

ABSTRACT
Complement component C3 is central to the complement system, a humoral effector mechanism of innate immune defense. When activated, C3 covalently binds to target particles, marking them for uptake and clearance by phagocytosis. We now show that C3 also exists within the cytosol where it interacts with ATG16L1, and is therefore involved in the intracellular clearance and recycling of material by macroautophagy/autophagy in pancreatic beta cells. C3 is highly expressed in isolated human islets, and its expression is upregulated in islets isolated from diabetic patients and rodents, and correlates with patient HBA1c and body mass index (BMI). Knockout of C3 in clonal beta cells leads to dysfunctional autophagy, and increased cell death after challenge with diabetogenic stresses, which are usually alleviated by increased autophagic turnover. However, autophagic degradation of INS (insulin) granules regulates total INS content, and increased autophagy due to C3 upregulation may deplete beta cell INS stores. C3 is therefore required for efficient autophagic turnover in beta cells, and is upregulated as a cytoprotective factor during diabetes.
KEYWORDS: ATG16L1, autophagy, complement C3, diabetes, intracellular complement
The complement system is an evolutionarily ancient component of innate immunity, and was discovered over a century ago as a heat-labile antimicrobial activity of serum, which ‘complemented’ the activity of antibodies. Today it is known to consist of a multitude of interacting proteins, which can be activated by pattern recognition molecules including C1Q, which recognizes bound IgG and IgM, hence complement activity being ‘complementary’ to antibodies. Complement activation leads to cleavage of C3, revealing a reactive thioester group, which then binds covalently to target particles, irreversibly marking them for uptake and destruction by phagocytosis.
We have been investigating roles of innate immune pathways in pancreatic inflammation in type 2 diabetes. Total RNA sequencing of isolated human pancreatic islets reveals high expression of C3, with significant upregulation in diabetic compared to healthy donors. This upregulation of C3 expression is also replicated in islets from various rodent models of diabetes. Furthermore, human islet expression of C3 correlates with HBA1c, BMI, and pro-inflammatory cytokine expression. Surprisingly, we found C3 present within the cytosolic fraction of human islets; C3 is well-known as a secreted protein and is indeed one of the most abundant proteins in human serum, although there is increasing evidence for non-canonical, intracellular roles of complement in metabolism and cell survival. We used protein interaction microarrays to identify novel cytosolic binding partners for C3, and found a strong interaction with ATG16L1, a core component of autophagy.
Autophagy is required both for compensatory expansion of beta cell mass during obesity, as well as for maintaining INS secretion and glucose tolerance. We generated CRISPR-Cas9-mediated C3 knockout INS-1 832/13 cells, a rat beta cell line popular for in vitro functional beta cell physiology studies. C3 knockout leads to dysfunctional autophagy, with a decrease in autophagic turnover, and accumulation of non-acidified autophagosome-like structures as identified by GFP-LC3 puncta assays and electron microscopy. These results were confirmed using isolated islets from C3 knockout mice. The C3-deficient INS-1 clones were more vulnerable to cell death when exposed to known diabetogenic autophagy-inducing cellular stresses. Autophagy dysregulation by C3 knockout also leads to increased cellular insulin content and secretion, as autophagy is involved in homeostatic degradation of INS granules. Upregulation of C3 and therefore autophagy may therefore decrease INS content by autophagic degradation. This may seem paradoxical, but is probably without significance for glucose homeostasis, as previous ultrastructural studies have demonstrated that INS granules are present in great excess, and that only a minor fraction (~1%) are released upon stimulation. The main outcome of C3-dependent autophagy upregulation will therefore by cytoprotective, and preserve of beta cell function.
C3 therefore plays a surprising intracellular role in cytoprotective autophagy regulation, being upregulated within beta cells under cellular stress during diabetes. One of the surprising features of this is the cytosolic presence of C3, given that C3 entry into the secretory pathway is so well demonstrated. We discovered that C3 cDNA contains several in-frame ATG codons just downstream of the encoded signal peptide, which, if used as non-canonical transcriptional start sites, would result in cytosolic translation of this protein. We tested this using cDNA constructs in which the canonical ATG start site was removed, resulting in efficient C3 protein production into the cytosol and a total lack of C3 secretion, showing that non-canonical translational start sites can be utilized to result in C3 and ATG16L1 being present in the same subcellular compartment. With C3 being such a highly abundant serum protein, we also tested whether its uptake from the extracellular space could rescue autophagy in C3 knockout cells, but this was not the case, demonstrating the importance of intrinsic expression of C3 within the cytosol, and highlighting the different roles of C3 in different cellular compartments.
C3 is already well-described as an extracellular opsonin, marking particles and pathogens for destruction by phagocytosis. Our results show that C3 also functions intracellularly, in the marking and destruction of intracellular material by autophagy. In the extracellular space, the activity of C3 is carefully controlled by cleavage; intact mature C3 is activated by cleavage into C3b, revealing the reactive thioester group by which it covalently attaches to targets, and C3b can be further cleaved and inactivated first into iC3b, then into C3dg, in well-defined and understood steps. The mechanism for intracellular C3 regulation now requires further investigation.
