

Abstract
Background/Aims:
Data from Western countries indicate that biliary atresia (BA) is the leading cause of end-stage liver disease in children and the most common indication for liver transplantation (LT) in the pediatric population. There is no data on the epidemiology and outcome of BA in Saudi Arabia. The main objective of our study was to understand the clinical and epidemiological characteristics and outcome of BA in the Saudi population.
Patients and Methods:
We retrospectively reviewed the database of infantile cholestasis cases that presented to our center from 2008 to 2015 and identified BA cases. Data on clinical, biochemical, imaging, and histopathological characteristics were collected by chart review. The two primary study outcomes were (1) successful Kasai portoenterostomy (KPE) defined as resolution of jaundice (total serum bilirubin <20 μmol/L) and (2) survival with native liver.
Results:
Over the study period, we evaluated 450 cases of infantile cholestasis. In all, 21 cases (11 males) were diagnosed with BA (4.7%). BA cases were first seen by pediatric gastroenterologists at a median age of 65 days. KPE was performed in 12 cases at a median age of 73 days. Successful KPE was achieved in four cases (33%). Five of the remaining eight cases had LT and three died before LT. Nine of the 21 BA cases were denied KPE and had primary LT at median 8 months of age. The native liver 4-year survival rate was 14.3% and the overall survival rate was 81%.
Conclusion:
BA is an uncommon cause of infantile cholestasis in Saudi Arabia. Our study provides a snapshot of the epidemiology of BA in Saudi Arabia that is characterized by late referral to pediatric gastroenterologists and poor outcome without LT.
Keywords: Biliary atresia, cholestasis, Kasai, late referral, liver transplant, outcome, portoenterostomy
INTRODUCTION
Biliary atresia (BA) is an idiopathic neonatal obstructive cholangiopathy characterized by a progressive fibrosclerosing obliteration of the large bile ducts that present uniquely in the first few months of life. Early diagnosis of BA and Kasai portoenterostomy (KPE) improves outcomes.[1,2,3,4,5,6,7] Data from Western countries indicate that BA is the most common cause of neonatal cholestasis (25%), the leading cause of end-stage liver disease in children, and the most common indication for liver transplantation (LT) in the pediatric population.[1,2,3,4,5,6,7] Successful KPE (defined as clearance of jaundice within the first 6 months after KPE) was reported in 70%–80% of the patients with BA, when performed before 60 days of age, and was associated with a 30%–40% 10-year survival with the most experienced centers in the West and approximately 60% in Japan.[8] If KPE is not performed, 50%–80% of BA cases will die (without LT) from biliary cirrhosis by 1 year of age, and 90%–100% will die by 3 years of age.[9,10] The age at performance of KPE is one of the most important factors determining surgical outcome. If KPE is performed between 60 and 90 days of life, approximately 40%–50% of patients show bile drainage; if performed after 90 days of life, up to 25% show drainage.[2,3,4,5,6]
Data on BA in Saudi Arabia is very limited. The only available published literature on prevalence of BA indicated that BA was the cause of cholestasis in 17 of 64 neonates.[11] Data from two liver transplant centers in Saudi Arabia indicated that BA and progressive familial intrahepatic cholestasis were the leading causes for pediatric LT; BA constituted 30%–33% of the cases.[12,13] These studies did not provide further details of the epidemiology and outcome of the BA cases. Being first in the rank of the need for pediatric LT in Saudi Arabia might indicate that BA is really a leading cause of pediatric liver disease in Saudi Arabia. Another possibility is that BA is over-represented as a cause of infantile cholestasis because LT centers are more likely to receive the severe cases that lead to advanced liver disease. We conducted this study to establish the current prevalence of BA as a cause of infantile cholestasis, to study the epidemiology, clinical characteristics, and outcome of BA in Saudi population, and to compare our results with those from the West and Far East of Asia.
