

ABSTRACT
Differentiated tissue is particularly vulnerable to alterations in protein and organelle homeostasis. The essential protein VCP, mutated in hereditary inclusion body myopathy, amyotrophic lateral sclerosis and frontotemporal dementia, is critical for efficient clearance of misfolded proteins and damaged organelles in dividing cells, but its role in terminally differentiated tissue affected by disease mutations is less clear. To understand the relevance of VCP in differentiated tissue, we inactivated it in skeletal muscle of adult mice. Surprisingly, knockout muscle demonstrated a necrotic myopathy with increased macroautophagic/autophagic proteins and damaged lysosomes. This was not solely due to a defect in autophagic degradation because age-matched mice with muscle inactivation of the autophagy essential protein, ATG5, did not demonstrate a myopathy. Notably, myofiber necrosis was preceded by upregulation of LGALS3/Galectin-3, a marker of damaged lysosomes, and TFEB activation, suggesting early defects in the lysosomal system. Consistent with that, myofiber necrosis was recapitulated by chemical induction of lysosomal membrane permeabilization (LMP) in skeletal muscle. Moreover, TFEB was activated after LMP in cells, but activation and nuclear localization of TFEB persisted upon VCP inactivation or disease mutant expression. Our data identifies VCP as central mediator of both lysosomal clearance and biogenesis in skeletal muscle.
Abbreviations: AAA: ATPases Associated with diverse cellular Activities; TUBA1A/α-tubulin: tubulin alpha 1a; ATG5: autophagy related 5; ATG7: autophagy related 7; ACTA1: actin alpha 1, skeletal muscle; CLEAR: coordinated lysosomal expression and regulation; CTSB/D: cathepsin B/D; Ctrl: control; DAPI: diamidino-2-phenylindole; EBSS: Earle’s balanced salt solution; ELDR: endolysosomal damage response; ESCRT: endosomal sorting complexes required for transport; Gastroc/G: gastrocnemius; H&E: hematoxylin and eosin; HSPA5/GRP78: heat shock protein family A (Hsp70) member 5; IBMPFD/ALS: inclusion body myopathy associated with Paget disease of the bone, frontotemporal dementia and amyotrophic lateral sclerosis; i.p.: intraperitoneal; LAMP1/2: lysosomal-associated membrane protein 1/2; LLOMe: Leu-Leu methyl ester hydrobromide; LGALS3/Gal3: galectin 3; LMP: lysosomal membrane permeabilization; MTOR: mechanistic target of rapamycin kinase; MYL1: myosin light chain 1; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MSP: multisystem proteinopathy; PBS: phosphate-buffered saline; PCR: polymerase chain reaction; Quad/Q: quadriceps; RHEB: Ras homolog, mTORC1 binding; SQSTM1: sequestosome 1; TFEB: transcription factor EB; TA: tibialis anterior; siRNA: small interfering RNA; SQSTM1/p62, sequestosome 1; TARDBP/TDP-43: TAR DNA binding protein; TBS: Tris-buffered saline; TXFN, tamoxifen; UBXN6/UBXD1: UBX domain protein 6; VCP: valosin containing protein; WT: wild-type.
KEYWORDS: Autophagy, lysosome, myopathy, skeletal muscle, TFEB, VCP
Introduction
VCP (valosin containing protein) is an essential and abundant AAA+ protein, which is best known for its role in facilitating ubiquitin-mediated protein degradation to ensure protein homeostasis, but also to regulate critical signaling pathways including cell cycle regulation [1]. This activity has made it an attractive therapeutic target in cancer chemotherapeutics since these therapies preferentially affect rapidly dividing cells with a high protein synthetic rate [2]. Consistent with this, germline knockout of VCP is embryonic lethal with lethality occurring prior to embryonic day 4.5 [3]. However, the role of VCP in non-rapidly dividing or differentiated tissues is less clear.
Dominantly inherited mutations in VCP lead to a late onset progressive degenerative disease affecting muscle, brain and bone [4]. This disorder is termed inclusion body myopathy associated with Paget disease of the bone, frontotemporal dementia, and amyotrophic lateral sclerosis (IBMPFD/ALS) and is more recently defined by the nomenclature, multisystem proteinopathy (MSP) as a means of including other degenerative phenotypes such as parkinsonism and peripheral neuropathy often associated with VCP disease mutations [5]. Although affecting disparate tissues, MSP pathology is unified by cellular degeneration and ubiquitinated inclusions in pathologic post-mitotic and terminally differentiated tissue [6]. While the phenotypic penetrance of MSP can vary from patient to patient, 90% will develop muscle weakness implying an important role for VCP in skeletal muscle [7]. Muscle from patients with VCP-associated myopathy accumulate ubiquitinated inclusions, autophagic debris and endolysosomal remnants [8–11].
VCP plays an important role in protein homeostasis. Knockdown of VCP or expression of a dominant negative VCP protein in differentiated skeletal muscle inhibited proteasomal and autophagic protein degradation [12]. Under conditions known to produce muscle atrophy, knockdown of VCP increased the levels of ubiquitinated myofibrillar proteins and blocked muscle atrophy [12]. Similarly under non-atrophic conditions, VCP knockdown increases myofiber diameter suggesting that VCP is responsible for maintaining myofiber integrity [12].
VCP also participates in organelle homeostasis and is essential for both mitochondrial and lysosomal dynamics [1]. VCP knockdown in drosophila indirect flight muscle leads to disruptions in mitochondrial fission and fusion via impaired degradation of mitofusin resulting in elongated mitochondria and myodegeneration [13]. Others have found that VCP knockdown in drosophila larval muscle leads to the collapse of a tubular lysosome network within myofibers [14]. This knockdown also led to impaired autophago-lysosomal degradation with the accumulation of ubiquitinated inclusions and damaged mitochondria [14]. These data suggest that VCP’s role in organelle homeostasis is particularly important in differentiated tissue.
Most studies exploring the role of VCP in skeletal muscle have focused on the pathomechanism of VCP disease mutations. >40 different mutations have been identified as causing disease in patients with VCP mutations [7]. The pathological features of autophagic debris and ubiquitinated inclusions has supported the findings that VCP disease mutations affect lysosomal degradation. Disease mutations cluster structurally at the N-D1 domain interface. Indeed, VCP disease mutants have compromised interaction with the specific VCP adaptor UBXN6/UBXD1 that binds at that interface, resulting in a loss of UBXN6 dependent functions [9,10,15,16]. Specifically, a VCP-UBXN6 complex is needed for the sorting of CAV (caveolin)-positive late endosomes [10]. VCP disease mutations lead to enlarged CAV- and ubiquitin-positive endosomes that are delocalized from the sarcolemma in patient muscle. More recently, we identified a VCP-UBXN6-dependent role in the endolysosomal damage response (ELDR) pathway and the clearance of damaged lysosomes via autophagy [9]. VCP and UBXN6 are recruited to damaged lysosomes that are marked with the sensor LGALS3/Galectin-3 and ubiquitin [17]. Loss of VCP, UBXN6 or expression of VCP disease mutations leads to accumulation of LGALS3-positive late endosomes that failed to be cleared via autophagy [9].
VCP’s pleiotropy in cellular functions has made it challenging to dissect its predominant role in specific cell types. It is conceivable that VCP may participate in unique or more specific cellular processes in distinct tissue types. For example tissue specific knockout of essential autophagic proteins such as ATG5 and ATG7 led to the unexpected observation that different mammalian tissues (e.g. skeletal muscle and neurons) have unique requirements for autophagy [18–20]. We have utilized a similar approach and performed targeted deletion of VCP in differentiated skeletal muscle. Our study offers a unique opportunity to understand the role of VCP in terminally differentiated tissue.
Results
Muscle-specific inactivation of VCP leads to a necrotic myopathy with autophagic pathology
To investigate the role of VCP in differentiated skeletal muscle, we developed a conditional VCP gene inactivation strategy utilizing the Cre-LoxP system. Vcpfl/fl mice were generated by inserting LoxP sites within intronic regions of the murine Vcp gene (Figure 1(a,b)). Vcpfl/fl mice were then crossed to Myl1p-Cre mice that express Cre recombinase under a Myl1p (myosin, light polypeptide 1) promoter leading to expression in differentiated skeletal muscle and are hereafter referred to as Myl1p-cre-vcp-/-. VCP deletion in Myl1p-cre-vcp-/- skeletal muscle was confirmed by immunoblot in 6 wk old mice (Figure 1(c)). Myl1p-cre-vcp-/- weighed less, developed progressive weakness, and had decreased mobility requiring euthanasia after 6 months of age as compared with littermate controls (Figure 1(d,g)). Histochemical staining of control or Myl1p-cre-vcp-/- mouse muscle at 6 and 9 wk demonstrated myopathic features with variation in fiber size and disorganized internal architecture associated with prominent degeneration and regeneration (Figure 2(a) and Figure S1A). Degenerating necrotic fibers were apparent with acid phosphatase staining as were dystrophic changes as evidenced by an increase in endomysial and perimysial connective tissue (Figure S1B). The degeneration in Myl1p-cre-vcp-/- was not solely due to a loss of autophagic function, since skeletal muscle pathology at 9 wk in Atg5fl/fl:Myl1p-Cre mice (Myl1p-cre-atg5-/-) was less pronounced (Figure 2(a) and Figure S1A). Muscle degeneration in Myl1p-cre-vcp-/- at 6 wk correlated with an increase in the autophagic markers SQSTM1 and LC3-I/II, ER stress protein HSPA5/GRP78 and high molecular weight ubiquitin conjugates (Figure 2(b,d)). Notably the ratio of LC3-II:LC3-I was not significantly changed in Myl1p-cre-vcp-/- mouse muscle (Figure 2(d,e)). Myl1p-cre-atg5-/- mouse muscle similarly accumulated SQSTM1, HSPA5, and ubiquitin conjugates but as expected with ATG5 inactivation, only LC3-I was present (Figure 2(c)). Quantitative PCR demonstrated a significant increase in the expression of autophagic and lysosomal proteins in 9-week-old Myl1p-cre-vcp-/- mouse muscle as compared with controls (Figure 2(f,g)).
Figure 1.
