

ABSTRACT
Cellular adaption to nutrient stress is exquisitely regulated, and its dysregulation could underlie human diseases including neurodegeneration. C9orf72 is linked to the most common forms of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) as well as rare cases of other neurological disorders. Recent studies have implicated C9orf72 functions in the autophagy-lysosome pathway, but the exact roles of C9orf72 remain unclear. We found that C9orf72 is required for the lysosomal targeting and degradation of CARM1, which is an important epigenetic regulator of macroautophagy/autophagy and lipid metabolism. In cells with C9orf72 deficiency including those derived from ALS-FTD patients, CARM1 is abnormally accumulated especially under glucose starvation stress, leading to dysregulated autophagy and lipid metabolism. These findings suggest that C9orf72 is a key regulator in the negative feedback control of the autophagy-lysosome pathway during nutrient stress responses.
KEYWORDS: ALS, autophagy, C9orf72, CARM1, fatty acid, FTD, lipid metabolism, lysosome, NOX2
Nutrient utilization and energy metabolism are exquisitely regulated under physiological conditions, and their dysregulation contributes to the pathology of many human diseases. ALS is a motor neuron degeneration disease clinically characterized by energy metabolism defects, including hypermetabolism and hyperlipidemia. The hexanucleotide repeat expansion in C9orf72 is the most common cause for both ALS and FTD. The reduction of C9orf72 protein in patients suggests that deficiency in C9orf72 functions could contribute to the pathogenesis of ALS-FTD. Although C9orf72 contains domains of DENN-like proteins and has been implicated in several cellular processes including autophagy, its functions remain poorly understood. In our recent study [1], we found that C9orf72 functions in lipid metabolism by controlling the lysosomal degradation of the epigenetic factor CARM1 under nutrient stress (Figs.1, 2).
Figure 1.
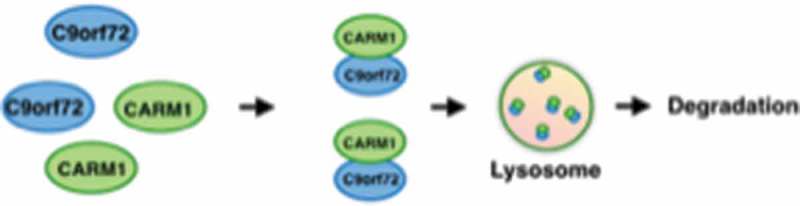
C9orf72 binds to CARM1 and promotes delivery of CARM1 to the lysosome for degradation. C9orf72 directly binds to CARM1, and under glucose starvation, C9orf72 causes localization of CARM1 to the lysosome by an unknown mechanism for degradation.
Figure 2.
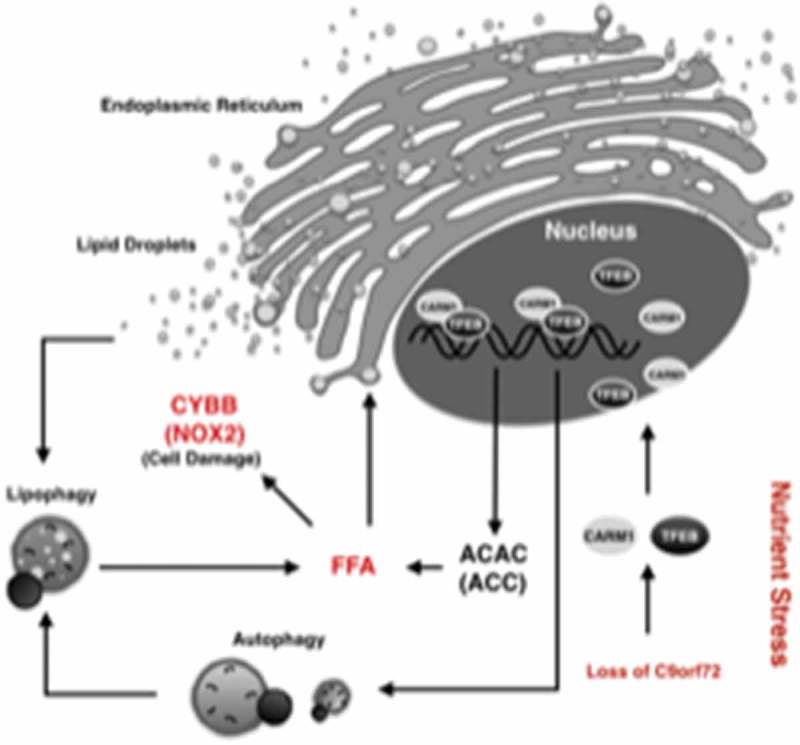
C9orf72 regulates autophagy and lipid metabolism via CARM1 under conditions of glucose starvation. During glucose starvation, loss of C9orf72 decreases the lysosomal degradation of CARM1, resulting in the nuclear enrichment of CARM1 and the activation of epigenetic regulation on transcription. Nuclear CARM1, an arginine methyltransferase for histones, epigenetically regulates gene transcription related to autophagy, lysosomes, and fatty acid de novo biogenesis. Overactivation of both autophagic digestion of lipid droplets and fatty acid de novo biogenesis results in increased levels of fatty acids, which leads to upregulation of CYBB/NOX2, an NADPH oxidase associated with oxidative stress. This pathway represents a new mechanism in the regulation of autophagy and lipid metabolism under nutrient stress, which could be dysregulated in human diseases. FFA, free fatty acids. ACAC/ACC, acetyl-CoA carboxylase.
