

ABSTRACT
Precise control of cell death in the nervous system is essential for development. Spatial and temporal factors activate the death of Drosophila neural stem cells (neuroblasts) by controlling the transcription of multiple cell death genes through a shared enhancer. The activity of this enhancer is controlled by abdominal A and Notch, but additional inputs are needed for proper specificity. Here, we show that the Cut DNA binding protein is required for neuroblast death, regulating reaper and grim downstream of the shared enhancer and of abdominal A expression. The loss of cut accelerates the temporal progression of neuroblasts from a state of low overall levels of H3K27me3 to a higher H3K27me3 state. This is reflected in an increase in H3K27me3 modifications in the cell death gene locus in the CNS on Cut knockdown. We also show that cut regulates the expression of the cohesin subunit Stromalin. Stromalin and the cohesin regulatory subunit Nipped-B are required for neuroblast death, and knockdown of Stromalin increases H3K27me3 levels in neuroblasts. Thus, Cut and cohesin regulate apoptosis in the developing nervous system by altering the chromatin landscape.
KEY WORDS: Developmental cell death, Drosophila, Neuroblast, Cut, Cohesin
Summary: Cut regulates the programmed death of neural stem cells by altering cohesin levels and promoting a more open chromatin conformation to allow cell death gene expression.
INTRODUCTION
Programmed cell death is important for normal nervous system development in organisms ranging from Caenorhabditis elegans to humans (Arya and White, 2015). Precise control of cell death in the nervous system requires the integration of spatial, temporal and cell identity signals from both cell intrinsic and extrinsic sources. Conserved signaling pathways that are instrumental in many developmental cell fate decisions also control the commitment of cells to death. Examining how these pathways interact to determine the cell death fate in a specific context is crucial not only for understanding normal development, but also to gain insight into how developmental pathways and homeostasis are disrupted in diseases such as cancer and neurodegeneration.
In the developing ventral nerve cord (VNC) of the fly, the majority of neural stem cells (neuroblasts; NBs) in the abdominal segments are eliminated by apoptosis late in embryonic development (Peterson et al., 2002; Truman and Bate, 1988; White et al., 1994). In the absence of this death, the VNC becomes massively hypertrophic, and adult longevity is compromised (Peterson et al., 2002). The cell death genes reaper (rpr), grim and sickle (skl) are required for NB death in the Drosophila embryo (Tan et al., 2011; White et al., 1994). These genes are part of the rpr hid grim (RHG) gene cluster of cell death activators. Transcription of the RHG genes is coordinately regulated by conserved intergenic enhancers to initiate cell death in specific developmental contexts (Arya and White, 2015; Bangs et al., 2000; Fuchs and Steller, 2011; Moon et al., 2008; Tan et al., 2011; Zhang et al., 2008).
We have previously described how the Hox gene abdominal A (abd-A) and Notch (N) are necessary for NB death in the abdominal segments of the embryonic VNC (Arya et al., 2015). N activation in NBs requires the expression of the Delta ligand on NB progeny, and activates a late pulse of abd-A in NBs. The late pulse of Abd-A could convey both spatial and temporal information about the specific NBs fated to die. Mis-expression of abd-A is sufficient to cause ectopic NB death (Arya et al., 2015; Prokop et al., 1998). abd-A regulates rpr, grim and skl expression through a regulatory element between rpr and grim called the NB regulatory region enhancer1 (enh1) (Arya et al., 2015). This element is required for full expression of rpr, grim and skl in NBs (Tan et al., 2011). Recent data indicate that abd-A and grainy head (grh) may be direct regulators of this NB cell death enhancer (Khandelwal et al., 2017). However, it is clear that not all cells that express abd-A or grh activate the cell death genes and undergo cell death. In particular, many cells outside of the central nervous system (CNS) express these genes, and are not fated to die (Abrams et al., 1993; Almeida and Bray, 2005; Bray et al., 1989; Karch et al., 1990). Furthermore, mis-expression of Abd-A is not sufficient to activate ectopic NB death until later stages of development (Arya et al., 2015), suggesting that there are additional temporal and tissue-specific factors required for the activation of NB death.
Here, we report that the DNA-binding protein Cut is required for NB death, acting through a mechanism that is distinct from Abd-A and enh1. Cut is a transcriptional regulator with four DNA binding domains: three CUT domains and a homeobox domain (Nepveu, 2001). Drosophila Cut is structurally and functionally homologous to CUX1 [also known as CCAAT displacement protein (CDP)] in human and Cux1/2 in mouse, and can act as either an enhancer or repressor of transcription. In the Drosophila embryo, cut is expressed in the embryonic central and peripheral nervous system, Malpighian tubules and anterior and posterior spiracles (Blochlinger et al., 1990; Zhai et al., 2012). Opposing roles for cut in cell death have been described: loss of cut in the fly can enhance tumor growth, and cut has also been implicated in promoting differentiation and cell survival in posterior spiracle and tracheal development (Pitsouli and Perrimon, 2013; Wong et al., 2014; Zhai et al., 2012). In mammals, the functions of the Cux1 and Cux2 homologs are equally complex. Loss of Cux1 in mouse results in reduced proliferation and organ hypoplasia (Sansregret and Nepveu, 2008), but Cux1 has also been implicated as a haploinsufficient tumor suppressor in myeloid malignancies, and is associated with poor prognosis (Wong et al., 2014). Paralleling our findings on the role of Drosophila cut in NB death, Cux2 is required to limit the expansion of neuronal precursors in mouse brain development (Cubelos et al., 2008), but conversely in the spinal cord it is required for the maintenance of neural progenitors (Iulianella et al., 2008).
To examine how cut regulates cell death, we placed it in the regulatory framework defined by our previous studies (Arya et al., 2015). Our data indicate that cut plays a permissive role in neural stem cell apoptosis, acting to modify the chromatin landscape and facilitating the expression of rpr and grim independently of the previously identified NB cell death enhancer. We show that there is normally a temporal increase in the overall levels of H3K27me3 in NBs, and H3K27me3 levels are enhanced throughout this temporal progression in the absence of cut. The H3K27me3 histone modification normally reflects repressed chromatin accessibility, and is strikingly lower in NBs than in more differentiated cells in the VNC. Within the cell death gene locus, loss of cut increases H3K27me3 levels and suppresses proapoptotic gene expression and NB death.
Importantly, we found that cut regulates expression of the cohesin subunit Stromalin (SA). Cohesin is important for sister chromatid cohesion and long-range enhancer/promoter interactions (Kagey et al., 2010; Rollins et al., 1999). We demonstrate that cohesin components are required for normal NB death, and that loss of cohesin results in increased numbers of NBs with high levels of H3K27me3, similar to that seen with the loss of cut. We propose a model for the regulation of NB death through the combinatorial control of chromatin accessibility, chromatin architecture, and the temporal and spatial activity of sequence-specific transcription factors.
RESULTS
cut is necessary and sufficient for abdominal NB death
The requirement for cut in NB apoptosis was identified in a previous RNAi screen for regulators of NB death (Arya et al., 2015). Knockdown of cut in the CNS results in a large increase of persistent NBs late in Drosophila embryogenesis (compare Fig. 1A with B,D), at a time when the majority of abdominal NBs have undergone apoptosis in the wild type. This phenotype was validated with multiple RNAi lines and a cut null mutant, cutC145 (Johnson and Judd, 1979; Micchelli et al., 1997) (Fig. 1C,D). Ectopic abdominal NB survival is also detected in third instar larvae, although many of the ectopic embryonic NBs do not survive into late larvae (Fig. S1). The rescue of NB death is not due to cut activity in neighboring glia, as cut knockdown in glia does not inhibit NB death (Fig. S1). Thus, cut is required cell-autonomously for the programmed death of NBs in late embryogenesis.
Fig. 1.

cut is necessary and sufficient for NB death. (A-C) Knockdown of cut in the nervous system (B), or cut loss in mutant embryos (C), results in ectopic abdominal NB survival, compared with control (A), as detected by Dpn staining in stage 17 embryos. Yellow bars mark the abdominal hemisegments. (D) Quantification of Dpn+ NBs in five abdominal hemisegments in late stage 17 embryos (n=4-6). (E-G) cut overexpression (F) results in premature NB loss by apoptosis compared with control (yw; E). Fewer NBs can be seen in each hemisegment, particularly in the anterior of each hemisegment (red bar) at stage 14. This NB loss is blocked by the broad-spectrum caspase inhibitor p35 (G). Yellow dashed lines mark parasegment boundaries. (H) Quantification of premature NB loss in three abdominal hemisegments of stage 14 embryos (n=3 for each genotype). Overexpression of cut results in premature NB death, which is partially inhibited by p35. Premature NB loss resulting from cut overexpression is not rescued by abd-A knockdown. For all embryos anterior is to the top. Data are mean±s.d. *P<0.05 versus control; unpaired t-test. Scale bars: 20 μm.