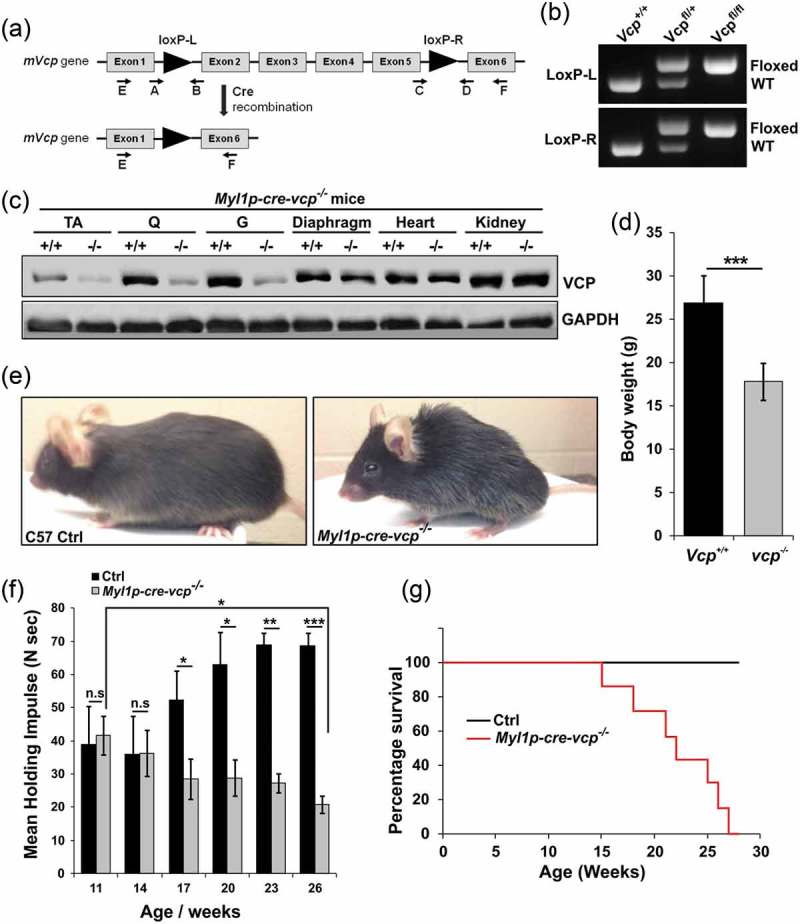
Muscle-specific inactivation of VCP leads to weakness. (a) VCP inactivation strategy. (b) PCR genotyping confirms germline transmission and homozygosity of double floxed allele. (c) Immunoblot of lysates from tibialis anterior (TA), quadriceps (Q), gastrocnemius (G), diaphragm muscle, heart and kidney of 6 wk old control (+/+) or Myl1p-cre-vcp-/- (-/-) mice using antibodies to VCP or GAPDH. (d) Average body weight of 23-week-old control (+/+) or Myl1p-cre-vcp-/- mice. N = 6 mice per group. (e) Image of C57 control and Myl1p-cre-vcp-/- mice at 24 wk of age. (f) Quantification of mean holding impulse for control or Myl1p-cre-vcp-/- mice from age 11–26 wk. N = 6 mice per group. (g) Kaplan-Meier survival curve of control and Myl1p-cre-vcp-/- mice. N = 7 per group. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant.
Figure 2.
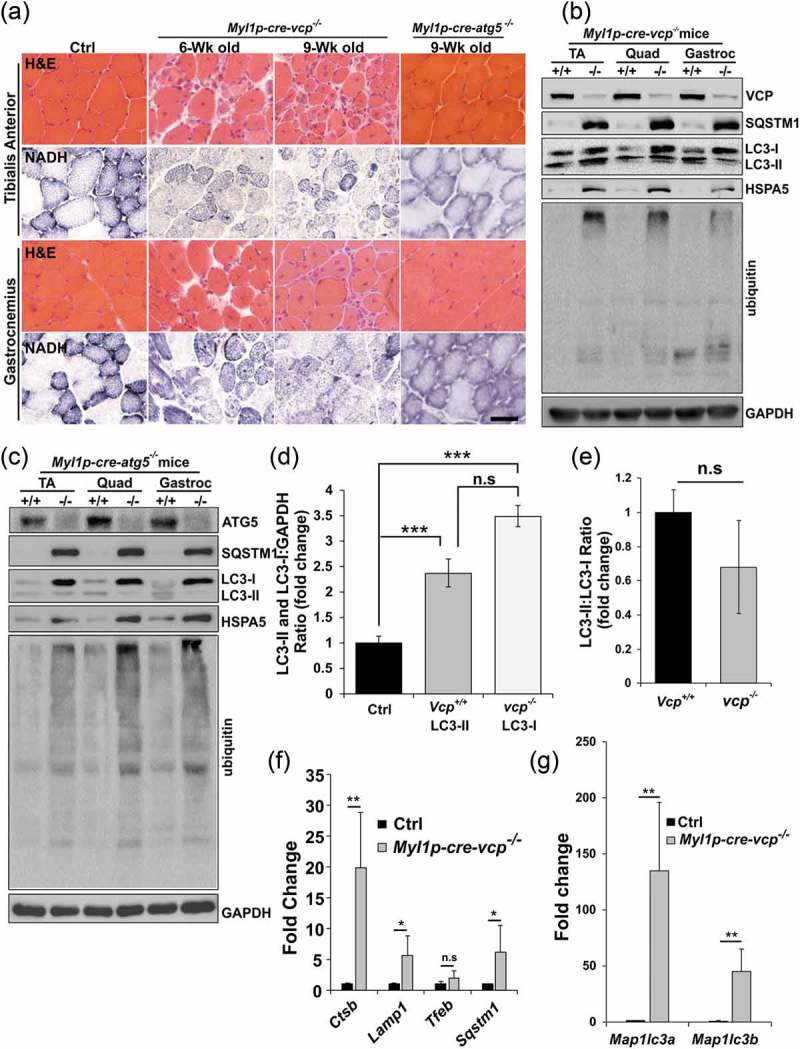
Muscle-specific inactivation of VCP leads to a myopathy. (a) Hematoxylin and eosin (H & E) or NADH staining of tibialis anterior and gastrocnemius from 6-week-old control, 6- and 9-week-old Myl1p-cre-vcp-/- or 9-week-old Myl1p-cre-atg5-/- mice. (b) Immunoblot of lysates from TA, quadriceps, or gastrocnemius of 6-week-old control (+/+) or Myl1p-cre-vcp-/- (-/-) mice using antibodies to VCP, SQSTM1, LC3, HSPA5, ubiquitin or GAPDH. (c) Immunoblot of lysates from TA, quadriceps or gastrocnemius muscle of 9-week-old control or Myl1p-cre-atg5-/- mice using antibodies to ATG5, SQSTM1, LC3, HSPA5, ubiquitin or GAPDH. (d) Densitometric analysis of LC3-I and LC3-II levels in control and Myl1p-cre-vcp-/- mouse muscle. N = 3 mice per group. Quantification was performed using western blots of TA’s, quadriceps, and gastrocnemius from each mouse. Comparison between groups was performed using a paired Student t-test. (e) Densitometric ratios of LC3-II:LC3-I levels in control and Myl1p-cre-vcp-/- mouse muscle. N = 3 mice per group. Quantification was performed using western blots of TA’s, quadriceps, and gastrocnemius from each mouse. Comparison between groups was performed using a paired Student t-test. (f) Graph of fold expression change of mRNA of lysosomal proteins from the TA muscle from 9-week-old control or Myl1p-cre-vcp-/- mice. (g) Graph of fold expression change of mRNA of Map1lc3a/b from the TA muscle from 9-week-old control or Myl1p-cre-vcp-/- mice. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant. Scale: 100 μm.
To more carefully define the time course of myofiber degeneration and necrosis in the setting of VCP inactivation, we crossed Vcpfl/fl mice with ACTA1p-cre/Esr1 mice that express a tamoxifen-dependent Cre recombinase under a human skeletal ACTA1 promoter (ACTA1p-cre/Esr1-vcp-/-). Treatment for 5 d with daily intraperitoneal injection (i.p.) of tamoxifen in adult ACTA1p-cre/Esr1-vcp-/- mice led to a significant reduction of VCP protein after 2 and 4 wk that returned to normal levels after 6 wk suggesting myofiber regeneration (Figure 3(a,b)). Notably, ACTA1p-cre/Esr1-vcp-/- mice express Cre recombinase only in the presence of tamoxifen and any newly formed or regenerated myofibers will express normal levels of VCP protein. Histochemical staining of control mice or ACTA1p-cre/Esr1-vcp-/- mice after 2, 4 and 6 wk post tamoxifen injection demonstrated prominent degeneration and myofiber necrosis at 4 wk with normal appearing muscle at two weeks and regenerated fibers at 6 wk as suggested by the return of VCP expression at that time point (Figure 3(c) and Figure S1C). Similar to Myl1p-cre-vcp-/- muscle, there was an increase in SQSTM1, LC3, HSPA5 and high molecular weight ubiquitin conjugates at 4 wk post tamoxifen treatment correlating with myodegeneration (Figure 3(a,b)). In addition, the increase in SQSTM1 and ubiquitin conjugates returned to normal levels after 6 wk and notably was only mildly increased at two weeks when VCP protein levels were significantly diminished (Figure 3(a,b)). Similarly, quantitative PCR for autophagic and lysosomal proteins demonstrated a dramatic increase at 4 wk that normalized by 6 wk (Figure 3(d,e)). For SQSTM1, an autophagic substrate, a subtle increase in SQSTM1 protein levels was found at 2 wk preceding the transcriptional increase at 4 wk (Figure 3(a,d)). Notably, there was a statistically significant increase in MAP1LC3A at one and two weeks suggesting that this may be an early feature (Figure 3(d)).
Figure 3.
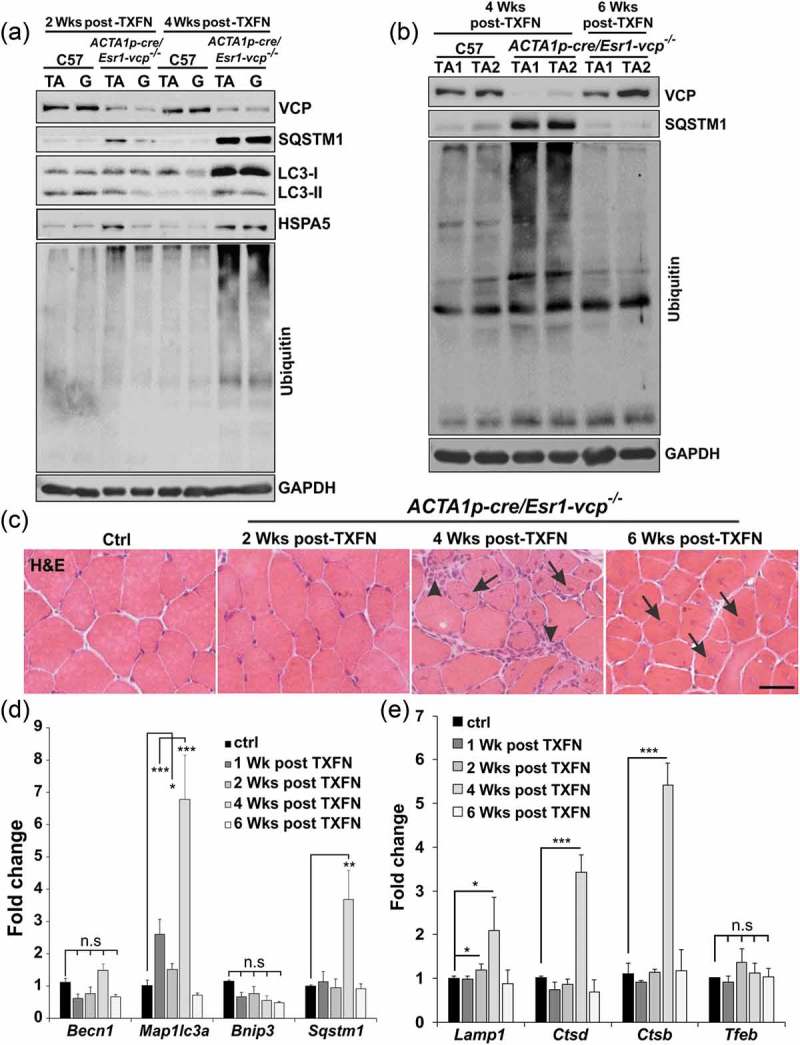
VCP is necessary for differentiated skeletal muscle survival. (a) Immunoblot of lysates from tibialis anterior (TA) or gastrocnemius (G) muscle of age-matched control (C57) or ACTA1p-cre/Esr1-vcp-/- mice after 2 and 4 wk post tamoxifen (TXFN) injection for 5 d using antibodies to VCP, SQSTM1, LC3, HSPA5, ubiquitin or GAPDH. (b) Immunoblot of lysates from TA of 2 age-matched control or 2 ACTA1p-cre/Esr1-vcp-/- mice after 4 and 6 wk post tamoxifen injection for 5 d using antibodies to VCP, SQSTM1, ubiquitin or GAPDH. (c) H & E of TA muscle from age-matched control or ACTA1p-cre/Esr1-vcp-/- mice after 2, 4 and 6 wk post tamoxifen injection for 5 d. Arrows point to regenerating fibers and arrowheads point to necrotic fibers. (d) Graph of fold expression change of mRNA of autophagic proteins from the TA muscle from control or ACTA1p-cre/Esr1-vcp-/- mice after 1, 2, 4 and 6 wk post tamoxifen injection for 5 d. (e) Graph of fold expression change of mRNA of lysosomal proteins from the TA muscle from control or ACTA1p-cre/Esr1-vcp-/- mice after 1, 2, 4 and 6 wk post tamoxifen injection for 5 d. *p < 0.05; **p < 0.01; ***p < 0.001; Scale: 100 μm.
