

Abstract
The isolation, characterization and total synthesis of the macrocyclic polyene mangrolide D is reported. A 16-step total synthesis relies on robust Suzuki and ring-closing metathesis reactions, and an iron-catalyzed hydroazidation of an exo-methylene substituted tetrahydropyran as a key step for the synthesis of the appended 4-epi-vancosamine sugar. Although mangrolide D did not display antibiotic activity, this work should prove enabling towards the synthesis of the antitubercular tiacumicins which display a virtually identical macrocyclic backbone.
Graphical Abstract

In 2013, Jamison and MacMillan disclosed the isolation, structural characterization, and biological activity of a novel marine-derived macrolide antibiotic termed mangrolide A (2, Fig. 1).1 The original sample of mangrolide A (2), isolated from the fermentation broth of the rare marine actinomycete strain Actinoalloteichus spitensis, was found to possess Gram-negative antibacterial activity and stimulated us and others to pursue a total synthesis of this material. Gademann and coworkers, based on their very elegant synthesis of the structurally related macrolide fidaxomicin (3),2 recently reported the total synthesis of mangrolide A (2).3 While this study fully corroborated the structure disclosed for the isolated natural product sample,1 synthetic mangrolide A was found to be devoid of any antibacterial activity.3 Through our own synthetic work, we had come to the same disconcerting conclusion that our synthetic mangrolide A (2),4,5 as well as ultra-purified re-isolated natural material, failed to register the Gram-negative activity reported for the original batch of fermented mangrolide A. Therefore, we postulated that the Gram-negative activity of the original sample could be derived from a minor contaminating or interfering metabolite, a phenomenon not uncommon in the field of natural product isolation and bioactivity.6 In this context, we reevaluated the founder strain leading to the isolation and structural characterization of a natural analog termed mangrolide D (1) as reported herein in conjunction with a total synthesis. Given the structural identity of the mangrolide macrocycle to the macrocyclic core of fidaxomicin (3, marketed as Dificid®), an antibiotic used clinically to treat the gastrointestinal infection caused by C. difficile,7 we were mindful that an efficient synthetic approach towards mangrolides could also be leveraged towards the synthesis of fidaxomicin analogs not available from fermentation.8,9
Figure 1.
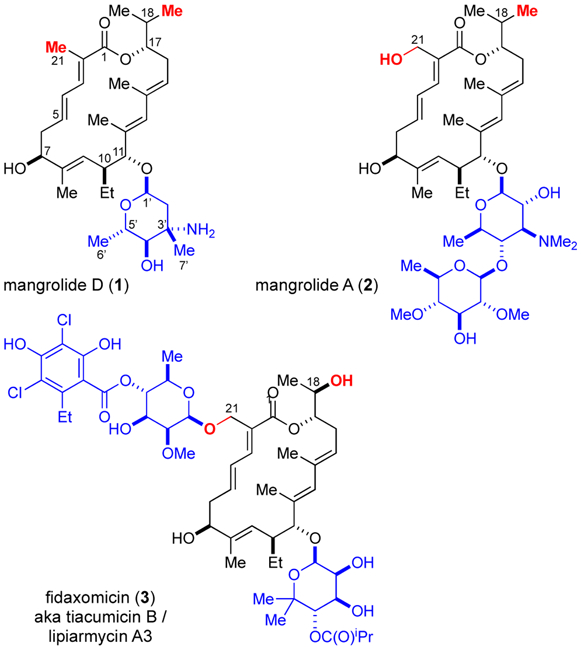
Mangrolide A (2) and D (1) and Fidaxomicin (3).