Cut is expressed in the CNS starting at early stage 12. Initial expression is very low, but by stage 15 Cut is strongly expressed in many cells of the CNS, including NBs (Fig. S2). Thus, Cut is expressed in NBs at a time when NB death begins (stage 14), and is expressed in most or all NBs at the time cell death peaks (stage 15/16). Widespread Cut expression in both NBs and neurons indicates that, at normal levels, cut is not sufficient to activate apoptosis in all cells. Rather, cut could be permissive for the activation of the cell death genes by additional spatial and temporal factors, including N, abd-A and grh (Almeida and Bray, 2005; Arya et al., 2015; Cenci and Gould, 2005; Khandelwal et al., 2017; Maurange et al., 2008).
Overexpression of cut in the CNS results in premature loss of NBs in abdominal segments at stage 14, before their normal death (Fig. 1E-H). This cut-induced NB loss can be inhibited by the baculovirus broad-spectrum caspase inhibitor p35 (Clem and Miller, 1994; Hay et al., 1994) (Fig. 1G,H), demonstrating that NB loss is due to caspase-dependent cell death, and not to alterations in NB fate. Overexpression of cut in the whole embryo with heatshock-gal4 also results in ectopic cell death (Fig. S3). Taken together, these findings demonstrate that cut is necessary for timely NB death, and may be temporally limiting for the activation of cell death.
cut acts upstream of rpr and grim and downstream of enh1
NB death requires the combined activity of the rpr, grim and skl genes (Tan et al., 2011). These genes are transcribed in doomed cells, and the Rpr, Grim and Skl proteins inhibit DIAP1 to activate caspases (Kornbluth and White, 2005). To determine whether cut regulates NB death through this pathway, we assessed rpr and grim transcript levels on knockdown or overexpression of cut. In stage 15 embryos both rpr and grim expression are clearly reduced when cut is knocked down in the CNS (Fig. 2A-D,I). Conversely, cut overexpression throughout the CNS with wor-gal4 increased levels of accumulated rpr and grim transcripts when cell death is inhibited with p35 (Fig. 2E-H,J). Interestingly, even when cut is overexpressed in many cells of the CNS with wor>cut, rpr and grim are hyperactivated only in a subset of cells in the CNS. This suggests that cut is permissive for rpr and grim expression, but requires additional inputs to fully regulate NB death.
Fig. 2.

cut alters rpr and grim expression independently of the NB regulatory region. (A-D) cut knockdown in the CNS of stage 15 embryos decreases rpr and grim expression in the VNC, as detected by FISH. (E-H) On cut overexpression, rpr and grim mRNA levels accumulate to greater levels in p35-rescued cells, compared with p35-rescue alone. (I) FISH intensity was measured in cells in ventral abdominal hemisegments in wor-gal4>+ and wor-gal4>cutRNAi (n=3-4). (J) In situ intensity was measured in cells in ventral abdominal hemisegments in wor-gal4>p35 and wor-gal4>cut (n=3-4). (K-M) Although cut regulates rpr and grim expression, cut knockdown does not reduce the expression of enhancer1-GFP in the VNC, but rather slightly enhances the number of enh1-GFP-expressing cells (n=3), suggesting that cut loss rescues cells downstream of enh1-GFP (M). For all embryos anterior is to the top. Dotted white line outlines the VNC. Data are mean±s.d. *P<0.05 versus control; unpaired t-test. Scale bars: 20 μm.
Our previous studies identified a regulatory region between rpr and grim, the NB regulatory region, which controls rpr, grim and skl expression to promote abdominal NB death (Tan et al., 2011). A 5 kb transgenic reporter generated from this region, enh1-GFP, is expressed in doomed abdominal NBs and is responsive to the levels of N and abd-A, which regulate NB death (Arya et al., 2015). We found that cut does not regulate enh1-GFP activity: knockdown of cut does not decrease enh1-GFP expression. Instead, we see a slight increase in the number of enh1-expressing cells on cut knockdown (Fig. 2K-M). This suggests that loss of cut blocks the death of enh1-GFP-expressing cells. Therefore, cut acts independently of enh1 to facilitate the activation of rpr and grim for NB death.
cut acts downstream of abdominal A
We, and others, have shown that abd-A is necessary and sufficient for abdominal NB apoptosis (Arya et al., 2015; Khandelwal et al., 2017; Prokop et al., 1998). Overexpression of abd-A results in premature NB death in abdominal segments and in ectopic enh1-GFP expression and NB death in thoracic segments (Arya et al., 2015) (Fig. 3B,F). If cut acts to regulate apoptosis downstream of abd-A, cut knockdown should block killing by ectopic abd-A. Indeed, we found that cut knockdown in the context of abd-A overexpression blocks ectopic NB death in both thoracic and abdominal segments of the VNC (Fig. 3A-D). Importantly, ectopic abd-A-activated enh1-GFP expression is still apparent in cut knockdown (Fig. 3E-H), indicating that cut knockdown does not prevent abd-A from activating the regulatory region, but blocks rpr and grim expression downstream of enhancer activation. We also found that abd-A knockdown does not rescue NB death induced by cut overexpression (Fig. 1H). These data support the conclusion that cut regulates NB death downstream of abd-A and the enh1 regulatory region, and upstream of the RHG genes.
Fig. 3.

cut acts downstream of abd-A. (A-C) Knockdown of cut inhibits NB killing by abd-A overexpression in both abdominal and thoracic domains (indicated by pale blue and pale orange brackets, respectively). NBs are detected using Dpn staining. (D) Quantification of NBs in thoracic and abdominal segments (n=3-4). (E-G) Loss of cut does not inhibit ectopic enh1-GFP expression in thoracic segments (pale orange bracket) induced by abd-A mis-expression. GFP is detected using FISH (green), wor>dsRed (red). (H) Quantification of enh1-GFP-expressing thoracic NBs in each genotype (n=3-4). Data are mean±s.d. *P<0.05; unpaired t-test. Scale bars: 50 μm.
cut does not inhibit NB death through a binding site in the IRER left barrier
Enhancer/promoter interactions can be temporally controlled by changes in chromatin accessibility (Uyehara et al., 2017). Loss of cut inhibits NB death downstream of enh1 activity, suggesting that cut could influence rpr and grim expression in NBs by influencing chromatin accessibility in the rpr/grim region. Another enhancer in the cell death gene locus, the irradiation responsive enhancer region (IRER), which is located 5′ of the rpr promoter, shows temporal changes in chromatin conformation that are responsible for the reduced sensitivity to irradiation in later stages of embryogenesis (Zhang et al., 2008). This finding is consistent with our hypothesis that changes in chromatin conformation at the cell death gene locus regulate competence to respond to apoptosis-inducing signals.
Previous studies showed that, in older embryos, there is an enrichment in the repressive histone modifications H3K27me3 and H3K9me3 in the IRER enhancer, whereas the rpr proximal promoter remains enriched for the active chromatin mark H3K4me3 (Fig. S4) (Lin et al., 2011). This study identified a chromatin barrier within the IRER (IRER left barrier element, or ILB) that could block the spreading of repressive chromatin into the promoter proximal region (Lin et al., 2011). This barrier contains a putative Cut binding site that is necessary for barrier function. We therefore asked whether the Cut binding site in the ILB was crucial to prevent heterochromatin spreading from the IRER into the rpr proximal promoter, allowing rpr activation and NB death in response to activation of enh1. We generated several deletions of the putative Cut binding site using CRISPR/Cas9 (Fig. S4A). We examined NB death in animals homozygous for these deletions, and found that NB death was normal (Fig. S4B). NB death is also normal in animals homozygous for the larger IRER deletion generated by L.Z.’s lab (data not shown). Thus, the predicted Cut binding site in the ILB is not required for NB death downstream of enh1 activation. This result suggests that additional cut-dependent mechanisms are important for communication between the NB enhancer and the rpr and grim promoters.
cut inhibition of NB death correlates with altered H3K27me3 in NBs
We hypothesized that an increase in repressive histone modifications at the cell death gene locus in NBs could limit NB death on cut knockdown. To test this, we used ChIP to assay repressive and activating histone modifications in the CNS of control embryos and after cut knockdown. To enrich for NB chromatin and to limit stress-induced changes in cell death gene expression, ChIP was carried out on chromatin that had been isolated from sorted fixed CNS nuclei (stage 13-17, based on wor>dsRed expression; Arya et al., 2015) from wor>dsRed (wor>+) and wor>dsRed cutRNAi (wor>cutRNAi) embryos (Bowman et al., 2013).