It has been suggested that loss of VCP activity in myofibers leads to muscle hypertrophy after 7 d inhibition due to a decrease in the degradation of myofibrillar protein components [12]. We quantified the cross-sectional area of myofibers from control or ACTA1p-cre/Esr1-vcp-/- mice treated with 5 d tamoxifen after 1, 2, 4 or 6 wk. In agreement with the prior study, there was a shift of myofiber cross sectional area toward larger fibers at 1 wk. This shift however moved toward smaller fibers at 4 wk (Figure S2A). Moreover, there was no consistent increase in myofibrillar protein in skeletal muscle from ACTA1p-cre/Esr1-vcp-/- mice four weeks after tamoxifen treatment or in 6 wk old Myl1p-cre-vcp-/- mice (Figure S2B-C).
vcp knockout muscle accumulates damaged endolysosomes
To further characterize autophagic and endolysosomal dysfunction in Myl1p-cre-vcp-/- skeletal muscle, we performed immunohistochemistry for LC3 and SQSTM1. As expected there was an increase in LC3 and SQSTM1 puncta that occasionally co-localized in myopathic muscle (Figure 4(a)). The increase in LC3 puncta was similarly seen in necrotic and non-necrotic fibers from ACTA1p-cre/Esr1-vcp-/- mouse muscle four weeks after tamoxifen treatment (Figure 4(b)). Electron microscopy from tibialis anterior muscle of Myl1p-cre-vcp-/- mice identified vacuolated structures consistent with autophagosomes, endosomes and lysosomes that accumulated within the myofiber (Figure 4(c)). We recently demonstrated that VCP is part of the endolysosomal damage response (ELDR) and is necessary for the clearance of damaged late endosomes via a process termed lysophagy [9]. In cells and tissue, damaged late endosomes are marked with LGALS3/galectin-3 [17]. To see if inactivation of VCP in skeletal muscle led to the accumulation of damaged endolysosomes, we immunostained skeletal muscle from control and 6 wk old Myl1p-cre-vcp-/- mice with antibodies to LGALS3 and LAMP2. Consistent with the accumulation of damaged lysosomes, there was an increase in LAMP2- and LGALS3-positive fibers in Myl1p-cre-vcp-/- mouse muscle but not in control muscle (Figure 5(a)). To more clearly understand the time course of accumulation of damaged lysosomes, we immunostained control muscle or muscle from ACTA1p-cre/Esr1-vcp-/- mice treated with 5 d tamoxifen after 2 and 4 wk. Muscle from ACTA1p-cre/Esr1-vcp-/- mice after 2 wk had a mild increase in LAMP2-LGALS3 puncta that was markedly increased after 4 wk (Figure 5(b)). Immunoblotting of the same muscle for LGALS3 demonstrated that control muscle had no LGALS3 expression whereas muscle from Myl1p-cre-vcp-/- and ACTA1p-cre/Esr1-vcp-/- skeletal muscle contained LGALS3 with the levels increasing from 2 wk to 4 wk in ACTA1p-cre/Esr1-vcp-/- mice (Figure 5(c,d)). While Myl1p-cre-atg5-/- mouse muscle did have an increase in LGALS3 proteins levels on immunoblot (Figure 5(e)), LGALS3 was diffuse in scattered myofibers as contrasted with the prominent SQSTM1 puncta seen by immunofluorescence in Myl1p-cre-atg5-/- mouse muscle (Figure 5(f,g)).
Figure 4.
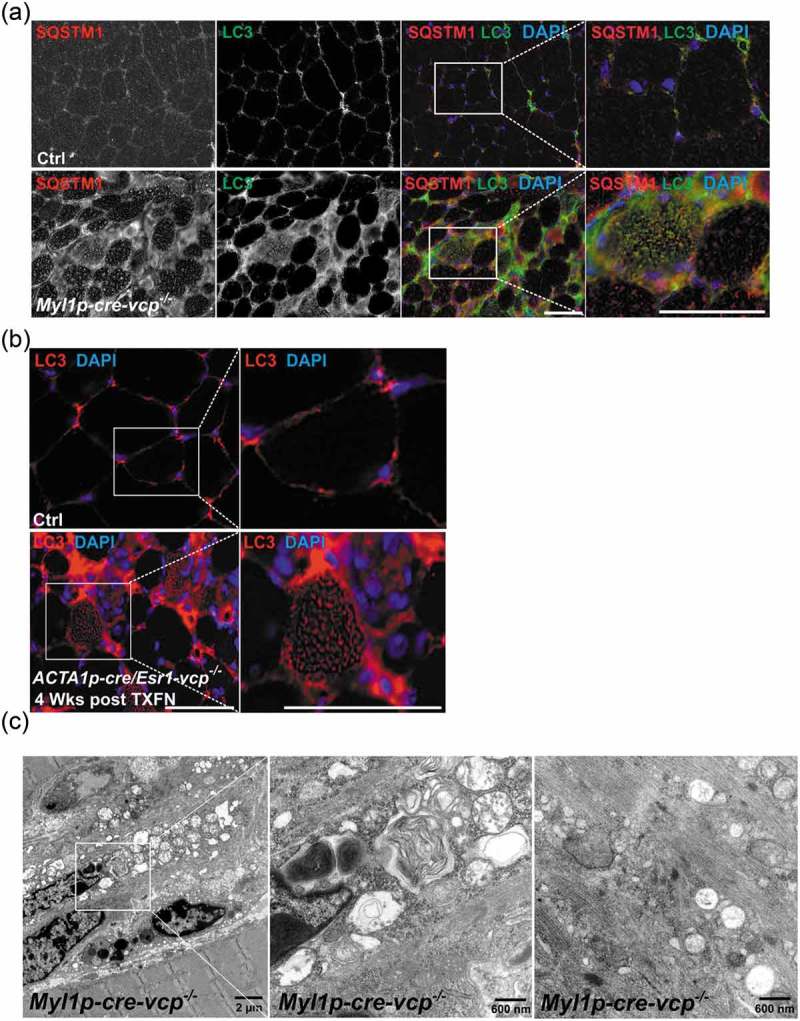
Muscle-specific inactivation of VCP leads to the accumulation of autophagic debris. (a) Co-immunofluorescence for LC3 (green) and SQSTM1 (red), in 6-week-old control or Myl1p-cre-vcp-/- mice. Merged images also contain DAPI nuclei staining (blue). Scale: 100 μm. (b) Immunofluorescence for LC3 (red) and DAPI-stained nuclei (blue) in ACTA1p-cre/Esr1-vcp-/- mice after 4 wk post tamoxifen injection for 5 d. Scale: 100 μm. (c) Electron micrograph images of the TA from Myl1p-cre-vcp-/- mice. Scale bars: 2 μm and 600 nm.
Figure 5.
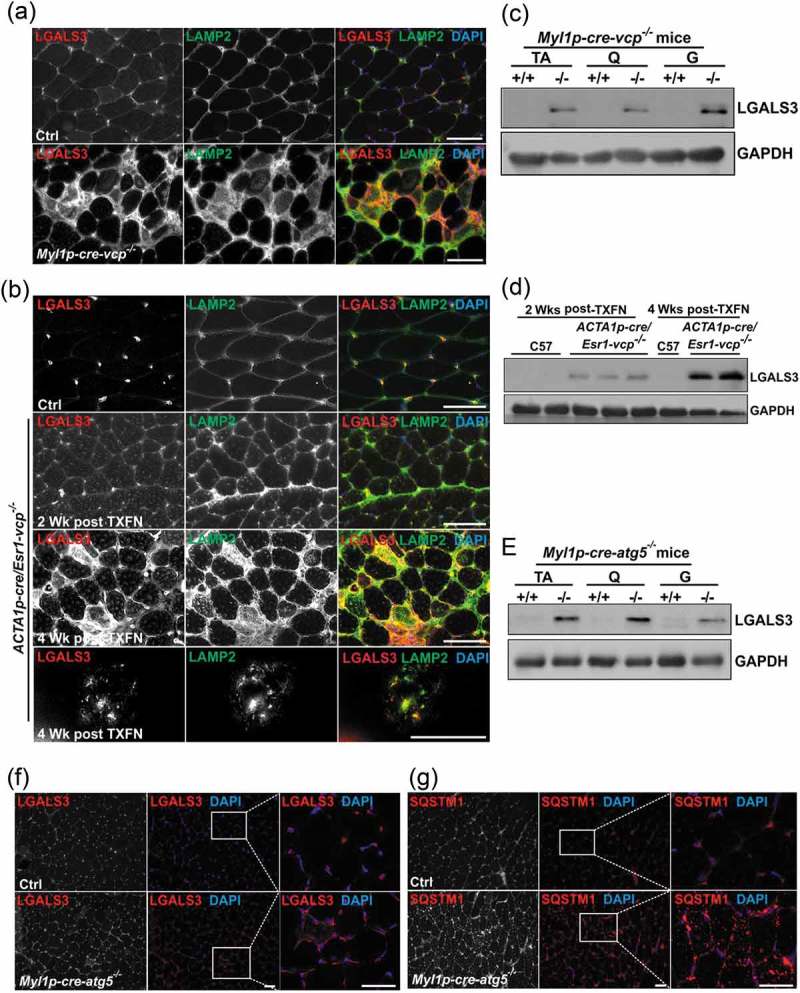
Damaged lysosomes are an early feature of VCP inactivation in muscle degeneration. (a) Co-immunofluorescence for LAMP2 (green) and LGALS3 (red), in 6-week-old control or Myl1p-cre-vcp-/- mice. (b) Co-immunofluorescence for LAMP2 (green) and LGALS3 (red) in control or ACTA1p-cre/Esr1-vcp-/- mice after 2 and 4 wk post tamoxifen injection for 5 d. (c) Immunoblot of lysates from TA, quadriceps or gastrocnemius muscle of 6-week-old control (+/+) or Myl1p-cre-vcp-/- (-/-) mice using antibodies to LGALS3 or GAPDH. (d) Immunoblot of lysates from TA of age-matched control or ACTA1p-cre/Esr1-vcp-/- mice after 2 and 4 wk post tamoxifen injection for 5 d using antibodies to LGALS3, or GAPDH. (e) Immunoblot of lysates from tibialis anterior, quadriceps or gastrocnemius muscle of 9-week-old control or Myl1p-cre-atg5-/- mice using antibodies to LGALS3 or GAPDH. f-g) Immunofluorescence for LGALS3 or SQSTM1 in control or 9-week-old Myl1p-cre-atg5-/- mouse muscle. DAPI (blue) stains nuclei. Scale: 100 μm.
We reasoned that accumulated damaged lysosomes may lead to myofiber degeneration and necrosis under conditions of VCP inactivation. To test this, we injected control mouse tibialis anterior muscle with the lysosomotropic agent LLOMe or vehicle and harvested muscle after 3 and 6 h. Six h post-LLOMe treatment, muscle had myofibers at varying stages of necrosis (Figure S3A). In addition, immunostaining for LAMP2 and LGALS3 demonstrated the presence of enlarged vesicular structures consistent with damaged endolysosomes (Figure S3B). Immunoblotting of muscle at 3 and 6 hours post-LLOMe treatment further demonstrated a rapid increase in LGALS3 and autophagic proteins (Figure S3C). These data suggest that lysosomal injury is a potent mediator of myofiber necrosis.
