

ABSTRACT
Recent studies suggest that defects in macroautophagy/autophagy contribute to the pathogenesis of systemic lupus erythamatosus (SLE), especially in adaptive immunity. The occurrence and progression of lupus nephritis (LN) is the end result of complex interactions between regulation of immune responses and pathological process by renal resident cells, but there is still a lot of missing information for establishing the role of autophagy in the pathogenesis of LN, and as a therapy target. In our recent study, we observed that autophagy is activated in LN, especially in podocytes. Based on in vitro assays, many of the most important mediators of the disease – patients’ sera, patients’ IgG and IFNA/IFN-α – can induce autophagy in both murine and human podocytes, by reactive oxygen species production or MTORC1 inhibition; autophagy activation negatively associates with podocyte injury. With regard to intervention, autophagy activators can protect against podocyte injury, whereas autophagy inhibitors aggravate injury. Taken together, our findings suggest that podocyte autophagy is involved in lupus renal protection and may be a therapeutic target. These data shed new light on the role of rapamycin and autophagy inducers in the treatment of SLE.
Abbreviations: ALB: albumin; ARHGDIB: Rho GDP dissociation inhibitor beta; APOL1: apolipoprotein L1; ATG5: autophagy related 5; ATG7: autophagy related 7; ATG16L2: autophagy related 16 like 2; BECN1: beclin 1; CDKN1B: cyclin dependent kinase inhibitor 1B; CLEC16A, C-type lectin domain containing 16A; CYBB: cytochrome b-245 beta chain; DC: dendritic cell; DRAM1: DNA damage regulated autophagy modulator 1; eQTL: expression quantitative trait loci; GWAS: genome-wide association study; IFNA: interferon alpha; IRGM: immunity related GTPase M; LRRK2: leucine rich repeat kinase 2; MAP1LC3B: microtubule associated protein 1 light chain 3 beta; MTMR3: myotubularin related protein 3; LAP” LC3-associated phagocytosis; LN: lupus nephritis; NOD: non-obese diabetic; NPHS2: NPHS2, podocin; PBMC: peripheral blood mononuclear cell; RUBCN: rubicon autophagy regulator; SLE: systemic lupus erythematosus.
KEYWORDS: Autophagy, lupus nephritis, MTORC1, podocyte, rapamycin
Autophagy is a highly conserved process that degrades certain intracellular contents in both physiological and pathological conditions [1–5]. The immunological roles of autophagy have become understood as well-established physiological functions in nearly all aspects of immunity [6–9].
Systemic lupus erythematosus (SLE) is a spectrum of autoimmune disease in which defects can occur at multiple points of the immune cascade, resulting in a striking heterogeneity of clinical presentations. The central pathology is the recognition of, and reaction to, self-antigens by the immune system, leading to type I interferon signaling and the production of pathogenic auto-antibodies, which mediate much of the disease [10–13].
In the past decade, the combined data from genome-wide association studies (GWAS) and inhibition assays have implicated autophagy in SLE. The direct association between autophagy and SLE was identified in hypothesis-free GWAS data. Several GWASs confirm the genetic associations between variants in/near ATG5 and SLE, in both Caucasians and Asians [14–16]. Further candidate gene/pathway analysis revealed that variants locating in additional autophagy related genes, including ATG7, IRGM, DRAM1, CDKN1B, APOL1, MTMR3, CLEC16A, LRRK2, ATG16L2, and MAP1LC3B are associated with susceptibility to SLE [14,17–25]. A small sub-phenotype analysis suggests risk alleles of ATG5 associated with anemia, the presence of anti-DNA autoantibodies and renal involvement [26]. In addition, APOL1 and MTMR3 alleles are more likely associated with renal injury in systemic lupus [22,24]. However, all the above reported associated variants are not located in coding regions. Some of the functional consequences of these polymorphisms are confirmed to be expression regulatory by cis-eQTL or trans-eQTL effects, causing a cytokine imbalance [14,17,20,21,27–30]. Our previous data also showed that PRDM1-ATG5 risk allelic variant is associated with increased ATG5 expression and IFNA signature genes in B cells [14]. Despite the fact that genetic replications and functional significance of the associated variants in disease pathogenesis are still limited, these data highly support the idea that autophagy is actively involved in the occurrence and/or progression of the disease.
