

ABSTRACT
The formation of protein aggregates is linked to several diseases collectively called proteinopathies. The mechanisms and the molecular players that control the turnover of protein aggregates are not well defined. We recently showed that TRIM16 acts as a key regulatory protein to control the biogenesis and degradation of protein aggregates. We show that TRIM16 interacts with, enhances K63-linked ubiquitination of, and stabilizes NFE2L2/NRF2 leading to its activation. The activated NFE2L2 upregulates the SQSTM1/p62 and ubiquitin pathway proteins, which interact with and ubiquitinate the misfolded proteins resulting in protein aggregate formation. TRIM16 is physically present around the protein aggregates and acts as a scaffold protein to recruit SQSTM1 and macroautophagy/autophagy initiation proteins for sequestration of the protein aggregates within autophagosomes, leading to their degradation. Hence, TRIM16 utilizes a two-pronged approach to safely dispose of the stress-induced misfolded proteins and protein aggregates, and protect cells from oxidative and proteotoxic stresses. This study could provide a framework for understanding the mechanisms of protein aggregate formation in neurodegeneration. The enhancement of TRIM16 activity could be a beneficial therapeutic approach in proteinopathies. On the flip side, cancer cells appear to hijack this machinery for their survival under stress conditions; hence, depleting TRIM16 could be a beneficial therapeutic strategy for treating cancer.
KEYWORDS: Aggrephagy, autophagy, cancer, neurodegeneration, NFE2L2/NRF2, oxidative stress, protein aggregates, protein homeostasis, protein quality control, SQSTM1/p62, TRIM16
Protein quality control mechanisms are vital for the maintenance of protein homeostasis (proteostasis) in the cell. Genetic mutations, cellular and environmental stresses, and aging can increase the protein misfolding beyond the capacity of the cell to properly fold them. In these conditions, misfolded proteins oligomerize into toxic aggregates leading to imbalanced proteostasis resulting in many pathological conditions collectively termed as proteinopathies (e.g., Alzheimer disease, Parkinson disease, amyotrophic lateral sclerosis). The recent literature suggests that the soluble oligomeric intermediate protein aggregates are much more toxic than fully formed insoluble protein aggregates (inclusion bodies). Thus, understanding the mechanisms of formation and degradation of protein aggregates will provide novel strategies to develop new and better therapies for the treatment of proteinopathies.
In our recent publication [1], we revealed a new mechanism by which protein aggregates are formed and degraded. We demonstrated that a TRIM family protein named TRIM16 orchestrates the SQSTM1-KEAP1-NFE2L2 axis to mediate stress-induced biogenesis of protein aggregates (Figure 1) . NFE2L2 (nuclear factor, erythroid 2 like 2) is a master transcription regulator of anti-oxidative and anti-proteotoxic responses. Under basal conditions, KEAP1 negatively regulates NFE2L2 by mediating its degradation; whereas in stress conditions, KEAP1 is degraded by SQSTM1-mediated selective autophagy, leading to release of NFE2L2 for a global transcriptional response. We found that TRIM16 interacts with SQSTM1, NFE2L2, and KEAP1. TRIM16 stabilizes SQSTM1 and NFE2L2 and destabilizes KEAP1, and hence positively regulate NFE2L2 activity. TRIM16 utilizes multiple mechanisms to stabilize NFE2L2. TRIM16 displaces KEAP1 from NFE2L2, and also it increases the SQSTM1-NFE2L2 interaction. The latter might happen at the SPRY domain of TRIM16 to which both NFE2L2 and SQSTM1 interact. TRIM16 also enhances the KEAP1-SQSTM1 interaction, which can lead to the sequestration and autophagic degradation of KEAP1. TRIM16 is an E3 ligase, and we found that TRIM16 via its SPRY domain mediates the K63-linked ubiquitination of NFE2L2 leading to its enhanced stability. We further found that TRIM16 regulates NFE2L2’s downstream oxidative and proteotoxic stress response. We discovered that one of the components of this NFE2L2 transcriptional response is the upregulation of ubiquitination machinery proteins. TRIM16 knockout cells are severely defective in the biogenesis of protein aggregates as are the NFE2L2-depleted cells. In addition, when we knock down the TRIM16-NFE2L2-regulated ubiquitination machinery proteins such as UBB and UBE2N, the total number of stress-induced protein aggregates are reduced significantly.
Figure 1.