TFEB is activated in vcp knockout muscle
Lysosomal integrity is important for several signaling pathways in cells and tissue. Specifically, the lysosome senses nutrients leading to MTORC1 activation [21]. We previously demonstrated that nutrient stimulated MTORC1 signaling is diminished in the setting of VCP disease mutations [22]. In contrast, the phosphorylation of the downstream MTORC1 target RPS6 (ribosomal protein S6) in 6-week-old Myl1p-cre-vcp-/- mouse muscle was increased (Figure 6(a) and S4A). This increase was similar to that seen in Myl1p-cre-atg5-/- mouse muscle (Figure 6(h)). To understand the time course of RPS6 phosphorylation, we looked at muscle from ER/HSA-VCP-/- treated with i.p. injection of tamoxifen for 5 d and then after one, two or four weeks VCP inactivation. Notably, although VCP was diminished at 1 and 2 wk, the increase in p-RPS6 occurred after 4 wk coincident with necrosis and autophagic dysfunction (Figure 6(b,c)). Consistent with lysosomal injury leading to MTORC1 activation, treatment of control muscle with LLOMe led to an acute increase in p-RPS6 and p-EIF4EBP1 (Figure S3C).
Figure 6.
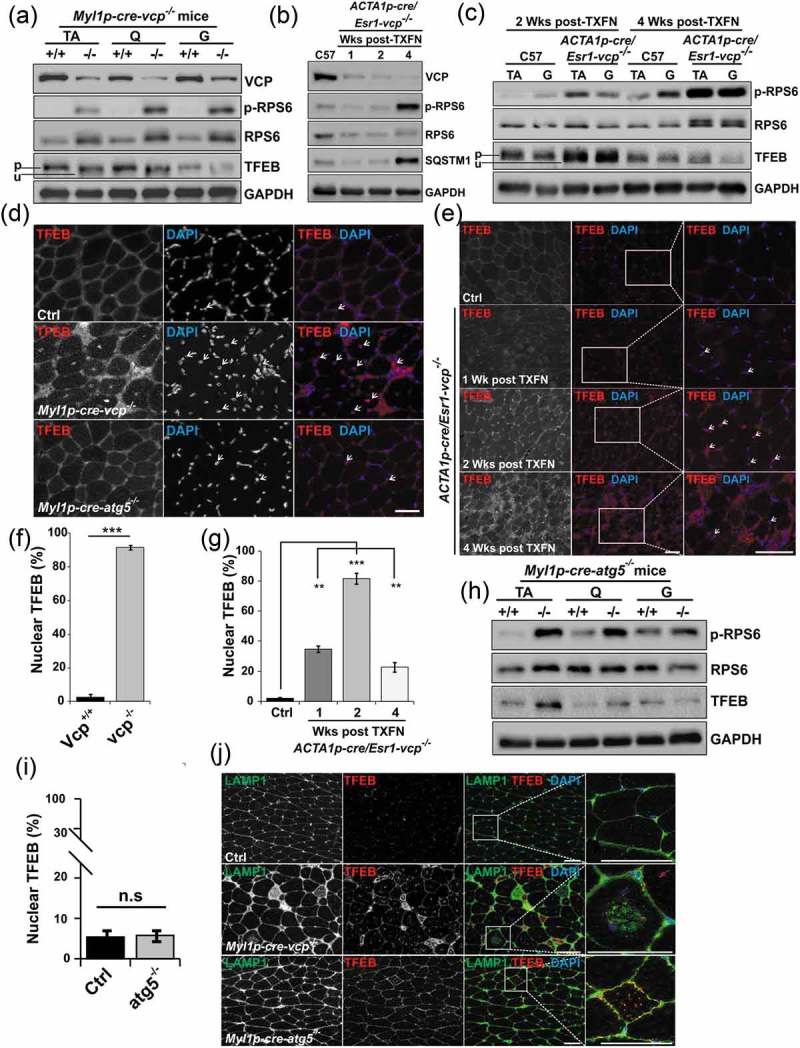
TFEB activation is an early feature of VCP inactivation in muscle degeneration. (a) Immunoblot of lysates from TA, quadriceps or gastrocnemius muscle of 6-week-old control (+/+) or Myl1p-cre-vcp-/- (-/-) mice using antibodies to VCP, p-RPS6, RPS6, TFEB or GAPDH. Lines adjacent to TFEB denote the higher migrating phosphorylated form (p) and the faster migrating unphosphorylated form (u). (b) Immunoblot of lysates from TA of age matched control or ACTA1p-cre/Esr1-vcp-/- mice after 1, 2 and 4 wk post tamoxifen injection for 5 d using antibodies to VCP, p-RPS6, RPS6, SQSTM1 or GAPDH. (c) Immunoblot of lysates from tibialis anterior or gastrocnemius muscle of age matched control or ACTA1p-cre/Esr1-vcp-/- mice after 2 and 4 wk post tamoxifen injection for 5 d using antibodies to p-RPS6, RPS6, TFEB or GAPDH. Lines adjacent to TFEB denote the higher migrating phosphorylated form (p) and the faster migrating unphosphorylated form (u). (d) Immunofluorescence with an antibody to TFEB on TA muscle from 6-week-old control, Myl1p-cre-vcp-/-, and 9-week-old Myl1p-cre-atg5-/-mice. Arrows point to nuclear-localized TFEB. (e) Immunofluorescence with an antibody to TFEB on TA muscle from age-matched control or ACTA1p-cre/Esr1-vcp-/- mice after 1, 2 and 4 wk post tamoxifen injection for 5 d. Arrows point to nuclear localized TFEB. (f) Bar graph of the percentage of TFEB-positive nuclei in muscle from 6-week-old control (+/+) and Myl1p-cre-vcp-/- (-/-) mice. Quantification was performed by counting the number of nuclei stained with TFEB in control (+/+) and Myl1p-cre-vcp-/- (-/-) mice using ImageJ software. At least 198 nuclei were counted per condition using 3 different mice. Comparison between groups was performed by paired Student t-test. (g) Bar graph of the percentage of TFEB-positive nuclei in muscle from control or ACTA1p-cre/Esr1-vcp-/- mice after 1, 2 and 4 wk post tamoxifen injection for 5 d. Quantification was performed by counting the number of nuclei stained with TFEB using ImageJ software. At least 236 nuclei were counted per condition from 3 different mice. Comparison between groups was performed using a paired Student t-test. (h) Immunoblot of lysates from tibialis anterior, quadriceps or gastrocnemius muscle of 9-week-old control or Myl1p-cre-atg5-/- mice using antibodies to p-RPS6, RPS6, TFEB or GAPDH. (i) Bar graph of the percentage of TFEB-positive nuclei in muscle from 9-week-old control and Myl1p-cre-atg5-/- mice. Quantification was performed by counting the number of nuclei stained with TFEB using ImageJ software. At least 1293 nuclei were counted per condition from 3 different mice. Comparison between groups was performed by paired Student t-test. (j) Co-immunofluorescence of LAMP1 and TFEB in control, Myl1p-cre-atg5-/-, or Myl1p-cre-vcp-/- mouse muscle. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant. DAPI (blue) stains nuclei. Scale: 100 μm.
The lysosome is also a site for TFEB (transcription factor EB) inhibition. TFEB is phosphorylated by MTORC1 on the lysosome leading to its inactivation and cytosolic retention [23]. Active TFEB is unphosphorylated and localizes to the nucleus where it induces transcription of lysosome and autophagy genes which contain the CLEAR (Coordinated Lysosomal Expression and Regulation) motif in their promoters [24]. Although MTORC1 was active in 6-week-old Myl1p-cre-vcp-/- muscle as demonstrated by increased phosphorylation of RPS6, TFEB migrated at a lower molecular weight on immunoblot, consistent with it being dephosphorylated (Figure 6(a)).
Immunohistochemical staining for TFEB confirmed it was myonuclear in Myl1p-cre-vcp-/- as compared to control muscle (Figure 6(d,f)). To further understand the time course of TFEB activation and see if it preceded the increase in MTORC1 activity, autophagic dysfunction and muscle pathology, we immunoblotted for TFEB and immunostained for TFEB in muscle from ACTA1p-cre/Esr1-vcp-/- mice treated with tamoxifen for 5 d and then after 1, 2 or 4 wk VCP inactivation (Figure 6(b,c,e,g)). TFEB dephosphorylation and myonuclear localization was an early feature of VCP inactivation in ACTA1p-cre/Esr1-vcp-/- mouse muscle and was detectable at one week post VCP inactivation, peaking at two weeks prior to any evidence of myopathology (Figure 6(c,e)). In Myl1p-cre-atg5-/- mouse muscle TFEB was not dephosphorylated (Figure 6(h)) and not myonuclear (Figure 6(d,i)) but instead sarcoplasmic where it localized to LAMP1-positive endolysosomes (Figure 6(j)). In contrast, Myl1p-cre-vcp-/- mouse muscle had an increase of LAMP1 and TFEB but they did not colocalize on accumulated lysosomes (Figure 6(j)). These data are also in agreement with the selective increase in expression of TFEB target genes such as Ctsb, Ctsd, Map1lc3a, Sqstm1 and Lamp1/2 as compared to genes without a CLEAR motif such as Bnip3 and Tfeb itself in Myl1p-cre-vcp-/- and ACTA1p-cre/Esr1-vcp-/- mouse muscle (Figure 2(f,g) and Figure 3(d,e)). TFEB activation under conditions of VCP knockout was not unique to skeletal muscle since siRNA knockdown of VCP in HeLa cells for 2 d similarly led to a significant increase in the number of TFEB-positive nuclei (Figure 7(a,b)).
Figure 7.
