 10.20 | 0.000 | 99.992 | 99.981 |
| Shotgun | None | 525,428 | KJ490012.1 | 0.62 | 3.19 | 0.70 | 2.20 | 10.150 | 0.950 | 0.000 | ||||
| Digitaria exilis | Mali | (no voucher) CM05784 – IRD Montpellier | Band, >20 kbp | Chloroplast enrichment | Oryza sativa | 141,248 | NC_024176.1 | 94.42 | 3311.87 | 0.29 | 11.40 | 0.000 | 100.000 | 99.700 |
| Shotgun | None | 8,665,102 | NC_024176.1 | 6.85 | 593.29 | 0.33 | 0.50 | 0.045 | 99.846 | 99.462 | ||||
| Podococcus acaulis | Gabon | Couvreur TLP 556 (WAG) | Band, 15–20 kbp | Chloroplast enrichment | Podococcus barteri | 249,416 | NC_027276.1 | 15.72 | 497.68 | 0.47 | 10.20 | 0.454 | 96.163 | 93.096 |
| Shotgun | None | 308,230 | NC_027276.1 | 0.63 | 1.14 | 2.63 | 4.60 | 56.858 | 0.597 | 0.085 | ||||
| Raphia textilis | Angola | Lautenschläger 1086 (K) | Slight degradation, 5–16 kbp | Chloroplast enrichment | Podococcus barteri | 83,832 | NC_020365.1 | 87.49 | 893.98 | 0.47 | 12.00 | 0.352 | 96.068 | 91.972 |
| Shotgun | None | 57,007,740 | NC_020365.1 | 0.92 | 381.41 | 3.77 | 6.20 | 0.200 | 99.018 | 98.258 | ||||
| Phytelephas aequatorialis | Ecuador | Couvreur 1191 (QCA) – TAGUA F1 | Smear, 1–10 kbp fragments | Chloroplast enrichment | Podococcus barteri | 202,924 | NC_029957.1 | 79.04 | 1713.58 | 0.53 | 10.30 | 0.004 | 99.522 | 96.383 |
| Shotgun | None | 699,918 | NC_029957.1 | 3.65 | 22.68 | 1.58 | 3.50 | 0.000 | 95.083 | 1.380 |
APPENDIX 2. Protocols for long‐fragment capture of plastomes.
Protocol 1: Construction of rice chloroplast based on biotinylated probes (adapted from Mariac et al., 2014)
Step 1: Long‐range PCR (LR‐PCR) is performed to amplify chloroplast regions of the selected species using 300 ng of DNA, 3 μL of 5 mM dNTPs, 4 μL of 10 μM forward/reverse primer mix, 10 μL of 5× reaction buffer, and 2 μL of LongAmp Taq DNA Polymerase (#M0323 [#E5200 for the whole kit]; New England BioLabs, Ipswich, Massachusetts, USA) in a final volume of 50 μL and incubated in a thermocycler*1 (T‐1 Thermoblock; Biometra, Dublin, Ireland) for an initial 30 s at 94°C; followed by 35 cycles of 30 s at 94°C, 1 min at annealing temperature (T a), and 18 min at 65°C; with a final extension of 10 min at 65°C. In our case, 11 fragments from ca. 7–18 kbp were amplified to cover a majority of the chloroplast. Amplicon size is checked on a TAE 1×, 0.8% agarose gel electrophoresis via a 30‐min run at 100 V using an adapted size control. Primers and elongation temperature are provided in Appendix S1.
Step 2: The amplicons resulting from LR‐PCR are purified using Agencourt AMPure XP magnetic beads (#A63881; Beckman Coulter, Beverly, Massachusetts, USA) by adding and mixing 1.8× volume of AMPure XP to the PCR products. After 5 min of incubation at room temperature, the supernatant is removed and two 70% ethanol washes are performed. Air‐dried beads are resuspended in 50 μL of DNase/RNase‐free water. Concentration measurement is realized on a NanoQuant Infinite M200 (Tecan Group Ltd., Männedorf, Switzerland) so purified amplicons can be equimolarly bulked.