We noted that the overall enrichment for the repressive histone modification H3K27me3 was generally low in the grim to rpr region in CNS chromatin in both genotypes, in contrast to data that were previously obtained from whole stage 16 embryos (14-16 h) (Nègre et al., 2011). In response to cut knockdown, we saw a very significant enrichment for this repressive mark at rpr (Fig. S5A), consistent with the decreased transcription of rpr that was detected in the absence of cut (Fig. 2). There was a slight enrichment of peaks elsewhere in the grim to rpr region. This suggests that cut may normally inhibit the formation of facultative heterochromatin in the rpr region in the developing CNS. In contrast to the relatively weak enrichment for H3K27me3 peaks in the rpr region, we detected strong enrichment in both genotypes in the bithorax complex (Fig. S5D), validating the quality of our library (Bowman et al., 2013).
To confirm changes that were detected by ChIP-seq, we assayed H3K27me3 enrichment at several positions in the rpr region by ChIP-qPCR using multiple independent chromatin preparations from fixed CNS nuclei. We found that cut knockdown led to enrichment for H3K27me3 within the rpr and grim open reading frames, in the region 5 kb upstream of rpr corresponding to the ILB (Lin et al., 2011) (Fig. S5B), and at the rpr promoter (Fig. S5C). Taken together, these assays indicate that cut knockdown increases repressive histone modifications in the RHG region in the developing CNS, and this could be responsible for decreased rpr and grim expression. However, the effect of cut knockdown on H3K27me3 levels is relatively limited, which could reflect redundant mechanisms underlying the activity of cut, or could be because of the low representation of the cells of interest (NBs) in our chromatin preparation.
Overall levels of repressive H3K27me3 increase in NBs during development, and this is inhibited by cut
To focus more precisely on cut-dependent changes in histone modifications in NBs, we stained control and cut knockdown embryos for H3K27me3. Strikingly, we found that in control embryos at stage 14, there was a clear difference in the overall levels of H3K27me3 in NBs, compared with other tissues in the embryo, and with other cells in the CNS (Fig. 4A). H3K27me3 levels were low or undetectable in the ventral NB layer, and much higher in the more dorsal layers of the CNS that contain the differentiated neurons and glia (Fig. S6). This is not because of a defect in histone antibody accessibility in these cells, as other histone modifications were not strikingly different in NBs compared with other neural cells (Fig. S6). Quantification showed that only ∼10% of NBs at stage 14 had high global levels of H3K27me3 (Fig. 4B,C). The lower levels of the repressive H3K27me3 modification in early NBs may reflect the increased plasticity of chromatin in these stem cells (Marshall and Brand, 2017). Furthermore, we found that levels of H3K27me3 increase in control NBs over time, so that by stage 16 ∼30% of NBs were scored as H3K27me3-positive (Fig. 4B). This increase in repressive histone modifications could reflect a restriction of stem cell potential over developmental time.
Fig. 4.

cut knockdown increases H3K27me3 levels in the rpr to grim interval. (A) H3K27me3 levels are lower in NBs than in the rest of the embryo. (B) The proportion of NBs with strong H3K27me3 labeling increases as embryos age. Knockdown of cut increases this proportion at each stage (n=3-6). (C) At stage 14 most NBs in control embryos lack strong H3K27me3 staining (white arrows), whereas a small proportion show higher overall H3K27me3 levels (yellow arrowheads). Knockdown of cut increases the number of NBs with higher H3K27me3 levels. Data are mean±s.d. *P<0.05 versus control; unpaired t-test.
Surprisingly, we found that cut knockdown increased the number of NBs with high H3K27me3 at stages 14 through 16 (Fig. 4B). This indicates that cut is required to inhibit the deposition of H3K27me3 in NBs. Lower global levels of H3K27me3 and a more open chromatin conformation could be required for normal rpr and grim activation during the period of cell death. Interestingly, there was still a temporal increase in H3K27me3 levels even on cut knockdown; the number of NBs with increased H3K27me3 was higher in older cut knockdown embryos than at earlier stages.
Cohesins operate downstream of cut to regulate NB death
Because cut has not been characterized as a histone modifier or as part of a histone modifier complex, we hypothesized that the effect of cut knockdown on H3K27me3 levels in NBs was likely to be indirect. We scanned our H3K27ac ChIP-seq data to identify potential cut-regulated genes that could restrain H3K27me3 levels in NBs. The H3K27ac histone modification is associated with active genes and enhancers, and altered H3K27ac could indicate a change in gene expression (Nègre et al., 2011). Most genes did not exhibit changes in H3K27ac peaks, including polycomb (Pc) complex components. However, two structural components of the cohesin complex, Stromalin (SA) and SMC1 showed decreased H3K27ac levels following cut knockdown, suggesting that they could be positively regulated by Cut (Fig. S7A). The cohesin complex is implicated in three-dimensional chromatin architecture, including enhancer/promoter interactions (Kagey et al., 2010). Cohesin also interacts with the polycomb repressive complex 1 (PRC1), and may sequester PRC1 from repressive chromatin, resulting in an overall mutually exclusive distribution of cohesin binding and repressive histone modifications (Misulovin et al., 2008; Schaaf et al., 2013).
We hypothesized that cut knockdown could decrease cohesin activity, leading to increased repressive chromatin and decreased cell death gene expression. A decrease in cohesin expression upon cut knockdown could interfere with communication between the NB enhancer and the rpr and grim promoters, altering their expression and inhibiting NB death. In addition, loss of cohesin could enhance the deposition of repressive chromatin to limit the expression of cell death genes.
To examine whether SA expression was controlled by cut, we assessed SA protein expression in cut knockdown. Loss of cut decreased SA protein levels in both NBs and neurons (Fig. 5A-C), indicating that Cut regulation of SA is not restricted to a single cell type. In addition, cut overexpression was sufficient to increase SA protein expression in NBs (Fig. 5D-E). We also assayed SA transcript levels using qPCR on RNA from sorted CNS nuclei from control and cut-RNAi embryos, and found that SA RNA levels were decreased on cut knockdown (Fig. S7B), indicating that Cut regulates SA at the transcriptional level.
Fig. 5.

cut controls the expression of the cohesin component SA. (A) SA protein levels are decreased in the CNS by cut knockdown. Lower panels are magnifications of the image above to facilitate the comparison of SA levels. Anterior is to the top. (B) Mean intensity of SA in stage 16 NBs is significantly decreased relative to non-CNS cells on cut knockdown (n=3). (C) Mean intensity of SA in neurons is significantly decreased relative to non-CNS cells at stage 15 and 16 on cut knockdown. (D,E) SA levels are significantly increased at stages 14-16 on cut overexpression (n=3). Data are mean±s.d. *P<0.05 versus control; unpaired t-test. Scale bars: 20 μm.
If cut regulates NB death by altering cohesin expression, then cohesin knockdown should phenocopy the loss of cut and inhibit NB death. Indeed, we found that NB survival was increased by a mild knockdown of SA (Fig. 6A,B,D, Fig. S7E-G). This was not due to a role for cohesin in NB proliferation, as mild knockdown of SA did not alter NB proliferation, NB formation or cut expression (Fig. S7C,D). In addition, knockdown of Nipped-B, part of the kollerin complex that is required for cohesin loading (Dorsett and Kassis, 2014), also resulted in ectopic NB survival (Fig. 6C,D). These data demonstrate a previously unknown requirement for cohesin in the regulation of NB death.
Fig. 6.