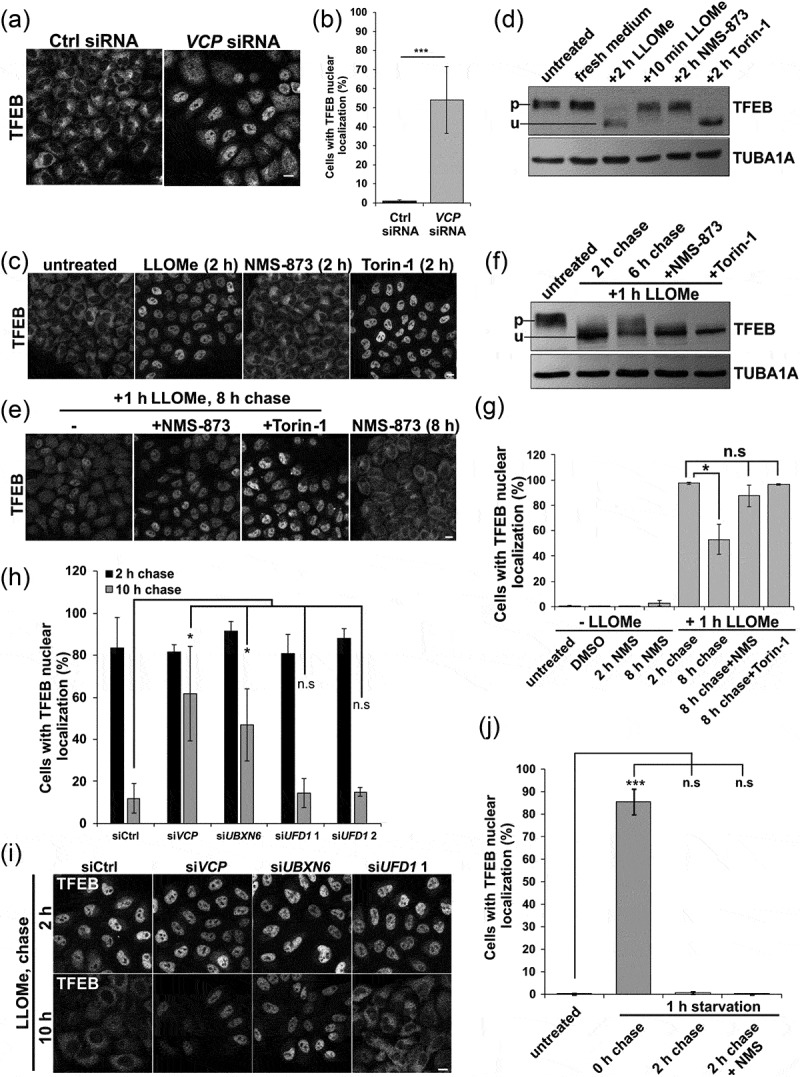
Inhibition of VCP and ELDR components lead to TFEB nuclear persistence following LMP. (a) TFEB immunolocalization in HeLa cells following control or VCP siRNA treatment for 48 h. (b) Bar graph of the percentage of TFEB-positive nuclei in HeLa cells following control or Vcp siRNA treatment. At least 249 cells were counted per condition and experiment. (c) TFEB immunolocalization from cells treated with LLOMe, NMS-873 or Torin-1 for 2 h. (d) Immunoblot for TFEB or TUBA1A/alpha-tubulin from HeLa cells treated with LLOMe, NMS-873 or Torin-1 for the indicated times. Lines adjacent to TFEB denote the higher migrating phosphorylated form (p) and the faster migrating unphosphorylated form (u). (e) TFEB immunolocalization from HeLa cells treated with LLOMe for one hour and allowed to recovery in the absence of LLOMe for 8 h. In some cases, cells were also incubated with NMS-873 or Torin-1 during recovery. (f) Immunoblot for TFEB or TUBA1A/α-tubulin from HeLa cells treated with LLOMe for 2 h or 1 h and then allowed to recover in the absence of LLOMe for 6 h. In some cases, cells were also incubated with NMS-873 or Torin-1 during recovery. Lines adjacent to TFEB denote the higher migrating phosphorylated form (p) and the faster migrating unphosphorylated form (u). (g) Bar graph of the percentage of cells with TFEB-positive nuclei from conditions in C and E. At least 644 cells were counted per condition and experiment. (h) Bar graph of the percentage of cells with TFEB-positive nuclei following control or VCP, YOD1, UBXN6, PLAA or UFD1 siRNA knockdown following 1 h of treatment with LLOMe and subsequent chase with media lacking LLOMe for 2 or 10 h. At least 251 cells were counted per condition and experiment. (i) TFEB immunolocalization in HeLa cells with control or VCP, UBXN6 or UFD1 siRNA knockdown following 1 h of treatment with LLOMe and subsequent chase with media lacking LLOMe for the 2 or 10 h. (j) Bar graph of TFEB-positive nuclei following 1 h of nutrient starvation and 2 h recovery nutrient-rich media with and without NMS-873. At least 631 cells were counted per condition and experiment. *p < 0.05; ***p < 0.001; n.s., not significant. Automated quantification with N = 3. Scale: 1 μm.
VCP inactivation leads to persistent TFEB activation
TFEB activation can occur with MTORC1 inhibition and more recently, TFEB activation was shown to be induced in response to lysosomal membrane permeabilization with LLOMe in cells [25,26]. LLOMe treatment in skeletal muscle also led to myonuclear redistribution of TFEB (Figure S3D-E). To see if acute VCP inhibition leads to dephosphorylation and nuclear localization of TFEB, we treated HeLa cells with LLOMe, NMS-873 (a VCP inhibitor) and Torin-1 (an MTOR inhibitor) for 2 h. Consistent with previous reports, Torin-1 and LLOMe treatment led to an increase in nuclear TFEB and a more rapid migration of TFEB suggesting it is dephosphorylated [23,25] (Figure 7(c,d,g)). Of note, acute inactivation of VCP with NMS-873 did not activate TFEB (Figure 7(c,d,g)).
Because LLOMe treatment increased nuclear TFEB and VCP participates in recovery from lysosomal injury, we treated Hela cells with LLOMe for one hour and then washed out the LLOMe allowing cells to recover. After 8 h of recovery, TFEB moves from the nucleus back to the cytoplasm (Figure 7(e,g)). In addition, an immunoblot for TFEB proteins showed an increase in a higher molecular weight TFEB band consistent with its phosphorylation (Figure 7(f)). Treatment with NMS-873 or Torin-1 resulted in TFEB persistence in the nucleus and a faster migration on immunoblot suggesting delayed phosphorylation during recovery (Figure 7(f,g)).
We reasoned that the persistence of TFEB in the setting of VCP inactivation may relate more globally to the persistence of damaged lysosomes. To discriminate between the different roles of VCP, we performed siRNA knockdown of VCP and UBXN6 that cooperates with VCP in the endolysosomal system. As a control, we performed knockdown with an established siRNA of the VCP adaptor, UFD1, that assists VCP in proteasomal degradation pathways and is unrelated to lysosomal integrity [27]. Similar to that seen with NMS-873 treatment, VCP and UBXN6 knockdown lead to TFEB persistence in the nucleus following LLOMe washout (Figure 7(h,i)).
To see if VCP dependent TFEB inactivation was specific to LMP, we activated TFEB by starving Hela cells of serum and amino acids for 60 min, which redistributes TFEB to the nuclei. TFEB then returns to the cytosol within 2 h of refeeding (Figure 7(j)). In contrast to LLOMe-induced lysosomal damage, NMS-873 or siRNA knockdown of VCP or the adaptors UBXN6 and UFD1 did not affect TFEB recovery after re-feeding (Figure 7(j) and S4B).
VCP disease-associated mutations lead to persistent TFEB activation in U2OS cells and muscle tissue
We have previously demonstrated that expression of VCP disease mutations in cell culture leads to the persistence of damaged LGALS3-positive endolysosomes [9]. To see if expression of VCP disease mutants causes persistent activation of TFEB similar to siRNA knockdown of VCP, we treated U2OS cells stably expressing a tetracycline inducible wild-type VCP or VCP disease mutations. TFEB was activated rapidly upon LLOMe treatment and localized to the nucleus by 90 min (Figure 8(a)). In addition, there was no difference in TFEB dephosphorylation in VCP WT- or VCP disease mutant-expressing cells as demonstrated via immunoblot (Figure 8(b)). Similar to knockdown of VCP, VCP disease mutations failed to redistribute TFEB from the nucleus to the cytoplasm following LLOMe treatment and subsequent recovery for 22 h (Figure 8(c,d)). The persistence of TFEB activity was not seen when similar cells were starved of serum and amino acids for 60 min and then refed for 3 h suggesting that lysosomal injury and not the mechanisms of TFEB inactivation or phosphorylation was the reason for the persistence (Figure 8(e,f)). In addition, TFEB persistence was MTORC1 independent since expression of a constitutively active RHEB did not lead to cytosolic redistribution of TFEB after LLOMe recovery in VCP mutant cells (Figure 8(g) and S4C).
Figure 8.
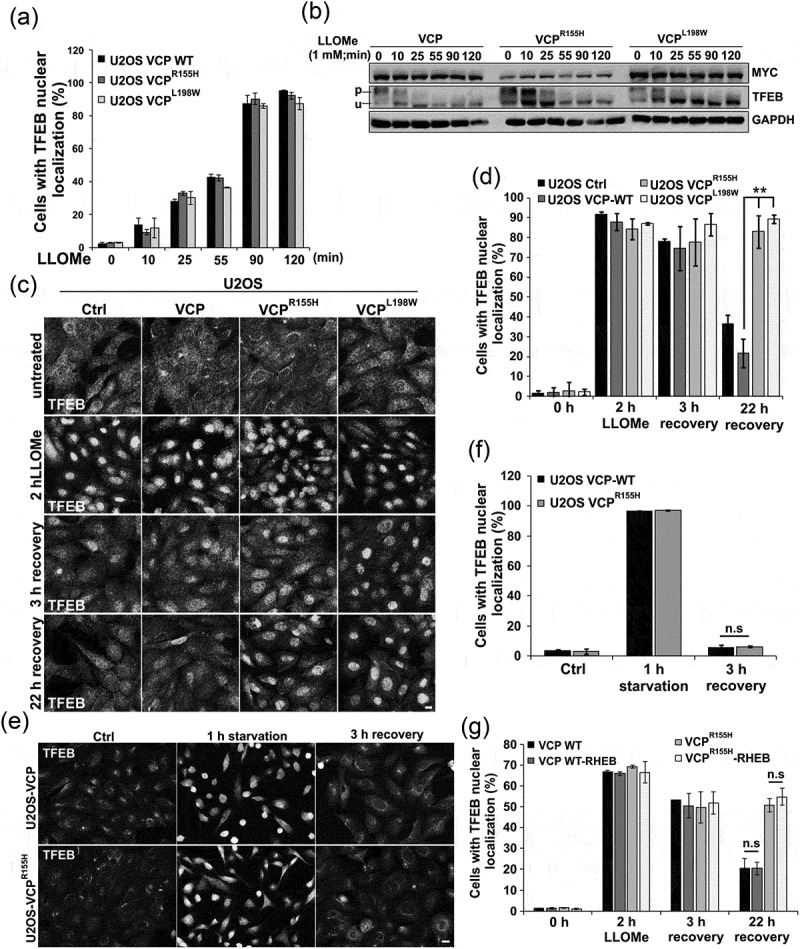
VCP mutant expression leads to TFEB nuclear persistence following LMP. (a) Bar graph of TFEB-positive nuclei following LLOMe treatment in VCP WT and 2 VCP disease mutant (VCPR155H or VCPL198W)-expressing cells. Quantification was performed by counting the number of cells with nuclear TFEB localization using ImageJ software. At least 304 cells were counted per group and condition. Comparison between groups was performed by paired Student t-test. (b) Immunoblot of lysates from U2OS cells stably expressing VCP WT, VCPR155H or VCPL198W following LLOMe treatment for the indicated times. Lines adjacent to TFEB denote the higher migrating phosphorylated form (p) and the faster migrating unphosphorylated form (u). (c) TFEB immunolocalization in U2OS cells expressing control, VCP WT or 1 of 2 VCP mutations following 2 h of treatment with LLOMe and subsequent chase with media lacking LLOMe for the 3 or 22 h. (d) Bar graph of TFEB-positive nuclei following LLOMe treatment and recovery in VCP WT- and mutant-expressing cells. Quantification was performed by counting the number of cells with nuclear TFEB localization using ImageJ software. At least 100 cells were counted per group and condition. Comparison between groups was performed by paired Student t-test. (e) TFEB immunolocalization in U2OS cells expressing control, VCP WT or VCPR155H following 1 h of nutrient starvation and subsequent chase with media containing serum and amino acids for 3 h. (f) Bar graph of TFEB-positive nuclei following starvation and recovery in VCP WT- and VCPR155H mutant-expressing U2OS cells. Quantification was performed by counting the number of cells with nuclear TFEB localization using ImageJ software. At least 568 cells were counted per group and condition. Comparison between groups was performed by paired Student t-test. (g) Bar graph of TFEB-positive nuclei following LLOMe treatment and recovery in VCP WT- and mutant-expressing U2OS cells with or without constitutively active RHEB. Quantification was performed by counting the number of cells with nuclear TFEB localization using ImageJ software. At least 248 cells were counted per group and condition. Comparison between groups was performed by paired Student t-test.**p < 0.01; n.s., not significant. N = 3. Scale: 10 μm.