PATIENTS AND METHODS
Diagnosis of biliary atresia
We retrospectively reviewed our database of infantile cholestasis cases that presented to our tertiary care center in Riyadh city, the capital of Saudi Arabia, from 2008 to 2015, and identified BA cases. All infants presenting to our center with infantile cholestasis underwent extensive work-up to exclude infectious, structural, metabolic, endocrine, infiltrative, and familial causes. Inclusion criteria: We included cases of infantile cholestasis with clinical, biochemical, radiological data, liver histology, and surgical findings that were all consistent with BA. Formalin-fixed biopsies were stained with hematoxylin and eosin and examined under light microscopy by a single pathologist certified in gastrointestinal and hepatic pathology. BA was suspected when there was presence of pale stool in combination with high gamma-glutamyl transferase (GGT), infantile cholestasis (GGT > 200 IUL) and demonstration of portal tracts fibrosis, proliferation of portal bile ducts, inflammatory cell infiltration, and bile plugs in liver biopsy. The definitive diagnosis was made when cholangiography failed to show a patent biliary tree. In cases where cholangiography was not undertaken, the diagnosis of BA was confirmed by operative findings at subsequent LT.
Data collection
Data on clinical, biochemical, imaging, and histopathological characteristics were collected by chart review. Data collected from each chart included demographic information, birth weight, date of onset of jaundice, date of referral, presenting signs and symptoms, diagnostic studies and laboratory values at the time of first evaluation for cholestasis, whether a Kasai operation was performed, date of Kasai operation, time to clear jaundice, postoperative course and complications after KPE (e.g., cholangitis, ascites, variceal hemorrhage), whether LT was performed, date of last follow-up, and final outcome. The postoperative medical and nutritional management was reasonably uniform during this 10-year period and included the use of fat-soluble vitamins, a medium-chain triglyceride milk formula, ursodeoxycholic acid, and perioperative intravenous antibiotics followed by prophylactic sulfamethoxazole–trimethoprim antibiotic for 3 months. Development of ascending cholangitis was an indication to give prophylactic antibiotic beyond 3 months.
Biliary atresia splenic malformation (BASM) syndrome was defined as a splenic malformation occurring together with another major malformation in infants diagnosed with biliary atresia. Clinically evident portal hypertension (PHT) was diagnosed when there was either (1) a history of a complication of PHT (esophageal or gastric variceal bleed, ascites, or hepatopulmonary syndrome) or (2) clinical findings consistent with PHT (both splenomegaly (spleen palpable >2 cm below the costal margin) and thrombocytopenia (platelet count <150,000/mL)). All children had at least 3 years follow-up after KPE.
Study outcomes
Several patients' outcomes were evaluated, including (1) successful KPE defined as resolution of jaundice with pigmented stools and serum total bilirubin level lower than 20 μmol/L; (2) failed KP defined as persistence of jaundice (>20 μmol/L) and pale stool post KPE; (3) overall survival rate (OSR) defined as starting at birth and ending at death or last follow-up; (4) native liver survival rate starting at birth and ending at death, transplant, or last follow-up.
RESULTS
During the study period, 450 cases of infantile cholestasis were referred to our center for evaluation. In all, 21 patients with BA were identified (11 males) which corresponds to 4.7% of the cases of infantile cholestasis.
Clinical and laboratory characteristics
Table 1 shows the clinical and laboratory characteristics of the 21 BA cases at the time of presentation to our hospital at a mean age of 66 ± 29.2 days (median 65 days, range: 30–159 days). All the 21 cases were product of full-term gestation except one preterm baby (gestation age 34 weeks). The mean birth weight was 3 kg (range: 2–4.1 kg); all were appropriate for age except one case with intrauterine growth retardation. About 43% of the parents were consanguineous. There was no family history of BA among the 21 cases. Twelve BA cases (57%) underwent KPE at a mean of 70 ± 13.5 days of life (median 73 days, range: 50–94 days). The mean time taken from first evaluation by a pediatric gastroenterologist to performance of KPE was 13.6 ± 4.14 days (median 13 days, range: 7–21 days).
Table 1.