Step 3: Approximately 2–10 μg of DNA bulk are diluted in 100 μL of water and sheared using 0.65‐mL microtubes and a Bioruptor Pico (#B01060001; Diagenode, Denville, New Jersey, USA) with the target peak set to 200 bp: 13 cycles of 30 s ON/30 s OFF. Sheared DNA size is checked on a QIAxcel 12‐channel capillary electrophoresis system using the QIAxcel DNA Screening Kit (2400) (#929004; QIAGEN, Hilden, Germany).
Step 4: Sheared DNA is blunted and 5′ phosphorylated using the NEBNext End Repair Module Kit (#E6050; New England BioLabs) by adding 5 μL of 10× End Repair Reaction Buffer and 5 μL of End Repair Enzyme Mix in a final volume of 50 μL. The mix is incubated for 30 min at room temperature (22°C), and the reaction is stopped with an AMPure XP 2× clean‐up step. DNA concentration is checked on NanoQuant Infinite M200 to ensure enough material is available for downstream steps.
Step 5: Adapter oligonucleotide hybridization is performed using 2 nM (20 μL/100 μM) of linker 1 (5′‐AGAAGCTTGAATTCGAGCAGTCAG‐3′ with 5′ phosphate modification), 2 nM (20 μL/100 μM) of linker 2 (5′‐CTGCTCGAATTCAAGCTTCT‐3′), 5 μL of 100 mM Tris‐HCl (pH 8), and 5 μL of 100 mM NaCl in a final volume of 50 μL and incubated in a thermocycler*1 for an initial 2 min at 97°C, followed by 72 cycles of 1 min at 97°C with −1°C for each cycle, and a final 5 min at 25°C. Hybridized adapters are stored overnight at 4°C to reach greater ligation efficiency thereafter. Then 0.8 pM (100 ng/200 bp) of DNA fragments are ligated with 16 pM (4 μL/4 μM) of the adapter and 2 μL of 5 U/μL T4 DNA ligase (#EL0011; Thermo Fisher Scientific, Vilnius, Lithuania) in a final volume of 20 μL for 2.5 h at 22°C, followed by a heat inactivation step at 65°C for 10 min. A clean‐up step is performed with a AMPure XP 2× purification.
Step 6: PCR biotinylation is performed using ca. 100 ng of adapter‐ligated DNA, 2 μL of 25 μM 5′ TEG‐biotinylated linker 2, 25 μL of 2× KAPA HiFi HotStart ReadyMix (#KM2605; KAPA Biosystems, Roche Sequencing and Life Science, Bâle, Switzerland) in a final volume of 50 μL and incubated in a thermocycler*1 for an initial 3 min at 95°C; followed by 35 cycles of 20 s at 98°C, 15 s at 55°C, and 10 s at 72°C; with a final extension of 10 min at 72°C. The amplicons are then purified by adding and mixing 2× volume of AMPure XP to the PCR products. After 5 min of incubation at room temperature, the supernatant is removed and two 70% ethanol washes are performed. Air‐dried beads are resuspended in 30 μL of DNase/RNase‐free water. Concentration and average size of amplicons are checked on NanoQuant Infinite M200 and QIAxcel. Effective biotinylation is controlled by using a streptavidin‐binding test: streptavidin‐linked probes will remain on magnetic beads while the supernatant is removed and checked on agarose gel electrophoresis in comparison with purified probes.
Step 7: The average probe length can be determined by adding and mixing 0.7× volume of AMPure XP to the biotinylated probes. After 5 min of incubation at room temperature, the beads are discarded and an additional 2× AMPure XP volume is added and mixed to the supernatant. After another 5 min at room temperature, the supernatant is removed and two 70% ethanol washes are performed. Air‐dried beads are then resuspended in 20–25 μL of DNase/RNase‐free water. Concentration and average size of amplicons are checked on NanoQuant Infinite M200 and QIAxcel.
Protocol 2: Construction of an enriched library based on Rohland and Reich (2012), adapted for long fragments (2–20 kbp)
Library preparation follows the published protocol of Rohland and Reich (2012) but with the inclusion of five additional steps (steps 6–10) for the enrichment procedure (biotinylated probe capture) and some modifications including fragment sizing and a final Nanopore MinION library multiplex preparation.
