

ABSTRACT
Moderate or severe traumatic brain injury (TBI) causes widespread neuronal cell death. Microglia, the resident macrophages of the brain, react to injury by migrating to the lesion site, where they phagocytose cellular debris. Microglial phagocytosis can have both beneficial (e.g. debris clearance) and detrimental (e.g. respiratory burst, phagoptosis) consequences. Hence, whether the overall effect of microglial phagocytosis after brain injury in vivo is neuroprotective or neurotoxic is not known. Here, we establish a system with which to carry out dynamic real-time analyses of the mechanisms regulating cell death after brain injury in vivo. We show that mechanical injury to the larval zebrafish brain induces distinct phases of primary and secondary cell death. Excitotoxicity contributes to secondary cell death in zebrafish, reflecting findings from mammals. Microglia arrive at the lesion site within minutes of injury, where they rapidly engulf dead cells. Importantly, the rate of secondary cell death is increased when the rapid removal of cellular debris by microglia is reduced pharmacologically or genetically. In summary, our results provide evidence that microglial debris clearance is neuroprotective after brain injury in vivo.
KEY WORDS: Brain injury, Secondary cell death, Microglia, Phagocytosis, In vivo imaging, Zebrafish
Summary: Zebrafish microglia help to prevent the spread of tissue damage in the aftermath of a brain injury by migrating to the lesion site and clearing it of dead cells.
INTRODUCTION
Traumatic brain injury (TBI) is a leading cause of death and disability worldwide (Maas et al., 2008). Neuronal cell death after TBI can occur through either primary or secondary cell death. Primary cell death happens as a direct result of physical forces acting during injury. In contrast, secondary cell death is an indirect consequence of the injury, and is caused by complex neurotoxic processes in the hours and days after the initial insult (Park et al., 2008). Secondary cell death is a major factor in the progressive neurological deterioration seen in many individuals with TBI (Loane et al., 2015). Neurotoxic processes such as excitotoxicity (Dorsett et al., 2017) and oxidative stress (Rodríguez-Rodríguez et al., 2014) have been found to drive secondary neuronal cell death in mammals. However, these findings have not translated into the clinic, and no medications are available for the prevention of secondary cell death (Chakraborty et al., 2016; Hawryluk and Bullock, 2016). Hence, further research into the mechanisms underlying secondary cell death is urgently required.
In mammals, brain injury elicits a rapid inflammatory response. Microglia, the resident macrophages of the brain, are important cellular effectors of injury-induced neuroinflammation. They migrate to the lesion site within minutes of brain injury, where they phagocytose cellular debris (Davalos et al., 2005; Hanisch and Kettenmann, 2007; Nimmerjahn et al., 2005). Whether microglial phagocytosis is beneficial or detrimental in the context of neuronal injury is the subject of ongoing debate (Diaz-Aparicio et al., 2016; Fu et al., 2014; Sierra et al., 2013). Microglial phagocytosis clears dead cells, which might otherwise release noxious substances into their environment and thereby exacerbate tissue damage. However, microglial phagocytosis can also have detrimental consequences. Phagocytosis stimulates the activation of NADPH oxidase in a so-called respiratory burst (Minakami and Sumimotoa, 2006). NADPH oxidase produces high levels of reactive oxygen species (ROS), which can kill neurons when released extracellularly. Consistent with a detrimental role of the phagocytosis-induced respiratory burst, the phagocytic uptake of neuromelanin (Wilms et al., 2003) or neural debris (Claude et al., 2013) by microglia leads to the production of ROS and neuronal death in neuron-microglia co-cultures. Furthermore, microglial phagocytosis can kill stressed-but-viable neurons through phagoptosis (Brown and Neher, 2014). This was demonstrated in neuron-microglia co-cultures, where the inflammatory stimulation of microglia leads to loss of viable neurons through phagocytosis (Neher et al., 2011). Since the functional consequences of microglial phagocytosis have predominantly been studied in vitro, it is not known whether its overall effect is beneficial or detrimental after brain injury in vivo.
Importantly, microglial reactions to injury and injury-induced cell death are both highly dynamic processes that evolve over time in the aftermath of an injury. Hence, mechanistic investigations into the regulation of secondary cell death critically depend on the ability to conduct dynamic real-time analyses of these processes in vivo. Due to their suitability for in vivo imaging, larval zebrafish represent an ideal model system for such studies. Importantly, microglial reactions to brain injury are conserved across vertebrate species, and microglia in the larval zebrafish brain respond to injury by migrating to the injury site where they phagocytose neural debris (Sieger et al., 2012), as do their mammalian counterparts.
Here we conduct real-time analyses of the dynamics of cell death after brain injury in larval zebrafish. We find that a peak in primary cell death immediately after injury is followed by a peak in secondary cell death with a delay of several hours. Pharmacological manipulation of excitotoxicity confirmed its detrimental role in secondary cell death, replicating key findings from mammals. We also observe that microglia appear at the lesion site within minutes of injury, and in vivo imaging and quantification of microglial phagocytosis showed that they engulf substantial amounts of neuronal debris. Blocking microglial phagocytosis pharmacologically or genetically led to an increase in the rate at which secondary cell death occurs. Hence, microglial debris phagocytosis plays a key role in limiting the spread of tissue damage in the aftermath of a brain injury.
RESULTS
Primary and secondary cell death occur in distinct phases after brain injury in larval zebrafish
To investigate the dynamics of cell death after brain injury in vivo, we established an experimental setup for induction of mechanical injury in the optic tectum of larval zebrafish at 4 days post fertilisation (dpf). The optic tectum's superficial location within the transparent larval brain makes it easily accessible for experimental manipulation and in vivo imaging. Mechanical lesions were induced by piercing the optic tectum with a fine metal pin mounted on a micromanipulator (Fig. 1A).
Fig. 1.

Mechanical injury induces two distinct phases of cell death in the optic tectum of larval zebrafish. (A) Bright-field image of the head of a larval zebrafish. Mechanical brain injury is induced by piercing the optic tectum with a fine metal pin. FB, forebrain; OT, optic tectum; HB, hindbrain. Scale bar: 100 µm. (B) Confocal images of the optic tectum of a H2A:GFP transgenic animal with PI before and immediately after mechanical injury. The damaged area of the cell body layer is outlined. Scale bar: 50 µm. (C) Quantification of injury volume at 0 hpi. n=15 animals. (D,E) Confocal images of PI (D) and H2A:GFP (E) from the animal shown in (B) at different time points after injury. Scale bars: 50 µm. (F,G) Higher magnifications of the boxed regions indicated in E. White arrows indicate pyknotic nuclei. Scale bars: 5 µm. (H) Quantification of PI+ cells and pyknotic nuclei over time after mechanical injury. Dashed line indicates time of injury. n≥9 animals per experimental group. (I) Heat maps illustrating the spatial distribution of pyknotic nuclei in H2A:GFP animals in the hours following injury.
First, we characterised the extent and variability of tissue damage thus induced. To achieve this, we conducted in vivo confocal imaging of H2A:GFP animals. In this transgenic line, all cell nuclei are labelled (Pauls et al., 2001), which allowed us to monitor tissue architecture after injury. Imaging was carried out in the presence of the cell death indicator propidium iodide (PI) before and immediately after injury (Fig. 1B). Mechanical injury displaced tectal cells from the injury site, creating a lesion cavity within the neural tissue. In addition, PI+ dead cells were present both within the lesion cavity and in the neural tissue bordering the injury site (Fig. 1B). To quantify injury volume, we measured the area of the tissue that showed disrupted architecture or contained dead cells across z-stacks of the tectum, and found that 7.2±0.2% of tectal tissue displayed signs of damage at 0 h post injury (hpi). Importantly, injury volume was consistent between different animals (Fig. 1C). The extent of injury is relatively small compared with other models of mechanical injury in the zebrafish brain, such as stab lesioning of the telencephalon, which damages about 40% of the injured hemisphere (Kroehne et al., 2011).
We then investigated the temporal dynamics of cell death after injury. The number of PI+ cells increased sharply immediately after the injury and started to decrease again as early as 1 hpi (Fig. 1D,H; Movie 1). Using the H2A:GFP transgenic line allowed us to assess pyknosis, the condensation of chromatin in the nucleus of a dying cell, as an additional readout for cell death. In contrast to PI+ cells, the number of pyknotic cells did not increase immediately after injury (Fig. 1E-H), but started to rise with a delay at 1 hpi and peaked at 6 hpi (Fig. 1E-H; Movie 1). There also was a small delayed increase in PI+ cells at 6 hpi (Fig. 1H). In contrast to those at 0 hpi, these PI+ cells were pyknotic (Fig. S1), and hence may represent a subset of pyknotic cells whose membrane has become permeable at the late stages of cell death. At 6 hpi, pyknotic nuclei were seen in cells near the injury site (Fig. 1F), but also at locations more distant from the injury site within both the ipsilateral and contralateral hemispheres (Fig. 1G). This is reminiscent of findings from mammals, where delayed cell death can be observed in regions remote from the lesion site after experimental brain injury (Rink et al., 1995). When we visualised the spatial distribution of pyknotic nuclei over time, we found that they first appeared near the injury site, and had spread through large parts of the optic tectum by 6 hpi (Fig. 1I). Both PI+ cells and pyknotic cells had been mostly cleared from the tissue at 24 hpi (Fig. 1E-I).
To validate these results, we used three additional methods of cell death detection. In the transgenic line NBT:secA5-BFP (Mazaheri et al., 2014; van Ham et al., 2012, 2010), fluorescently labelled secreted annexin A5 binds to phosphatidylserine exposed on the plasma membrane of dying cells. Live imaging of NBT:secA5-BFP animals showed no increase in annexin A5+ cells at 0 hpi but revealed a rise in their number across the tectum at 6 hpi, which had subsided again at 24 hpi (Fig. S2A,B). Using caspase 3 immunohistochemistry, we observed no difference between sham and injured animals at 0 hpi, but found an increase in the number of caspase 3+ cells in both tectal hemispheres at 6 hpi (Fig. S2C,D). TUNEL staining revealed TUNEL+ dead cells at the injury site at 0 hpi, and in higher numbers across the tectum at 6 hpi (Fig. S2E,F). Together, these results provided further evidence for both immediate and delayed cell death taking place after brain injury in larval zebrafish. In addition, we tested whether different cell death markers would label distinct populations of dying cells. We found partial but not complete overlap between annexin A5+ and PI+ cells at 6 hpi (133±9 annexin A5+/PI− cells; 45±4 annexin A5+/PI+ cells; 64±6 annexin A5−/PI+ cells; n=11 animals), showing that not all dying cells express all markers simultaneously.
We next investigated whether tectal cells were dying through primary or secondary cell death. Given the appearance of PI+ cells immediately after the injury and their localisation at or near the injury site (Fig. 1D,H), we hypothesised that these cells died as a direct result of structural damage resulting from physical injury, and hence underwent primary cell death. To test this hypothesis, we followed individual tectal neurons over time after an injury. To do this, we generated a construct where the neuronal promoter elavl3 (Park et al., 2000) drives the expression of membrane-tagged TdTomato. Injection of elavl3:memTdTomato plasmid DNA into one-cell stage H2A:GFP embryos resulted in mosaic labelling of one or a few tectal neurons at 4 dpf (Fig. S3A,C,E). A neuron residing at the prospective injury site showed normal morphology before the injury. Immediately after injury, drastic structural damage to the neuron was apparent, and its remains had been cleared from the tissue by 6 hpi (Fig. S3B). We observed several neurons located at the prospective injury site that followed this pattern (n=30 neurons in 21 animals). These findings support the notion that these cells died through primary cell death.