Large-scale fermentation of Alloteichus spitensis strain SNA-018 and subsequent extraction with XAD-7 resin, followed by flash and HPLC C18 purification led to 1 (1.1 mg from 20 L) as a white solid; [α]D +XX ; UV (MeOH) 204, 234, and 260. A molecular formula of C33H53NO6 was derived from the HRMS ESI-TOF with an [M+H]+ of m/z 560.3949 (calcd for C42H70NO12, 560.3951). Analysis of the 1H and 13C chemical shifts indicated a polyketide structure with five olefins, four vinyl methyls, and a sugar moiety.10 Briefly, the aglycon structure shows similarities to the aglycon of tiacumicin B (3) and mangrolide A (2). However, its macrocyclic core differs from 3 by virtue of the oxidation state at C21 (Me vs CH2OR) and C18 (Me vs OH).11 The configuration at C7, C10, C11 and C17 were assigned based on homology to mangrolide A (2) and 3.1,2b The relative configuration of the α-linked 4-epi-vancosamine at C11 was assigned based on detailed analysis of the coupling constants as well as NOE measurements.10 A small coupling of 2.5 Hz from the anomeric proton H1’ to H2’-axial establishes the equatorial position for H1’. NOE correlations from the axial methyl protons H7’ to H6’ and to H2’equatorial, establish the relationship between C1’ and C3’ as well as the C3’/C4’ and C4’/C5’ configurations as anti, anti. This is consistent with previous reports of 4-epi-vancosamine.12,13
Our strategy towards mangrolide D (1), originally conceived for mangrolide A (2) as disclosed in 2013,4 is depicted in Scheme 1.14 With future analog studies in mind, we advanced a flexible and convergent approach that would unite fragments 4-6 via robust and interchangeable bond-forming reactions (cross-coupling, olefin metathesis, esterification). In the event, we settled on the olefin metathesis as the macrocycle-forming step.
Scheme 1.
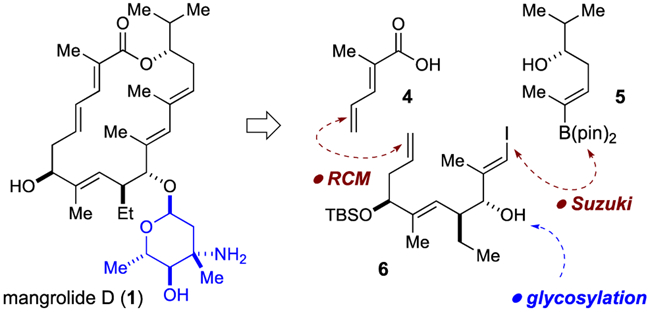
Synthetic Strategy
The synthesis of the top ester fragment is shown in Scheme 2. Similar to Gademann,3 Lewis acid-assisted opening of (R)-isopropyloxirane (7)15 with propynyllithium, followed by Cu(I)-catalyzed boration of homopropargylic alcohol 83 yielded multigram quantities of enantiomerically pure vinylborane 5.3 Esterification of this alcohol 5 with dienoic acid 4 (prepared from olefination of acrolein with phosphorane 10)16 provided the desired northern coupling partner 9 in three steps longest linear sequence from commercially available material.
Scheme 2. Synthesis and Coupling of Fragments 4 and 5a.

aReaction conditions: (a) propyne, n-BuLi (1.4 equiv), BF3·OEt2 (1.1 equiv), THF, −78 °C, 2 h, 77%; (b) B2(pin)2 (1.2 equiv), CuCl (5 mol%), PPh3 (6 mol%), tBuOK (0.2 equiv), THF/MeOH (5/1), 0 °C, 4 h, 70%; (c) 4 (1 equiv), TCBC (2 equiv), NEt3 (5 equiv), DMAP (3 equiv), PhMe, rt, 4 h, 62%; (d) acrolein (1.2 equiv), CH2Cl2, reflux, 3.5 h; (e) 6% aq. NaOH in EtOH, reflux, 1 h, 63% (2 steps). Pin, pinacolate; TCBC, trichlorobenzoyl chloride.
The synthesis of southern fragment 6 commenced with a highly diastereoselective aldol reaction of bornanesultam 12 with (E)-3-iodomethacrolein derived from MnO2-mediated oxidation of known alcohol 112b,8b (Scheme 3).17 The syn-aldol product 13 was obtained in 82% yield and protected as silyl ether 14 (91% yield). Reduction of 14 (DIBAL-H, CH2Cl2, −78 °C)18 delivered the corresponding aldehyde (82%) which was homologated to unsaturated ester 15 via treatment with phosphorane 10 in 78% yield. Reduction of the ester, followed by oxidation of the resulting primary alcohol with Dess-Martin periodinane yielded aldehyde 16 (92%, 2 steps). A highly diastereoselective Brown allylation (→ 17) was followed by silylation of the homoallylic alcohol (→ 18) and selective deprotection of the triethylsilyl ether to provide southern fragment 6 in 10 steps and 30% overall yield from vinyl iodide 11.
Scheme 3. Synthesis of Fragment 6a.