Step 1 (optional): For each individual, approximately 1–4 μg of total DNA are diluted in 80 μL of water and sheared using a g‐TUBE (Covaris, Woburn, Massachusetts, USA) by a two‐way centrifugation 2 × 1 min at 6000 rpm, making sure that the entire volume passes through the thin membrane both ways. The rotor speed selection may be modified depending on the targeted size and the DNA mass. Sheared DNA size is checked on a TAE 1×, 0.8% agarose gel electrophoresis via a 30‐min run at 100 V using an adapted size control. Sheared DNA size is checked on a QIAxcel 12‐channel capillary electrophoresis system using the QIAxcel DNA Screening Kit (2400) (#929004; QIAGEN).
Step 2: If needed or for short and/or degraded samples, a single fragment size selection can be performed using Agencourt AMPure XP magnetic beads by adding and mixing 0.4× volume of AMPure XP to the DNA. After 5 min of incubation at room temperature, the supernatant is removed and two 70% ethanol washes are performed. Air‐dried beads are resuspended in 50 μL of DNase/RNase‐free water.
Step 3: Sheared DNA is blunted and 5′ phosphorylated using the NEBNext End Repair Module Kit (#E6050; New England BioLabs) by adding 5 μL of 10× End Repair Reaction Buffer and 5 μL of End Repair Enzyme Mix in a final volume of 50 μL. The mix is incubated for 30 min at room temperature (20°C), and the reaction is stopped with an AMPure XP 0.5× clean‐up step. DNA concentration is checked on NanoQuant Infinite M200 to ensure enough material is available for downstream steps.
Step 4: A total of 50–100 fM (300 ng/5–10 kbp) of DNA fragments are ligated with 8 pM (2 μL/4 μM) of PE‐P7, 8 pM (2 μL/4 μM) of barcoded P5 adapters using hexamer barcodes (Rohland and Reich, 2012), and 2 μL of 5 U/μL T4 DNA ligase and 2 μL of 10× T4 Buffer (#EL0011 and #B69; Thermo Fisher Scientific) in a final volume of 20 μL for 2.5 h at 22°C followed by a heat inactivation step at 65°C for 10 min. PE‐P7 can be replaced by MPE‐P7 in order to add indices subsequently. A clean‐up step is performed with an AMPure XP 1× purification.
Step 5: A nick fill‐in step is performed using 2 μL of 8 U/μL Bst DNA polymerase (#M0275; New England BioLabs), 3 μL of 10× ThermoPol Reaction Buffer (New England BioLabs), and 1.5 μL of 5 mM dNTPs in a final volume of 30 μL. The mix is incubated in a thermocycler*1 for 15 min at 37°C and the reaction is stopped with an AMPure XP 1× clean‐up step. DNA concentration is checked on NanoQuant Infinite M200 to ensure enough material is available for downstream steps.
Enrichment steps
Step 6: A real‐time pre‐hybridization LR‐PCR is performed using 20 μL of DNA, 25 μL of KAPA HiFi HS Real‐Time Master Mix (2×) (#KM2702; KAPA Biosystems), 25 pM (2.5 μL/10 μM) of PreHyb‐PE_F primer (CTTTCCCTACACGACGCTCTTC), and 25 pM (2.5 μL/10 μM) of PreHyb‐PE_R primer (CTCGGCATTCCTGCTGAACC) following Rohland and Reich (2012) in a final volume of 50 μL and incubated in a LightCycler 480 Instrument II (Roche Molecular Systems, Bâle, Switzerland) for an initial 45 s at 98°C, followed by 55 cycles of 15 s at 98°C, 30 s at 62°C, and 10 min at 72°C, and ended by a single point fluorescence acquisition. PCR is monitored and stopped after optimal cycles are reached just before the plateau phase as recommended by the manufacturer. In our case, this varied around 5–12 cycles, depending on the DNA quantity. The amplicons are then purified and size‐selected by adding and mixing 0.38× volume of AMPure to the PCR products. Particular care must be taken with the pipetting accuracy as it is critical that the volume be correctly measured. After 5 min of incubation at room temperature, the supernatant is removed and two 70% ethanol washes are done. Air‐dried beads are resuspended in 25 μL of DNase/RNase‐free water. PreHyb‐PE‐R has to be replaced by PreHyb‐MPE‐R primer (TGACTGGAGTTCAGACGTGTG) if subsequent index addition is intended. DNA concentration is checked on NanoQuant Infinite M200 to ensure enough material is available for downstream steps.