After injury, pyknotic cells appeared across the tectum with a delay of several hours (Fig. 1E-I). The delay in their death raises the possibility that these cells escaped structural damage and died as an indirect result of the injury through neurotoxic processes such as excitotoxicity or oxidative stress; this would be considered secondary cell death. Alternatively, pyknotic cells might have been directly structurally damaged during mechanical injury but not enough to kill them immediately, so instead died at a later time point; this would be considered primary cell death, despite the delay. When following neurons residing more than 50 µm from the injury site in the ipsilateral or contralateral hemisphere (n=67 neurons in 21 animals), we found that they displayed no signs of structural damage at 0 hpi (Fig. S3D,F). Although most of these neurons survived until 6 hpi (Fig. S3D), some of them (n=8 neurons in 21 animals) subsequently underwent cell death, showing a pyknotic nucleus and disrupted dendrite morphology at 6 hpi (Fig. S3F). The absence of structural damage immediately after injury, followed by delayed cell death at 6 hpi, strongly suggests that these cells underwent secondary cell death.
Taken together, we established an experimental setup that enables the real-time visualisation of injury-induced primary and secondary cell death in real time in a living animal. This provides an ideal platform for mechanistic investigations into the regulation of secondary cell death in vivo.
Most of the tectal cells undergoing injury-induced cell death are neurons
Next, we sought to determine which types of tectal cells die after injury. At 4 dpf, the majority of tectal cells are neurons, which can be visualised in the elavl3:GFP (Park et al., 2000) and NBT:dsRed (Peri and Nüsslein-Volhard, 2008) neuronal reporter lines (Fig. 2A,E). The tectal ventricle is lined with radial glia, which express the Notch target gene her4.3 and are labelled in the her4.3:GFP (Yeo et al., 2007) and her4.3:mCherry (Knopf et al., 2010) transgenic lines (Fig. 2C,G). Using the oligodendrocyte reporter line mbp:memGFP (Almeida et al., 2011), we detected very few oligodendrocytes in the tectum at 4 dpf (Fig. S4), consistent with previous research (Brösamle and Halpern, 2002). We found that the majority of PI+ cells at 0 hpi were elavl3+ neurons (Fig. 2B) rather than her4.3+ radial glia (Fig. 2D). This was confirmed by quantification, which showed that 92±2% of PI+ cells were neurons, whereas only 3±1% were radial glia (Fig. 2I). Likewise, the majority of pyknotic cells at 6 hpi in NBT:dsRed;H2A:GFP animals were NBT+ neurons (Fig. 2F), and only a few pyknotic cells in her4.3:mCherry;H2A:GFP animals were her4.3+ radial glia (Fig. 2H). Quantification showed that 92±1% of pyknotic cells were neurons and 6±1% were radial glia (Fig. 2J). These results indicate that the majority of tectal cells dying after brain injury through either primary or secondary cell death are neurons.
Fig. 2.

The majority of cells dying after brain injury are neurons. (A,C,E,G) Confocal images of the optic tectum of the neuronal reporter lines elavl3:GFP (A) and NBT:dsRed (E), and the radial glia reporter lines her4.3:GFP (C) and her4.3:mCherry (G). Scale bars: 50 µm. (B,D,F,H) Live imaging of PI+ cells in elavl3:GFP (B) or her4.3:GFP (D) animals at 0 hpi, and of pyknotic nuclei in NBT:dsRed;H2A:GFP (F) or her4.3:mCherry;H2A:GFP (H) animals at 6 hpi. Filled and empty arrows indicate colocalisation, or lack thereof, of cell death and cell type markers, respectively. Scale bars: 5 µm. (I,J) Quantification of elavl3+/PI+ and her4.3+/PI+ cells at 0 hpi (I) and of NBT+ and her4.3+ pyknotic nuclei at 6 hpi (J). n≥10 animals per experimental group. Each circle represents one set of results. Data are mean±s.e.m.
Excitotoxicity contributes to secondary neuronal cell death
Glutamate-mediated excitotoxicity has been identified as an important neurotoxic mechanism driving secondary cell death after brain injury in mammals (Dorsett et al., 2017). Excitotoxic cell death occurs when glutamate released from damaged neurons overactivates glutamate receptors on neighbouring cells, leading to an influx of pathologically high levels of calcium (Arundine and Tymianski, 2003). To determine whether this holds true in our model system, we first visualised and quantified calcium dynamics in tectal cells after brain injury in vivo. For this, we generated a transgenic line expressing the calcium reporter GCaMP6f (Chen et al., 2013) under the β-actin promoter (Fig. 3A). Confocal time-lapse imaging of β–actin:GCaMP6f transgenic larvae revealed spontaneous calcium transients in tectal cells of sham animals (Fig. 3A,C,E), consistent with previous research (Avitan et al., 2017). After mechanical injury, the number of calcium transients in tectal cells was increased (Fig. 3A-E). This increase was detectable immediately after injury and persisted until at least 6 hpi (Fig. 3E). Hence, the increase in calcium transients coincided with the time window during which secondary cell death takes place in the optic tectum (Fig. 1).
Fig. 3.
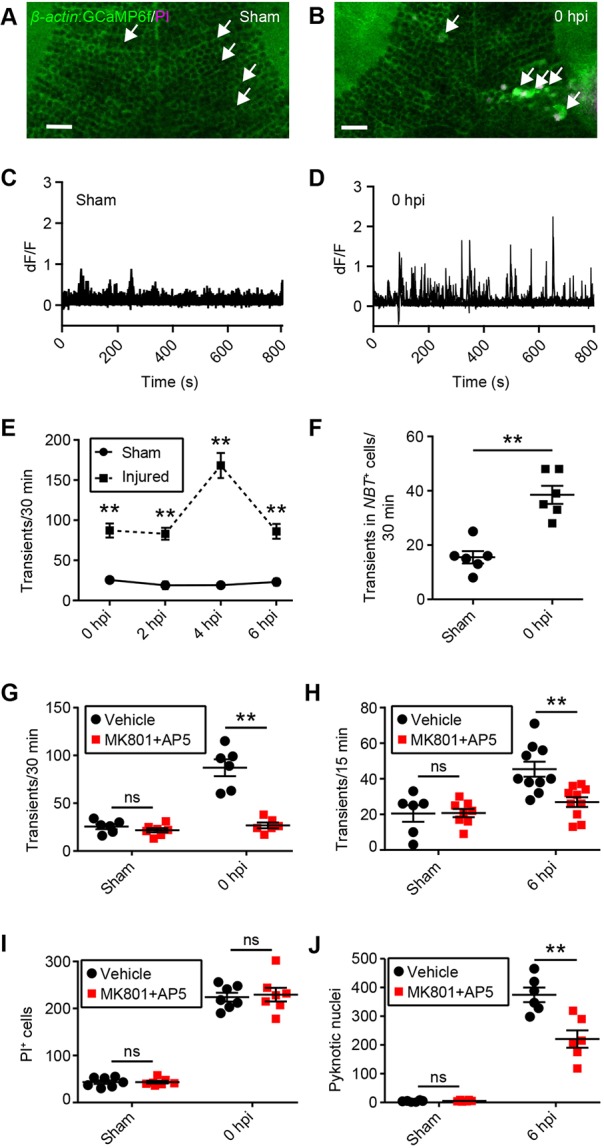
Excitotoxicity contributes to secondary cell death after brain injury in larval zebrafish. (A,B) Live imaging of calcium dynamics in the optic tectum of β-actin:GCaMP6f larvae in a sham animal (A) and an injured animal (B) at 0 hpi. White arrows indicate individual tectal cells. Scale bars: 20 µm. (C,D) Calcium traces from the individual tectal cells highlighted in A and B. (E) Quantification of calcium transients in sham and injured larvae during the time in which secondary cell death occurs. n=6 animals per experimental group. **P<0.01 using two-way ANOVA. (F) Quantification of calcium transients in tectal neurons, as shown by live imaging of β-actin:GCaMP6f;NBT:dsRed larvae. n=6 animals per experimental group. **P<0.01 using a Mann–Whitney test. (G,H) Quantification of calcium transients in tectal cells at 0 hpi (G) and 6 hpi (H) with MK801 and AP5. n≥6 animals per experimental group. ns, not significant and **P<0.01 in two-way ANOVA. (I,J) Quantification of PI+ cells at 0 hpi (I) and pyknotic nuclei at 6 hpi (J) with MK801 and AP5. n≥6 animals per experimental group. ns, not significant and **P<0.01 using two-way ANOVA. Data are mean±s.e.m.
If excitotoxicity contributed to secondary cell death, we would expect a spatial correlation between the distribution of calcium transients and the regions containing pyknotic nuclei. To test this hypothesis, we monitored calcium transients from 2 to 3 hpi in β-actin:GCaMP6f larvae and generated spatial maps of calcium dynamics within the tectum. The same animals were then fixed and their nuclei stained, allowing us to map regions containing pyknotic nuclei at 3 hpi. Comparing the spatial distribution of calcium transients with that of pyknotic nuclei did indeed reveal a correlation between these patterns (Fig. S5). Interestingly, elevated levels of calcium transients were observed in somewhat broader regions than pyknotic nuclei, possibly indicating that a certain level of calcium transients must be exceeded before cell death occurs.
Since the calcium indicator GCaMP6f is expressed ubiquitously in β-actin:GCaMP6f transgenic larvae, we next sought to confirm that calcium transients were increased in tectal neurons. For this we crossed β-actin:GCaMP6f animals with the neuronal reporter line NBT:dsRed. We then quantified calcium transients occurring in NBT+ cells through time-lapse imaging of β-actin:GCaMP6f; NBT:dsRed double transgenic larvae. This showed that calcium transients were indeed increased in tectal neurons after injury (Fig. 3F).
The overactivation of NMDA-type glutamate receptors contributes to excitotoxicity-induced calcium overload and cell death in mammalian neurons (Arundine and Tymianski, 2003). We used a combination of the non-competitive NMDA receptor antagonist MK801 and the competitive antagonist AP5 to determine whether this would also be the case in our model system. Indeed, the increase in calcium transients usually seen in injured animals was abolished in the presence of 100 µM MK801 and 10 µM AP5 both at 0 hpi (Fig. 3G) and at 6 hpi (Fig. 3H). We then investigated whether preventing the injury-induced increase in calcium transients would affect either PI+ cells at 0 hpi or pyknotic nuclei at 6 hpi. The number of PI+ cells was unchanged by MK801 and AP5 at 0 hpi (Fig. 3I). This provides additional evidence that PI+ cells die through primary cell death because their death cannot be prevented by blocking a known neurotoxic process such as excitotoxicity. In contrast, the number of pyknotic nuclei at 6 hpi was reduced when MK801 and AP5 were present (Fig. 3J). The fact that pyknotic cells could be rescued by blocking excitotoxicity indicates that these cells would otherwise have died from secondary cell death. We noted that while treatment with NMDA receptor antagonists virtually completely suppressed the injury-induced increase in calcium transients both at 0 hpi (Fig. 3G) and 6 hpi (Fig. 3H), the increase in pyknotic nuclei at 6 hpi was reduced by about 40%, rather than abolished, by MK801 and AP5 (Fig. 3J). This indicates that other neurotoxic processes such as oxidative stress (Rodríguez-Rodríguez et al., 2014) likely also contribute to secondary cell death.