Clinical and biochemical characteristics of the 21 patients with biliary atresia at time of presentation to our hospital
| Variable | Mean±SD | Median | (Range) |
|---|---|---|---|
| Onset of jaundice (days) | 10.36±9.52 | 7.00 | (7-14) |
| Age at referral (days) | 66±29.2 | 65 | (30-159) |
| Age at performance of KPE (days) | 70±13.5 | 73 | (50-94) |
| Onset of liver failure post KPE (months) | 13.50±5.51 | 13.00 | (8-20) |
| Primary LT (without KPE) (months) | 8.7±2.4 | 8.00 | (4-12) |
| LT post KPE (months) | 8.20±3.35 | 8.00 | (6-12) |
| TSB μmol/L) | 178.29±63.34 | 163.00 | (92-353) |
| Direct bilirubin μmol/L) | 149.90±51.76 | 134.00 | (87-279) |
| ALT (IU/L) | 182.57±102.60 | 174.00 | (62-440) |
| AST (IU/L) | 296.43±147.22 | 245.00 | (105-613) |
| ALP (IU/L) | 961.19±435.13 | 920.00 | (381-1951) |
| GGT (IU/L) | 735.43±576.70 | 730.00 | (90-2328) |
| Serum bile acids μmol/L) | 143.16±47.57 | 143.00 | (78-244) |
SD: Standard deviation; KPE: Kasai portoenterostomy; LT: Liver transplant; TSB: Total serum bilirubin; ALT: Alanine aminotransferase; AST: Aspartate transaminase; ALP: Alkaline phosphatase; GGT: Gamma-glutamyl transferase
Congenital anomalies/systemic involvement
Among the 21 patients, 6 (28%) had congenital anomalies identified. BASM was diagnosed in two cases (9.5%) associated with absent tail and body of pancreas and interrupted inferior vena cava in one case. Congenital heart disease was seen in four cases (19%): ventricular septal defect in three cases, atrial septal defect in two cases, patent ductus arteriosus in two cases, and pulmonary valve stenosis in one case. Skeletal abnormality was observed in two cases: one with lumbar spine kyphosis and the other had butterfly vertebra. One case had neonatal-onset diabetes mellitus.
Imaging studies
Ultrasound was obtained in all cases. The gall bladder was reported to be hypoplastic/atrophic in 17 (81%) cases and normal in 4 (19%). The triangular cord sign was visualized in four cases (19%). Magnetic resonance cholangiopancreatography was performed in 13 cases; 12 studies showed hypoplastic/atrophic gall bladder and one study showed normal gall bladder. Hepatobiliary scintigraphy (HIDA scan) was performed for two patients early during the study period; both were nonexcretory.
Outcome of the 21 BA cases
Figure 1 illustrates the treatment and outcome of the 21 patients with BA.
Figure 1.
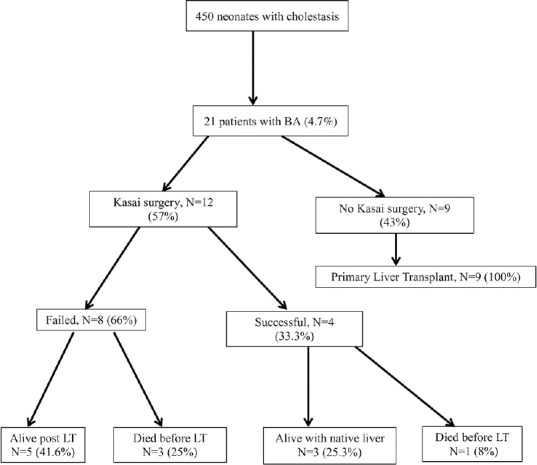
Management and outcome of the 21 biliary atresia cases (LT: Liver transplant; BA: Biliary atresia)
Of 12 patients who underwent KPE, 4 had successful KPE (33.3%). Those four BA cases were seen first by a pediatric gastroenterologist at a median age of 57 days (range: 40–68 days) and KPE was performed at a median age of 69 days (range: 50–73 days) by four pediatric surgeons in our center. The jaundice resolved at a median age of 18 weeks (range: 15–20 weeks). One child succumbed at age 14 months due to fulminant sepsis. The remaining three children are still alive with their native liver at a median age of 4.3 years (range: 3.8–10 years); two of them are jaundice-free. A 10-year-old girl has compensated cirrhosis with portal hypertension. A 4.3-year-old girl has been referred for LT because of decompensated cirrhosis. A 3.8-year-old girl is healthy without any complication
Eight cases failed to clear jaundice after KPE (66.6%). They were seen first by a pediatric gastroenterologist at a median age of 58 days (range: 39–81 days) and KPE was performed at a median age of 76.5 days (range: 51–94 days). Three cases died at a mean age of 7.3 months due to progressive liver failure. The remaining five patients had successful LT at a median age of 8 months (range: 6–12 months)
Post-KPE course was complicated by ascending cholangitis in seven patients (58%), one episode of ascending cholangitis each. Portal hypertension developed in two of the three BA cases surviving so far
Nine patients did not undergo KPE and had successful primary LT (43%) (at a median age of 8 months, range: 4–12 months) because of late presentation in six patients (median 90 days, range: 70–159 days) and parents' willingness in the remaining three patients
The native liver 4-year survival rate was 14.3% and the OSR was 81% after a median follow-up period of 4.3 years (range: 3.8–10 years).