Further cellular and model animal studies indicate that autophagy is upregulated in. various immune cells in SLE, including B cells, T cells and macrophages [29,31–44]. Activation of autophagy is observed in B cells, especially naïve B cells from both the lupus NZB/W mouse model and human patients [31]. Autophagy deficiency leads to impaired plasmablast differentiation, which may be a mechanism in auto-antibody production [31,34,45]. However, the combined loss of autophagy in both dendritic cells (DCs) and B cells in Tlr7 transgenic (lupus) mice leads to a lethal sepsis [37]. The autophagic marker LC3-II is also increased in lupus T cells; however, the precise activity and role of autophagy are still not well understood. Gros et al observed that both lupus-prone mice (MRLlpr/lpr and NZB/NZW F1) and human lupus patients have increased autophagic flux compared to normal controls, and the flux is also partially blocked in lupus T cells [38]. They hypothesized that deregulated autophagy could contribute to the survival of autoreactive T cell in lupus. However, Alessandri et al observed a blockade of the autophagic flux instead of activation. Factors present in the serum of SLE patients, probably antibodies like anti-ARHGDIB/D4GDI autoantibodies, could induce autophagy in T lymphocytes from healthy donors instead of T cells from patients with SLE [40,46]. Autophagy-resistant T cells from patients with SLE display an upregulation of genes negatively regulating autophagy and an increase of apoptosis [40], which may be partly caused by increased endoplasmic reticulum stress [39,41]. These data support the concept that T cell autophagy is deregulated in lupus, and there is an imbalance between autophagosome generation and their degradation. These two-way imbalances may be caused by differences in T cell subtypes, time period, activation state and stress severity. The difference in the types of injuries and severity of injuries may produce different outcome of autophagy, in that a certain degree of autophagic activity can maintain tissue homeostasis, whereas excessive autophagic activity results in cell death, especially in chronic inflammation and aging circumstances. Furthermore, it also possible that in some populations, such as memory cells, macroautophagy contributes to survival and chronic activation; in others such as naive cells, or Treg cells [47], the continuous generation of autophagosomes is not balanced by their degradation and leads to cell death [48,49]. Future investigations, for instance by using cell-specific and targeted autophagic gene knockout mice, are necessary to elucidate and clarify the precise functional role of autophagy in lupus. For example, Clec16a knockdown mice are protected from autoimmunity in the non-obese diabetic (NOD) model for type 1 diabetes. Loss of Clec16a diminishes the pathogenicity of NOD CD4+ T cells. The effects are not T cell intrinsic but are imparted during thymic selection owing to its role in thymic epithelial autophagy [25]. LC3-associated phagocytosis (LAP) is a novel form of non-canonical autophagy where LC3 is conjugated to phagosome membranes using a portion of the canonical autophagy machinery, which bridges the phagocytic and autophagic pathways. Core autophagy or LAP-specific genes (ATG5, ATG7, BECN1, RUBCN and CYBB/NOX2) deficiency in myeloid cells (i.e., macrophages, monocytes, and neutrophils) lead to a decreased capacity to clear dying cells, and the establishment of a lupus-like autoimmune disease in mice [29]. Mice display reduced body weight, accumulation of circulating lymphocytes, activated CD8+ T cells, increased spleen proliferation, development of autoantibodies (including anti-double-stranded DNA and nuclear antibodies), increased expression of Ifna signature genes, and, importantly, signs of kidney damage (Figure 1) [29]. Thus, LAP is engaged in the protective mechanism against SLE. As can be concluded from in vitro assays, autophagy is activated and deregulated in various immune cells involved in both innate immune and adaptive immune responses in SLE. In autophagy gene knockout mouse models, loss of autophagy often results in a markedly increased susceptibility to lupus-like autoimmunity [25,29,34,37]. In a more recent trial of rapamycin (sirolimus, the best-known trigger for autophagy, which acts by inhibiting MTOR [mechanistic target of rapamycin kinase]) in patients with active and resistant SLE [50,51], it was observed that a progressive improvement in disease activity is associated with expansion of CD4+ IL2RA/CD25+ FOXP3+ regulatory T cells and CD8+ memory T-cell populations. Thus, rapamycin, which has been approved by the US Food and Drug Administration for prevention of organ transplant rejection, could be a novel and attractive therapeutic option for patients with SLE. Accordingly, better understanding the mechanisms of autophagy surely will be also of high interest.