Interestingly, there was no difference in PI+ cells at 6 hpi in the presence of MK801 and AP5 (sham vehicle, 42±1 PI+ cells; sham MK801+AP5, 42±3 PI+ cells; injured vehicle, 101±19 PI+ cells; injured MK801+AP5, 105±18 PI+ cells; n=7 animals per experimental group). This suggests that MK801 and AP5 do not change the number of cells entering the late stages of cell death, but instead affect earlier stages, consistent with previous literature (Arundine and Tymianski, 2003).
To confirm that excitotoxicity can drive secondary cell death in our model system, we tested whether application of 10 µM L-glutamate would exacerbate injury-induced cell death. Indeed, the number of pyknotic nuclei at 6 hpi was increased in the presence of L-glutamate (Fig. S6). Taken together, these results indicate that glutamate-mediated excitotoxicity contributes to secondary cell death after brain injury in larval zebrafish.
Microglia rapidly accumulate at the lesion site within the brain
Next, we analysed the dynamics and cellular effectors of injury-induced inflammation in our model system. In mammals, brain-resident microglia accumulate at the lesion site after brain injury (Davalos et al., 2005). In addition, the disruption of the blood-brain barrier allows for infiltration of peripheral immune cells, including monocyte-derived macrophages, neutrophils, dendritic cells and T cells (Jin et al., 2012). As the zebrafish's adaptive immune system has not yet developed at 4 dpf (Meijer and Spaink, 2011), we were able to focus our analysis on cells of the innate immune system. First, we carried out in vivo confocal imaging of mpeg1:GFP transgenic larvae (Ellett et al., 2011), where macrophage-lineage cells, including microglia are labelled. Mpeg1+ cells started to accumulate at the lesion site within the brain shortly after injury, and the number of mpeg1+ cells continued to increase in the following hours (Fig. 4A,B). To analyse the injury responses of mpeg1+ cells at the single-cell level, we analysed the 3D spatial trajectories of individual cells over time. Tracking of mpeg1+ cells in intact animals (Fig. 4C; Movie 2) showed low average speed (Fig. 4D) and little net displacement of their cell bodies (Fig. 4E). After injury, mpeg1+ cells rapidly migrated towards the lesion site (Fig. 4C; Movie 3), which was reflected in marked increases in their average speed (Fig. 4D) and net displacement (Fig. 4E). Interestingly, mpeg1+ cells both within the brain and on the skin migrated towards the lesion site, and mpeg1+ cells also accumulated within the wounded skin (Fig. 4C; Movie 3).
Fig. 4.

Microglia and skin macrophages rapidly migrate to the injury site, but skin macrophages rarely enter the brain. (A) Live imaging of the microglia/macrophage reporter line mpeg1:GFP at different time points after injury. Scale bar: 50 µm. (B) Quantification of mpeg1+ cells at the injury site within the brain. n=6 animals per experimental group. (C) Traces visualising cell body displacement of mpeg1+ cells for 30 min before injury or in the 2 h after injury, in maximum intensity projections of z-stacks of the optic tectum. Scale bar: 50 µm. (D,E) Quantification of migration speed (D) and net displacement (E) of mpeg1+ cell bodies before and after injury. n=8 animals per experimental group. **P<0.01 using Student's t-test. (F) Confocal images of the optic tectum of a p2y12:GFP;mpeg1:mCherry animal at 6 hpi. Yellow arrows indicate p2y12+/mpeg1+ cells. Light-blue arrows indicate p2y12−/mpeg1+ cells. Scale bar: 40 µm. (G,H) Quantification of p2y12+/mpeg1+ and p2y12−/mpeg1+ cells at the entire injury site (G) or within the brain (H). n=12 animals per experimental group. (I) Traces visualising cell body displacement of mpeg1+ skin macrophages in the hour after injury in a p2y12:GFP;mpeg1:mCherry animal in a maximum intensity projection of a z-stack of the optic tectum. Scale bar: 40 µm. (J) Quantification of the number of transitions of mpeg1+ skin macrophages into the brain in p2y12:GFP;mpeg1:mCherry animals within 6 h of injury. n=6 animals. Data are mean±s.e.m.
To distinguish microglia from skin macrophages and other peripheral macrophages, we next crossed p2y12:GFP transgenic animals (Sieger et al., 2012) with the mpeg1:mCherry line (Ellett et al., 2011). P2Y12 is a purinergic receptor that is expressed in microglia but not in peripheral macrophages in mammals (Sasaki et al., 2003). In intact animals, we detected P2Y12 expression in virtually all tectal mpeg1+ cells (Fig. S7A,B) and in a small subset of mpeg1+ skin macrophages (Fig. S7C,D), but not in mpeg1+ cells in the trunk (Fig. S7E,F). Hence, P2Y12 expression can differentiate between microglia and peripheral macrophages, but not microglia and skin macrophages.
Live imaging of p2y12:GFP;mpeg1:mCherry double transgenic larvae between 0 and 6 hpi showed recruitment of both p2y12+/mpeg1+ and p2y12−/mpeg1+ cells to the injury site spanning the brain and skin (Fig. 4F,G). Interestingly, p2y12−/mpeg1+ cells were mostly confined to the skin (Fig. 4F), whereas virtually all mpeg1+ cells within the brain expressed P2Y12 (Fig. 4F,H). These findings are consistent with recruitment of microglia to the injury site within the brain and of skin macrophages to the skin wound, but do not rule out migration of skin macrophages into the brain.
To address this, we carried out time-lapse imaging of p2y12:GFP;mpeg1:mCherry larvae for 6 h after injury, and analysed the 3D spatial trajectories of individual skin macrophages over time (Fig. 4I). This revealed a small number of skin macrophages that entered the brain, typically fewer than 10 (Fig. 4J). These skin macrophages stayed relatively close to the surface rather than migrating deep into the brain (Fig. 4I; penetration depth at 6 hpi: 15±1 μm, n=6 animals). Invading macrophages can switch on expression of P2Y12 after entering the brain (Chia et al., 2018), but we did not observe this in our experiments (n=6 animals). Furthermore, we did not detect any p2y12−/mpeg1+ cells entering the tectum from a ventral direction (n=6 animals), arguing against substantial recruitment of peripheral macrophages from elsewhere in the body.
We next used the 4C4 antibody, which has previously been used to label microglia in zebrafish (Becker and Becker, 2001; Chia et al., 2018; Ohnmacht et al., 2016), in mpeg1:GFP animals. The 4C4 antibody labelled nearly all tectal mpeg1+ cells (Fig. S8A,B) and a subset of mpeg1+ skin macrophages (Fig. S8C,D), but very few mpeg1+ cells in the trunk (Fig. S8E,F) of intact animals. Thus, like P2Y12 expression, 4C4 labelling can differentiate between microglia and peripheral macrophages, but not microglia and skin macrophages. At 6 hpi, we detected both 4C4+/mpeg1+ and 4C4−/mpeg1+ cells at the injury site spanning the brain and skin (Fig. S9A,B), with 4C4− cells mostly residing on the skin (Fig. S9A) and almost all mpeg1+ cells within the brain labelled with 4C4 (Fig. S9C). Although these findings cannot rule out recruitment of skin macrophages into the brain, they suggest that recruitment of substantial numbers of peripheral macrophages from elsewhere in the body is unlikely.
We also attempted to use the microglia reporter line apoE:GFP (Peri and Nüsslein-Volhard, 2008). However, about one-third of tectal mpeg1+ cells were apoE− even in uninjured animals (Fig. S10). Hence, we could not use ApoE to distinguish these cells from potential invading apoE− peripheral macrophages.
We then explored whether neutrophils were recruited to the lesion site using live imaging of the neutrophil reporter line mpo:GFP (Renshaw et al., 2006). Very few neutrophils were present in the optic tectum of uninjured animals (Fig. S11A,B). Between 0 and 6 hpi, neutrophils appeared at the injury site spanning the brain and skin (Fig. S11A,B). A few of these were found within the brain, typically no more than three or four cells at 6 hpi (Fig. S11B). Reminiscent of skin macrophages, these cells resided relatively close to the surface (Fig. S11A; penetration depth at 6 hpi: 33±5 μm, n=8 animals).
Taken together, our data indicate that microglia are the main effectors of the inflammatory response to injury within the brain, with a small contribution from skin macrophages and neutrophils. Injury also led to the recruitment of skin macrophages and neutrophils to the skin wound.
Microglial phagocytosis rapidly removes PI+ cells after brain injury
Given the prompt appearance of microglia at the site of brain injury (Fig. 4) and their well-known role as the professional phagocytes of the brain (Hanisch and Kettenmann, 2007), we hypothesised that microglia are involved in the rapid removal of PI+ cells after brain injury (Fig. 1). To test this, we conducted in vivo time-lapse imaging of microglial phagocytosis in intact and injured mpeg1:GFP larvae. A microglial cell in an intact animal actively surveyed its environment as shown by the high motility of its cellular processes (Fig. 5A; Movie 4). Quantification of the rate of phagocytosis of PI+ cells by microglia detected virtually no phagocytic events (Fig. 5E), presumably reflecting the low number of PI+ cells in the optic tectum of intact animals (Fig. 1). Conversely, after an injury a microglial cell approached and engulfed a PI+ cell as early as 0.5 hpi (Fig. 5B; Movie 5). This was reflected in a marked increase in the rate of phagocytosis (Fig. 5E). At 2 hpi, individual microglial cells had taken up substantial amounts of neuronal debris (Fig. S12).
Fig. 5.