aReaction conditions: (a) MnO2 (8.5 equiv), 3Å MS, CH2Cl2, rt, 1 h; (b) 12 (1.15 equiv), Bu2BOTf (1.3 equiv), iPr2NEt (1.6 equiv), CH2Cl2, −78 °C, 30 min; then add crude (E)-3-iodo-2-methylacrylaldehyde, −78 °C, 1.5 h, then 0 °C, 1.5 h, 82% (2 steps), dr = 20:1; (c) TESOTf (1.5 equiv), 2,6-lutidine (2 equiv), CH2Cl2, 0 °C, 30 min, 91%; (d) DIBAL-H, CH2Cl2, −78 °C, 1 h, 82%; (e) 10 (1.6 equiv), PhMe, reflux, 8 h, 78%; (f) DIBAL-H, THF, −78 °C, 1 h; (g) Dess-Martin periodinane (1.6 equiv), NaHCO3 (2 equiv), CH2Cl2, rt, 20 min, 92% (2 steps); (h) (−)-Ipc2Ballyl (1.3 equiv), Et2O, −78 °C, 2 h, 91%; (i) TBSOTf (3 equiv), 2,6-lutidine (4 equiv), CH2Cl2, 0 °C, 20 min, 95%; (j) py·HF, MeCN/H2O) (1/1), 0 °C, 1.5 h, 80%. Ipc, isopinocampheyl.
Next, we focused on the synthesis of the 4-epi-vancosamine sugar fragment,19,20 which we plan to introduce via the activated 2-deoxy-azido glycosyl donor 27, the synthesis of which begins with a selective oxidation of the diol derived from commercially available 3,4-di-O-acetyl-6-deoxy-L-glucal (19, Scheme 4).21 Oxa-Michael addition of methanol to the resulting enone 20 yielded tetrahydropyranone 21 (75%).22 Extensive experimentation revealed that methylenation of 21 was best achieved using Peterson olefination conditions, providing 22 in 54% yield.23 Gratifyingly, the key hydroazidation of 22 using conditions first described by Boger and coworkers provided azide 23 in good yield and with desired stereochemistry (10:1).24,25 Hydrolysis of the anomeric methoxy substituent (→ 24) and peracetylation (→ 25) set the stage for selective hydrazine-mediated removal of the primary acetate (→ 26) and activation of the anomeric position as the trichloroacetimidate 27. The latter was not purified and used as such for coupling with fragment 6 as described in Scheme 5.
Scheme 4. Synthesis of Activated Azidosugar 27a.

aReaction conditions: (a) K2CO3 (3 equiv), MeOH, 0 °C, 45 min, 95%; (b) MnO2 (6 equiv), CH2Cl2, rt, 12 h, 57% (20% starting material recovered); (c) NaOMe (0.3 equiv), MeOH, rt, 1.3 h, 75%; (d) TMSCH2MgCl (3 equiv), PhMe, 0 °C to rt, 2 h, 71%; (e) KH (3 equiv), THF, rt, 1 h, 76%; (f) Fe2(ox)3 (5 equiv), NaBH4 (3 equiv), NaN3 (8 equiv), H2O/THF (1/1), 0 °C, 30 min, 70%; (g) aq. H2SO4 (0.1 M), 40 °C, 10 h, 85%; (h) Ac2O (10 equiv), NEt3 (20 equiv), DMAP (1 equiv), CH2Cl2, rt, 2 h, 86%; (i) N2H4·HOAc (.1 equiv), DMF, rt, 14 h, 64%; (j) Cl3CCN (10 equiv), DBU (0.8 equiv), CH2Cl2, rt, 10 h. ox, oxalate.
Scheme 5. Fragment Coupling and Final Stepsa.

aReaction conditions: (a) 27 (2 equiv), BF3·OEt2 (2 equiv), 4Å MS, CH2Cl2, 0 °C, 1 h, 78%; (b) Me3P (2.5 equiv), H2O (5 equiv)), THF, 0 °C to rt, 3 h, 70%; (c) 9 (2 equiv), Pd(PPh3)4 (1.2 equiv), TlOEt (1.5 equiv), THF/H2O (3/1), rt, 0 min, 74%; (d) Grubbs II (20 mol%), PhMe, reflux, 20 h, 65%; (e) py·HF, THF, rt, 36 h; (f) Et3N, MeOH, rt, 12 h, 42% (2 steps).