Figure 1.
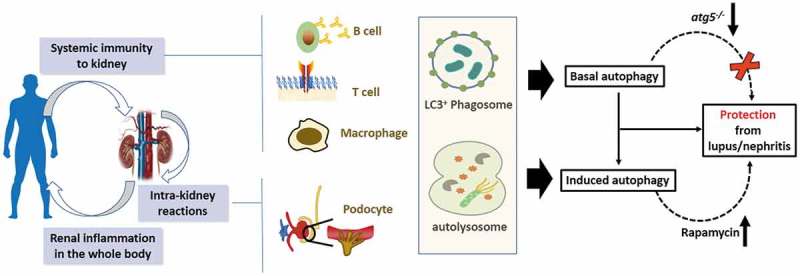
Friend or foe? Is there a protective role of autophagy in SLE/nephritis?. Lupus nephritis (LN) is one of the most serious manifestations of SLE and is an important predictor of poor outcome. The occurrence and progression of LN is the end result of complex interactions between the regulation of immune responses and pathological processes involving renal resident cells. More recent data from Atg5 gene deletion mouse models (B cell, and myeloid cells) support a protective role of autophagy and LAP in SLE. The deletion of core autophagy or LAP-specific genes results in signs of kidney damage. In our recent paper, we reported that podocyte autophagy can protect against renal injury in lupus and might be a therapeutic target. Based on in vivo models and in vitro assays, it seems that autophagy is a renoprotective factor in the development and progression of lupus nephritis.
Lupus nephritis (LN) is one of the most serious manifestations of SLE and is an important predictor of poor outcome. Occurrence and progression of LN is the end result of complex interactions between regulation of immune responses and pathological process involving renal resident cells. Similar to other diseases, autophagy is also implicated in the pathological process of renal diseases, in which autophagy interfaces with most cellular stress-response pathways, including those involved in controlling immune response and inflammation [52–55]. At the current stage, the precise role of autophagy in renal injury response and the pathogenesis of kidney disease is not well understood. There have been studies that provide evidence to support a cytoprotective role of autophagy, and others that support deleterious effects of autophagy. Likewise, rapamycin shows beneficial effects in lupus nephritis [56–60], but no controlled studies have been reported in human patients. Thus, there is still a lot of missing information necessary for a full establishment on the role of autophagy in disease pathogenesis and as a therapy target.
Thus, we aimed to reveal the role and function of autophagy in kidney pathology in LN, checking whether modulating autophagic activity in certain renal resident cells may be therapeutically beneficial [61].
We first confirmed that autophagy is involved in pathogenesis of lupus nephritis and is activated. In lupus-prone MRLlpr/lpr.mice, we observed that the levels of autophagy vary in different immune organ homogenates of MRLlpr/lpr compared with C57BL mice. In kidney tissues, the levels of autophagy are significantly elevated in MRLlpr/lpr mice with renal injury compared with MRLlpr/lpr mice without renal injury and C57BL control mice. In addition, in LN patients, we observed that the expression of autophagy-related genes is increased in PBMC and urinary sediments. These observations reinforce the notion that both aberrant systemic immunity and intrarenal immunopathology contribute to lupus nephritis, in which autophagy may exert an important effect. However, as PBMC and urinary sediments are all mixtures of different cell types, they may be not optimal specimens, but they both shed light on autophagy upregulation, which is also consistent with all the previous reports in SLE. To understand the precise role of autophagy activation in renal injury response and the pathogenesis of LN, we further checked relations between autophagy induction and podocyte injury, including apoptosis, NPHS2/podocin derangement, ALB (albumin) filtration, and wound healing. We observed that many of the most important mediators of the disease – patients’ sera, patients’ IgG and IFNA – can induce autophagy in both murine and human podocytes. In addition, autophagy activation negatively associates with podocyte injury, which is responsible for proteinuria and progression of glomerular diseases. Patients’ IgG regulates the generation of reactive oxygen species and podocyte apoptosis whereas, IFNA treatment links MTORC1 inhibition and the filtration barrier function of podocytes [61]. We only observed autophagosomes in the podocytes, but not in other glomerular-resident cells (such as mesangial cells or endothelial cells), suggesting the regulation and function of autophagy is likely cell-type and context specific. In interventional studies, we observed that an autophagy activator can protect against podocyte injury, whereas an autophagy inhibitor aggravates podocyte injury (Figure 1).