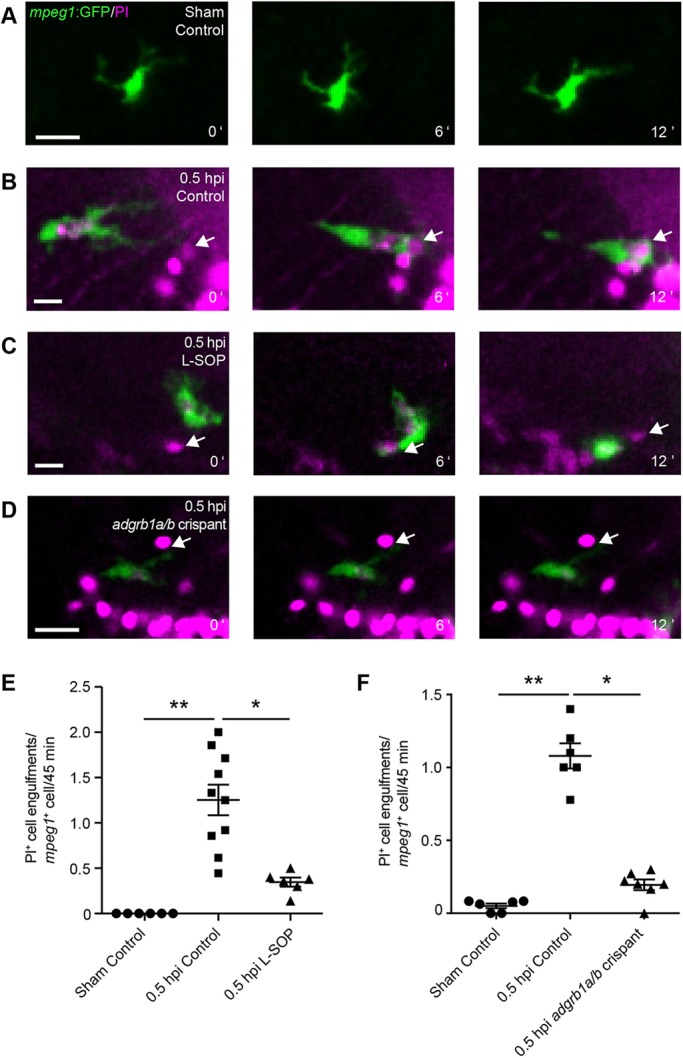
Microglia rapidly phagocytose PI+ cells after injury. (A-D) Time-lapse imaging of microglial phagocytosis in mpeg1:GFP larvae in a sham animal (A), an injured animal (B), an injured animal treated with L-SOP (C) and an injured adgrb1a/b crispant (D). White arrows indicate PI+ cells. Scale bars: 15 µm. (E,F) Quantification of phagocytosis of PI+ cells per mpeg1+ cell from 30 to 75 min after injury in animals treated with L-SOP (E) or in adgrb1a/b crispants (F). Control animals were treated with vehicle (E) or uninjected (F). n≥6 animals per experimental group. *P<0.05 and **P<0.01 using Kruskal–Wallis test with Dunn's post-hoc test. Data are mean±s.e.m.
To be able to investigate the functional consequences of microglial phagocytosis in vivo, we sought to establish tools with which to reduce the rate of phagocytosis in microglia. Phagocytosis is a complex process regulated by ‘eat-me’ signals on the target cell and their cognate receptors on the phagocyte. The best studied ‘eat-me’ signal is phosphatidylserine (PS), a phospholipid exposed on the plasma membrane of dead cells (Brouckaert et al., 2004; Fadok et al., 1992; Hirt and Leist, 2003). Importantly, the engulfment of dead cells by microglia in the optic tectum of larval zebrafish has been shown to be PS dependent (Mazaheri et al., 2014).
To pharmacologically inhibit PS-dependent phagocytosis, we used O-phospho-L-serine (L-SOP), a small molecule that mimics the PS headgroup and thereby prevents the interaction between PS and its receptors. L-SOP has previously been used to inhibit phagocytosis of dead neurons in vitro (Witting et al., 2000) and in the zebrafish CNS in vivo (Bailey et al., 2010). Indeed, in vivo time-lapse imaging showed a microglial cell that approached, but did not engulf, a PI+ cell in the presence of 1 µM L-SOP (Fig. 5C; Movie 6). Consequently, the rate of phagocytosis of PI+ cells was markedly reduced by L-SOP (Fig. 5E).
To investigate whether L-SOP affected microglial recruitment to the lesion site, we conducted in vivo confocal imaging of mpeg1:GFP animals treated with vehicle or L-SOP. This did not reveal a difference in mpeg1+ cells at the injury site within the brain at any time point until 6 hpi (Fig. S13A). As L-SOP can modulate intracellular calcium dynamics through activation of group III metabotropic glutamate receptors in vitro (Koulen et al., 1999), we asked whether it also affected calcium dynamics in our model system in vivo. To test this, we conducted time-lapse imaging of calcium dynamics in β-actin:GCaMP6f animals treated with vehicle or L-SOP. Our results did not show a difference in the number of calcium transients in either sham animals or injured animals at 0 hpi (Fig. S13B).
We next sought to genetically disrupt PS-dependent phagocytosis. The PS receptor BAI1 mediates engulfment of dead cells in both mammals (Park et al., 2007) and zebrafish (Mazaheri et al., 2014). Zebrafish have two BAI1 paralogues, named adgrb1a and adgrb1b (Harty et al., 2015). To disrupt their function, we used CRISPR/Cas9-mediated gene editing. In zebrafish, the injection of a guide RNA (gRNA) targeting a gene of interest together with Cas9 enzyme into one-cell stage embryos efficiently generates mutations in the targeted gene in a high proportion of cells (Jao et al., 2013). This allows for analysis of gene function in gRNA-injected F0 animals, which we refer to as crispants (Jao et al., 2013). Indeed, injection of gRNAs targeting adgrb1a and adgrb1b efficiently disrupted both genes in F0 embryos, as shown by loss of targeted endonuclease restriction sites (Fig. S14A). This was confirmed by sequencing, which revealed a 54% mutation rate in adgrb1a (Table S1) and a 93% mutation rate in adgrb1b (Table S2). Importantly, RT-qPCR indicated that mRNA levels in crispants were reduced by more than 50% for adgrb1a and more than 60% for adgrb1b, demonstrating that CRISPR/Cas9-induced mutations activate RNA surveillance pathways (Fig. S14B).
To investigate the effect of CRISPR/Cas9-mediated gene knockdown on microglial phagocytosis, we co-injected adgrb1a and adgrb1b gRNAs into mpeg1:GFP larvae, and then conducted in vivo time-lapse imaging of microglial cells. In an adgrb1a/b crispant, a microglial cell resided next to a PI+ cell but did not engulf it (Fig. 5D; Movie 7). Quantification confirmed that the rate of phagocytosis of PI+ cells was markedly reduced in adgrb1a/b crispants (Fig. 5F). Live imaging of microglial recruitment in mpeg1:GFP animals showed no difference in mpeg1+ cells at the injury site within the brain between uninjected animals and adgrb1a/b crispants (Fig. S15). These results show that L-SOP and adgrb1a/b knockdown can be used to reduce microglial phagocytosis in our model system.
Rapid clearance of cellular debris reduces secondary cell death after mechanical injury
Having established tools for pharmacological and genetic reduction of phagocytosis, we next investigated whether the overall effect of microglial phagocytosis was beneficial or detrimental with regard to secondary cell death. In an initial experiment, we assessed the number of pyknotic nuclei at 6 hpi in H2A:GFP animals treated with L-SOP or injected with adgrb1a/b gRNAs (Fig. 6A). Interestingly, we found that the number of pyknotic nuclei was increased by L-SOP (Fig. 6A,B) and in adgrb1a/b crispants (Fig. 6A,C). This finding is consistent with an overall neuroprotective effect of microglial phagocytosis after brain injury. However, this increase could also be a direct result of reduced phagocytic clearance of debris (Fig. 5), which would lead to an accumulation of dead cells over time.
Fig. 6.

Microglial phagocytosis of cellular debris reduces secondary cell death. (A) Confocal images of pyknotic nuclei (white arrows) in H2A:GFP animals treated with L-SOP and in adgrb1a/b crispants at 6 hpi. Scale bar: 10 µm. (B,C) Quantification of pyknotic nuclei at 6 hpi in animals treated with L-SOP (B) or in adgrb1a/b crispants (C). Control animals were treated with vehicle (B) or uninjected (C). n≥6 animals per experimental group. ns, not significant and **P<0.01 in two-way ANOVA. (D) Live imaging of pre-existing (white arrows) and newly appearing (magenta arrows) pyknotic nuclei in H2A:GFP animals treated with L-SOP and in adgrb1a/b crispants. Scale bar: 15 µm. (E,F) Quantification of newly appearing pyknotic nuclei between 4.5 and 5 hpi in animals treated with L-SOP (E) or in adgrb1a/b crispants (F). Control animals were treated with vehicle (E) or uninjected (F). n≥6 animals per experimental group. ns, not significant and **P<0.01 in two-way ANOVA. Data are mean±s.e.m.
To exclude this possibility, we conducted an additional experiment where we followed individual pyknotic nuclei in H2A:GFP animals treated with L-SOP or injected with adgrb1a/b gRNAs (Fig. 6D). Importantly, this enabled us to quantify the rate at which new dead cells appeared, and hence provided us with a direct measure of the rate of secondary cell death. If phagocytosis had an overall neuroprotective effect, we would expect to see an increase in the rate of secondary cell death when phagocytosis is reduced pharmacologically or genetically. Importantly, our results did indeed show an increase in the rate of secondary cell death after brain injury both in the presence of L-SOP (Fig. 6D,E) and in adgrb1a/b crispants (Fig. 6D,F).
Microglia are the professional phagocytes of the brain. Nonetheless, other cell types, such as glial cells and neuroblasts, can also show phagocytic activity (Abiega et al., 2016; Lu et al., 2011), albeit at lower efficiency than professional phagocytes (Abiega et al., 2016; Magnus et al., 2002; Parnaik et al., 2000). Hence, we sought to test whether cell types other than microglia made a major contribution towards debris clearance after brain injury in our model system. We focussed our analysis on the her4.3+ radial glia that line the tectal ventricle (Fig. 2C,G). Confocal live imaging did not reveal any radial glial cells with ingested PI+ cells either in sham animals or in injured animals at 1 hpi (Fig. S16A). In addition, when we quantified the rate of phagocytosis of PI+ cells by radial glia, we detected virtually no phagocytic events either in sham animals or in injured animals at 0.5 hpi (Fig. S16B), indicating that these cells do not make a major contribution towards dead cell clearance.
Taken together, these findings indicate that the rapid phagocytosis of PI+ cells by microglia has a neuroprotective effect after brain injury in vivo.
DISCUSSION
In the present study, we took advantage of the larval zebrafish's amenability to in vivo imaging to establish a novel model system for imaging-based mechanistic investigations of secondary neuronal cell death. To achieve this, we set up a rapid, simple and cost-effective method for mechanical brain injury. By piercing the optic tectum with a fine metal pin, we can reliably induce two phases of cell death: an immediate phase, characterised by the appearance of PI+ cells at or near the injury site, and a delayed phase, the hallmark of which is an increase in the number of pyknotic cells both at the injury site and across the tectum.
The key distinction between primary and secondary cell death is that the former is caused directly by structural damage from the injury, whereas the latter is caused indirectly by neurotoxic processes. We used several approaches to distinguish primary from secondary cell death. First, tracking individual tectal neurons over time showed that cells at the injury site exhibit drastic structural damage at 0 hpi, while neurons further away from the injury site show no signs of structural damage immediately after injury but nonetheless can undergo cell death at 6 hpi (Fig. S3). Second, when excitotoxicity is pharmacologically reduced, the number of PI+ cells at 0 hpi is unchanged, whereas that of pyknotic cells at 6 hpi is decreased (Fig. 3I,J). These results suggest that PI+ cells at 0 hpi die through primary cell death, whereas pyknotic cells at 6 hpi die through secondary cell death. However, it is possible that some of the pyknotic cells present at the injury site at 6 hpi were directly structurally damaged by the injury but died at a later time point through delayed primary cell death. Previous studies had shown an increase in cell death after central nervous system injury in larval zebrafish, but did not address whether cells were dying from primary or secondary cell death (Morsch et al., 2015; Ohnmacht et al., 2016).