To start with, C3 expressed within the cytosol bypasses post-translational modifications that occur within the ER lumen, which includes cleavage of the pro-C3 polypeptide into separate beta and alpha chains, which remain connected by a disulfide bond, as well as N-linked glycosylation. Cytosolic C3 translated from non-canonical start sites appears unprocessed into separate chains. We found that ER-processed mature C3 has a much higher affinity for ATG16L1 than C3b or iC3b, suggesting that C3 cleavage could be a mechanism for regulation of the C3-ATG16L1 interaction. Our results show that in beta cells, C3 knockout allows autophagosome formation, but that maturation is inhibited, suggesting a 2-step process for the C3-ATG16L1 interaction and autolysosome maturation (Figure 1). The exact nature of this interaction and the factors that are involved in intracellular C3 cleavage are under current investigation. It has now also been shown that pathogens that invade into the cytosol, and carry with them C3 deposited onto their surface from the extracellular space, are similarly targeted for destruction by autophagy due to recruitment of ATG16L1, showing that C3 is important not only for homeostatic autophagy, but also for induced xenophagic destruction of microbes.
Figure 1.
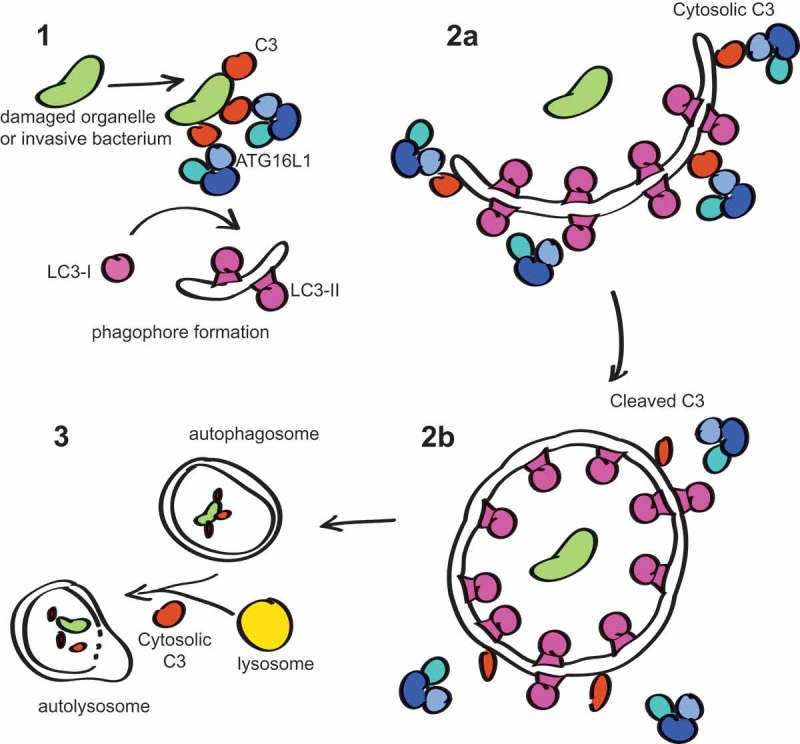
Proposed model of C3 involvement in autophagy: (1) Extracellular C3 opsonised onto invasive bacteria has been shown to aid initiation of xenophagy to combat infection. Similarly, C3 deposited onto damaged membranes or organelles could initiate their removal. (2a) Cytosolic C3 may be involved in recruitment of the ATG16L1 complex during extension of the phagophore membrane. (2b) C3 function is regulated by cleavage, leading to loss of affinity to ATG16L1, which may be required for dissociation of ATG16L1 from the completed autophagosome. ATG protein dissociation is required for their recycling and for autophagosome maturation; otherwise autophagy becomes stalled. (3) Cytosolic C3 may be involved in fusion of lysosomes with autophagosomes via interaction with additional factors. These models are not mutually exclusive.
Complement is evolutionarily ancient, having evolved far earlier than the antibodies to which its complementary activity lent its discovery and name. It has been recently suggested that C3 evolved initially as a defender of the intracellular space, and our findings fit this narrative, showing a cytoprotective role for a novel cytosolic form of the C3 protein. This is reflected by findings from Drosophila, which like many protostomes has lost the complement system, but which still expresses a complement-related protein, which covalently binds targets via a thioester bond, and which recently was found to regulate autophagy in macrophage-like cells. Given this apparently conserved function in distantly related species, it now remains to be investigated how far this function of C3 reaches in humans, beyond beta cells, and into other tissue types, and in which contexts or cell types C3 is a limiting factor for homeostatic or responsive autophagy.
Funding Statement
This work was supported by the Knut and Alice Wallenberg foundation.
Disclosure statement
No potential conflict of interest was reported by the authors.