Cohesin is required for normal abdominal NB death. (A-C) Dpn staining reveals ectopic NB survival in stage 17 embryos after SA or Nipped-B knockdown. Yellow bars mark the abdominal hemisegments. (D) Dpn-positive NBs were counted in three abdominal segments, and are significantly increased in stage 17 embryos on cohesin knockdown (n=5-6). (E) Knockdown of SA results in an increased proportion of H3K27me3-positive NBs at stage 16 (n=3-5). (F,G) Single confocal section from stage 16 embryos to illustrate H3K27me3-positive NBs. F″ and G″ show enlargement of boxed areas in F′ and G′, respectively, to indicate H3K27me3 staining. (H,I) FISH analysis reveals a significant decrease in grim expression in NBs on SA knockdown. Single confocal section of the VNC with enlarged view of boxed area on right. (J,K,L) Expression of grim in NBs is significantly altered at stages 14-16, whereas expression of rpr and skl are not significantly affected (n=2-4). Data are mean±s.d. *P<0.05 versus control; unpaired t-test. Scale bars: 20 μm.
Cohesin knockdown increases Pc binding at the majority of H3K27me3-marked genes (Schaaf et al., 2013). We examined overall H3K27me3 levels in NBs after cohesin knockdown. We found that SA knockdown increased the number of NBs with high levels of H3K27me3 in embryos (Fig. 6E-G), suggesting that higher H3K27me3 levels in NBs on cut knockdown could be caused by decreased cohesin. Furthermore, we found that SA knockdown suppressed the levels of grim mRNA in NBs, but did not significantly alter rpr and skl expression (Fig. 6H-L). Thus, cohesin is required for abdominal NB death, regulating cell death gene expression and altering overall levels of repressive chromatin in NBs. We propose that the role of Cut in regulating NB death involves its control of cohesin component expression levels, which in turn regulate chromatin architecture at the cell death genes to promote their expression in the appropriate context (Fig. 7).
Fig. 7.
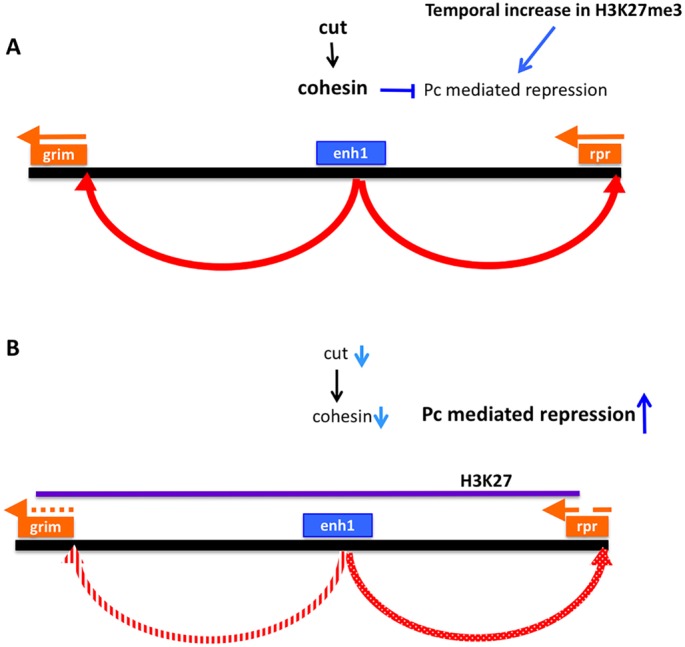
Model of cut action in NB death. (A) As neural stem cells age, they show an overall increase in repressive chromatin, marked by H3K27me3. Expression of cut inhibits this increase, at least in part through enhancement of cohesin expression. A more open configuration in the grim/rpr region allows the NB enhancer, activated by additional cell type-specific spatial and temporal factors, to turn on the transcription of grim and rpr leading to properly programmed cell death. (B) When cut expression is suppressed, SA and possibly other cohesin subunits decline, allowing a premature increase in H3K27me3 in NBs. At the grim/rpr locus, this blocks expression in response to upstream apoptosis regulatory factors.
DISCUSSION
In this work, we report that cut plays a previously unknown role in the regulation of NB death. cut is permissive for the expression of rpr and grim, acting downstream of the previously identified NB regulatory region (Arya et al., 2015; Tan et al., 2011). We show that cut loss increases the number of NBs with high levels of H3K27me3, indicating a role for cut in maintaining open chromatin in NBs. At the RHG locus, this is reflected in higher levels of H3K27me3, associated with lower rpr and grim expression. Importantly, we find that cut regulates the levels of the cohesin subunit SA in the CNS, and we show that cohesin is required for NB death. This work demonstrates a novel connection between cut and cohesin in controlling the chromatin landscape and cell death in the developing CNS.
Is cut the ‘cell identity’ signal that is permissive for NB death?
Our previous work has identified the Hox gene abd-A as an important spatial signal for NB death in the embryo (Arya et al., 2015). A late pulse of Abd-A in NBs is regulated by Notch activation, which is dependent on Delta ligand expression in NB progeny. abd-A is necessary and sufficient for NB death, and has been shown to bind to enh1 in the NB regulatory region (Khandelwal et al., 2017). However, abd-A is clearly expressed in many cells that do not die (Arya et al., 2015; Karch et al., 1990). Furthermore, mis-expression of abd-A does not activate ectopic NB death before stage 13 of embryogenesis (Arya et al., 2015; Prokop et al., 1998), which suggests that there are additional temporal and cell identity signals that regulate the competence of cells to respond to abd-A.
Here, we identify cut as a novel regulator of NB death. Expression of cut in the embryonic CNS increases as NB death begins. However, most cells that normally express cut do not die, indicating that other factors coordinate with cut to regulate NB death. We find that loss of cut inhibits rpr and grim transcription, but in contrast to abd-A and N, cut does not act on enh1, as detected by enh1-GFP. In addition, cut knockdown blocks NB killing in response to abd-A mis-expression, despite an expansion of enh1 expression. These data indicate that cut acts downstream of enh1, and suggests that cut acts in the nervous system as a permissive factor that regulates the competence of NBs to respond to other cell death signals.
cut alters the chromatin landscape in the nervous system
We found that cut functions in the CNS to restrict overall levels of repressive H3K27me3-marked chromatin. We demonstrate that NBs have a significantly lower level of overall H3K27me3 than other tissues in the embryo, possibly associated with stem-cell plasticity (Zhu et al., 2013). These data support the findings of Marshall and Brand, who detected low levels of Polycomb repressed genes in larval NBs (Marshall and Brand, 2017). As embryos age, the number of NBs with higher overall levels of H3K27me3 increases. The cause and consequences of this transition are unknown, but could be related to a gradual restriction of NB fate (Marshall and Brand, 2017; Yuzyuk et al., 2009; Zhu et al., 2013).
We observed that loss of cut promotes more NBs to acquire an H3K27me3 high state at a given stage of embryogenesis. Interestingly, in both control and cut knockdown there is a temporal increase in the proportion of NBs with high H3K27me3. This suggests that additional temporal factors control this maturation of NBs to a more repressed state, but cut restrains the number of H3K27me3 high cells throughout this transition.
Our data indicate that cut overexpression is sufficient to cause increased rpr and grim expression and premature apoptosis in NBs. This is not because of hyperactivation of enh1, as ectopic cut does not increase enh1-GFP expression. In addition, cut overexpression causes increased cell death in other cells that normally survive, as seen with heat shock-gal4. Our data support the hypothesis that ectopic cut activates rpr and grim expression through increased cohesin levels, possibly decreasing repressive H3K27me3 modifications or facilitating enhancer/promoter interactions in the RHG locus.
The role of cut in activating NB death is in contrast to previous work that has suggested cut inhibits cell death in the developing posterior spiracle by directly inhibiting rpr expression (Zhai et al., 2012). In the developing spiracle, cut is also required for normal differentiation. Several other tissues also require cut for normal differentiation, such as the bristle cells in the eye and the developing trachea. In these tissues, cell death is also increased in the absence of cut (Pitsouli and Perrimon, 2010; Zhai et al., 2012). The role of cut in promoting cell survival in these tissues differs from its role in facilitating cell death in the CNS. This may reflect the diverse activities of cut as a transcriptional regulator, or could be due to cut’s regulation of cohesin, which acts as a chromatin organizer, altering the landscape for binding by both activators and repressors of RHG gene transcription. Both pro-differentiation and pro-apoptotic roles of cut are consistent with its role as a potential tumor suppressor (Wong et al., 2014; Zhai et al., 2012).