We also sought to distinguish types of cell death such as necrosis, apoptosis and necroptosis. Given their prompt appearance at or near the lesion site, it seems likely that PI+ cells at 0 hpi die through accidental necrosis caused by physical forces acting during the injury. Conversely, dead cells at 6 hpi can be labelled by immunohistochemistry for caspase 3, a key enzyme in the execution phase of apoptosis (Wyllie, 1997). Pyknosis, PS exposure and TUNEL staining can occur in both necrotic and apoptotic cells, and hence do not differentiate between these types of cell death (Fink and Cookson, 2005). The gene encoding MLKL, the central effector of necroptotic cell death, is absent from zebrafish (Czabotar and Murphy, 2015). Hence, our results indicate that primary cell death is predominantly necrotic, whereas secondary cell death can occur through apoptosis. Mammalian neurons can die through either necrosis or apoptosis after brain injury (Stoica and Faden, 2010), highlighting the relevance of our model system for the study of injury-induced neuronal death.
We noticed that different cell death markers yielded somewhat different results regarding levels of cell death at 6 hpi. We usually detected about 500 pyknotic nuclei (Fig. 1H), 100 PI+ cells (Fig. 1H), 150 annexin A5+ cells (Fig. S2B), 100 caspase 3+ cells (Fig. S2D) and 200 TUNEL+ cells (Fig. S2F). This discrepancy is most likely due to a combination of factors. Differences in sensitivity may lead to levels of cell death appearing higher with some markers than with others (Zhao et al., 2003). In addition, some markers, such as PS exposure, are predominantly associated with the early stages of cell death (Martin et al., 1995), whereas others, such as TUNEL staining, do not appear until the later stages (Negoescu et al., 1996). Hence, not all cell death markers would be expected to be present in all dying cells at the same time, leading to apparent differences in cell numbers when different markers are used. This is consistent with our finding of partial but not complete overlap between the populations of annexin A5+ and PI+ cells at 6 hpi.
While fast calcium waves occurring within seconds of injury are crucial for initiation of wound repair in many tissues, including larval zebrafish epidermis (Yoo et al., 2012) and brain (Sieger et al., 2012), the influx of pathologically high levels of calcium into neurons through overactivation of glutamate receptors can lead to excitotoxic cell death (Arundine and Tymianski, 2003). Consequently, the NMDA receptor antagonists MK801 and AP5 can protect mammalian neurons from excitotoxic cell death (Foster et al., 1988; Wang et al., 1997). In our model system, exogenous glutamate exacerbates cell death at 6 hpi (Fig. S6), whereas MK801 and AP5 decrease it (Fig. 3). This is consistent with a previous study that used exogenous glutamate to induce excitotoxicity in larval zebrafish and showed that application of MK801 reduced excitotoxicity-induced cell death (McCutcheon et al., 2016). Interestingly, we did not detect an increase in cell death in intact animals upon glutamate exposure (Fig. S6), probably owing to a relatively short incubation time (McCutcheon et al., 2016). Thus, an important mammalian neurotoxic mechanism also exacerbates secondary cell death in our model system, again highlighting its potential translational relevance.
In mammals, the inflammatory response to brain injury is brought about by a range of different cell types, including brain-resident microglia and peripheral immune cells such as monocyte-derived macrophages, neutrophils, T cells and dendritic cells (Hanisch and Kettenmann, 2007; Jin et al., 2012). Interestingly, we found that microglia were the main cell type at the lesion site within the brain in our model system, alongside a small number of skin macrophages and neutrophils (Fig. 4; Fig. S9; Fig. S11). We also observed accumulation of skin macrophages and neutrophils in the skin wound, consistent with previous research (Ellett et al., 2011; Renshaw et al., 2006). Microglia can reduce neutrophil infiltration after neuronal injury in mammalian brain slices (Neumann et al., 2008). Similarly, macrophages residing at the wound site repel approaching neutrophils after tail transection in larval zebrafish (Tauzin et al., 2014). Whether microglia actively reduce the migration of skin macrophages and neutrophils into the brain or whether there is a lack of factors attracting these cells remains to be determined.
Once at the site of injury, one of the key functions of microglia is the clearance of dead cells and cellular debris through phagocytosis (Hanisch and Kettenmann, 2007). Microglial phagocytosis in the context of neuronal injury has traditionally been studied mostly in in vitro systems, and very little is known about its functional consequences in vivo. Consistent with an overall beneficial effect of debris uptake by microglia, it has been shown that fractalkine-mediated microglial clearance of damaged neurons attenuates neuronal cell death in neuron-microglia co-cultures (Noda et al., 2011). Likewise, genetic deletion of the phagocytic receptor TREM2, which attenuates microglial phagocytosis in vitro, is associated with exacerbated tissue damage in a rodent model of ischemic stroke (Kawabori et al., 2015). Direct evidence supporting a beneficial effect of microglial phagocytosis in vivo was thought to come from a mouse model of Rett syndrome, where mutation of the MECP2 gene is associated with a deficit in phagocytosis that may underlie the neuronal dysfunction seen in mutant animals (Derecki et al., 2012). However, the results from this study were contradicted by subsequent research (Wang et al., 2015), leaving the question of the overall effect of microglial phagocytosis in vivo still unanswered. The data presented here show that when the rate of microglial phagocytosis is reduced pharmacologically or genetically after brain injury in vivo, secondary cell death is increased. Hence, our data suggest that the net effect of microglial phagocytosis in the context of neuronal injury in vivo is beneficial. These findings highlight the significance of microglial phagocytosis as a potential target for therapeutic interventions after TBI. Furthermore, it is important to emphasise that with the advent of high-throughput automated screening technologies for zebrafish larvae (Pardo-Martin et al., 2010) and rapid CRISPR/Cas9-mediated gene editing (Jao et al., 2013), our model system can be used as a powerful platform for the rapid identification of pharmacological compounds and genes that modulate secondary cell death after brain injury in vivo.
MATERIALS AND METHODS
Fish husbandry
Zebrafish (Danio rerio) of both sexes were used in this study. All animals were maintained under standard conditions (Westerfield, 2007), and experiments were performed in accordance with British Home Office regulations and European Commission guidelines. Wild-type animals used were of the WIK strain. The following zebrafish lines were used: H2A.F/Z:GFP (Pauls et al., 2001), referred to as H2A:GFP; NBT:DLexPR:secA5-BFP (Mazaheri et al., 2014), referred to as NBT:secA5-BFP; elavl3:GFP (Park et al., 2000); NBT:dsRed (Peri and Nüsslein-Volhard, 2008); her4.3:GFP (Yeo et al., 2007); her4.3:mCherry (Knopf et al., 2010); mbp:GFP-CAAX (Almeida et al., 2011), referred to as mbp:memGFP; mpeg1:GFP (Ellett et al., 2011); mpeg1:mCherry (Ellett et al., 2011); p2y12:P2Y12-GFP (Sieger et al., 2012), referred to as p2y12:GFP; apoE:lyn-GFP (Peri and Nüsslein-Volhard, 2008), referred to as apoE:GFP; and mpo:GFP (Renshaw et al., 2006). If necessary, larvae were treated with 100 µM N-phenylthiourea (PTU) to inhibit melanogenesis. All chemicals were supplied by Sigma unless otherwise stated.
Generation of β-actin:GCaMP6f transgenic line
The transgenic line β-actin:GCaMP6f was generated by placing the GCaMP6f sequence (Chen et al., 2013) under the zebrafish β-actin promoter, flanked by Tol2 sites (Kwan et al., 2007). The construct was injected into one-cell stage embryos together with Tol2 transposase. Larvae from F2 or subsequent generations were used for this study.
Mosaic labelling of tectal neurons
To generate the elavl3:TdTomato-CAAX plasmid, referred to as elavl3:memTdTomato, we combined the entry clones p5E-elavl3 (Don et al., 2017), pME-TdTomato-CAAX (Walton et al., 2015) and p3E-polyA (Kwan et al., 2007) with the destination vector pDestTol2pA2 (Kwan et al., 2007) using Gateway LR Clonase II enzyme (Life Technologies) according to manufacturer's instructions. For mosaic labelling of tectal neurons, 1 nl of elavl3:memTdTomato plasmid DNA at a concentration of 25 ng/µl was injected into one-cell stage zebrafish embryos, and animals were allowed to develop until 4 dpf.
Induction of brain injury
Zebrafish larvae at 4 dpf were anaesthetised using 0.01% ethyl 3-aminobenzoate methanesulfonate (MS-222), and mounted in 1% low melting point agarose (Life Technologies) with their dorsal side facing upward. The optic tectum was then injured under visual guidance of a stereomicroscope using a fine metal pin with a diameter of ∼80 µm (Fine Science Tools) mounted on a micromanipulator (Narishige). The tip of the metal pin was slightly bevelled to facilitate penetration of the skin, and the angle between the pin and the horizontal plane was ∼20-30°. For induction of injury, the tip of the metal pin was inserted into the optic tectum to a depth of ∼200 µm and subsequently retracted. The entire injury procedure usually took less than 20 s. After injury, larvae were carefully released from the agarose and allowed to recover for varying amounts of time depending on experimental requirements. Sham animals were anaesthetised, embedded in agarose and subsequently released as described, but not injured.
Drug treatments
Pharmacological agents were delivered by bath application. Animals were pre-incubated with compounds for 1 h before the start of the experiment, and incubation continued until the time of experimental readout. The following drugs were used: 100 µM MK801 (Abcam); 10 µM AP5 (Abcam); 10 µM L-glutamate (Abcam); and 1 µM O-phospho-L-serine, referred to as L-SOP. All drugs were solubilised in double distilled H2O. Working stocks were prepared immediately before the start of every experiment by adding an appropriate volume of solubilised drug to the embryo medium. For vehicle treatment, the same volume of double distilled H2O without drug was added.
CRISPR/Cas9-mediated gene editing
Guide RNAs were designed manually to target BslI restriction enzyme sites in adgrb1a (ENSDART00000188431.1) and adgrb1b (ENSDART00000143118.4) exonic sequences. The target sequences were 5′-CAAGGGGGGGCTGCTGGATA-3′ for adgrb1a and 5′-CCCTCATACGGCGGCTCCGAGTG-3′ for adgrb1b. For generation of adgrb1a/b crispants, 1 nl injection solution containing adgrb1a and adgrb1b crRNAs and tracrRNA (all at 250 ng/µl; Merck) together with Cas9 enzyme (NEB) was injected into one-cell stage embryos.
The efficiency of mutagenesis was analysed through restriction fragment length polymorphism analysis. PCR was performed on genomic DNA extracted from individual larvae at 24 hpf using the following primers: adgrb1a fw, 5′-CACTTTCTCATCGTTGTGTCTCC-3′; adgrb1a rev, 5′-GGCAGTGGGAGTCTTGCTC-3′; adgrb1b fw, 5′-AGTTGATGGATTCTGGAACGACT-3′; and adgrb1b rev: 5′-GGTGTTTAGTGTACCAGGGCA-3′. PCR products were digested using BslI, and fragment length was analysed by agarose gel electrophoresis.
CRISPR/Cas9-induced mutagenesis was analysed in detail by genomic DNA sequencing. PCR products from eight injected embryos were pooled and subcloned using the Strataclone PCR Cloning Kit (Life Technologies) according to manufacturer's instructions. The ligated plasmids were then transformed and 100 colonies were sequenced.