Evidence from multiple studies suggests that C9orf72 has functions in the autophagy-lysosome pathway. Here, we found that C9orf72 is required for the lysosomal degradation of CARM1, a histone arginine methyltransferase known to function as an epigenetic regulator of autophagy. C9orf72 directly interacts with CARM1, and C9orf72 promotes delivery of CARM1 to the lysosome for degradation under nutrient stress conditions (Figure 1). CARM1 was reported to be degraded by the proteasome in the nucleus via AMPK-SKP2-dependent ubiquitination; however, we have uncovered a previously unrecognized route for CARM1 degradation through the lysosome. C9orf72 is required for the lysosomal, but not proteasomal, degradation of CARM1. Upon glucose starvation, both C9orf72 and CARM1 relocalize to the lysosome. Remarkably, in the absence of C9orf72, the recruitment of CARM1 to the lysosome is impaired especially under stress conditions. Therefore, C9orf72 emerges an important regulator for lysosomal targeting. It is possible that C9orf72 is required for the lysosomal targeting and degradation of other proteins. The mechanisms through which C9orf2 mediates lysosomal targeting remains to be elucidated.
Because C9orf72 is required for the lysosomal degradation of CARM1, reduction of C9orf72 would lead to accumulation of CARM1, which was consistently observed under multiple settings including in mouse fibroblasts lacking C9orf72, in motor neurons differentiated from ALS-FTD patient-derived stem cells, and in ALS-FTD patients’ spinal cord tissues. When CARM1 is accumulated in the cytoplasm, it translocates to the nucleus via a nuclear localization sequence identified in this study. Once inside the nucleus, CARM1 acts as a transcriptional co-activator by methylating histones and epigenetically regulating gene transcription (Figure 2). Consistent with previous reports, we identified CARM1 as the epigenetic regulator of autophagic and lysosomal genes such as Atg14 and Map1lc3b. Interestingly, CARM1 was also found to epigenetically regulate de novo fatty acid synthesis because it is enriched at the promotor region of the gene encoding ACAC/ACC, the rate-limiting enzyme for fatty acid synthesis. The enrichment of CARM1 at the promotors of these genes is significantly increased in cells lacking C9orf72, which could explain the observations that loss of C9orf72 leads to dysregulation of gene expression involved in autophagy and lipid metabolism.