Treatment of homoallylic alcohol 6 with the above prepared crude imidate 27 in the presence of BF3-etherate selectively provided the α-anomer of glycosylated product 28 (78%).26 Staudinger reduction of the azide 28 yielded primary amine 29, a material that underwent uneventful Suzuki cross-coupling with top fragment 9.2,27 Ring-closing metathesis of 30 with second generation Grubbs catalyst provided protected macrocycle 31 as an inseparable mixture of E/Z-isomers (1:1).2 Fluoride-mediated removal of the silylether was followed by mild acetate removal to yield mangrolide D (1) and the corresponding Z-isomer (42% combined) that were separated via reversed phase HPLC. The proton and carbon NMR spectra of synthetic mangrolide D fully matched those of the natural product, whereas significant chemical shift differences were observed for the corresponding mangrolide diastereomer containing the enantiomeric 4-epi-vancosamine derived from 3,4-di-O-acetyl-6-deoxy-D-glucal (ent-19).21 The optical rotations for synthetic and natural mangrolide D were of the same sign and similar magnitude (synthetic: [α]D = −67.5 (c = 0.08, CHCl3); natural: [α]D = −50.1 (c = 0.08, CHCl3)). Alas, synthetic or natural mangrolide D was poorly active when tested against the Gram-positive strain B. subtilis (MIC = 32 μg/mL) and inactive against the Gram-negative strain P. aeruginosa PA01 (MIC > 256 μg/mL). The control compound streptomycin killed B. subtilis with an MIC of 0.125 μg/mL and gentamicin’s antibacterial activity registered with an MIC of 1 μg/mL against P. aeruginosa PA01. These results imply that mangrolide D was not responsible for the antibacterial activity of the original mangrolide A fermentation sample.
In summary, our search to identify a potential metabolite responsible for the Gram-negative activity of a fermentation sample originally assigned to mangrolide A led to the isolation and structural characterization of mangrolide D. A convergent, efficient total synthesis of mangrolide D (16 total steps longest linear sequence) fully confirmed its structure and assignment of the absolute configuration of the attached 4-epi-vancosamine. Other highlights are the efficient approach to both enantiomers of 4-epi-vancosamine, a natural aminosugar for which there were virtually no reported synthetic protocols described.13,20 In particular, a highly stereoselective iron-catalyzed hydroazidation developed by Boger (22 → 23) was key to a successful assembly of the 3-deoxy-4-azido sugar. Although mangrolide D did not register antibacterial activity, our synthetic efforts are nevertheless highly relevant in the context of synthetic macrolide antibiotics.28 The macrocyclic core of mangrolide is virtually identical to the core of the clinically useful fidaxomicin (3). Analogs of the latter could find significance in the context of multidrug-resistant tuberculosis if systemic bioavailable analogs can be identified.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (GM111328) and the Robert A. Welch Foundation (I-1422) for financial support. Jef K. De Brabander holds the Julie and Louis Beecherl, Jr., Chair in Medical Science.
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
Isolation, structural characterization and analytical data for mangrolide D, and experimental procedures and analytical data for new synthetic compounds (PDF)
REFERENCES
- 1.Jamison MT Mangrolide A, A Novel Marine Derived Polyketide with Selective Antibiotic Activity. Ph.D. Thesis, UT Southwestern Medical Center, May 2013. [Google Scholar]
- 2. (a).Kaufmann E; Hattori H; Miyatake-Ondozabal H; Gademann K Total Synthesis of the Glycosylated Macrolide Antibiotic Fidaxomicin. Org. Lett 2015, 17, 3514–3517; [DOI] [PubMed] [Google Scholar]; (b) Hattori H; Kaufmann E; Miyatake-Ondozabal H; Berg R; Gademann K Total Synthesis of Tiacumicin A. Total Synthesis, Relay Synthesis, and Degradation Studies of Fidaxomicin (Tiacumicin B, Lipiarmycin A3). J. Org. Chem 2018, 83, 7180–7205. [DOI] [PubMed] [Google Scholar]
- 3.Hattori H; Roesslein J; Caspers P; Zerbe K; Miyatake-Ondozabal H; Ritz D; Rueedi G; Gademann K Total Synthesis and Biological Evaluation of the Glycosylated Macrocyclic Antibiotic Mangrolide A. Angew. Chem. Int. Ed 2018, 57, 11020–11024. [DOI] [PubMed] [Google Scholar]
- 4.For our synthesis of the mangrolide A aglycon and disaccharide, see: De Brabander J Small Molecule Compounds Selective Against Gram-negative Bacterial Infections. U.S. Patent Appl. No. 14/711,545, filed May 13, 2015 (provisional Appl. No. 61/992,642 filed May 13, 2014), Pub. No. US 2015/0329582 A1, Pub. Date November 19, 2015.