Step 7: In‐solution hybridization capture is carried out using 200 ng of heat‐denatured DNA for 5 min at 95°C, 100 ng of biotinylated probes, 2 μL of 1% SDS, 12 μL of 20× SSC, 1.3 μL of 400 ng/μL BSA, 0.5 μL of 100 μM Univ_Block_P7 primer (AGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG), and 0.5 μL of 100 μM Univ_Block_P5+INOSINE primer (CTTTCCCTACACGACGCTCTTCCGATCTiiiiii) in a final volume of 40 μL and incubated at 65°C for 16–20 h at 800 rpm in a ThermoMixer C (#5382000015; Eppendorf, Hamburg, Germany). Univ_Block_P7 has to be replaced by MPE‐P7 primer (TGACTGGAGTTCAGACGTGTGCTCTTCCGATCT) for further indexation.
Step 8: Biotinylated probes annealed to the DNA targets are then immobilized with 100 μg of streptavidin‐coated beads from the streptavidin‐coupled Dynabeads M‐280 kilobaseBINDER Kit (#60101; Invitrogen, ThermoFisher Scientific) previously washed and resuspended in 40 μL of the manufacturer's binding solution and heated for 5 min at 65°C. Specific care must be used when mixing beads with libraries, and it is advised to homogenize by flicking the tubes instead of pipetting. The complex is then incubated for 2–3 h at 1000 rpm in the ThermoMixer C. Samples are then placed on a magnetic plate to remove the supernatant.
Step 9: Beads are then subjected to two washes with the washing solution and one wash with 1 M Tris‐HCl (pH 8), each wash including a 5‐min incubation step at 65°C. The supernatant is removed, and samples are placed on a magnetic plate as recommended by the manufacturer. Particular care must be taken when pipetting supernatants because beads can easily be caught in the pipette tip.
Step 10: Beads are resuspended in 20 μL of DNase/RNase‐free water and incubated for 10 min at room temperature and 5 min at 95°C to release hybridized DNA fragments. Samples are then immediately placed on a magnetic plate, and the supernatant containing the captured targeted DNA is retained.
Library preparation (end)
Step 11: A real‐time post‐hybridization LR‐PCR is undertaken to extend the adapter sequence and enrich library fragments using 20 μL of DNA, 25 μL of 2× KAPA HiFi HS Real‐Time Master Mix (#KM2702; KAPA Biosystems), 25 pM (2.5 μL/10 μM) of Sol‐PE‐PCR_F primer (AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTC), and 25 pM (2.5 μL/10 μM) of Sol‐PE‐PCR_R primer (CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACC) following Rohland and Reich (2012) in a final volume of 50 μL and incubated in a LightCycler 480 Instrument II (Roche Molecular Systems) for an initial 45 s at 98°C, followed by 55 cycles of 15 s at 98°C, 30 s at 62°C, and 10 min at 72°C, and ended by a single point fluorescence acquisition. PCR is monitored and stopped after optimal cycles are reached just before the plateau phase as recommended by the manufacturer. In our case, this varied 14–22 cycles depending on the DNA quantity after capture. The amplicons are then purified by adding and mixing 2× volume of AMPure to the PCR products. After 5 min of incubation at room temperature, the supernatant is removed and two 70% ethanol washes are performed. Air‐dried beads are resuspended in 25 μL of DNase/RNase‐free water. Sol‐PE‐PCR_R can be replaced here by any desired index primer, such as Sol‐MPE‐IND1_R primer (CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGT). Amplicon size is checked on a TAE 1×, 0.8% agarose gel electrophoresis via a 30‐min run at 100 V using an adapted size control. DNA concentration is checked on NanoQuant Infinite M200 to ensure enough material is available for downstream steps.
Barcoded libraries were then sequenced using the MinION library preparation protocol steps (Oxford Nanopore Technologies, Oxford, United Kingdom) as: 1D Amplicon by ligation (SQK‐LSK108) or 1D Native barcoding genomic DNA (with EXP‐NBD103 and SQK‐LSK108) and sequenced on a MinION flow cell device.