DISCUSSION
This is the first local study that describes the prevalence, various characteristics, and outcome of BA in Saudi Arabia. Our study showed several important findings: first, BA is an uncommon cause of infantile cholestasis in Saudi Arabia (4.7%), when compared with 20–25% in European and North American data.[1,2,3,4,5,6,7,8,9] The high percentage (30–33%) of BA reported from the LT centers in Saudi Arabia[12,13] likely over-represents the BA as a cause of severe liver disease leading to end-stage liver disease and need for LT. Second, an alarming feature in our data is the late referral of BA cases to pediatric gastroenterologists (median 65 days) and subsequent delay in performance of KPE (median 73 days). As a result, the outcome of BA cases in our center was poor when compared with the data from Western countries and Japan [Table 2]: the success rate of KPE was low (33%) and the short-term survival rate without LT was poor (14.3%). However, the OSR of 81% is comparable to Western data due to the success of the LT programs in Saudi Arabia.
Table 2.
Series of biliary atresia published from different countries
| Country (year) | Number of patients | Median age at KPE (days) | Survival with native liver |
|---|---|---|---|
| Japan (1989-1999)[8] | 1381 | 61-70 | 5 years: 59.7% |
| United Kingdom and Ireland (1993-1995)[7] | 93 | 54 | 5 years: 30.1% |
| United States (1997-2002)[3] | 104 | 61 | 2 years: 55.8%» |
| France (1997-2002)[5] | 271 | 57 | 4 years: 42.7% |
| Canada (1985-2002)[2] | 349 | 65 | 4 years: 33% |
| England and Wales (1999-2002)[4] | 148 | 54 | 4 years: 51% |
| Brazil (1982-2008)[12] | 517 | 82.6* | 4 years: 36.8% |
| Germany (2001-2005)[13] | 183 | 57* | 2 years: 20.3% |
| Switzerland (1994-2004)[14] | 48 | 68 | 4 years: 37.4% |
| Hong Kong (1980-2015)[15] | 141 | 65* | 20 years: 51% |
KPE: Kasai portoenterostomy; *Mean
Most of the literature on the outcome of KPE originated from Europe, North America, and Far East of Asia [Table 2].[1,2,3,4,5,6,7,8,9,10,14,15,16,17]. Although late referral of BA cases is a problem all over the world, it is more pronounced in Saudi Arabia. These studies have identified several factors for the delay at referral: (1) lack of knowledge on the importance of an early diagnosis by pediatricians and families; (2) physicians may not recognize the importance of investigating prolonged jaundice in neonates greater than 2 weeks of age thinking it is breast milk jaundice which is very common in this age; (3) limited access to healthcare system especially by families living in the peripheries away from cities. In Saudi Arabia, after birth, a newborn is seen in the well-baby clinic at 2 months of age for vaccination and checkup, and therefore family physicians miss the opportunity of early detection of prolonged neonatal jaundice within the first month of life. Other factors that have been reported to determine the success of KPE include the following: (a) patient's age at the time of surgery, with best results (70–80% success rate) reported when KPE was performed before 60 days of age, (b) surgeon's experience as reflected by the number of KPE performed per year, and (c) occurrence of postoperative cholangitis.[1,2,3,4,5,6,7,8,9] We believe that these factors in combination were major reasons for poor outcome of KPE in our data. In addition, the wide gap between the referral date and age of surgery (median 13 days, range: 7–21 days) was another factor for the poor outcome of KPE in our study. This factor has been recognized and addressed in some pediatric liver units around the world by adopting practical protocols to accelerate the diagnostic work-up so that the diagnosis of BA can be ascertained within 5 days from admission. Another relevant strong indicator of good long-term outcome of KPE and thus avoidance of LT is the postoperative clearance of jaundice. All the successful KPE cases, except one, survived with native liver at a median age of 4.3 years (range: 3.8–10 years).