To see if lysosomal injury and TFEB activation was a feature of VCP disease in a mouse model, we utilized a previously generated VCPR155H/+ knockin mouse line [28]. These mice are reported to develop features similar to VCP disease patients at 19–24 months of age that include weakness and accumulation of ubiquitinated and TARDBP/TDP-43 inclusions in skeletal muscle [28]. To see if there was accumulation of damaged endolysosomes in these mice at earlier time points, we analyzed muscle by histochemistry, immunohistochemistry and immunoblot at one year of age. There was no evidence of myopathology or immunohistochemical features consistent with disrupted autophagy or lysosomal injury at this time point (Figure 9(a)). However, immunoblot of muscle lysates from these same mice did find an increase in LGALS3 and TARDBP but no increase in any autophagic proteins, ER stress marker HSPA5 or ubiquitinated proteins (Figure 9(b,c)). Fluorescence microscopy of mouse muscle following electroporation of the tibialis anterior muscle with plasmids expressing an mCherry tagged LGALS3 and a GFP-tagged LAMP1 protein identified LGALS3 and LAMP1-positive puncta in VCPR155H/+ mouse muscle that were not present in control mouse muscle (Figure 9(d,e)). The accumulation of LGALS3-positive endosomes was similar to what we have previously described in VCP patient muscle [9]. To see if TFEB nuclear localization was an early feature of VCP associated disease in skeletal muscle, we immunostained one year old control and VCPR155H/+ mouse muscle for TFEB. Notably one year old muscle had an increase in myonuclear TFEB as compared with 6 wk old mouse muscle (compare Figure 9(g) with (Figure 6(f)). The number of TFEB-positive nuclei in VCPR155H/+ knockin mice was further increased as compared with littermate age matched controls (Figure 9(f,g)).
Figure 9.
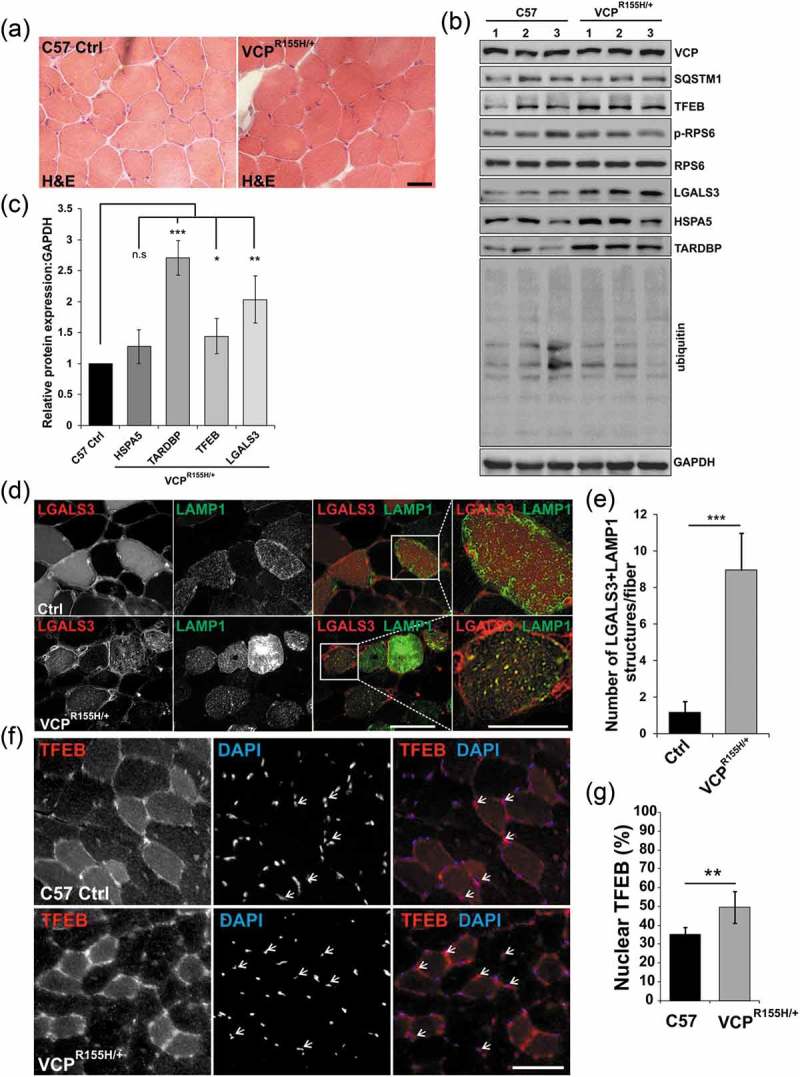
Lysosomal homeostasis is dysregulated in VCP diseased skeletal muscle. (a) H & E of TA from 13-month-old control (C57) or VCPR155H/+ heterozygous knockin mice. (b) Immunoblot of lysates from the TA from 13-month-old control or VCPR155H/+ knockin mice with antibodies to VCP, SQSTM1, TFEB, p-RPS6, RPS6, LGALS3, HSPA5, TARDBP, ubiquitin and GAPDH. N = 3 per group. (c) Densitometric analysis of TFEB, LGALS3, HSPA5, and TARDBP protein levels in VCPR155H/+ mice as compared to controls. N = 3 per group. (d) Fluorescence microscopy of control or VCPR155H/+ mouse TA muscle electroporated with plasmids expressing LAMP1-GFP (green) or mCherry-LGALS3 (red). (e) Quantification of the average number of LGALS3+ LAMP1 puncta in control or VCPR155H/+ mouse TA muscle electroporated with plasmids expressing LAMP1-GFP or mCherry-LGALS3. N = 30 co-electroporated fibers were counted per condition. (f) Immunofluorescence for TFEB (red) in TA from 13-month-old control or VCPR155H/+ knockin mice. Arrows point to nuclear-localized TFEB. (g) Quantification of the percent of TFEB-positive nuclei in tibialis anterior muscle from 13-month-old control or VCPR155H knockin mice. Quantification was performed by counting the number of nuclei stained with TFEB using ImageJ software. At least 1028 nuclei were counted per group from 3 different mice. Comparison between groups was performed by paired Student t-test. DAPI (blue) stains nuclei. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant. Scale: 100 μm.
Discussion
While diverse functions have been assigned to VCP and speculated to contribute to disease pathogenesis, the role of VCP in terminally differentiated post-mitotic tissue such as skeletal muscle has been largely unexplored. In the current study, we inactivated VCP in differentiated skeletal muscle and find that it is essential for muscle survival. Specifically, we identify an important role for VCP in lysosomal homeostasis that when dysregulated leads to myofiber necrosis. VCP and UBXN6 recognize damaged endolysosomes, that are marked by LGALS3 and ubiquitin, facilitating their selective autophagic degradation termed lysophagy [9]. Our data supports that the lysophagic degradation of damaged lysosomes is impaired in skeletal muscle when VCP is inactivated resulting in the accumulation of LGALS3-positive lysosomes. Moreover, this lysosomal dysfunction leads to the prominent accumulation of autophagic substrates. In addition, we identify a new role for VCP in the transcriptional regulation of newly synthesized lysosomal components, which compensates for the reduced lysosomal capacity following lysosomal injury. This process is triggered by LMP, which activates TFEB, thus allowing for the re-establishment of lysosomal homeostasis. The impaired clearance of damaged lysosomes with VCP knockout subsequently leads to persistent TFEB activation. Our findings emphasize the critical connection between lysosomal clearance and lysosomal biogenesis in the maintenance of lysosomal and skeletal muscle homeostasis.
Loss of VCP and ATG5 in skeletal muscle leads to the accumulation of autophagic substrates, ubiquitinated proteins and ER stress markers. However in sharp contrast to Atg5 deletion, vcp knockout muscle demonstrated myofiber necrosis with the accumulation of LGALS3-positive damaged lysosomes and myonuclear TFEB suggesting that loss of autophagy does not solely explain the myopathy phenotype. Moreover, the selective macroautophagic degradation of lysosomes via lysophagy cannot exclusively explain the phenotype seen with VCP inactivation since lysophagy is impaired in the setting of ATG5 knockdown following LMP [9]. TFEB localization in VCP knockout muscle was predominantly myonuclear but when sarcoplasmic did not co-localize with lysosomes. Interestingly, while TFEB did accumulate within scattered fibers from ATG5 knockout muscle it was not myonuclear but instead sarcoplasmic co-localizing with the lysosomal marker LAMP1 suggesting that intact lysosomes are necessary to maintain TFEB in the sarcoplasm [23]. These data suggest that the persistence of damaged lysosomal membranes is sufficient to alter TFEB signaling.
By utilizing a tamoxifen inducible system, we identified that the time course of myofiber necrosis with VCP knockout is rapid, occurring four weeks after gene inactivation. Notably, the accumulation of LGALS3-positive damaged lysosomes, TFEB myonuclear redistribution and its activation preceded autophagic dysfunction and tissue necrosis suggesting that these were early events contributing to muscle degeneration. In line with that, we demonstrate that LMP is a potent mediator of muscle necrosis as demonstrated by the application of the lysosomotropic agent, LLOMe, to skeletal muscle. Indeed, we have previously demonstrated that cells are more sensitive to LMP associated toxicity when VCP is inhibited or inactivated [9]. Moreover, LMP leads to the rapid activation and nuclear localization of TFEB.
LMP with LLOMe has been previously reported to activate TFEB in tissue culture cells [25,26]. Prolonged treatment of cells with LLOMe redistributed TFEB to the nucleus and inhibited MTORC1 activity as measured by a decrease in the phosphorylation of MTORC1 targets suggesting that TFEB activation in response to LMP is MTORC1 dependent [25,26]. This is consistent with current models of TFEB activation, where MTORC1 inhibition with chemical inhibitors or serum starvation potently leads to nuclear localization of unphosphorylated TFEB [23]. In contrast, other studies report that acute treatment with LLOMe activates MTORC1 even under conditions when LMP is apparent [29,30]. Moreover, MTORC1 independent activation of TFEB has been reported [31]. Our data supports an MTORC1 independent mechanism of TFEB activation following LMP with LLOMe in skeletal muscle. Specifically, treatment of skeletal muscle with LLOMe leads to a rapid increase in the phosphorylation of MTORC1 targets such as EIF4EBP1 and TFEB myonuclear localization.
An alternative mechanism of TFEB activation occurs via the release of lysosomal calcium through MCOLN1 during nutrient deprivation or exercise in skeletal muscle [32]. Lysosomal calcium release activates the phosphatase, PPP3/calcineurin which dephosphorylates TFEB enabling its nuclear translocation. LMP may similarly lead to lysosomal calcium release and TFEB activation via PPP3/calcineurin. More intriguing is the possibility that stressors such as exercise in skeletal muscle can lead to TFEB activation via lysosomal calcium release. Indeed, acute LLOMe treatment leads to the release of lysosomal calcium stores that recruit endosomal sorting complexes required for transport (ESCRTs) which perform lysosomal membrane repair [33]. Notably, ESCRT recruitment to damaged lysosomes temporally precedes both LGALS3 incorporation and lysophagic degradation suggesting that LGALS3 may not mark all permeabilized lysosomes [33]. We suggest that LGALS3 likely indicates late-stage damaged lysosomes and may explain why TFEB nuclear localization precedes LGALS3 accumulation in both VCP inactivated muscle and cells.