Cell death genes are highly sensitive to altered chromatin accessibility
This study, and previous work from L.Z.’s lab, indicates that the rpr region is particularly sensitive to alterations in chromatin conformation, reflecting the need for rapid and robust transcription of the cell death genes in cells fated to die (Zhang et al., 2008). Other factors that control histone modifications are involved in cell death. For example, the dUTX H3K27me3 demethylase is required for Ecdysone Receptor-mediated activation of rpr expression in salivary gland death (Denton et al., 2013). This supports our finding that a more open chromatin conformation is particularly important for cell death gene activation. Expression of other components of the cell death pathway may also be controlled by changes in chromatin conformation. For example, treatment of Drosophila larvae with HDAC inhibitors, or HDAC1 knockdown, increases sensitivity to cell death activation through altered expression of caspases (Kang et al., 2017). Conversely, loss of Pc-mediated suppression is associated with loss of postembryonic NBs, although this may be due to ectopic abd-A expression (Bello et al., 2007). There is also evidence for epigenetic regulation of genes that are important for cell death in the mammalian nervous system and in cancer (Song et al., 2011; Wright et al., 2007). Here, we provide evidence that control of histone modifications in the rpr region is an important aspect of developmental cell death regulation.
Cohesin as a regulator of cell death
Given the lack of evidence for a direct histone-modifying role of Cut, we investigated alternative indirect mechanisms for cut’s role in suppressing overall H3K27me3 levels in developing NBs. We determined that cut promotes expression of the cohesin subunit SA. We found that, similar to loss of cut, downregulation of SA or Nipped-B results in ectopic NB survival, and SA knockdown results in increased numbers of NBs with high levels of H3K27me3 at stage 16. Cohesins are involved in sister chromatid cohesion, formation of topologically associated domains and in long-range enhancer/promoter interactions (Kagey et al., 2010; Newkirk et al., 2017). Furthermore, cohesin binding can inhibit repressive histone modifications (Misulovin et al., 2008; Schaaf et al., 2013) and cohesin knockdown can increase repressive chromatin in hematopoietic stem and progenitor cells (Chen et al., 2018). Our findings in NBs strongly support a role for cohesin in controlling the chromatin landscape. Both long-range enhancer/promoter interactions and overall chromatin accessibility may be important in regulating Drosophila developmental cell death. In the RHG genomic region, multiple cell death genes are activated in different tissues in response to overlapping signals impinging on distinct regulatory enhancers, which act at considerable distance from the targeted promoters (Arya et al., 2015; Jiang et al., 2000; Khandelwal et al., 2017; Lohmann et al., 2002; Zhang et al., 2008). This suggests that three-dimensional chromatin interactions, including those mediated by cohesin, are crucial for facilitating precise gene activation in the RHG region. In addition, we show here that slight alterations in repressive chromatin in the RHG genomic region are associated with altered cell death gene expression.
Loss of one copy of the human Nipped-B homolog NIPBL, and of other cohesin components, is associated with Cornelia de Lange syndrome, a developmental disorder that affects growth, cognitive function, and facial and limb morphology (Newkirk et al., 2017; Wu et al., 2015). This is likely owing to the downregulation of developmentally important genes, as detected in MEFs with one copy of NIPBL deleted (Newkirk et al., 2017). Nipped-B heterozygous flies also exhibit reduced growth, learning and memory deficits, abnormal brain morphology and reduced expression of many genes (Wu et al., 2015). Interestingly, Nipped-B heterozygotes are resistant to dMyc-induced apoptosis, a phenotype that is also seen when the IRER is deleted (Wu et al., 2015; Zhang et al., 2015), suggesting that cohesin also regulates cell death gene expression activated by the IRER enhancer. Our data suggest that control of cell death in the nervous system could also contribute to the Cornelia de Lange syndrome phenotype. Additional studies are needed to understand how cohesin activity is targeted towards regulating the expression of specific genes under specific circumstances.
Precise control of apoptotic gene expression is particularly important in the nervous system, the site of the majority of developmental cell death in flies, worms and mammals, and the tissue most affected by the absence of cell death (Arya and White, 2015). Our work has led to a greater understanding of the temporal, spatial and tissue specific control of this death in flies through developmentally important transcription factors as well as regulation of chromatin accessibility and architecture. Given the conserved functions of the pathways we have identified, it is likely that these studies will provide insight into the regulation of cell death in human nervous system development and disease.
MATERIALS AND METHODS
Fly stocks and genotypes
Flies were raised at 25°C, and RNAi crosses were done at 29°C. Wild-type fly lines used in this study are yw67c23 and wor-Gal4/+. The following lines were obtained from the Bloomington Drosophila Stock Center (BL) and the Vienna Drosophila Resource Center (VDRC) and by personal communications: wor-gal4 (Albertson et al., 2004), repo-Gal4 (BL 7415), UAS-nls-dsRed (BL 8546), abd-A-RNAi (BL 35644), cut-RNAi (BL 33967, VDRC GD5687), SA RNAi (BL 33395), Nipped-B RNAi (BL 32406), UAS-Cut::UAS-mcd8-GFP (BL 36496; Norbert Perrimon, Harvard Medical School, MA, USA), cutc145 (BL6946), cut-RNAi; cut-RNAi (provided by Y.N. Jan, University of California, San Francisco, CA, USA; Zhai et al., 2012), UAS-abd-A.HA (provided by Y. Graba, CNRS, Marseille, France) and dpn-GFP (dpn[MI00051-GFSTF.0] BL 59755). The enh1-GFP transgenic line was previously generated in our lab (Arya et al., 2015).
Immunostaining and fluorescent in situ hybridization
Staining of whole embryos and larval CNS, and TUNEL staining, were carried out as described previously (Arya et al., 2015). The following primary antibodies were used in various combinations: rat anti-Dpn (1:150, ab195173, Abcam), goat anti-Abd-A (1:500, dH17, Santa Cruz Biotechnology), rabbit or mouse anti-GFP antibody (1:1000, rabbit A-11122, Invitrogen, or mouse jl8, Clontech), mouse anti-Cut (1:700, 2B10, Developmental Studies Hybridoma Bank), rabbit anti-H3K27me3 (1:500, 39157, Active Motif), rabbit anti-H3K9me3 (1:500, ab8898, Abcam), rabbit anti-H3K27ac (1:500, 39136, Active Motif), rabbit anti-H3K4me3 (1:500, 39159, Active Motif) and rabbit anti-Stromalin (1:700, a gift from Dale Dorsett, Saint Louis University School of Medicine, MO, USA). Secondary antibodies (Molecular Probes, Alexa Fluor 488, 568 or 647 conjugates to antibodies of appropriate specificity; for SA, anti-guinea pig 647 conjugate was obtained from Jackson ImmunoResearch) were used at 1:200 dilution. All antibody staining results were found to be consistent in repeat experiments. Experimental/control pairs processed simultaneously were used for quantification.
Fluorescent in situ hybridization (FISH) was performed as previously described (Arya et al., 2015). FISH/antibody staining for quantification of NB expression was carried out by including dpn-GFP in the wor-gal4 strain. Double labeling was carried out essentially as described in Lécuyer et al. (2008), except the anti-Dig (1:1000, 11207733910, Roche) and anti-GFP (1:200, A-11122, Molecular Probes) antibodies were left on overnight, and the anti-rabbit secondary antibody was incubated for 48 h after the tyramide signal amplification step. Digoxigenin (DIG)-labeled probes for grim, rpr and GFP were used. To allow comparison of expression levels, in situs were processed in parallel, and imaged with matched confocal settings and image processing. Embryos were imaged with a Nikon A1SiR confocal microscope. Image processing was carried out using Nikon Elements, ImageJ or Adobe Photoshop. FISH signals for rpr and grim in NBs were quantified using ImageJ, measuring the average in situ signal in at least 15 Dpn+ abdominal NBs and subtracting average background in three abdominal hemisegments in at least three embryos.
The intensity of histone marks was quantified with ImageJ software: average fluorescence intensity within a hemisegment of the nervous system was calculated and normalized to the intensity of a corresponding area within the epidermis. This calculation was performed across 11 confocal slices beginning with the ventral-most slice of the nervous system, as determined by Deadpan (Dpn) staining. Data in Fig. S6 are presented as the ratio of the average intensity of signal within the nervous system to the average intensity of signal within the epidermis at a given confocal slice. H3K27me3-high NBs were counted in abdominal NBs, based on visual inspection. In some cases, H3K27me3 signal was measured in abdominal NBs, background was subtracted, and a cutoff was set as H3K27me3-high. This cutoff was verified by an independent visual analysis, and the two methods provided very similar results.