Levels of adgrb1a and adgrb1b mRNA in crispants were assessed through RT-qPCR. For this, 10 larvae per biological replicate were pooled and RNA was extracted using the RNEasy Micro kit (Qiagen). This was transcribed into cDNA using the iScript cDNA Synthesis kit (Bio-Rad). RT-qPCR was run on a Roche LightCycler 96 using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad), and relative quantities of transcript levels were calculated using the Livak method (Livak and Schmittgen, 2001). RT-qPCR primers were designed to span an exon-exon junction and were validated to lie within 5% of 100% efficiency at 1 µM. The following primers were used: adgrb1a fw, 5′-TGTGTGTCCAGAACATGGGG-3′; adgrb1b rev, 5′-CTCCCGAATTCGCTGCATTG-3′; adgrb1b fw, 5′-AACGGAGCCTGGGATGAATG-3′; adgrb1b rev: 5′-CACACGGCAATGTTGCAGAA-3′.
Immunohistochemistry
Larvae were euthanised in 0.4% MS-222 and fixed in 4% paraformaldehyde with 1% DMSO for 12-16 h at 4°C. After fixation, larvae were washed in PBS. If necessary, pigment was removed by incubation in bleaching solution (3% H2O2, 5% formamide, 0.5×SSC in double distilled H2O) for 30-40 min. Larvae were then permeabilised using 2 mg/ml collagenase in PBS for 25 min, and post-fixed in 4% paraformaldehyde with 1% DMSO for 30 min. Blocking was carried out for 2 h in blocking buffer (1% normal goat serum, 1% BSA, 1% DMSO, 1% Triton X-100, 0.01% sodium azide in PBS). Larvae were then incubated in primary antibody in blocking buffer for 12-16 h at 4°C. Primary antibodies used were rabbit anti-cleaved caspase 3 (Kratz et al., 2006) at 1:250 (BD Pharmingen 559565), mouse anti-4C4 (Becker and Becker, 2001) at 1:50 (courtesy of the Becker lab, University of Edinburgh, UK) and chicken anti-GFP (Kuscha et al., 2012) at 1:400 (Abcam ab13970). Larvae were washed in PBS with 1% Triton X-100, and treated with 100 μg/ml RNAse A in PBS for 30 min at 37°C. They were then incubated in secondary antibody with 1 µg/ml PI in blocking buffer for 12-16 h at 4°C. Secondary antibodies used were Alexa Fluor 488 goat anti-rabbit at 1:500 (Thermo Fisher Scientific A-11008), Alexa Fluor 647 donkey anti-mouse at 1:500 (Jackson ImmunoResearch 715-605-151) and Alexa Fluor 488 donkey anti-chicken at 1:500 (Jackson ImmunoResearch 703-545-155). Larvae were then washed in PBS with 1% Triton X-100 and subsequently in PBS before mounting and confocal imaging.
TUNEL staining
Larvae were euthanised, fixed, bleached and permeabilised as described above. They were then incubated with reaction mix from the In Situ Cell Death Detection Kit TMR Red (Roche) according to manufacturer's instructions, and washed in PBS before mounting and imaging.
Image acquisition
For live confocal imaging, zebrafish larvae were anaesthetised using 0.01% MS-222 and mounted in 1% low melting point agarose, with their dorsal side facing upward for imaging of the optic tectum and their left side facing upward for imaging of the trunk. The agarose was then covered in embryo medium to prevent desiccation during imaging. For visualisation of PI+ dead cells, 1 µg/ml PI was added to both agarose and embryo medium. For imaging of fixed samples from immunohistochemistry or TUNEL staining, larvae were mounted in 1% low melting point agarose, and overlaid with PBS to prevent desiccation during imaging.
For z-stacks of the optic tectum in living or fixed samples, images with 3.6-6 µm intervals between optical planes were acquired to a depth of 100-150 µm starting at the dorsal side of the tectum. In injured animals, z-stacks were chosen such as to encompass the entire injury site. For z-stacks of the trunk, images with 3.6 µm intervals between optical planes were acquired to a depth of 80-120 µm starting beneath the skin.
For time-lapse imaging of the dynamics of primary and secondary cell death after injury, z-stacks of the tectum of H2A:GFP animals were acquired in the presence of PI every 25 min. Time-lapse imaging of calcium signalling was conducted by acquisition of images from a single optical plane at the level of the injury every 2 s in β-actin:GCaMP6f or β-actin:GCaMP6f;NBT:dsRed larvae. Tracking of the movements of microglia or epidermal macrophages was carried out from z-stacks of the optic tectum of mpeg1:GFP or p2y12:GFP;mpeg1:mCherry animals acquired every 5 min. In vivo analysis of the phagocytic behaviour of individual microglial cells was conducted from z-stacks of the optic tectum acquired every 6 min in mpeg1:GFP larvae in the presence of PI. Bright-field images were acquired on a GXM-XTL3TV1 stereomicroscope (GT Vision). All confocal imaging was conducted on Zeiss LSM 710 or Zeiss LSM 880 laser scanning confocal microscopes.
Image processing and analysis
For quantification of injury volume, the region of tectal tissue that displayed signs of damage such as disrupted tissue architecture or presence of PI+ dead cells was manually outlined in ImageJ (https://imagej.nih.gov/ij). The area of the injury site was measured across all consecutive images in z-stacks of the optic tectum. To obtain the approximate volume of the injury, each area was then multiplied by the distance between optical planes in the z-stack. The total volume of the tectum was obtained by the same methodology except that the entire tectum rather than just the damaged region was outlined.
PI+ cells, pyknotic nuclei in H2A:GFP animals, annexin A5+ or annexin A5+/PI+ cells in NBT:secA5-BFP animals, caspase 3+ cells, TUNEL+ cells, mbp+ cells in mbp:memGFP animals and apoE+ cells in apoE:GFP animals were manually counted in ImageJ across both tectal hemispheres and across all consecutive images in z-stacks of the optic tectum. Viable radial glial cells that took up low levels of PI after injury (James and Butt, 2002) were excluded from quantification of PI+ cells. For quantification of P2Y12 expression or 4C4 labelling in mpeg1+ cells in the tectum, skin or trunk of intact animals, p2y12+/mpeg1+ cells in p2y12:GFP;mpeg1:mCherry animals or 4C4+/mpeg1+ cells in mpeg1:GFP animals after 4C4 and GFP immunohistochemistry were manually counted across both tectal hemispheres and all consecutive images of z-stacks of the tectum, across the entire skin of the head in z-stacks of the tectum or all consecutive images of z-stacks of the trunk. Elavl3+/PI+ cells in elavl3:GFP animals, her4.3+/PI+ cells in her4.3:GFP animals, NBT+ pyknotic nuclei in NBT:dsRed;H2A:GFP animals and her4.3+ pyknotic nuclei in her4.3:mCherry;H2A:GFP animals were manually counted across both tectal hemispheres in one optical plane at the level of the injury, in at least 30 cells per animal. The diameter of PI+ and PI− cells at 0 hpi and 6 hpi was measured manually across both tectal hemispheres in one optical plane at the level of the injury, in at least 10 cells per animal.
For generation of heatmaps illustrating the density of apoptotic cells, pyknotic nuclei were manually selected in ImageJ from z-stacks of the optic tectum of H2A:GFP animals. The xy coordinates of all pyknotic nuclei within the stack were identified, projected onto a 2D surface, and used to generate heatmaps through a custom-written script in MATLAB using a modified scattercloud function (Eilers and Goeman, 2004).
For quantitative analysis of calcium signalling, time series of confocal images were registered using the StackReg plugin in ImageJ to correct for xy drift. For each animal, 250-300 tectal cells were manually selected such that the chosen cells were evenly distributed across the entire cellular layer of the tectum. Fluorescence intensities for the selected cells over time were extracted from imaging data; the baseline fluorescence for each cell over time was calculated using a time-averaging sliding window method; dF/F traces for each cell were determined; and the number of calcium transients for each cell was calculated. All steps of the analysis were performed using custom-written scripts in MATLAB (Herrgen et al., 2014). One transient was defined as an event with dF/F greater than 4.25 SD of baseline fluorescence, and with a duration of longer than two time frames. The total number of transients for each larva was calculated by summation of all transients detected in individual cells.
For generation of heatmaps illustrating the spatial distribution of calcium transients, a matrix was created based on the location and number of transients in individual tectal cells using the accumarray function, smoothed by 15 pixels for illustrative purposes and plotted using the imagesc function. All steps were performed in MATLAB. Scripts for analysis of calcium signalling and generation of heatmaps are available upon request.
To quantify the number of microglia, skin macrophages, other peripheral macrophages and neutrophils at the site of brain injury, mpeg1+ cells in mpeg1:GFP animals, p2y12+/mpeg1+ cells in p2y12:GFP;mpeg1:mCherry animals, 4C4+/mpeg1+ in mpeg1:GFP animals after 4C4 and GFP immunohistochemistry, and mpo+ cells in mpo:GFP animals were manually counted in ImageJ in a region of interest measuring 200 µm horizontally and 200 µm vertically, and centred on the injury site, across all consecutive images in z-stacks of the optic tectum. Cells residing on the skin overlying the tectum were included for quantification of cells at the entire injury site spanning the brain and skin, but excluded for quantification of cells at the injury site within the brain.
Tracking of individual microglial cells within the brain and of macrophages on the skin were carried out using IMARIS 8.2.1 software. For this, time-lapse movies of mpeg1:GFP or p2y12:GFP;mpeg1:mCherry larvae were visualised in IMARIS, and individual cells were tracked using an autoregressive motion algorithm under manual supervision. Speed and net displacement of microglial cell bodies were obtained as outputs of the algorithm.
To determine the frequency of phagocytic events, time-lapse movies of mpeg1:GFP animals were visualised in IMARIS, individual microglial cells were followed over time and phagocytic events were identified manually. A phagocytic event was defined as the formation of a phagocytic cup around PI+ cellular debris, followed by movement of engulfed cargo towards the cell body. The same protocol was used to determine the frequency of phagocytic events in radial glial cells, except that her4.3:GFP transgenic larvae were used.
For quantification of newly appearing pyknotic nuclei in H2A:GFP animals, z-stacks of the optic tectum were transformed into interpolated 3D projections in ImageJ using the brightest point projection method with 100% inferior depth cueing. Individual pyknotic nuclei in the hemisphere contralateral to the injury were traced over time and newly appearing nuclei were identified manually.
Experimental design and statistical analysis
Animals were randomly allocated to experimental groups before the start of each experiment. Researchers were blinded to experimental group for data analysis.
All data presented are from at least two independent experiments. All population data are presented as mean±s.e.m. In some graphs, error bars are not visible because they are shorter than the height of the symbol. Statistical analysis was performed using Prism (GraphPad). Briefly, data sets were assessed for normality, and appropriate statistical tests were carried out as stated in the figure legends.
Figures were assembled in Adobe Illustrator CS6 and Adobe Illustrator CC 2017. Movies were generated in IMARIS and ImageJ.
Supplementary Material
Acknowledgements
The authors thank the BVS zebrafish facility (QMRI, University of Edinburgh) for maintenance and care of the zebrafish. We are grateful to Ryan Kelly for help with generating the elavl3:memTdTomato construct; to the Becker, Lyons, Rossi and Sieger labs for sharing reagents and protocols; and to Robert Stewart for help with generating heatmaps. We thank David Lyons and Veronique Miron for critical reading of the manuscript.
Footnotes
Competing interests
M.K.'s salary was paid through a collaborative grant from Biogen; this did not have any influence on study design or interpretation. The other authors declare no competing or financial interests.