To improve the outcome of KPE, some European countries have undertaken several practical steps that included (1) awareness campaigns to alert family physicians and pediatricians about alarming signs and importance of early referral of neonatal cholestasis cases to pediatric gastroenterologists; (2) centralization of the care of BA to a very limited number of tertiary care centers to increase the case load/center/year to improve surgeons' experience;[4,5] and (3) development of appropriate screening tests for BA.[8] A clear limitation of our study was the small number of BA patients included. Insufficient numbers of patients in the subgroups to be compared precluded a meaningful statistical analysis to determine the factors predictive of outcomes. Hence, we need a large multicenter study and a national BA registry to study the incidence of BA and to investigate the hurdles that prevent prompt referral of suspected BA cases and timely performance of KPE to enable us to make appropriate recommendations to improve the KPE outcome. Until we have enough data from a large multicenter study, efforts are needed to raise awareness of parents, general practitioners and pediatricians, for the necessity of prompt investigations of any case with prolonged jaundice (jaundice persisting >2 weeks of life) and early referral of cases with cholestasis to a pediatric gastroenterologist. On the other hand, pediatric gastroenterologists need to adopt a practical protocol to accelerate the diagnostic work-up so that the diagnosis of BA can be established within 5–7 days from admission.
In conclusion, in comparison to data from the Western countries and Japan, BA is an uncommon cause of infantile cholestasis in Saudi Arabia. Although our study's sample size is small, it gives a snapshot of the epidemiology of BA in Saudi Arabia that is characterized by late referral to pediatric gastroenterologists and poor outcome without LT during late infancy.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Sun LY, Yang YS, Zhu ZJ. Outcomes in children with biliary atresia following liver transplantation. Hepatobiliary Pancreat Dis Int. 2013;12:143–8. doi: 10.1016/s1499-3872(13)60023-5. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber RA, Barker CC, Roberts EA, Martin SR, Alvarez F, Smith L, et al. Canadian Pediatric Hepatology Research Group. Biliary atresia: The Canadian experience. J Pediatr. 2007;151:659–65. doi: 10.1016/j.jpeds.2007.05.051. [DOI] [PubMed] [Google Scholar]
- 3.Shneider BL, Brown MB, Haber B, Whitington PF, Schwarz K, Squires R, et al. A multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J Pediatr. 2006;148:467–74. doi: 10.1016/j.jpeds.2005.12.054. [DOI] [PubMed] [Google Scholar]
- 4.Davenport M, De Ville de Goyet J, Stringer MD, Mieli-Vergani G, Kelly DA, McClean P, et al. Seamless management of biliary atresia in England and Wales (1999-2002) Lancet. 2004;363:1354–7. doi: 10.1016/S0140-6736(04)16045-5. [DOI] [PubMed] [Google Scholar]
- 5.Serinet MO, Broue P, Jacquemin E, Lachaux A, Sarles J, Gottrand F, et al. Management of patients with biliary atresia in France: Results of a decentralized policy 1986-2002. Hepatology. 2006;44:75–84. doi: 10.1002/hep.21219. [DOI] [PubMed] [Google Scholar]