SA levels in NBs and neurons were quantified in ImageJ. SA intensity was measured in ten NBs or neurons in A3 to A6 segments of three embryos. To account for embryo-to-embryo variability in staining, SA levels in NBs or neurons was normalized to the average SA intensity outside of the nervous system in the same z-section.
Embryo collection and nuclei preparation
Embryos were collected for 18 h at 25°C. Dechorionated embryos were fixed in 1:1 solution of 1.8% formaldehyde and heptane for 15 min at room temperature. The fixative was quenched with a 2 min wash with 125 mM glycine in PBS [130 mM NaCl, 7 mM Na2HPO4, 3 mM KH2PO4 (pH 8.0)] with 0.1% Triton X-100, and then briefly rinsed with PBS 0.1% Triton X-100. Embryos were snap frozen in liquid nitrogen, and stored at −80°C. About 1 g of embryos of each genotype was used for nuclei isolation as described by Bowman et al. (Bowman et al., 2014, 2013).
Nuclear sorting and ChIP-seq
Fixed nuclei from wor>dsRed embryos were enriched using a Bio-Rad S3e cell sorter with 561 nm excitation. Nuclei were sorted at 4°C in 100 μl of PBS, with 1% bovine serum albumin, 0.1% Triton X-100 and 1× protease inhibitor. wor-gal4 is expressed in the nervous system from stage 11 onwards (Arya et al., 2015). The sorted nuclei represent ∼0.5-3% of total embryonic nuclei and were at least 50% pure, based on post-isolation assessment of dsRed by confocal microscopy. About a million nuclei were used for chromatin preparation. After isolation, chromatin was fragmented with 15 U of micrococcal nuclease (MNase, Worthington Biochemical Corporation) followed by 3 min sonication in a Diagenode Bioruptor 377 (Bowman et al., 2013). Immunoprecipitation was carried out in 500 μl volume using 2 μg of H3K27me3 antibody (39136, Active Motif) or 1 μg of H3K27Ac antibody (39136, Active Motif). Input DNA was used as a control. Single end tag libraries were prepared and sequenced on an Illumina, Hiseq2500 in high output mode at the Massachusetts General Hospital Next Generation Sequencing Core.
ChIP-seq data analysis
High-throughput sequence data were processed and analyzed for quality. Samples with reasonable ChIP strength were further analyzed. A single ChIP-seq prep of sufficient quality was available for H3K27me3 and H3K27Ac control and experimental pairs. Reads were mapped to the genome (dm6) using Bowtie2 (Langmead and Salzberg, 2012). The resulting sequence alignment map files were used to identify enrichment using SICER, which is particularly appropriate for analysis of histone modifications (Xu et al., 2014). For SICER peak calls a window size of 200 bp was used, as suggested by the developer for analysis of histone modifications. The fragment size parameter was set to 150 bp, based on our empirical data. A gap size of 600 bp and an effective genome fraction of 0.74 was used, with a false discovery rate of 0.01. The resulting BED file of enriched genomic regions and the normalized BedGraph files were loaded to the University of California, Santa Cruz, Genome Browser for comparison and analysis. For differential peak analysis we used MAnorm with standard parameters (Shao et al., 2012). With significance set at P<0.001, there were 1230 total peaks with significant H3K27me3 enrichment, of which 709 are unique (Table S1). Of these peaks, 348 were unique to control and 361 to wor>cutRNAi. For H3K27ac, there were 1555 total significant peaks, of which 643 were unique (Table S2). Of these peaks, 395 were unique to control and 248 were unique to cutRNAi.
ChIP-RT-qPCR analysis
For the validation of changes detected in ChIP-seq data, ChIP-qPCR was performed. Independent nuclei sorts were used for each chromatin prep. Immunoprecipitation was conducted as described above, using the H3K27me3 antibody. The immunoprecipitated DNA was processed for qPCR analysis using iTaq universal SYBER green supermix (Bio-Rad) on an Applied Biosystems 7000 Real-Time System. Data was analyzed using the delta delta CT method. The following primers were used in the study: Rpr2-F: TGGGTTGGCTCATGCTTATT; Rpr2-R: ATCCGAAGACCGGAAGAAAG; Rpr-5kb1-F: CCGTCTACGGCCTTTGTTTA; Rpr-5kb1-R: AGTGGAAGAACCAACCTGACA; Rpr-5kb2-F: TTTTCGGAATGGGTTTTCAG; Rpr-5kb2-R: ACACACACGAACCGAATGAA; GRIM2-F: TTATGCCAACAACCAACCAA; GRIM2-R: CCCCCTTTCTAGTTCCGAAG; AbdbChip2-F: TCTACTCCACCGGTTTGCTC; AbdbChip2-R: ACAGGCGGTCCTTATTGATG; intergenicSKB-F: TCAAGCCGAACCCTCTAAAAT; intergenicSKB-R: AACGCCAACAAACAGAAAATG; rpr_pro_F: AGAAGGCCAAAATGAGCAGC; rpr_pro_R: GCGCACACACTTTTCTTTCG; Act5C_f: ATGTGTGTGTGAGAGAGCGA; Act5C_b: AAACCGACTGAAAGTGGCTG.
Nuclear RNA preparation
After isolation of nuclei, as described above, proteins were digested and crosslinking reversed in 20 mM Tris/1 mM CaCl2/0.5% SDS with 1 U/μl RNAse inhibitor and 500 μg/ml proteinase K (Roche) at 55°C for 3 h. Approximately 600,000 nuclei were used for RNA purification with RNAzol (Sigma-Aldrich). A mix of oligodT and random hexamer primers were used for reverse transcription, and qPCR was carried out using an Applied Biosystems 7000 Real-Time System. DNA contamination was assessed with a no reverse transcriptase control, and data were analyzed using the delta delta CT method. The following primers were used for qPCR: dRP49-F: 5′ CTC ATG CAG AAC CGC GTT TA 3′; dRP49-R: 5′ ACA AAT GTG TAT TCC GAC CA 3′; SA-F: GGACAAGATAATACCACCCGC; SA-R: CGCTTGATCAGTTTCGCCAT.
Generation of CRISPR deletion
Small deletions (120-130 bp) of the Cut binding site upstream of the rpr promoter were generated using CRISPR/Cas9 (Fig. S4). Two guide RNAs were cloned in pCFD4 Vector (Addgene plasmid #49411) as previously described (Port et al., 2014). Stable gRNA transgenics were made by BestGene and crossed with nos–cas9 (BL 54591). The progeny were screened for deletions using PCR, and breakpoints were confirmed by sequencing. Two lines, CRISPR_ILB_2.9 and 431CRISPR_ILB_3.1, were used for phenotypic analysis.
Supplementary Material
Acknowledgements
We thank Norbert Perrimon, Y.N. Jan, the Bloomington Drosophila Stock Center and the Vienna Drosophila Resource Center for fly stocks; Antoine Borensztejn for generating and validating the wor-gal4 dpn-GFP line; Dale Dorsett and the Developmental Studies Hybridoma Bank for antibodies; Sarah Bowman, Katia Georgopoulos and Georgopoulos lab members for advice on ChIP studies and data analysis; and the White lab and Katia Georgopoulos for comments on the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: R.A., L.Z., K.W.; Formal analysis: R.A., S.G., K.H., L.Z., K.W.; Investigation: R.A., S.G., K.H., T.S., Y.L.; Writing - original draft: K.W.; Writing - review & editing: R.A., K.H., K.W.; Visualization: K.W.; Supervision: K.W.; Project administration: K.W.; Funding acquisition: K.W.
Funding
This work was funded by the National Institutes of Health R01GM110477 (K.W. and L.Z.), R03NS092063 (K.W.), R01GM106174 (L.Z.), a Massachusetts General Hospital Fund for Medical Discovery award (R.A.) and a Department of Biotechnology, Ministry of Science and Technology, Ramalingaswami Fellowship (R.A.). Deposited in PMC for release after 12 months.