Author contributions
Conceptualization: C.H., L.H.; Methodology: C.H., L.H.; Formal analysis: C.H., L.P.G., M.K., L.H.; Investigation: C.H., L.P.G., M.K., L.H.; Resources: D.G., C.M., F.P.; Writing - original draft: C.H., L.H.; Writing - review & editing: C.H., L.H.; Supervision: L.H.; Project administration: L.H.; Funding acquisition: L.H.
Funding
This work was supported by a University of Edinburgh Chancellor's Fellowship to L.H.; by a Wellcome Trust/The University of Edinburgh Institutional Strategic Support Fund to L.H.; by a Carnegie Trust for the Universities of Scotland Research Incentive Grant (70457 to L.H.); by a Rosetrees Trust/Stoneygate Trust Seedcorn Award (M602 to L.H.); and by a Wellcome Trust Seed Award in Science (207701/Z/17/Z to L.H.). Deposited in PMC for release after 6 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.174698.supplemental
References
- Abiega O., Beccari S., Diaz-Aparicio I., Nadjar A., Layé S., Leyrolle Q., Gómez-Nicola D., Domercq M., Pérez-Samartín A., Sánchez-Zafra V. et al. (2016). Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol. 14, e1002466 10.1371/journal.pbio.1002466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida R. G., Czopka T., Ffrench-Constant C. and Lyons D. A. (2011). Individual axons regulate the myelinating potential of single oligodendrocytes in vivo. Development 138, 4443-4450. 10.1242/dev.071001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arundine M. and Tymianski M. (2003). Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34, 325-337. 10.1016/S0143-4160(03)00141-6 [DOI] [PubMed] [Google Scholar]
- Avitan L., Pujic Z., Mölter J., Van De Poll M., Sun B., Teng H., Amor R., Scott E. K. and Goodhill G. J. (2017). Spontaneous activity in the zebrafish tectum reorganizes over development and is influenced by visual experience. Curr. Biol. 27, 2407-2419. 10.1016/j.cub.2017.06.056 [DOI] [PubMed] [Google Scholar]
- Bailey T. J., Fossum S. L., Fimbel S. M., Montgomery J. E. and Hyde D. R. (2010). The inhibitor of phagocytosis, O-phospho-L-serine, suppresses Müller glia proliferation and cone cell regeneration in the light-damaged zebrafish retina. Exp. Eye Res. 91, 601-612. 10.1016/j.exer.2010.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T. and Becker C. G. (2001). Regenerating descending axons preferentially reroute to the gray matter in the presence of a general macrophage/microglial reaction caudal to a spinal transection in adult zebrafish. J. Comp. Neurol. 433, 131-147. 10.1002/cne.1131 [DOI] [PubMed] [Google Scholar]
- Brösamle C. and Halpern M. E. (2002). Characterization of myelination in the developing zebrafish. Glia 39, 47-57. 10.1002/glia.10088 [DOI] [PubMed] [Google Scholar]
- Brouckaert G., Kalai M., Krysko D. V., Saelens X., Vercammen D., Ndlovu M. N., Ndlovu M., Haegeman G., D'Herde K. and Vandenabeele P. (2004). Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol. Biol. Cell 15, 1089-1100. 10.1091/mbc.e03-09-0668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G. C. and Neher J. J. (2014). Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 15, 209-216. 10.1038/nrn3710 [DOI] [PubMed] [Google Scholar]
- Chakraborty S., Skolnick B. and Narayan R. K. (2016). Neuroprotection trials in traumatic brain injury. Curr. Neurol. Neurosci. Rep. 16, 29 10.1007/s11910-016-0625-x [DOI] [PubMed] [Google Scholar]
- Chen T.-W., Wardill T. J., Sun Y., Pulver S. R., Renninger S. L., Baohan A., Schreiter E. R., Kerr R. A., Orger M. B., Jayaraman V. et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295-300. 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia K., Mazzolini J., Mione M. and Sieger D. (2018). Tumor initiating cells induce Cxcr4-mediated infiltration of pro-tumoral macrophages into the brain. Elife 7, e31918 10.7554/eLife.31918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claude J., Linnartz-Gerlach B., Kudin A. P., Kunz W. S. and Neumann H. (2013). Microglial CD33-related Siglec-E inhibits neurotoxicity by preventing the phagocytosis-associated oxidative burst. J. Neurosci. 33, 18270-18276. 10.1523/JNEUROSCI.2211-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar P. E. and Murphy J. M. (2015). A tale of two domains-a structural perspective of the pseudokinase, MLKL. FEBS J. 282, 4268-4278. 10.1111/febs.13504 [DOI] [PubMed] [Google Scholar]
- Davalos D., Grutzendler J., Yang G., Kim J. V., Zuo Y., Jung S., Littman D. R., Dustin M. L. and Gan W.-B. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752-758. 10.1038/nn1472 [DOI] [PubMed] [Google Scholar]
- Derecki N. C., Cronk J. C., Lu Z., Xu E., Abbott S. B. G., Guyenet P. G. and Kipnis J. (2012). Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484, 105-109. 10.1038/nature10907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Aparicio I., Beccari S., Abiega O. and Sierra A. (2016). Clearing the corpses: regulatory mechanisms, novel tools, and therapeutic potential of harnessing microglial phagocytosis in the diseased brain. Neural Regen Res. 11, 1533-1539. 10.4103/1673-5374.193220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Don E. K., Formella I., Badrock A. P., Hall T. E., Morsch M., Hortle E., Hogan A., Chow S., Gwee S. S. L., Stoddart J. J. et al. (2017). A Tol2 gateway-compatible toolbox for the study of the nervous system and neurodegenerative disease. Zebrafish 14, 69-72. 10.1089/zeb.2016.1321 [DOI] [PubMed] [Google Scholar]
- Dorsett C. R., McGuire J. L., DePasquale E. A. K., Gardner A. E., Floyd C. L. and McCullumsmith R. E. (2017). Glutamate neurotransmission in rodent models of traumatic brain injury. J. Neurotrauma 34, 263-272. 10.1089/neu.2015.4373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers P. H. C. and Goeman J. J. (2004). Enhancing scatterplots with smoothed densities. Bioinformatics 20, 623-628. 10.1093/bioinformatics/btg454 [DOI] [PubMed] [Google Scholar]
- Ellett F., Pase L., Hayman J. W., Andrianopoulos A. and Lieschke G. J. (2011). mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood 117, e49-e56. 10.1182/blood-2010-10-314120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok V. A., Voelker D. R., Campbell P. A., Cohen J. J., Bratton D. L. and Henson P. M. (1992). Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207-2216. [PubMed] [Google Scholar]
- Fink S. L. and Cookson B. T. (2005). Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73, 1907-1916. 10.1128/IAI.73.4.1907-1916.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster A. C., Gill R. and Woodruff G. N. (1988). Neuroprotective effects of MK-801 in vivo: selectivity and evidence for delayed degeneration mediated by NMDA receptor activation. J. Neurosci. 8, 4745-4754. 10.1523/JNEUROSCI.08-12-04745.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu R., Shen Q., Xu P., Luo J. J. and Tang Y. (2014). Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 49, 1422-1434. 10.1007/s12035-013-8620-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch U.-K. and Kettenmann H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387-1394. 10.1038/nn1997 [DOI] [PubMed] [Google Scholar]
- Harty B. L., Krishnan A., Sanchez N. E., Schiöth H. B. and Monk K. R. (2015). Defining the gene repertoire and spatiotemporal expression profiles of adhesion G protein-coupled receptors in zebrafish. BMC Genomics 16, 62 10.1186/s12864-015-1296-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawryluk G. W. J. and Bullock M. R. (2016). Past, present, and future of traumatic brain injury research. Neurosurg. Clin. N. Am. 27, 375-396. 10.1016/j.nec.2016.05.002 [DOI] [PubMed] [Google Scholar]
- Herrgen L., Voss O. P. and Akerman C. J. (2014). Calcium-dependent neuroepithelial contractions expel damaged cells from the developing brain. Dev. Cell 31, 599-613. 10.1016/j.devcel.2014.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt U. A. and Leist M. (2003). Rapid, noninflammatory and PS-dependent phagocytic clearance of necrotic cells. Cell Death Differ. 10, 1156-1164. 10.1038/sj.cdd.4401286 [DOI] [PubMed] [Google Scholar]
- James G. and Butt A. M. (2002). P2Y and P2X purinoceptor mediated Ca2+ signalling in glial cell pathology in the central nervous system. Eur. J. Pharmacol. 447, 247-260. 10.1016/S0014-2999(02)01756-9 [DOI] [PubMed] [Google Scholar]
- Jao L.-E., Wente S. R. and Chen W. (2013). Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. USA 110, 13904-13909. 10.1073/pnas.1308335110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X., Ishii H., Bai Z., Itokazu T. and Yamashita T. (2012). Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS ONE 7, e41892 10.1371/journal.pone.0041892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabori M., Kacimi R., Kauppinen T., Calosing C., Kim J. Y., Hsieh C. L., Nakamura M. C. and Yenari M. A. (2015). Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci. 35, 3384-3396. 10.1523/JNEUROSCI.2620-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopf F., Schnabel K., Haase C., Pfeifer K., Anastassiadis K. and Weidinger G. (2010). Dually inducible TetON systems for tissue-specific conditional gene expression in zebrafish. Proc. Natl. Acad. Sci. USA 107, 19933-19938. 10.1073/pnas.1007799107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulen P., Kuhn R., Wässle H. and Brandstätter J. H. (1999). Modulation of the intracellular calcium concentration in photoreceptor terminals by a presynaptic metabotropic glutamate receptor. Proc. Natl. Acad. Sci. USA 96, 9909-9914. 10.1073/pnas.96.17.9909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz E., Eimon P. M., Mukhyala K., Stern H., Zha J., Strasser A., Hart R. and Ashkenazi A. (2006). Functional characterization of the Bcl-2 gene family in the zebrafish. Cell Death Differ. 13, 1631-1640. 10.1038/sj.cdd.4402016 [DOI] [PubMed] [Google Scholar]
- Kroehne V., Freudenreich D., Hans S., Kaslin J. and Brand M. (2011). Regeneration of the adult zebrafish brain from neurogenic radial glia-type progenitors. Development 138, 4831-4841. 10.1242/dev.072587 [DOI] [PubMed] [Google Scholar]
- Kuscha V., Frazer S. L., Dias T. B., Hibi M., Becker T. and Becker C. G. (2012). Lesion-induced generation of interneuron cell types in specific dorsoventral domains in the spinal cord of adult zebrafish. J. Comp. Neurol. 520, 3604-3616. 10.1002/cne.23115 [DOI] [PubMed] [Google Scholar]
- Kwan K. M., Fujimoto E., Grabher C., Mangum B. D., Hardy M. E., Campbell D. S., Parant J. M., Yost H. J., Kanki J. P. and Chien C.-B. (2007). The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 236, 3088-3099. 10.1002/dvdy.21343 [DOI] [PubMed] [Google Scholar]