The mechanisms by which TFEB is inactivated once it is nuclear are less clear but likely occur with the return of metabolic homeostasis [24]. For example, following recovery from starvation, TFEB moves from the nucleus back to the cytoplasm coincident with MTORC1 reactivation. This is not affected by VCP inactivation. Similarly, as cells recover from LMP, TFEB moves from the nucleus to the cytoplasm. This is also MTORC1 dependent but can be inhibited in the setting VCP knockdown or inhibition. These data suggest that TFEB nuclear persistence in the setting of VCP dysfunction is specific for LMP associated TFEB activation. TFEB persistent activation in the setting of LMP and VCP inactivation was MTORC1 independent since expression of a constitutively active RHEB which increases MTORC1 activity under conditions of VCP disease mutant expression [22] was unable to correct TFEB inactivation.
VCP gene inactivation results in a severe myopathy that resembles some of the features seen in VCP disease patient muscle. VCP disease mutations have diminished interactions with its adaptor UBXN6 but maintain binding to other adaptors such as Ufd1 [10]. Consistent with this, VCP disease mutations preferentially affect UBXN6 dependent processes. Indeed, while VCP knockout in skeletal resulted in the accumulation of ubiquitinated proteins, autophagosomes and damaged endolysosomes, VCPR155H/+ mouse muscle had a mild phenotype without changes in the levels of ubiquitinated proteins or SQSTM1. However, VCPR155H/+ mouse muscle did accumulate LGALS3 and had enhance TFEB activity consistent with accumulation of damaged lysosomes; a UBXN6 dependent process. These data are consistent with a recent study that inactivated VCP in the vertebrate model, zebrafish [34]. Loss of VCP caused cardiac and skeletal muscle degeneration with defects in both the ubiquitin-proteasome (UPS) and autophagy [34]. In contrast, targeted inactivation of the VCP cofactor Washc4 in zebrafish caused muscle degeneration, and only autophagic degradation appeared defective [34]. These data highlight the importance of VCP cofactor specific functions in disease pathogenesis.
In U2OS cells, a VCP-UBXN6 complex is necessary for the endolysosomal trafficking of ubiquitinated and CAV-positive endosomes [10]. VCP disease mutations impair VCP-UBXN6 binding and lead to the accumulation of enlarged late endosomes with reduced intraluminal vesicles [10]. Expression of VCP disease mutations also impairs the autophagic degradation of damaged lysosomes [9]. This leads to the accumulation of LGALS3-positive lysosomes in patient muscle [9]. Other cellular process affected in VCP disease mutation-expressing cells include defective autophagosome maturation and nutrient sensing [8,22,35]. Interestingly, all of these processes (endocytosis, autophagy and nutrient sensing) require an intact lysosome and suggest that an impairment in VCP-UBXN6-dependent interactions leads to the dysfunctional lysosome that underlies VCP disease pathogenesis.
Methods
Generation of Vcp conditional knockout mice
Vcp conditional knockout mice were generated using the CRISPR-Cas9 system to introduce 2 loxP sites, flanking exons 2–5, into the same allele of the Vcp gene. We designed gRNAs targeting introns 1 and 5 of the Vcp gene and oligonucleotides, containing loxP sites and BamHI or HindIII restriction sites, flanked by 60 bp homologous to the targeted intronic region. Zygotes were microinjected with a mixture of Cas9 mRNA, gRNAs, and oligos and transferred to 4 pseudo pregnant female mice. Genotyping of 35 F0 born pups using tail-extracted DNA and Vcp-specific primer sets flanking the loxP sites led to the identification of two founder mice carrying the loxP sites. The founder mice were healthy, normal in size, and did not display any phenotype compared to their wild-type littermates.
Animals and experimental protocols
Control (C57BL/6, Stock No: 000664), VCP-R155H (B6;129S-Vcptm1Itl/J, Stock No: 021968), Myl1p-Cre (Myl1tm1(cre)Sjb/J, Stock No: 024713), ACTA1p-cre/Esr1 (Tg(ACTA1-cre/Esr1*)2Kesr/J, Stock No: 025750 HSA-MCM) mice were purchased from Jackson Laboratories. Atg5 floxed mice were a gift from Noburu Mizushima (University of Tokyo). All animal experimental protocols were approved by the Animal Studies Committee of Washington University School of Medicine. Mice were housed in a temperature-controlled environment with 12-h light-dark cycles where they received food and water ad libitum.
Genotyping of mice
Genomic DNA was extracted from mutants and control mice tails using KAPA Express DNA extraction Kit (Kapabiosystems, KK7103) and standard PCR was performed using KAPA2G Fast Genotyping Kit (Kapabiosystems, KK7302) and gene-specific primers listed in Table S1. PCR products were subjected to electrophoresis on a 2.5% agarose gel.
Cell culture, RNA interference, and immunostaining
U2OS were cultured as described previously [9]. To induce lysosomal damage, U2OS cells were treated with 1 mM Leu-Leu methyl ester hydrobromide (Sigma-Aldrich, L7393) followed by washout of the drug and chase in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, 11965–084) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals, S10350).
HeLa cells were cultured in DMEM (PAN-Biotech, P04-03590) supplemented with 10% FBS (PAN-Biotech, P30-2102) in the presence of penicillin/streptomycin (PAN-Biotech, P06-07100). HeLa Cells were transfected for 48 h with siRNA (12.5 nM) using Lipofectamine RNAiMax (Life technologies, 13778150) according to the manufacturer’s instructions (Table S2). To induce lysosomal damage, HeLa cells were treated with 250 μM LLOMe (Sigma-Aldrich, L7393). Cells were also treated with 5 μM NMS-873 VCP/p97 inhibitor (Sigma-Aldrich, SML1128) or 1 μM Torin-1 (Tocris Bioscience, 4247) dissolved in DMSO (Sigma-Aldrich, D2650). Starvation experiments in HeLa cells utilized MEM Eagle with EBSS, without L-Glutamine (PAN-Biotech, P04-08050).
For U2OS cell immunostaining, cells were washed with Dulbecco’s Phosphate-Buffered Saline PBS (Gibco, 14190–136) and fixed in 3.7% paraformaldehyde for 10 min, blocked in PTB (1x PBS, 0.1% Triton X-100 [Sigma-Aldrich,T8787], 0.1% bovine serum albumin [Fisher Scientific, M-9501]) for 1 h, incubated in primary antibodies at a 1:200 dilution at 4°C overnight, and washed with PTB buffer. Cells were incubated with Alexa Fluor 488 goat anti-mouse (Invitrogen, A32723) or Alexa Fluor 555 goat anti-rabbit (Invitrogen, A32732) secondary antibodies at a 1:1,000 dilution and washed as above. Coverslips were mounted on slides using Mowiol reagent containing 4ʹ,6-diamidino-2-phenylindole (1:1,000). Images were acquired with a CoolSNAP EZ camera on a Nikon Eclipse 80i microscope with NIS-Elements AR 3.00 software (Nikon, Tokyo, Japan). Antibodies used were anti-TFEB (Bethyl Laboratories Inc., A303-673A), anti-LGALS3/Galectin-3 (Santa Cruz Biotechnology, rabbit H-160), anti-LAMP1 (Santa Cruz Biotechnology, H4A3), anti-LC3B (NanoTools, 0231–100), anti-SQSTM1 (Proteintech, 18420–1-AP), and anti-ubiquitin FK2 (Enzo Life Sciences, BML-PW8810).
For HeLa cells immunostaining, cells were fixed with 4% paraformaldehyde (Sigma-Aldrich, P6148), permeabilized with 0.1% Triton X-100 (Diagonal, A1388) in PBS, blocked by 3% bovine serum albumin (Diagonal, A1391) in PBS with 0.1% Triton X-100 and 0.1% saponin (Sigma-Aldrich, 47036), stained by indirect immunofluorescence for TFEB (rabbit; Cell Signaling Technology, 4240) and with Alexa Fluor® 488 (goat anti-rabbit; Life Technologies, A11034), Hoechst and HCS CellMask Deep Red Stain (Life Technologies, H32721) and mounted in ProLong Gold antifade mountant (Thermo Fisher Scientific, P36930). Automated confocal laser scanning microscopy was performed on a TCS SP5 AOBS system equipped with standard PMT detectors as well as sensitive HyD detectors (Leica Microsystems). Images were acquired using an HC PL APO 20x/0.75 NA dry objective. Lasers used for excitation were HeNe 633 nm (CellMask Deep Red), Ar 488 nm (Alexa Fluor® 488, EGFP) and Diode 405 nm (Hoechst).
For U2OS cell starvation and nutrient stimulation, growth medium was removed and the cells were rinsed twice with PBS (Invitrogen, 14190–136). The cells were serum starved (no serum in the growth media) in Hanks’ Balanced Salt Solution (HBSS; Invitrogen, 14025076) for 1 h then restimulated with the DMEM-FBS full medium containing 10% FBS.
For transfection, U2OS cells were cultured on coverslips and transfected with 1 µg each of pcDNA3-FLAG-RHEB-N153T (Addgene, plasmid 19997; depositing Lab: Fuyuhiko Tamanoi) and a GFP control plasmid pEGFPN1 (Clontech, 6085–1) using Lipofectamine 2000 (Invitrogen, 11668–019) according to the manufacturer’s instructions.
Immunoblot analysis
Muscle tissues and U2OS cells were homogenized in Radioimmunoprecipitation assay lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% NP-40 [Sigma, I3021], 0.25% Na deoxycholate [Sigma-Aldrich, 30970], 1 mm ethylenediaminetetraacetic acid) supplemented with protease inhibitor cocktail (Sigma-Aldrich, S8820). Lysates were centrifuged at 14,000g for 10 min. Aliquots of the supernatant were solubilized in Laemmli sample buffer and equal amounts were separated on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels, transferred to nitrocellulose, and blocked with 5% nonfat dry milk in TBST (Tris-buffered saline, 0.1% Tween 20 [Sigma-Aldrich, 30970]). Membrane was incubated with primary antibody at 1:500 dilution overnight followed by incubation with secondary antibody conjugated with horseradish peroxidase at a 1:5,000 dilution. Amersham ECL Western Blotting Detection Reagents Kit (GE Healthcare, RPN3244) was used for protein detection and immunoblots were visualized with G:Box Chemi XT4, Genesys version 1.1.2.0 (Syngene, Cambridge, UK). Antibodies used: anti-GAPDH (Cell Signaling Technology, 2118), anti-ubiquitin P4D1 (Cell Signaling Technology, 3936), anti-TFEB (Bethyl Laboratories Inc., A303-673A), anti-VCP (Fitzgerald, 10R-P104A), anti-SQSTM1 (Proteintech, 18420–1-AP), anti-phospho-RPS6 (Cell Signaling Technology, Ser240/244, 5364), anti-RPS6 (Cell Signaling Technology, 2217L), anti-HSPA5/GRP78/BIP (BD Transduction Labs, 610979), anti-TARDBP (Proteintech,10782–2-AP), anti-DES (abcam, ab8592), anti-ACTN (abcam, ab68167), anti-MYH (Sigma, M1570), anti-MYC (Cell Signaling Technology, 2276), and anti-TUBA/α-tubulin (Sigma, T5168).
Measurement of the fibers cross-sectional area
Cross sectional area of each muscle fiber of tibialis anterior (TA) was measured manually using ImageJ software. The Frequency histogram shows the distribution of cross-sectional areas (μm2) of at least 200 fibers in TA of control or ER/HSA-VCP mice treated with tamoxifen (TXFN) for 5 d and sacrificed 1 wk, 2 wk, 4 wk and 6 wk post the last TXFN injection.