Data availability
ChIP-seq data have been deposited in Gene Expression Omnibus under accession number GSE124998.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.166603.supplemental
References
- Abrams J. M., White K., Fessler L. I. and Steller H. (1993). Programmed cell death during Drosophila embryogenesis. Development 117, 29-43. [DOI] [PubMed] [Google Scholar]
- Albertson R., Chabu C., Sheehan A. and Doe C. Q. (2004). Scribble protein domain mapping reveals a multistep localization mechanism and domains necessary for establishing cortical polarity. J. Cell Sci. 117, 6061-6070. 10.1242/jcs.01525 [DOI] [PubMed] [Google Scholar]
- Almeida M. S. and Bray S. J. (2005). Regulation of post-embryonic neuroblasts by Drosophila Grainyhead. Mech. Dev. 122, 1282-1293. 10.1016/j.mod.2005.08.004 [DOI] [PubMed] [Google Scholar]
- Arya R. and White K. (2015). Cell death in development: signaling pathways and core mechanisms. Semin. Cell Dev. Biol. 39, 12-19. 10.1016/j.semcdb.2015.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya R., Sarkissian T., Tan Y. and White K. (2015). Neural stem cell progeny regulate stem cell death in a Notch and Hox dependent manner. Cell Death Differ. 22, 1378-1387. 10.1038/cdd.2014.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangs P., Franc N. and White K. (2000). Molecular mechanisms of cell death and phagocytosis in Drosophila. Cell Death Differ. 7, 1027-1034. 10.1038/sj.cdd.4400754 [DOI] [PubMed] [Google Scholar]
- Bello B., Holbro N. and Reichert H. (2007). Polycomb group genes are required for neural stem cell survival in postembryonic neurogenesis of Drosophila. Development 134, 1091-1099. 10.1242/dev.02793 [DOI] [PubMed] [Google Scholar]
- Blochlinger K., Bodmer R., Jan L. Y. and Jan Y. N. (1990). Patterns of expression of cut, a protein required for external sensory organ development in wild-type and cut mutant Drosophila embryos. Genes Dev. 4, 1322-1331. 10.1101/gad.4.8.1322 [DOI] [PubMed] [Google Scholar]
- Bowman S. K., Simon M. D., Deaton A. M., Tolstorukov M., Borowsky M. L. and Kingston R. E. (2013). Multiplexed Illumina sequencing libraries from picogram quantities of DNA. BMC Genomics 14, 466 10.1186/1471-2164-14-466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman S. K., Deaton A. M., Domingues H., Wang P. I., Sadreyev R. I., Kingston R. E. and Bender W. (2014). H3K27 modifications define segmental regulatory domains in the Drosophila bithorax complex. eLife 3, e02833 10.7554/eLife.02833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray S. J., Burke B., Brown N. H. and Hirsh J. (1989). Embryonic expression pattern of a family of Drosophila proteins that interact with a central nervous system regulatory element. Genes Dev. 3, 1130-1145. 10.1101/gad.3.8.1130 [DOI] [PubMed] [Google Scholar]
- Cenci C. and Gould A. P. (2005). Drosophila Grainyhead specifies late programmes of neural proliferation by regulating the mitotic activity and Hox-dependent apoptosis of neuroblasts. Development 132, 3835-3845. 10.1242/dev.01932 [DOI] [PubMed] [Google Scholar]
- Chen Z., Amro E. M., Becker F., Holzer M., Rasa S. M. M., Njeru S. N., Han B., Di Sanzo S., Chen Y., Tang D. et al. (2018). Cohesin-mediated NF-kappaB signaling limits hematopoietic stem cell self-renewal in aging and inflammation. J. Exp. Med. 216, 152-175. 10.1084/jem.20181505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem R. J. and Miller L. K. (1994). Control of programmed cell death by the baculovirus genes p35 and iap. Mol. Cell. Biol. 14, 5212-5222. 10.1128/MCB.14.8.5212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubelos B., Sebastián-Serrano A., Kim S., Moreno-Ortiz C., Redondo J. M., Walsh C. A. and Nieto M. (2008). Cux-2 controls the proliferation of neuronal intermediate precursors of the cortical subventricular zone. Cereb. Cortex 18, 1758-1770. 10.1093/cercor/bhm199 [DOI] [PubMed] [Google Scholar]
- Denton D., Aung-Htut M. T., Lorensuhewa N., Nicolson S., Zhu W., Mills K., Cakouros D., Bergmann A. and Kumar S. (2013). UTX coordinates steroid hormone-mediated autophagy and cell death. Nat. Commun. 4, 2916 10.1038/ncomms3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett D. (2016). The Drosophila melanogaster model for Cornelia de Lange syndrome: implications for etiology and therapeutics. Am. J. Med. Genet. C Semin. Med. Genet. 172, 129-137. 10.1002/ajmg.c.31490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett D. and Kassis J. A. (2014). Checks and balances between cohesin and polycomb in gene silencing and transcription. Curr. Biol. 24, R535-R539. 10.1016/j.cub.2014.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs Y. and Steller H. (2011). Programmed cell death in animal development and disease. Cell 147, 742-758. 10.1016/j.cell.2011.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay B. A., Wolff T. and Rubin G. M. (1994). Expression of baculovirus P35 prevents cell death in Drosophila. Development 120, 2121-2129. [DOI] [PubMed] [Google Scholar]
- Iulianella A., Sharma M., Durnin M., Vanden Heuvel G. B. and Trainor P. A. (2008). Cux2 (Cutl2) integrates neural progenitor development with cell-cycle progression during spinal cord neurogenesis. Development 135, 729-741. 10.1242/dev.013276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C. A., Lamblin A.-F. J., Steller H. and Thummel C. S. (2000). A steroid-triggered transcriptional hierarchy controls salivary gland cell death during Drosophila metamorphosis. Mol. Cell 5, 445-455. 10.1016/S1097-2765(00)80439-6 [DOI] [PubMed] [Google Scholar]
- Johnson T. K. and Judd B. H. (1979). Analysis of the cut locus of Drosophila melanogaster. Genetics 92, 485-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey M. H., Newman J. J., Bilodeau S., Zhan Y., Orlando D. A., van Berkum N. L., Ebmeier C. C., Goossens J., Rahl P. B., Levine S. S. et al. (2010). Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430-435. 10.1038/nature09380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y., Marischuk K., Castelvecchi G. D. and Bashirullah A. (2017). HDAC inhibitors disrupt programmed resistance to apoptosis during Drosophila development. G3 (Bethesda) 7, 1985-1993. 10.1534/g3.117.041541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch F., Bender W. and Weiffenbach B. (1990). abdA expression in Drosophila embryos. Genes Dev. 4, 1573-1587. 10.1101/gad.4.9.1573 [DOI] [PubMed] [Google Scholar]
- Khandelwal R., Sipani R., Govinda Rajan S., Kumar R. and Joshi R. (2017). Combinatorial action of Grainyhead, extradenticle and Notch in regulating Hox mediated apoptosis in Drosophila larval CNS. PLoS Genet. 13, e1007043 10.1371/journal.pgen.1007043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornbluth S. and White K. (2005). Apoptosis in Drosophila: neither fish nor fowl (nor man, nor worm). J. Cell Sci. 118, 1779-1787. 10.1242/jcs.02377 [DOI] [PubMed] [Google Scholar]
- Langmead B. and Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357-359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lécuyer E., Parthasarathy N. and Krause H. M. (2008). Fluorescent in situ hybridization protocols in Drosophila embryos and tissues. Methods Mol. Biol. 420, 289-302. 10.1007/978-1-59745-583-1_18 [DOI] [PubMed] [Google Scholar]
- Lin N., Li X., Cui K., Chepelev I., Tie F., Liu B., Li G., Harte P., Zhao K., Huang S. et al. (2011). A barrier-only boundary element delimits the formation of facultative heterochromatin in Drosophila melanogaster and vertebrates. Mol. Cell. Biol. 31, 2729-2741. 10.1128/MCB.05165-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann I., McGinnis N., Bodmer M. and McGinnis W. (2002). The Drosophila Hox gene deformed sculpts head morphology via direct regulation of the apoptosis activator reaper. Cell 110, 457-466. 10.1016/S0092-8674(02)00871-1 [DOI] [PubMed] [Google Scholar]
- Marshall O. J. and Brand A. H. (2017). Chromatin state changes during neural development revealed by in vivo cell-type specific profiling. Nat. Commun. 8, 2271 10.1038/s41467-017-02385-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurange C., Cheng L. and Gould A. P. (2008). Temporal transcription factors and their targets schedule the end of neural proliferation in Drosophila. Cell 133, 891-902. 10.1016/j.cell.2008.03.034 [DOI] [PubMed] [Google Scholar]