- Livak K. J. and Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402-408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Loane D. J., Stoica B. A. and Faden A. I. (2015). Neuroprotection for traumatic brain injury. Handb Clin Neurol 127, 343-366. 10.1016/B978-0-444-52892-6.00022-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Elliott M. R., Chen Y., Walsh J. T., Klibanov A. L., Ravichandran K. S. and Kipnis J. (2011). Phagocytic activity of neuronal progenitors regulates adult neurogenesis. Nat. Cell Biol. 13, 1076-1083. 10.1038/ncb2299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas A. I. R., Stocchetti N. and Bullock R. (2008). Moderate and severe traumatic brain injury in adults. Lancet Neurol. 7, 728-741. 10.1016/S1474-4422(08)70164-9 [DOI] [PubMed] [Google Scholar]
- Magnus T., Chan A., Linker R. A., Toyka K. V. and Gold R. (2002). Astrocytes are less efficient in the removal of apoptotic lymphocytes than microglia cells: implications for the role of glial cells in the inflamed central nervous system. J. Neuropathol. Exp. Neurol. 61, 760-766. 10.1093/jnen/61.9.760 [DOI] [PubMed] [Google Scholar]
- Martin S. J., Reutelingsperger C. P., McGahon A. J., Rader J. A., van Schie R. C., LaFace D. M. and Green D. R. (1995). Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 182, 1545-1556. 10.1084/jem.182.5.1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazaheri F., Breus O., Durdu S., Haas P., Wittbrodt J., Gilmour D. and Peri F. (2014). Distinct roles for BAI1 and TIM-4 in the engulfment of dying neurons by microglia. Nat. Commun. 5, 4046 10.1038/ncomms5046 [DOI] [PubMed] [Google Scholar]
- McCutcheon V., Park E., Liu E., Wang Y., Wen X.-Y. and Baker A. J. (2016). A model of excitotoxic brain injury in larval zebrafish: potential application for high-throughput drug evaluation to treat traumatic brain injury. Zebrafish 13, 161-169. 10.1089/zeb.2015.1188 [DOI] [PubMed] [Google Scholar]
- Meijer A. H. and Spaink H. P. (2011). Host-pathogen interactions made transparent with the zebrafish model. Curr. Drug Targets 12, 1000-1017. 10.2174/138945011795677809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakami R. and Sumimotoa H. (2006). Phagocytosis-coupled activation of the superoxide-producing phagocyte oxidase, a member of the NADPH oxidase (nox) family. Int. J. Hematol. 84, 193-198. 10.1532/IJH97.06133 [DOI] [PubMed] [Google Scholar]
- Morsch M., Radford R., Lee A., Don E. K., Badrock A. P., Hall T. E., Cole N. J. and Chung R. (2015). In vivo characterization of microglial engulfment of dying neurons in the zebrafish spinal cord. Front. Cell Neurosci. 9, 321 10.3389/fncel.2015.00321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negoescu A., Lorimier P., Labat-Moleur F., Drouet C., Robert C., Guillermet C., Brambilla C. and Brambilla E. (1996). In situ apoptotic cell labeling by the TUNEL method: improvement and evaluation on cell preparations. J. Histochem. Cytochem. 44, 959-968. 10.1177/44.9.8773561 [DOI] [PubMed] [Google Scholar]
- Neher J. J., Neniskyte U., Zhao J.-W., Bal-Price A., Tolkovsky A. M. and Brown G. C. (2011). Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 186, 4973-4983. 10.4049/jimmunol.1003600 [DOI] [PubMed] [Google Scholar]
- Neumann J., Sauerzweig S., Rönicke R., Gunzer F., Dinkel K., Ullrich O., Gunzer M. and Reymann K. G. (2008). Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J. Neurosci. 28, 5965-5975. 10.1523/JNEUROSCI.0060-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A., Kirchhoff F. and Helmchen F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314-1318. 10.1126/science.1110647 [DOI] [PubMed] [Google Scholar]
- Noda M., Doi Y., Liang J., Kawanokuchi J., Sonobe Y., Takeuchi H., Mizuno T. and Suzumura A. (2011). Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J. Biol. Chem. 286, 2308-2319. 10.1074/jbc.M110.169839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnmacht J., Yang Y., Maurer G. W., Barreiro-Iglesias A., Tsarouchas T. M., Wehner D., Sieger D., Becker C. G. and Becker T. (2016). Spinal motor neurons are regenerated after mechanical lesion and genetic ablation in larval zebrafish. Development 143, 1464-1474. 10.1242/dev.129155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo-Martin C., Chang T.-Y., Koo B. K., Gilleland C. L., Wasserman S. C. and Yanik M. F. (2010). High-throughput in vivo vertebrate screening. Nat. Methods 7, 634-636. 10.1038/nmeth.1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.-C., Kim C.-H., Bae Y.-K., Yeo S.-Y., Kim S.-H., Hong S.-K., Shin J., Yoo K.-W., Hibi M., Hirano T. et al. (2000). Analysis of upstream elements in the HuC promoter leads to the establishment of transgenic zebrafish with fluorescent neurons. Dev. Biol. 227, 279-293. 10.1006/dbio.2000.9898 [DOI] [PubMed] [Google Scholar]
- Park D., Tosello-Trampont A.-C., Elliott M. R., Lu M., Haney L. B., Ma Z., Klibanov A. L., Mandell J. W. and Ravichandran K. S. (2007). BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450, 430-434. 10.1038/nature06329 [DOI] [PubMed] [Google Scholar]
- Park E., Bell J. D. and Baker A. J. (2008). Traumatic brain injury: can the consequences be stopped? CMAJ 178, 1163-1170. 10.1503/cmaj.080282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnaik R., Raff M. C. and Scholes J. (2000). Differences between the clearance of apoptotic cells by professional and non-professional phagocytes. Curr. Biol. 10, 857-860. 10.1016/S0960-9822(00)00598-4 [DOI] [PubMed] [Google Scholar]
- Pauls S., Geldmacher-Voss B. and Campos-Ortega J. A. (2001). A zebrafish histone variant H2A.F/Z and a transgenic H2A.F/Z:GFP fusion protein for in vivo studies of embryonic development. Dev. Genes Evol. 211, 603-610. 10.1007/s00427-001-0196-x [DOI] [PubMed] [Google Scholar]
- Peri F. and Nüsslein-Volhard C. (2008). Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 133, 916-927. 10.1016/j.cell.2008.04.037 [DOI] [PubMed] [Google Scholar]
- Renshaw S. A., Loynes C. A., Trushell D. M. I., Elworthy S., Ingham P. W. and Whyte M. K. B. (2006). A transgenic zebrafish model of neutrophilic inflammation. Blood 108, 3976-3978. 10.1182/blood-2006-05-024075 [DOI] [PubMed] [Google Scholar]
- Rink A., Fung K. M., Trojanowski J. Q., Lee V. M., Neugebauer E. and McIntosh T. K. (1995). Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am. J. Pathol. 147, 1575-1583. [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Rodríguez A., Egea-Guerrero J. J., Murillo-Cabezas F. and Carrillo-Vico A. (2014). Oxidative stress in traumatic brain injury. Curr. Med. Chem. 21, 1201-1211. 10.2174/0929867321666131217153310 [DOI] [PubMed] [Google Scholar]
- Sasaki Y., Hoshi M., Akazawa C., Nakamura Y., Tsuzuki H., Inoue K. and Kohsaka S. (2003). Selective expression of Gi/o-coupled ATP receptor P2Y12 in microglia in rat brain. Glia 44, 242-250. 10.1002/glia.10293 [DOI] [PubMed] [Google Scholar]
- Sieger D., Moritz C., Ziegenhals T., Prykhozhij S. and Peri F. (2012). Long-range Ca2+ waves transmit brain-damage signals to microglia. Dev. Cell 22, 1138-1148. 10.1016/j.devcel.2012.04.012 [DOI] [PubMed] [Google Scholar]
- Sierra A., Abiega O., Shahraz A. and Neumann H. (2013). Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front. Cell Neurosci. 7, 6 10.3389/fncel.2013.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica B. A. and Faden A. I. (2010). Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 7, 3-12. 10.1016/j.nurt.2009.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauzin S., Starnes T. W., Becker F. B., Lam P.-Y. and Huttenlocher A. (2014). Redox and Src family kinase signaling control leukocyte wound attraction and neutrophil reverse migration. J. Cell Biol. 207, 589-598. 10.1083/jcb.201408090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham T. J., Mapes J., Kokel D. and Peterson R. T. (2010). Live imaging of apoptotic cells in zebrafish. FASEB J. 24, 4336-4342. 10.1096/fj.10-161018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham T. J., Kokel D. and Peterson R. T. (2012). Apoptotic cells are cleared by directional migration and elmo1- dependent macrophage engulfment. Curr. Biol. 22, 830-836. 10.1016/j.cub.2012.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton E. M., Cronan M. R., Beerman R. W. and Tobin D. M. (2015). The macrophage-specific promoter mfap4 allows live, long-term analysis of macrophage behavior during mycobacterial infection in Zebrafish. PLoS ONE 10, e0138949 10.1371/journal.pone.0138949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Jia W. and Cynader M. (1997). Comparison of the neuroprotective effects of APV and bcl-2 in glutamate-induced cell death. Neuroreport 8, 3323-3326. 10.1097/00001756-199710200-00026 [DOI] [PubMed] [Google Scholar]
- Wang J., Wegener J. E., Huang T.-W., Sripathy S., De Jesus-Cortes H., Xu P., Tran S., Knobbe W., Leko V., Britt J. et al. (2015). Wild-type microglia do not reverse pathology in mouse models of Rett syndrome. Nature 521, E1-E4. 10.1038/nature14444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. (2007). The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio Rerio). Eugene, Oregon: University of Oregon Press. [Google Scholar]
- Wilms H., Rosenstiel P., Sievers J., Deuschl G., Zecca L. and Lucius R. (2003). Activation of microglia by human neuromelanin is NF-kappaB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson's disease. FASEB J. 17, 500-502. 10.1096/fj.02-0314fje [DOI] [PubMed] [Google Scholar]
- Witting A., Müller P., Herrmann A., Kettenmann H. and Nolte C. (2000). Phagocytic clearance of apoptotic neurons by Microglia/Brain macrophages in vitro: involvement of lectin-, integrin-, and phosphatidylserine-mediated recognition. J. Neurochem. 75, 1060-1070. 10.1046/j.1471-4159.2000.0751060.x [DOI] [PubMed] [Google Scholar]
- Wyllie A. H. (1997). Apoptosis: an overview. Br. Med. Bull. 53, 451-465. 10.1093/oxfordjournals.bmb.a011623 [DOI] [PubMed] [Google Scholar]
- Yeo S.-Y., Kim M., Kim H.-S., Huh T.-L. and Chitnis A. B. (2007). Fluorescent protein expression driven by her4 regulatory elements reveals the spatiotemporal pattern of Notch signaling in the nervous system of zebrafish embryos. Dev. Biol. 301, 555-567. 10.1016/j.ydbio.2006.10.020 [DOI] [PubMed] [Google Scholar]
- Yoo S. K., Freisinger C. M., LeBert D. C. and Huttenlocher A. (2012). Early redox, Src family kinase, and calcium signaling integrate wound responses and tissue regeneration in zebrafish. J. Cell Biol. 199, 225-234. 10.1083/jcb.201203154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Schmid-Kotsas A., Gross H.-J., Gruenert A. and Bachem M. G. (2003). Sensitivity and specificity of different staining methods to monitor apoptosis induced by oxidative stress in adherent cells. Chin. Med. J. 116, 1923-1929. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.