Bethune, K. , Mariac C., Couderc M., Scarcelli N., Santoni S., Ardisson M., Martin J.‐F., Montúfar R., Klein V., Sabot F., Vigouroux Y., and Couvreur T. L. P.. 2019. Long‐fragment targeted capture for long‐read sequencing of plastomes. Applications in Plant Sciences 7(5): e1243.
DATA ACCESSIBILITY
The FASTQ sequences for all individuals are available in GenBank’s Sequence Read Archive under BioProject number PRJNA526996 (https://www.ncbi.nlm.nih.gov/sra/PRJNA526996).
LITERATURE CITED
- Bleidorn, C. 2016. Third generation sequencing: Technology and its potential impact on evolutionary biodiversity research. Systematics and Biodiversity 14: 1–8. [Google Scholar]
- Cronn, R. , Knaus B. J., Liston A., Maughan P. J., Parks M., Syring J. V., and Udall J.. 2012. Targeted enrichment strategies for next‐generation plant biology. American Journal of Botany 99: 291–311. [DOI] [PubMed] [Google Scholar]
- Dapprich, J. , Ferriola D., Mackiewicz K., Clark P. M., Rappaport E., D'Arcy M., Sasson A., et al. 2016. The next generation of target capture technologies: Large DNA fragment enrichment and sequencing determines regional genomic variation of high complexity. BMC Genomics 17: 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert, S. E. , Chan J. Z.‐M., Houniet D., the PATHSEEK consortium , Breuer J., and Speight G.. 2016. Enrichment by hybridisation of long DNA fragments for Nanopore sequencing. Microbial Genomics 2: e000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faye, A. , Deblauwe V., Mariac C., Richard D., Sonké B., Vigouroux Y., and Couvreur T. L. P.. 2016. Phylogeography of the genus Podococcus (Palmae/Arecaceae) in Central African rain forests: Climate stability predicts unique genetic diversity. Molecular Phylogenetics and Evolution 105: 126–138. [DOI] [PubMed] [Google Scholar]
- Gasc, C. , and Peyret P.. 2017. Revealing large metagenomic regions through long DNA fragment hybridization capture. Microbiome 5: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giolai, M. , Paajanen P., Verweij W., Percival‐Alwyn L., Baker D., Witek K., Jupe F., et al. 2016. Targeted capture and sequencing of gene‐sized DNA molecules. BioTechniques 61: 315–322. [DOI] [PubMed] [Google Scholar]
- Giolai, M. , Paajanen P., Verweij W., Witek K., Jones J. D. G., and Clark M. D.. 2017. Comparative analysis of targeted long read sequencing approaches for characterization of a plant's immune receptor repertoire. BMC Genomics 18: 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin, S. , McPherson J. D., and McCombie W. R.. 2016. Coming of age: Ten years of next‐generation sequencing technologies. Nature Reviews Genetics 17: 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, W.‐B. , and Schneeberger K.. 2017. The impact of third generation genomic technologies on plant genome assembly. Current Opinion in Plant Biology 36: 64–70. [DOI] [PubMed] [Google Scholar]
- Jones, M. R. , and Good J. M.. 2016. Targeted capture in evolutionary and ecological genomics. Molecular Ecology 25(1): 185–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov, M. , Yuan J., Lin Y., and Pevzner P.. 2019. Assembly of long, error‐prone reads using repeat graphs. Nature Biotechnology 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- Kurtz, S. , Phillippy A., Delcher A. L., Smoot M., Shumway M., Antonescu C., and Salzberg S. L.. 2004. Versatile and open software for comparing large genomes. Genome Biology 5: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver, T. W. , Caswell R. C., Moore K. A., Poschmann J., Johnson M. B., Owens M. M., Ellard S., et al. 2016. Pitfalls of haplotype phasing from amplicon‐based long‐read sequencing. Scientific Reports 6: 21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. , Gurtowski J., Yoo S., Nattestad M., Marcus S., Goodwin S., McCombie W. R., and Schatz M.. 2016. Third‐generation sequencing and the future of genomics. BioRxiv 048603 [preprint] 13 April 2016 [cited 2 February 2017]. Available from 10.1101/048603. [DOI] [Google Scholar]