We systematically checked autophagy in both lupus-prone mice and human patients, confirming the involvement of autophagy in LN. We found that the exact affect that autophagy might exert in renal disease is multifaceted and complex: depending on different stimulations, types of disease or cells. We thus further examined multiple mediators of the disease, checking their effect in 2 different podocyte cell lines, and by combining different autophagy detection system, different types of injuries and different drug interventions. Combining assays for multiple confirmation, we concluded that increased autophagy is cytoprotective against podocyte injury induced by antibody and IFNA in lupus nephritis.
As autoantibodies and IFNA play an important role in a variety of cell populations, autophagy may also be relevant as a biomarker in translational and therapeutic studies in several immune-related diseases. New insights into the molecular mechanisms of podocyte autophagy in LN may shed light on promising therapeutic strategies.
Funding Statement
Support was provided by the Training Program of the Major Research Plan of the National Natural Science Foundation of China [91642120]; National Science Foundation of China [Grant 81570629]; National Key Research and Development Program of China [2016YFC0904102]; Beijing Nova Program [Z171100001117023]; Beijing Youth Top-notch Talent Support Program [2017000021223ZK31]; Natural Science Foundation for Innovation Research Group of China (81621092); Beijing Natural Science Foundation [7152148]; the University of Michigan Health System–Peking University Health Science Center Joint Institute for Translational and Clinical Research [BMU2017JI007]; Chinese Society of Nephrology [15020030591]; and NIH grant GM053396;the University of Michigan Health System–Peking University Health Science Center Joint Institute for Translational and Clinical Research [BMU2017JI007].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Kroemer G. Autophagy: a druggable process that is deregulated in aging and human disease. J Clin Invest. 2015;125(1):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. [DOI] [PubMed] [Google Scholar]
- [3].Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. Embo J. 2017;36(13):1811–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20(3):233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Takahama M, Akira S, Saitoh T. Autophagy limits activation of the inflammasomes. Immunol Rev. 2018;281(1):62–73. [DOI] [PubMed] [Google Scholar]
- [7].Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016;16(11):661–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Netea-Maier RT, Plantinga TS, van de Veerdonk FL, et al. Modulation of inflammation by autophagy: consequences for human disease. Autophagy. 2016;12(2):245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhou XJ, Zhang H. Autophagy in immunity: implications in etiology of autoimmune/autoinflammatory diseases. Autophagy. 2012;8(9):1286–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Muskardin T, Niewold TB. Type I interferon in rheumatic diseases. Nat Rev Rheumatol. 2018;14(4):214–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Deng Y, Tsao BP. Updates in lupus genetics. Curr Rheumatol Rep. 2017;19(11):68. [DOI] [PubMed] [Google Scholar]
- [12].Kaul A, Gordon C, Crow MK, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2:16039. [DOI] [PubMed] [Google Scholar]
- [13].Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–2121. [DOI] [PubMed] [Google Scholar]
- [14].Zhou XJ, Lu XL, Lv JC, et al. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis. 2011;70(7):1330–1337. [DOI] [PubMed] [Google Scholar]
- [15].Gateva V, Jk S, Hom G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41(11):1228–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Han JW, Zheng HF, Cui Y, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41(11):1234–1237. [DOI] [PubMed] [Google Scholar]