- 6.Serinet MO, Wildhaber BE, Broué P, Lachaux A, Sarles J, Jacquemin E, et al. Impact of age at Kasai operation on its results in late childhood and adolescence: A rational basis for biliary atresia screening. Pediatrics. 2009;123:1280–6. doi: 10.1542/peds.2008-1949. [DOI] [PubMed] [Google Scholar]
- 7.McKiernan PJ, Baker AJ, Kelly DA. The frequency and outcome of biliary atresia in the UK and Ireland. Lancet. 2000;355:25–9. doi: 10.1016/S0140-6736(99)03492-3. [DOI] [PubMed] [Google Scholar]
- 8.Nio M, Ohi R, Miyano T, Saeki M, Shiraki K, Tanaka K. Five- and 10-year survival rates after surgery for biliary atresia: A report from the Japanese Biliary Atresia Registry. J Pediatr Surg. 2003;38:997–1000. doi: 10.1016/s0022-3468(03)00178-7. [DOI] [PubMed] [Google Scholar]
- 9.Chardot C, Carton M, Spire-Bendelac N, Le Pommelet C, Golmard J, Reding R, et al. Is the Kasai operation still indicated in children older than 3 months diagnosed with biliary atresia? J Pediatr. 2001;138:224–8. doi: 10.1067/mpd.2001.111276. [DOI] [PubMed] [Google Scholar]
- 10.Karrer FM, Price MR, Bensard DD, Sokol RJ, Narkewicz MR, Smith DJ, et al. Long-term results with the Kasai operation for biliary atresia. Arch Surg. 1996;131:493–6. doi: 10.1001/archsurg.1996.01430170039006. [DOI] [PubMed] [Google Scholar]
- 11.Abdullah AM, al Fadel Saleh M, al Madan M, el Mouzan M, Olasope B. Infantile cholestasis in the Central-Eastern Province Saudi Arabia. J Trop Pediatr. 1997;43:138–42. doi: 10.1093/tropej/43.3.138. [DOI] [PubMed] [Google Scholar]
- 12.Khan I, Al-Shaqrani MA, Arain ZB, Al-Hebbi HA, Wali SH, Bassas AF. One hundred and thirty-seven living donor pediatric liver transplants at Riyadh Military Hospital. Results and outlook for future. Saudi Med J. 2009;30:403–8. [PubMed] [Google Scholar]
- 13.Fayyad A, Shagrani M, AlGoufi T, ElSheikh Y, Murray J, Elgohary A, et al. Progress and outcomes of the first high-volume pediatric liver transplantation program in Saudi Arabia. Clin Transpl. 2013:77–83. [PubMed] [Google Scholar]
- 14.Queiroz TC, Ferreira AR, ED Fagundes, ML Roquete, Penna FJ. Biliary atresia: Evaluation on two distinct periods at a reference pediatric service. Arq Gastroenterol. 2014;51:53–9. doi: 10.1590/s0004-28032014000100011. [DOI] [PubMed] [Google Scholar]
- 15.Leonhardt J, Kuebler JF, Leute PJ, Turowski C, Becker T, Pfister ED, et al. Lessons learned from the voluntary German Registry. Eur J Pediatr Surg. 2011;21:82–7. doi: 10.1055/s-0030-1268476. [DOI] [PubMed] [Google Scholar]
- 16.Wildhaber BE, Majno P, Mayr J, Zachariou Z, Hohlfeld J, Schwoebel M. Biliary atresia: Swiss national study, 1994–2004. J Pediatr Gastroenterol Nutr. 2008;46:299–307. doi: 10.1097/MPG.0b013e3181633562. [DOI] [PubMed] [Google Scholar]
- 17.Wong CWY, Chung PHY, Tam PKH, Wong KKY. Long-term results and quality of life assessment in biliary atresia patients: A 35-year experience in a tertiary hospital. J Pediatr Gastroenterol Nutr. 2018;66:570–4. doi: 10.1097/MPG.0000000000001854. [DOI] [PubMed] [Google Scholar]