- Micchelli C. A., Rulifson E. J. and Blair S. S. (1997). The function and regulation of cut expression on the wing margin of Drosophila: Notch, Wingless and a dominant negative role for Delta and Serrate. Development 124, 1485-1495. [DOI] [PubMed] [Google Scholar]
- Misulovin Z., Schwartz Y. B., Li X.-Y., Kahn T. G., Gause M., MacArthur S., Fay J. C., Eisen M. B., Pirrotta V., Biggin M. D. et al. (2008). Association of cohesin and Nipped-B with transcriptionally active regions of the Drosophila melanogaster genome. Chromosoma 117, 89-102. 10.1007/s00412-007-0129-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon N.-S., Di Stefano L., Morris E. J., Patel R., White K. and Dyson N. J. (2008). E2F and p53 induce apoptosis independently during Drosophila development but intersect in the context of DNA damage. PLoS Genet. 4, e1000153 10.1371/journal.pgen.1000153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nègre N., Brown C. D., Ma L., Bristow C. A., Miller S. W., Wagner U., Kheradpour P., Eaton M. L., Loriaux P., Sealfon R. et al. (2011). A cis-regulatory map of the Drosophila genome. Nature 471, 527-531. 10.1038/nature09990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepveu A. (2001). Role of the multifunctional CDP/Cut/Cux homeodomain transcription factor in regulating differentiation, cell growth and development. Gene 270, 1-15. 10.1016/S0378-1119(01)00485-1 [DOI] [PubMed] [Google Scholar]
- Newkirk D. A., Chen Y.-Y., Chien R., Zeng W., Biesinger J., Flowers E., Kawauchi S., Santos R., Calof A. L., Lander A. D. et al. (2017). The effect of Nipped-B-like (Nipbl) haploinsufficiency on genome-wide cohesin binding and target gene expression: modeling Cornelia de Lange syndrome. Clin. Epigenet. 9, 89 10.1186/s13148-017-0391-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson C., Carney G. E., Taylor B. J. and White K. (2002). reaper is required for neuroblast apoptosis during Drosophila development. Development 129, 1467-1476. [DOI] [PubMed] [Google Scholar]
- Pitsouli C. and Perrimon N. (2010). Embryonic multipotent progenitors remodel the Drosophila airways during metamorphosis. Development 137, 3615-3624. 10.1242/dev.056408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitsouli C. and Perrimon N. (2013). The homeobox transcription factor cut coordinates patterning and growth during Drosophila airway remodeling. Sci. Signal. 6, ra12 10.1126/scisignal.2003424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Port F., Chen H.-M., Lee T. and Bullock S. L. (2014). Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc. Natl. Acad. Sci. USA 111, E2967-E2976. 10.1073/pnas.1405500111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokop A., Bray S., Harrison E. and Technau G. M. (1998). Homeotic regulation of segment-specific differences in neuroblast numbers and proliferation in the Drosophila central nervous system. Mech. Dev. 74, 99-110. 10.1016/S0925-4773(98)00068-9 [DOI] [PubMed] [Google Scholar]
- Rollins R. A., Morcillo P. and Dorsett D. (1999). Nipped-B, a Drosophila homologue of chromosomal adherins, participates in activation by remote enhancers in the cut and Ultrabithorax genes. Genetics 152, 577-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins R. A., Korom M., Aulner N., Martens A. and Dorsett D. (2004). Drosophila nipped-B protein supports sister chromatid cohesion and opposes the stromalin/Scc3 cohesion factor to facilitate long-range activation of the cut gene. Mol. Cell. Biol. 24, 3100-3111. 10.1128/MCB.24.8.3100-3111.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansregret L. and Nepveu A. (2008). The multiple roles of CUX1: insights from mouse models and cell-based assays. Gene 412, 84-94. 10.1016/j.gene.2008.01.017 [DOI] [PubMed] [Google Scholar]
- Schaaf C. A., Kwak H., Koenig A., Misulovin Z., Gohara D. W., Watson A., Zhou Y., Lis J. T. and Dorsett D. (2013). Genome-wide control of RNA polymerase II activity by cohesin. PLoS Genet. 9, e1003382 10.1371/journal.pgen.1003382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z., Zhang Y., Yuan G.-C., Orkin S. H. and Waxman D. J. (2012). MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 13, R16 10.1186/gb-2012-13-3-r16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S.-H., Han S.-W. and Bang Y.-J. (2011). Epigenetic-based therapies in cancer: progress to date. Drugs 71, 2391-2403. 10.2165/11596690-000000000-00000 [DOI] [PubMed] [Google Scholar]
- Tan Y., Yamada-Mabuchi M., Arya R., St Pierre S., Tang W., Tosa M., Brachmann C. and White K. (2011). Coordinated expression of cell death genes regulates neuroblast apoptosis. Development 138, 2197-2206. 10.1242/dev.058826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman J. W. and Bate M. (1988). Spatial and temporal patterns of neurogenesis in the central nervous system of Drosophila melanogaster. Dev. Biol. 125, 145-157. 10.1016/0012-1606(88)90067-X [DOI] [PubMed] [Google Scholar]
- Uyehara C. M., Nystrom S. L., Niederhuber M. J., Leatham-Jensen M., Ma Y., Buttitta L. A. and McKay D. J. (2017). Hormone-dependent control of developmental timing through regulation of chromatin accessibility. Genes Dev. 31, 862-875. 10.1101/gad.298182.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K., Grether M. E., Abrams J. M., Young L., Farrell K. and Steller H. (1994). Genetic control of programmed cell death in Drosophila. Science 264, 677-683. 10.1126/science.8171319 [DOI] [PubMed] [Google Scholar]
- Wong C. C., Martincorena I., Rust A. G., Rashid M., Alifrangis C., Alexandrov L. B., Tiffen J. C., Kober C., Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium, Green A. R. et al. (2014). Inactivating CUX1 mutations promote tumorigenesis. Nat. Genet. 46, 33-38. 10.1038/ng.2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright K. M., Smith M. I., Farrag L. and Deshmukh M. (2007). Chromatin modification of Apaf-1 restricts the apoptotic pathway in mature neurons. J. Cell Biol. 179, 825-832. 10.1083/jcb.200708086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Gause M., Xu D., Misulovin Z., Schaaf C. A., Mosarla R. C., Mannino E., Shannon M., Jones E., Shi M. et al. (2015). Drosophila Nipped-B mutants model cornelia de lange syndrome in growth and behavior. PLoS Genet. 11, e1005655 10.1371/journal.pgen.1005655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S., Grullon S., Ge K. and Peng W. (2014). Spatial clustering for identification of ChIP-enriched regions (SICER) to map regions of histone methylation patterns in embryonic stem cells. Methods Mol. Biol. 1150, 97-111. 10.1007/978-1-4939-0512-6_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzyuk T., Fakhouri T. H. I., Kiefer J. and Mango S. E. (2009). The polycomb complex protein mes-2/E(z) promotes the transition from developmental plasticity to differentiation in C. elegans embryos. Dev. Cell 16, 699-710. 10.1016/j.devcel.2009.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Z., Ha N., Papagiannouli F., Hamacher-Brady A., Brady N., Sorge S., Bezdan D. and Lohmann I. (2012). Antagonistic regulation of apoptosis and differentiation by the Cut transcription factor represents a tumor-suppressing mechanism in Drosophila. PLoS Genet. 8, e1002582 10.1371/journal.pgen.1002582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Lin N., Carroll P. M., Chan G., Guan B., Xiao H., Yao B., Wu S. S. and Zhou L. (2008). Epigenetic blocking of an enhancer region controls irradiation-induced proapoptotic gene expression in Drosophila embryos. Dev. Cell 14, 481-493. 10.1016/j.devcel.2008.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Casas-Tintó S., Li G., Lin N., Chung M., Moreno E., Moberg K. H. and Zhou L. (2015). An intergenic regulatory region mediates Drosophila Myc-induced apoptosis and blocks tissue hyperplasia. Oncogene 34, 2385-2397. 10.1038/onc.2014.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Adli M., Zou J. Y., Verstappen G., Coyne M., Zhang X., Durham T., Miri M., Deshpande V., De Jager P. L. et al. (2013). Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152, 642-654. 10.1016/j.cell.2012.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.