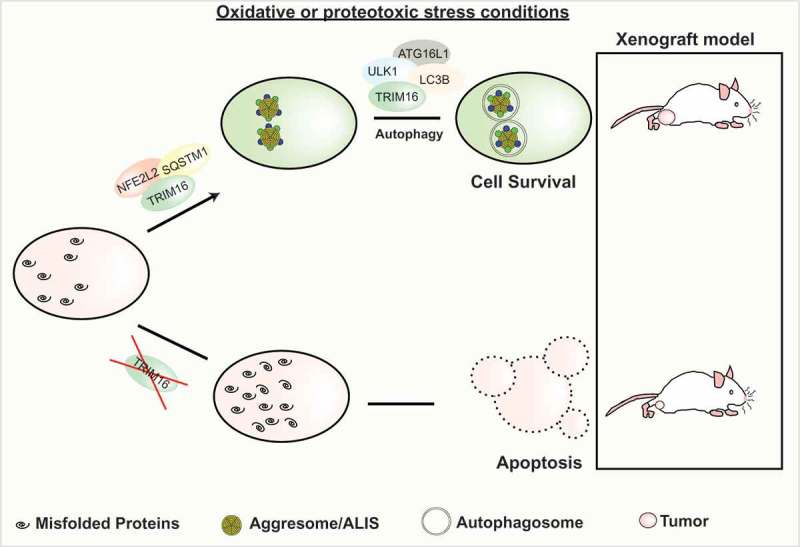
Graphical representation of the work. TRIM16 acts as key regulatory protein to mount a comprehensive response during oxidative stress conditions. We found that TRIM16, by regulating the SQSTM1/p62-NFE2L2/NRF2 axis, controls the formation of protein aggregates. Further, TRIM16 acts as a scaffold protein and recruits autophagy proteins, ULK1, ATG16L1, and LC3B to the protein aggregates to degrade them. Under stress conditions, TRIM16-depleted cells undergo apoptosis due to the toxicity of the increased number of misfolded proteins. In a xenograft model, the wild-type tumors are protected from oxidative stress, whereas a rapid regression of TRIM16-depleted tumors is observed. ALIS, aggresome-like induced structures.
In summary, we found that TRIM16 via an NFE2L2-mediated upregulation of a set of genes governs the biosynthesis of stress-induced protein aggregates. Interestingly, TRIM16 is also a transcriptional target of NFE2L2. Thus, TRIM16 and SQSTM1 are positive regulators of NFE2L2, and, in turn, both of them are positively regulated by NFE2L2 at transcriptional levels resulting in a regulatory positive feedback loop essential for oxidative and proteotoxic stress-induced formation of protein aggregates.
TRIM16 appears to be a multifunctional protein. Previously, TRIM16 was shown to recognize damaged endomembranes via LGALS3 (galectin 3), leading to assembly of the autophagy machinery over the damaged membrane for repair. In another study, TRIM16 was suggested to play a critical role in the autophagic secretion of IL1B/IL-1β. We found that TRIM16 is physically present around the protein aggregates. TRIM16 acts as a scaffold protein and its presence around the protein aggregates is required to recruit the autophagy receptor protein, SQSTM1, and also the autophagy initiation and elongation proteins including ULK1, ATG16L1, and MAP1LC3B, leading to selective degradation of protein aggregates. Hence, TRIM16 induces 2 different cell protective mechanisms during stress conditions. Along these lines, we found that TRIM16 CRISPR knock out HeLa cells show increased apoptotic cell death when exposed to oxidative and proteotoxic stresses.
This study could have implications for understanding the mechanisms of neurodegeneration. Although the protective role of NFE2L2 is well recognized in neurodegenerative disorders, its function in the formation of stress-induced protein aggregates was never established prior to this study. Even though the mechanism that is discovered in our study is not tested in neuronal cell lines; it is tempting to speculate that a TRIM16-NFE2L2-autophagy axis could be important for the turnover of neurodegeneration-related protein aggregates. Further, studies are required to establish whether increasing the activity of TRIM16 could be important for therapeutic intervention of neurodegenerative diseases. One line of evidence that supports this notion is a recent work where TRIM16 is shown to interact with and stabilize TARDBP/TDP-43 (TAR DNA binding protein), an aggregate-prone protein that is identified as the major component of the neuronal cytoplasmic inclusion bodies deposited in amyotrophic lateral sclerosis.
There is another interesting edge to our study; we found that in a xenograft model, the tumor formed by TRIM16 knockout HeLa cells regresses very rapidly (no significant regression in control tumors) when the mice are injected with arsenic trioxide (a known oxidative stress inducer). The data presented in this work suggest that TRIM16 protects cancer cells from the cytotoxicity of oxidative stress-induced protein aggregates by safely disposing of them utilizing both NFE2L2 and autophagy. Our work supports a study by Chen et al. (2017) showing that TRIM proteins-mediate an augmented capacity to degrade misfolded proteins, which alleviates the oxidative stress associated with cancerous growth and is required for both the initiation and maintenance of malignant phenotypes.
Funding Statement
This work is supported by the Wellcome Trust/Department of Biotechnology (DBT) India Alliance [IA/I/15/2/502071] fellowship, ILS core funding (Department of Biotechnology, India), and Early Career Reward [SERB, ECR/2016/000478] to Santosh Chauhan. Subhash Mehto is supported by a fellowship from SERB [NPDF, PDF/2016/001697]. Swati Chauhan is supported by fellowship from DST [SR/WOS-A/LS-9/2016].
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Jena KK, Kolapalli SP, Mehto S, et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. Embo J. 2018. September 14;37(18):pii: e98358 Epub 2018 Aug 24. [DOI] [PMC free article] [PubMed] [Google Scholar]