- 5.For our total synthesis, see: Yu X Total Synthesis of Mangrolide A. Ph.D. Thesis, UT Southwestern Medical Center, February 2018. [Google Scholar]
- 6.(a) For some recommended perspectives, see: Bisson J; McAlpine JB; Friesen JB; Chen S-N; Graham J; Pauli GF Can Invalid Bioactives Undermine Natural Product-Based Drug Discovery? J. Med. Chem 2016, 59, 1671–1690; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pauli GF; Chen S-N; Friesen JB; McAlpine JB; Jaki BU Analysis and Purification of Bioactive Natural Products: The AnaPurNa Study. J. Nat. Prod 2012, 75, 1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For selected reviews, see: Erb W; Zhu J From natural product to marketed drug: the tiacumicin odyssey. J. Nat. Prod 2013, 30, 161–174 [DOI] [PubMed] [Google Scholar]
- 8.(a) For syntheses of the fidaxomicin aglycon, see: Miyatake-Ondozabal H; Kaufmann E; Gademann K Total Synthesis of the Protected Aglycon of Fidaxomicin (Tiacumicin B, Lipiarmycin A3). Angew. Chem. Int. Ed 2015, 54, 1933–1936; [DOI] [PubMed] [Google Scholar]; (b) Glaus F; Altmann K-H Total Synthesis of the Tiacumicin B (Lipiarmycin A3/Fidaxomicin) Aglycone. Angew. Chem. Int. Ed 2015, 54, 1937–1940; [DOI] [PubMed] [Google Scholar]; (c) Erb W; Grassot J-M; Linder D; Neuville L; Zhu J Enantioselective Synthesis of Putative Lipiarmycin Aglycon Related to Fidaxomicin/Tiacumicin B. Angew. Chem. Int. Ed 2015, 54, 1929–1932; [DOI] [PubMed] [Google Scholar]; (d) Jeanne-Julien L; Masson G; Astier E; Genta-Jouve G; Servajean V; Beau J-M; Norsikian S; Roulland E Synthesis of a Tiacumicin B Protected Aglycon. Org. Lett 2017, 19, 4006–4009; [DOI] [PubMed] [Google Scholar]; (e) Jeanne-Julien L; Masson G; Astier E; Genta-Jouve G; Servajean V; Beau J-M; Norsikian S; Roulland E Study of the Construction of the Tiacumicin B Aglycone. J. Org. Chem 2018, 83, 921–929. [DOI] [PubMed] [Google Scholar]
- 9.Interests in this area stem from the profound anti-tubercular activity of fidaxomicin (3), which unfortunately has no systemic bioavailability.7 Analogs to improve bioavailability might thus open new doors towards antitubercular compound development.
- 10.Fermentation and isolation conditions, full structural elucidation details, and NMR chemical shifts can be found in the supporting information.
- 11.Tiacumicin A, like mangrolide D, also bears a C21-Me but lacks a branching C18-substituent (H vs Me), see: ref. 2b and references cited therein.
- 12.Hunt AH; Molloy RM; Debono M; Occolowitz JL Isolation and characterization of 4-epi-vancosamine. Tetrahedron Lett 1988, 29, 1223–1226. [Google Scholar]
- 13. For the synthesis of 4-epi-vancosamine from vancomycin degradation: (a).Losey HC; Peczuh MW; Chen Z; Eggert US; Dong SD; Pelczer I; Kahne D; Walsh CT Tandem Action of Glycosyltransferases in the Maturation of Vancomycin and Teicoplanin Aglycones: Novel Glycopeptides. Biochem 2001, 40, 4745–4755; [DOI] [PubMed] [Google Scholar]; (b) Lu W; Oberthür M; Leimkuhler C; Tao J; Kahne D; Walsh CT Characterization of a regiospecific epivancosaminyl transferase GtfA and enzymatic reconstitution of the antibiotic chloroeremomycin. Proc. Natl. Ac. Sci. USA 2004, 101, 4390–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Around the same time, starting in 2015, several groups reported syntheses of the structurally related fidaxomicin. Unsurprisingly, these various approaches toward fidaxomicin,2,8 the Gademann mangrolide A synthesis,3 and our4,5 mangrolide synthesis explore several common synthetic aspects and strategic bond disconnections.