Histological analysis
Muscle tissues were processed as previously described [8]. Cryostat sections of frozen muscle were processed and fixed in acetone. Antibodies at 1:200 dilution used were anti-LGALS3/Galectin-3 (Santa Cruz Biotechnology, H-160), anti-TFEB (Bethyl Laboratories Inc., A303-673A), anti-LAMP1 (Santa Cruz Biotechnology, H4A3), anti-LAMP2 (Sigma-Aldrich, L7543), anti-LC3B (NanoTools, 0231–100), anti-SQSTM1 (Sigma-Aldrich, P0067), anti-ubiquitin FK2 (Enzo Life Sciences, BML-PW8810). Sections were then incubated with Alexa Fluor 488 goat anti-mouse or Alexa Fluor 555 goat anti-rabbit secondary antibodies (Invitrogen, 21428) at a 1:1,000 dilution and washed as above. Images were acquired with a CoolSNAP EZ camera on a Nikon Eclipse 80i microscope with NIS-Elements AR 3.00 software (Nikon, Tokyo, Japan). Quantifications were performed using ImageJ software. Graphs and statistical analyses were performed using Excel (Microsoft Corporation).
qPCR analysis
RNA was isolated from tibialis anterior muscle of control or mutant mice using SV Total RNA isolation kit Z3100 (Promega). Complementary DNA (cDNA) was prepared by reverse transcription PCR using Transcriptor First Strand cDNA Synthesis Kit (Roche, 04 379 012 001) according to the manufacturer’s instructions. Quantitative PCR was performed using the primers listed in Table S3. Gene expression levels were analyzed by real-time PCR on an Applied Biosystems model 7500 (software v2.0.5) using the Faststart universal SYBR Green master ROX qPCR mastermix (Roche, 04913850001). The values were normalized to Gapdh and represented as fold change.
LLOMe treatment and electroporation in mice
For LLOMe treatment, mice were anesthetized using inhaled isoflurane. The skin overlying the TA muscle was shaved, and the animals were injected with 50 µl sterile phosphate-buffered saline PBS (Gibco, 14190–136) or LLOMe (2 mM in PBS) into the left and right TA, respectively, using a 0.5-ml syringe fitted with a 29-gauge needle. Animals were allowed to recover for 3, 6, and 24 h prior to muscle isolation.
For the electroporation, mice TA muscle was injected with 25 µg plasmids expressing an mCherry-tagged LGALS3 and a GFP-tagged LAMP1 protein diluted in sterile PBS and the electroporation was performed as previously described [8].
Wire screen holding test
The 4-limb hanging test was performed as previously described [36]. Briefly, mice were placed on a grid where it stood using all 4 limbs. Subsequently, the grid was inverted and held approximately 15 cm above the cage. Latency for the mouse to release the grid was recorded and the highest time of 3 trials was used as an outcome measure.
Statistical analysis
Densitometry of western blots protein bands was analyzed using the Gel Analyzer of ImageJ software, and quantification of protein levels normalized to GAPDH. Data are presented as the mean ± standard deviation of the mean with the exception of wire screen holding test, which was presented as the mean ± standard error. Significance was considered by a P value of <0.05 utilizing a paired student T-test. Quantification of nuclear TFEB in Hela cells was performed using automated acquisition of 36 fields per condition per experiment and was controlled by the HCS A module in the LAS AF software (Leica Microsystems). Images were processed using ImageJ software (http://rsbweb.nih.gov/ij/) and automated quantifications were performed using Cell Profiler 3.0 [37].
Funding Statement
CCW was supported by NIH (AG031867, AG042095, and AR068797); the Muscular Dystrophy Association; the Myositis Association; and the Hope Center for Neurological Disorders. KA was supported by NIH T32 NS007205. HM was supported by DFG grants Me1626/4-2 and Me1626/5-1.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data can be accessed here.
References
- [1].Meyer H, Weihl CC.. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci. 2014. September 15;127(Pt 18):3877–3883. PubMed PMID: 25146396; PubMed Central PMCID: PMCPMC4163641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lan B, Chai S, Wang P, et al. VCP/p97/Cdc48, a linking of protein homeostasis and cancer therapy. Curr Mol Med. 2018. March 7;17:608–618. PubMed PMID: 29521227. [DOI] [PubMed] [Google Scholar]
- [3].Muller JM, Deinhardt K, Rosewell I, et al. Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem Biophys Res Commun. 2007. March 9;354(2):459–465. PubMed PMID: 17239345. [DOI] [PubMed] [Google Scholar]
- [4].Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004. April;36(4):377–381. PubMed PMID: 15034582. [DOI] [PubMed] [Google Scholar]
- [5].Taylor JP. Multisystem proteinopathy: intersecting genetics in muscle, bone, and brain degeneration. Neurology. 2015. August 25;85(8):658–660. PubMed PMID: 26208960. [DOI] [PubMed] [Google Scholar]
- [6].Weihl CC, Pestronk A, Kimonis VE. Valosin-containing protein disease: inclusion body myopathy with Paget’s disease of the bone and fronto-temporal dementia. Neuromuscul Disord. 2009. May;19(5):308–315. PubMed PMID: 19380227; PubMed Central PMCID: PMCPMC2859037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Al-Obeidi E, Al-Tahan S, Surampalli A, et al. Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin Genet. 2018. January;93(1):119–125. PubMed PMID: 28692196; PubMed Central PMCID: PMCPMC5739971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ju JS, Fuentealba RA, Miller SE, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009. December 14;187(6):875–888. PubMed PMID: 20008565; PubMed Central PMCID: PMCPMC2806317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Papadopoulos C, Kirchner P, Bug M, et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. Embo J. 2017. January 17;36(2):135–150. PubMed PMID: 27753622; PubMed Central PMCID: PMCPMC5242375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ritz D, Vuk M, Kirchner P, et al. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat Cell Biol. 2011. August 7;13(9):1116–1123. PubMed PMID: 21822278; PubMed Central PMCID: PMCPMC3246400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Weihl CC, Temiz P, Miller SE, et al. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2008. October;79(10):1186–1189. PubMed PMID: 18796596; PubMed Central PMCID: PMCPMC2586594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Piccirillo R, Goldberg AL. The p97/VCP ATPase is critical in muscle atrophy and the accelerated degradation of muscle proteins. Embo J. 2012. August 1;31(15):3334–3350. PubMed PMID: 22773186; PubMed Central PMCID: PMCPMC3411080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang T, Mishra P, Hay BA, et al. Valosin-containing protein (VCP/p97) inhibitors relieve Mitofusin-dependent mitochondrial defects due to VCP disease mutants. Elife. 2017. March 21;6 DOI: 10.7554/eLife.17834 PubMed PMID: 28322724; PubMed Central PMCID: PMCPMC5360448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Johnson AE, Shu H, Hauswirth AG, et al. VCP-dependent muscle degeneration is linked to defects in a dynamic tubular lysosomal network in vivo. Elife. 2015. July 13;4 DOI: 10.7554/eLife.07366 PubMed PMID: 26167652; PubMed Central PMCID: PMCPMC4574298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schutz AK, Rennella E, Kay LE. Exploiting conformational plasticity in the AAA+ protein VCP/p97 to modify function. Proc Natl Acad Sci U S A. 2017. July 31;114:E6822–E6829. PubMed PMID: 28760999; PubMed Central PMCID: PMCPMC5565461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Trusch F, Matena A, Vuk M, et al. The N-terminal Region of the Ubiquitin Regulatory X (UBX) Domain-containing Protein 1 (UBXD1) modulates interdomain communication within the valosin-containing protein p97. J Biol Chem. 2015. December 4;290(49):29414–29427. PubMed PMID: 26475856; PubMed Central PMCID: PMCPMC4705944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Papadopoulos C, Meyer H. Detection and clearance of damaged lysosomes by the endo-lysosomal damage response and lysophagy. Curr Biol. 2017. December 18;27(24):R1330–R1341. PubMed PMID: 29257971. [DOI] [PubMed] [Google Scholar]
- [18].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006. June 15;441(7095):880–884. PubMed PMID: 16625205. [DOI] [PubMed] [Google Scholar]
- [19].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006. June 15;441(7095):885–889. PubMed PMID: 16625204. [DOI] [PubMed] [Google Scholar]
- [20].Masiero E, Agatea L, Mammucari C, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009. December;10(6):507–515. PubMed PMID: 19945408. [DOI] [PubMed] [Google Scholar]
- [21].Perera RM, Zoncu R. The lysosome as a regulatory hub. Annu Rev Cell Dev Biol. 2016. October 6;32:223–253. PubMed PMID: 27501449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ching JK, Elizabeth SV, Ju JS, et al. mTOR dysfunction contributes to vacuolar pathology and weakness in valosin-containing protein associated inclusion body myopathy. Hum Mol Genet. 2013. March 15;22(6):1167–1179. PubMed PMID: 23250913; PubMed Central PMCID: PMCPMC3657474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012. June 12;5(228):ra42 PubMed PMID: 22692423; PubMed Central PMCID: PMCPMC3437338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016. July 1;129(13):2475–2481. PubMed PMID: 27252382; PubMed Central PMCID: PMCPMC4958300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chauhan S, Kumar S, Jain A, et al. TRIMs and galectins globally cooperate and TRIM16 and Galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016. Oct 10;39(1):13–27. PubMed PMID: 27693506; PubMed Central PMCID: PMCPMC5104201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hao F, Kondo K, Itoh T, et al. Rheb localized on the Golgi membrane activates lysosome-localized mTORC1 at the Golgi-lysosome contact site. J Cell Sci. 2018. January 29;131(3). DOI: 10.1242/jcs.208017 PubMed PMID: 29222112. [DOI] [PubMed] [Google Scholar]
- [27].Dobrynin G, Popp O, Romer T, et al. Cdc48/p97-Ufd1-Npl4 antagonizes Aurora B during chromosome segregation in HeLa cells. J Cell Sci. 2011. May 1;124(Pt 9):1571–1580. PubMed PMID: 21486945. [DOI] [PubMed] [Google Scholar]
- [28].Nalbandian A, Llewellyn KJ, Badadani M, et al. A progressive translational mouse model of human valosin-containing protein disease: the VCP(R155H/+) mouse. Muscle Nerve. 2013. February;47(2):260–270. PubMed PMID: 23169451; PubMed Central PMCID: PMCPMC3556223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rebecca VW, Nicastri MC, McLaughlin N, et al. A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov. 2017. November;7(11):1266–1283. PubMed PMID: 28899863; PubMed Central PMCID: PMCPMC5833978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Manifava M, Smith M, Rotondo S, et al. Dynamics of mTORC1 activation in response to amino acids. Elife. 2016. October 11;5 DOI: 10.7554/eLife.19960 PubMed PMID: 27725083; PubMed Central PMCID: PMCPMC5059141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li Y, Xu M, Ding X, et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat Cell Biol. 2016. October;18(10):1065–1077. PubMed PMID: 27617930. [DOI] [PubMed] [Google Scholar]
- [32].Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015. March;17(3):288–299. PubMed PMID: 25720963; PubMed Central PMCID: PMCPMC4801004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Skowyra ML, Schlesinger PH, Naismith TV, et al. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018;360(6384):eaar5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kustermann M, Manta L, Paone C, et al. Loss of the novel Vcp (valosin containing protein) interactor Washc4 interferes with autophagy-mediated proteostasis in striated muscle and leads to myopathy in vivo. Autophagy. 2018. July 16;14:1911–1927. PubMed PMID: 30010465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tresse E, Salomons FA, Vesa J, et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010. February;6(2):217–227. PubMed PMID: 20104022; PubMed Central PMCID: PMCPMC2929010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Carlson CG, Rutter J, Bledsoe C, et al. A simple protocol for assessing inter-trial and inter-examiner reliability for two noninvasive measures of limb muscle strength. J Neurosci Methods. 2010. February 15;186(2):226–230. PubMed PMID: 19917311. [DOI] [PubMed] [Google Scholar]
- [37].Carpenter AE, Jones TR, Lamprecht MR, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7(10):R100 PubMed PMID: 17076895; PubMed Central PMCID: PMCPMC1794559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.