- Li, H. , and Durbin R.. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamanova, L. , Coffey A. J., Scott C. E., Kozarewa I., Turner E. H., Kumar A., Howard E., et al. 2010. Target‐enrichment strategies for next‐generation sequencing. Nature Methods 7: 111–118. [DOI] [PubMed] [Google Scholar]
- Mariac, C. , Luong V., Kapran I., Mamadou A., Sagnard F., Deu M., Chantereau J., et al. 2006. Diversity of wild and cultivated pearl millet accessions (Pennisetum glaucum [L.] R. Br.) in Niger assessed by microsatellite markers. Theoretical and Applied Genetics 114(1): 49–58. [DOI] [PubMed] [Google Scholar]
- Mariac, C. , Scarcelli N., Pouzadou J., Barnaud A., Billot C., Faye A., Kougbeadjo A., et al. 2014. Cost‐effective enrichment hybridization capture of chloroplast genomes at deep multiplexing levels for population genetics and phylogeography studies. Molecular Ecology Resources 14: 1103–1113. [DOI] [PubMed] [Google Scholar]
- Martin, M. 2011. Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.Journal 17(1): 10–12. [Google Scholar]
- Milne, I. , Bayer M., Cardle L., Shaw P., Stephen G., Wright F., and Marshall D.. 2010. Tablet—Next generation sequence assembly visualization. Bioinformatics 26: 401–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mower, J. P. , and Vickrey T. L.. 2018. Structural diversity among plastid genomes of land plants In Chaw S.‐M. and Jansen R. K. [eds.], Advances in botanical research, Vol. 85: Plastid genome evolution, 263–292. Academic Press, Cambridge, Massachusetts, USA. [Google Scholar]
- Quinlan, A. R. , and Hall I. M.. 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohland, N. , and Reich D.. 2012. Cost‐effective, high‐throughput DNA sequencing libraries for multiplexed target capture. Genome Research 22: 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarcelli, N. , Barnaud A., Eiserhardt W., Treier U. A., Seveno M., d'Anfray A., Vigouroux Y., and Pintaud J.‐C.. 2011. A set of 100 chloroplast DNA primer pairs to study population genetics and phylogeny in monocotyledons. PLoS ONE 6: e19954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn, J. , and Nam J.‐W.. 2018. The present and future of de novo whole‐genome assembly. Briefings in Bioinformatics 19(1): 23–40. [DOI] [PubMed] [Google Scholar]
- Straub, S. C. K. , Parks M., Weitemier K., Fishbein M., Cronn R. C., and Liston A.. 2012. Navigating the tip of the genomic iceberg: Next‐generation sequencing for plant systematics. American Journal of Botany 99: 349–364. [DOI] [PubMed] [Google Scholar]
- Stull, G. W. , Moore M. J., Mandala V. S., Douglas N. A., Kates H.‐R., Qi X., Brockington S. F., et al. 2013. A targeted enrichment strategy for massively parallel sequencing of angiosperm plastid genomes. Applications in Plant Sciences 1: 1200497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyford, A. D. , and Ness R. W.. 2017. Strategies for complete plastid genome sequencing. Molecular Ecology Resources 17(5): 858–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C. , Chu J., Warren R. L., and Birol I.. 2017. NanoSim: Nanopore sequence read simulator based on statistical characterization. GigaScience 6: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, T. , Furihata H. Y., and Kawabe A.. 2014. Patterns of genomic integration of nuclear chloroplast DNA fragments in plant species. DNA Research 21: 127–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1. Both sets of long‐range PCR primers, elongation times, and annealing temperature values used.
APPENDIX S2. Raw output data of the three MinION runs undertaken in our study.
APPENDIX S3. Detailed list of materials and costs to implement the capture protocol.
APPENDIX S4. Bioinformatic codes used for analyses of MinION data.
APPENDIX S5. Mummer visualization of assembled long reads for Cenchrus americanus.
APPENDIX S6. Mummer visualization of assembled long reads for Phytelephas aequatorialis.
APPENDIX S7. Mapping of Phytelephas aequatorialis–like simulated reads (using LAST software [http://last.cbrc.jp/]) to the reference plastome NC_029957.1.
Data Availability Statement
The FASTQ sequences for all individuals are available in GenBank’s Sequence Read Archive under BioProject number PRJNA526996 (https://www.ncbi.nlm.nih.gov/sra/PRJNA526996).