- [17].Qi YY, Zhou XJ, Nath SK, et al. A rare variant (rs933717) at FBXO31-MAP1LC3B in Chinese is associated with systemic lupus erythematosus. Arthritis Rheumatol. 2018;70(2):287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Molineros JE, Yang W, Zhou XJ, et al. Confirmation of five novel susceptibility loci for systemic lupus erythematosus (SLE) and integrated network analysis of 82 SLE susceptibility loci. Hum Mol Genet. 2017;26(6):1205–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang YM, Zhou XJ, Cheng FJ, et al. Autophagy-related gene LRRK2 is likely a susceptibility gene for systemic lupus erythematosus in northern Han Chinese. Oncotarget. 2017;8(8):13754–13761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhang YM, Cheng FJ, Zhou XJ, et al. Rare variants of ATG5 are likely to be associated with chinese patients with systemic lupus erythematosus. Medicine (Baltimore). 2015;94(22):e939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang YM, Cheng FJ, Zhou XJ, et al. Detecting genetic associations between ATG5 and lupus nephritis by trans-eQTL. J Immunol Res. 2015;2015:153132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhou XJ, Nath SK, Qi YY, et al. Brief report: identification of MTMR3 as a novel susceptibility gene for lupus nephritis in northern Han Chinese by shared-gene analysis with IgA nephropathy. Arthritis Rheumatol. 2014;66(10):2842–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yang W, Tang H, Zhang Y, et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. Am J Hum Genet. 2013;92(1):41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Freedman BI, Langefeld CD, Andringa KK, et al. End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol. 2014;66(2):390–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schuster C, Gerold KD, Schober K, et al. The autoimmunity-associated gene CLEC16A modulates thymic epithelial cell autophagy and alters T cell selection. Immunity. 2015;42(5):942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ciccacci C, Perricone C, Alessandri C, et al. Evaluation of ATG5 polymorphisms in Italian patients with systemic lupus erythematosus: contribution to disease susceptibility and clinical phenotypes. Lupus. 2018;27(9):1464–1469. [DOI] [PubMed] [Google Scholar]
- [27].Zhou M, Xu W, Wang J, et al. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-kappaB pathway quenches intestinal inflammation and oxidative stress injury. EBioMedicine. 2018;35:345–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lee JP, Foote A, Fan H, et al. Loss of autophagy enhances MIF/macrophage migration inhibitory factor release by macrophages. Autophagy. 2016;12(6):907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Martinez J, Cunha LD, Park S, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533(7601):115–119. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [30].Lopez P, Alonso-Perez E, Rodriguez-Carrio J, et al. Influence of Atg5 mutation in SLE depends on functional IL-10 genotype. PLoS One. 2013;8(10):e78756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Clarke AJ, Ellinghaus U, Cortini A, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. 2015;74(5):912–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Page N, Gros F, Schall N, et al. HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann Rheum Dis. 2011;70(5):837–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wilhelm M, Wang F, Schall N, et al. Lupus regulator peptide P140 represses B cell differentiation by reducing HLA class II molecule overexpression. Arthritis Rheumatol. 2018;70(7):1077–1088. [DOI] [PubMed] [Google Scholar]
- [34].Weindel CG, Richey LJ, Bolland S, et al. B cell autophagy mediates TLR7-dependent autoimmunity and inflammation. Autophagy. 2015;11(7):1010–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Page N, Gros F, Schall N, et al. A therapeutic peptide in lupus alters autophagic processes and stability of MHCII molecules in MRL/lpr B cells. Autophagy. 2011;7(5):539–540. [DOI] [PubMed] [Google Scholar]
- [36].Dang J, Bian X, Ma X, et al. ORMDL3 facilitates the survival of splenic B cells via an ATF6alpha-endoplasmic reticulum stress-beclin1 autophagy regulatory pathway. J Immunol. 2017;199(5):1647–1659. [DOI] [PubMed] [Google Scholar]
- [37].Weindel CG, Richey LJ, Mehta AJ, et al. Autophagy in dendritic cells and B cells is critical for the inflammatory state of TLR7-mediated autoimmunity. J Immunol. 2017;198(3):1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gros F, Arnold J, Page N, et al. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy. 2012;8(7):1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Colasanti T, Vomero M, Alessandri C, et al. Role of alpha-synuclein in autophagy modulation of primary human T lymphocytes. Cell Death Dis. 2014;5:e1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Alessandri C, Barbati C, Vacirca D, et al. T lymphocytes from patients with systemic lupus erythematosus are resistant to induction of autophagy. FASEB J. 2012;26(11):4722–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lee WS, Sung MS, Lee EG, et al. A pathogenic role for ER stress-induced autophagy and ER chaperone GRP78/BiP in T lymphocyte systemic lupus erythematosus. J Leukoc Biol. 2015;97(2):425–433. [DOI] [PubMed] [Google Scholar]