In accordance with these findings, mammalian cells lacking C9orf72 or ALS-FTD patient-derived motor neurons show altered levels of free fatty acids or total lipids (Figure 2). These cells exhibit higher levels of lipid droplets, which are the main cellular organelle for lipid storage. The biogenesis of lipid droplets in the endoplasmic reticulum is enhanced as a result of the overloaded free fatty acids. The increased levels of free fatty acids in the cell can be explained by a combination of enhanced de novo fatty acid biosynthesis and overactivated autophagic digestion of lipid droplets, or lipophagy. The increased lipophagy in fibroblasts lacking C9orf72 is consistent with the increased autophagic flux and the enhanced biogenesis of lipid droplets in the endoplasmic reticulum. Together, these data suggest an intrinsic defect in lipid metabolism in cells deficient in C9orf72 including the disease-relevant cells from patients.
Although lipids are crucial energy resources for cells under nutrient stress, overloaded lipids could activate CYBB/NOX2 and damage mitochondria. CYBB/NOX2 is the most studied NADPH oxidase family member and was reported to be upregulated in the nervous tissues of ALS patients. We found that CYBB/NOX2 is increased in C9orf72-linked patient-derived motor neurons and spinal cords, suggesting that the upregulation of CYBB/NOX2 is a consequence of the dysregulated lipid metabolism that is relevant to the ALS-FTD pathology.
To directly investigate the role of CARM1 in C9orf72-dependent regulation of autophagy and lipid metabolism, we generated C9orf72 and CARM1 double-knockout fibroblasts and examined the impact of CARM1 deletion on C9orf72-dependent phenotypes. The removal of CARM1 reverses several phenotypes of the dysregulated lipid metabolism, such as abnormal lipophagy and de novo fatty acid synthesis, confirming the C9orf72-CARM1 axis in the regulation of lipid metabolism.
We observed that the role of C9orf72 in lipid metabolism is especially important under nutrient stress conditions. Upon glucose starvation, the relevant phenotypes in C9orf72-deficient cells, including the accumulation of CARM1 and the aberrant lipid metabolism, are more pronounced than under normal conditions. We propose that the C9orf72-CARM1 axis is an important mechanism for metabolic regulation under nutrient stress. CARM1 is the transcription coactivator that enhances autophagy and lipid metabolism, which provide alternative energy sources in time of nutrient scarcity. By negatively regulating CARM1 through its lysosomal degradation, C9orf72 appears to act as a key regulator in the negative feedback control of the starvation-induced activation of autophagy and lipid metabolism. During the starvation-induced stress response, C9orf72 serves to dampen the metabolic processes and prevent their overactivation. When C9orf72 is deficient in the cell, the metabolic balance during stress responses could be compromised. Considering that the nervous system might be particularly vulnerable to perturbation in metabolic balances, C9orf72 deficiency may underlie age-dependent decline in the health of neurons and glia. This is reminiscent of some lysosomal storage disorders, in which lysosomal functional deficiency leads to metabolic perturbation and neurodegenerative symptoms. Taken together, our results uncover a function of C9orf72 in the regulation of autophagy and lipid metabolism, via the epigenetic factor CARM1, under nutrient stress. With the evidence for the dysregulation of the C9orf72-CARM1 cascade in ALS-FTD, this regulatory mechanism could be implicated in other related neurodegenerative diseases.
Funding Statement
This work was supported by grants from NIH (NS074324, NS089616, NS110098), Packard Center for ALS Research at Johns Hopkins, Muscular Dystrophy Association, ALS Association, and Maryland Stem Cell Research Fund.
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Liu Y, Wang T, Ji YJ, et al. A C9orf72-CARM1 axis regulates lipid metabolism under glucose starvation-induced nutrient stress. Genes Dev. 2018;32(21–22):1380–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]