- 15.Enantiopure oxirane 7 is commercially available, but we prepared it from racemic oxirane via Jabobsen HKR: Tokunaga M; Larrow JF; Kakiuchi F; Jacobsen EN Asymmetric Catalysis with Water: Efficient Kinetic Resolution of Terminal Epoxides by Means of Catalytic Hydrolysis. Science 1997, 277, 936–938. See Supporting Information for details. [DOI] [PubMed] [Google Scholar]
- 16.Xu T; Li C-C; Yang Z A Concise Approach for the Total Synthesis of Pseudolaric Acid A. Org. Lett 2011, 13, 2630–2633. [DOI] [PubMed] [Google Scholar]
- 17.Oppolzer W; Blagg J; Rodriguez I; Walther E Bornanesultam-Directed Asymmetric Synthesis of Crystalline, Enantiomerically Pure Syn Aldols. J. Am. Chem. Soc 1990, 112, 2767–2772. [Google Scholar]
- 18.Over-reduction of bornanesultam-based amides (to a primary alcohol) can be prevented when performed with DIBAL-H in dichloromethane at −78 °C, see: Oppolzer W; Darcel C; Rochet P; Rosset S; De Brabander J Non-Destructive Removal of the Bornanesultam Auxiliary in α-substituted N-Acylbornane-10,2-sultams Under Mild Conditions: An Efficient Synthesis of Enantiomerically Pure Ketones and Aldehydes. Helv. Chim. Act 1997, 80, 1319–1337. [Google Scholar]
- 19.For the synthesis of methyl N,O-dibenzoyl-L-4-epi-vancosamine: Avenoza A; Busto JH; Corzana F; Peregrina JM; Sucunza D; Zurbano MM Diastereoselective synthesis of protected 4-epi-vancosamine from (S)-N-Boc-N,O-isopropylidene-α-methylserinal. Tetrahedron Asymm 2003, 14, 1037–1043. [Google Scholar]
- 20.Review on 3-deoxy-4-aminosugars: Ding F; Cai S; William R; Liu X-W Pathways leading to 3-amino- and 3-nitro-2,3-dideoxy sugars: strategies and synthesis. RSC Adv 2013, 3, 13594–13621. [Google Scholar]
- 21.Given that we could not confidently asses the absolute configuration of the 4-epi-vancosamine unit, we also executed a synthesis of the enantiomer of 27 (ent-27) starting from 3,4-di-O-acetyl-6-deoxy-D-glucal (ent-19). See Supporting Information for details.
- 22.Greven R; Jütten P; Scharf H-D A New Stereoselective Route to Branched-Chain Nitro and Amino Sugars: Synthesis of Both Enantiomers of Decilonitrose and Avidinosamine. J. Org. Chem 1993, 58, 3742–3747. [Google Scholar]
- 23.This identical Peterson olefination of 21 to 22 was first reported by: Giuliano RM Stereoselective synthesis of L-olivomycose. Carbohyd. Res 1984, 131, 341–345. Wittig, Petasis, or Tebbe methylenation uniformly led to elimination of the anomeric methoxy substituent. [Google Scholar]
- 24.Leggans EK; Barker TJ; Duncan KK; Boger DL Iron(III)/NaBH4-Mediated Additions to Unactivated Alkenes: Synthesis of Novel 20’-Vinblastine Analogues. Org. Lett 2012, 14, 1428–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lower selectivities were obtained when the allylic alcohol was protected as an acetate (dr 4:1) or TBS-ether (dr 2.5:1). Also, the corresponding endocyclic Δ3,4-olefin isomer proved resistant to identical hydroazidation conditions.
- 26.For a review on various glycosylation strategies, see: Zhu X; Schmidt RR New Principles for Glycoside-Bond Formation. Angew. Chem. Int. Ed 2009, 48, 1900–1934. [DOI] [PubMed] [Google Scholar]
- 27.More than 1 equivalent of Pd catalyst was required, presumably due to inactivating complexation with the primary amine in the sugar moiety.
- 28.For a seminal contribution towards the first efficient fully synthetic macrolide antibiotics, see: Seiple IB; Zhang Z; Jakubec P; Langlois-Mercier A; Wright PM; Hog DT; Yabu K; Allu SR; Fukuzaki T; Carlsen PN; Kitamura Y; Zhou X; Condakes ML; Szczypiński Green, W. D.; Myers AG A platform for the discovery of new macrolide antibiotics. Nature 2016, 533, 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.