- [42].Li X, Liu F, Zhang X, et al. Notch-Hes-1 axis controls TLR7-mediated autophagic death of macrophage via induction of P62 in mice with lupus. Cell Death Dis. 2016;7(8):e2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li B, Yue Y, Dong C, et al. Blockade of macrophage autophagy ameliorates activated lymphocytes-derived DNA induced murine lupus possibly via inhibition of proinflammatory cytokine production. Clin Exp Rheumatol. 2014;32(5):705–714. [PubMed] [Google Scholar]
- [44].Majai G, Kiss E, Tarr T, et al. Decreased apopto-phagocytic gene expression in the macrophages of systemic lupus erythematosus patients. Lupus. 2014;23(2):133–145. [DOI] [PubMed] [Google Scholar]
- [45].Arbogast F, Arnold J, Hammann P, et al. ATG5 is required for B cell polarization and presentation of particulate antigens. Autophagy. 2018. September 22:1–15. Epub ahead of print DOI: 10.1080/15548627.2018.1516327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Barbati C, Alessandri C, Vomero M, et al. Autoantibodies specific to D4GDI modulate Rho GTPase mediated cytoskeleton remodeling and induce autophagy in T lymphocytes. J Autoimmun. 2015;58:78–89. [DOI] [PubMed] [Google Scholar]
- [47].Kato H, Perl A. Blockade of treg cell differentiation and function by the interleukin-21-mechanistic target of rapamycin axis via suppression of autophagy in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2018;70(3):427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Arbogast F, Gros F. Lymphocyte autophagy in homeostasis, activation, and inflammatory diseases. Front Immunol. 2018;9:1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Vomero M, Barbati C, Colasanti T, et al. Autophagy and rheumatoid arthritis: current knowledges and future perspectives. Front Immunol. 2018;9:1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fernandez DR, Crow MK. CD8 T cells and mTOR: new concepts and targets for systemic lupus erythematosus. Lancet. 2018;391(10126):1126–1127. [DOI] [PubMed] [Google Scholar]
- [51].Lai ZW, Kelly R, Winans T, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet. 2018;391(10126):1186–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lenoir O, Tharaux PL, Huber TB. Autophagy in kidney disease and aging: lessons from rodent models. Kidney Int. 2016;90(5):950–964. [DOI] [PubMed] [Google Scholar]
- [53].Kimura T, Isaka Y, Yoshimori T. Autophagy and kidney inflammation. Autophagy. 2017;13(6):997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fougeray S, Pallet N. Mechanisms and biological functions of autophagy in diseased and ageing kidneys. Nat Rev Nephrol. 2015;11(1):34–45. [DOI] [PubMed] [Google Scholar]
- [55].Wang Z, Choi ME. Autophagy in kidney health and disease. Antioxid Redox Signal. 2014;20(3):519–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Reddy PS, Legault HM, Sypek JP, et al. Mapping similarities in mTOR pathway perturbations in mouse lupus nephritis models and human lupus nephritis. Arthritis Res Ther. 2008;10(6):R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chan TM. Treatment of severe lupus nephritis: the new horizon. Nat Rev Nephrol. 2015;11(1):46–61. [DOI] [PubMed] [Google Scholar]
- [58].Stylianou K, Petrakis I, Mavroeidi V, et al. The PI3K/Akt/mTOR pathway is activated in murine lupus nephritis and downregulated by rapamycin. Nephrol Dial Transplant. 2011;26(2):498–508. [DOI] [PubMed] [Google Scholar]
- [59].Lui SL, Tsang R, Chan KW, et al. Rapamycin attenuates the severity of established nephritis in lupus-prone NZB/W F1 mice. Nephrol Dial Transplant. 2008;23(9):2768–2776. [DOI] [PubMed] [Google Scholar]
- [60].Gu Z, Tan W, Ji J, et al. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging (Albany NY). 2016;8(5):1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Qi YY, Zhou XJ, Cheng FJ, et al. Increased autophagy is cytoprotective against podocyte injury induced by antibody and interferon-α in lupus nephritis. Ann Rheum Dis. 2018;77(12):1799–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]