

ABSTRACT
Macroautophagy/autophagy is a conserved ubiquitous pathway that performs diverse roles in health and disease. Although many key, widely expressed proteins that regulate autophagosome formation followed by lysosomal fusion have been identified, the possibilities of cell-specific elements that contribute to the autophagy fusion machinery have not been explored. Here we show that a macrophage-specific isoform of the vacuolar ATPase protein ATP6V0D2/subunit d2 is dispensable for lysosome acidification, but promotes the completion of autophagy via promotion of autophagosome-lysosome fusion through its interaction with STX17 and VAMP8. Atp6v0d2-deficient macrophages have augmented mitochondrial damage, enhanced inflammasome activation and reduced clearance of Salmonella typhimurium. The susceptibility of atp6v0d2 knockout mice to DSS-induced colitis and Salmonella typhimurium-induced death, highlights the in vivo significance of ATP6V0D2-mediated autophagosome-lysosome fusion. Together, our data identify ATP6V0D2 as a key component of macrophage-specific autophagosome-lysosome fusion machinery maintaining macrophage organelle homeostasis and, in turn, limiting both inflammation and bacterial infection.
Abbreviations: ACTB/β-actin: actin, beta; ATG14: autophagy related 14; ATG16L1: autophagy related 16-like 1 (S. cerevisiae); ATP6V0D1/2: ATPase, H+ transporting, lysosomal V0 subunit D1/2; AIM2: absent in melanoma 2; BMDM: bone marrow-derived macrophage; CASP1: caspase 1; CGD: chronic granulomatous disease; CSF1/M-CSF: colony stimulating factor 1 (macrophage); CTSB: cathepsin B; DSS: dextran sodium sulfate; IL1B: interleukin 1 beta; IL6: interleukin 6; IRGM: immunity-related GTPase family M member; KO: knockout; LAMP1: lysosomal-associated membrane protein 1; LC3: microtubule-associated protein 1 light chain 3; LPS: lipo-polysaccaride; NLRP3: NLR family, pyrin domain containing 3; PYCARD/ASC: PYD and CARD domain containing; SNARE: soluble N-ethylmaleimide-sensitive factor attachment protein receptor; SNAP29: synaptosomal-associated protein 29; SQSTM1/p62: sequestosome 1; STX17: syntaxin 17; TLR: toll-like receptor; TNF: tumor necrosis factor ; TOMM20: translocase of outer mitochondrial membrane 20; ULK1: unc-51 like kinase 1; VAMP8: vesicle-associated membrane protein 8; WT: wild type; 3-MA: 3-methyladenine
KEYWORDS: ATP6V0D2, autophagosome-lysosome fusion, IL1B, inflammasome activation, Salmonella typhimurium
Introduction
Autophagy is a vital eukaryotic process that degrades cytoplasmic contents such as aggregated proteins and compromised organelles, to maintain cellular homeostasis [1,2]. In the innate immune system, autophagy controls infection by restricting the growth of intracellular bacteria either in the cytosol or in vacuoles [3]. In addition, autophagy clears various inflammasome agonists and components [4], such as damaged mitochondria, an upstream activating signal for NLRP3 inflammasome activation [5], thus regulating inflammation. Defects of autophagy result in excess IL1B secretion [6,7]. Furthermore, polymorphisms in autophagy-associated genes, such as ATG16L1 and IRGM, are associated with autoimmune and inflammatory diseases, particularly inflammatory bowel disease [8,9].
Classical autophagy begins with formation of a double-membrane phagophore that engulfs cytosolic materials and seals to form an autophagosome [10,11]. The process culminates in fusion of an autophagosome with a lysosome to generate the autolysosome, allowing lysosomal hydrolases to break down the contents within this organelle [12]. Potentially many factors regulate this fusion: proteins that associate with the initial development of the phagophore including ATG16L1 and ULK1 are missing from autophagosomes at the time of their fusion with lysosomes. Conversely, a set of SNARE proteins, including STX17, SNAP29 and VAMP8, are essential for the fusion between autophagosomes and lysosomes [13]. Yet these critical components are only part of a larger complex of proteins required for efficient and regulated autophagosome-lysosome fusion [14].
The activities of lysosomal hydrolases require an acidic environment, which is achieved by the proton pump activity of V-ATPase. V-ATPase is a large multiple subunit complex consisting of a cytosolic V1 domain with 8 subunits that mediates ATP hydrolysis, and an integral membrane V0 domain with 6 subunits in yeast and the additional Ac45 protein in more complex eukaryotes that has proton pump activity [15]. The ATP6V0D2/subunit d2, together with ATP6V1D/subunit D and ATP6V1F, forms a central stalk of the V-ATPase that rotates with the associated Atp6V0C/proteolipid ring and transports proton across the membrane; this rotation is driven by ATP hydrolysis within the V1 domain [15]. There are 2 isoforms of ATP6V0D/subunit d, ATP6V0D1 and ATP6V0D2, which share 82% sequence identity [16]. While ATP6V0D1 is ubiquitously expressed, ATP6V0D2 has been reported to have more restricted tissue expression, in osteoclasts and kidney tubules [17–19].
In the present study, we provide data showing that ATP6V0D2 was expressed in macrophages and was regulated by stimulation with lipopolysaccaride (LPS). Given the restricted expression pattern of Atp6v0d2, we speculated that this member of the V-type H+-ATPase complex may play a specific role in macrophages where there is a need for specialized autophagy machinery. Using a genetic approach and in vivo models, we found that ATP6V0D2 plays a complementary but separate role to ATP6V0D1 in autophagy. We find that deficiency of ATP6V0D2 resulted in enhanced inflammasome activation and accumulation of damaged mitochondria in murine macrophages. Furthermore, deficiency of atp6v0d2 led to a reduced ability to clear Salmonella and heightened inflammation in DSS-induced colitis in vivo. Together, these data identify a nonredundant role for ATP6V0D2 in the formation of the autolysosome that enables efficient clearing of damaged organelles and ingested bacteria, which in turn limits inflammation.
Results
Genetic deletion of Atp6v0d2 leads to enhanced inflammasome activation in macrophages and in LPS-induced peritonitis
To explore the role of novel genes in LPS-driven inflammation, we analyzed the transcriptome data of LPS-treated macrophages from Noubade et al and found that many genes suppressed by LPS, control lysosome function [20] (Figure S1(a)). Within this list we found that expression of ATP6V0D2 was most inhibited by the addition of LPS (Figure 1(a)). ATP6V0D2 is highly expressed in bone marrow-derived macrophages (BMDMs), and modestly expressed in mast cells but not in lymphoid cells (Figure S1(b,c)). Confirming this, we found that bacteria-derived LPS strongly inhibited the basal Atp6v0d2 mRNA and protein expression in BMDMs in a dose- and time-dependent manner (Figure 1(b, c). Furthermore, other TLR ligands had a similar effect on Atp6v0d2 expression (Figure S1(d–f)).
Figure 1.
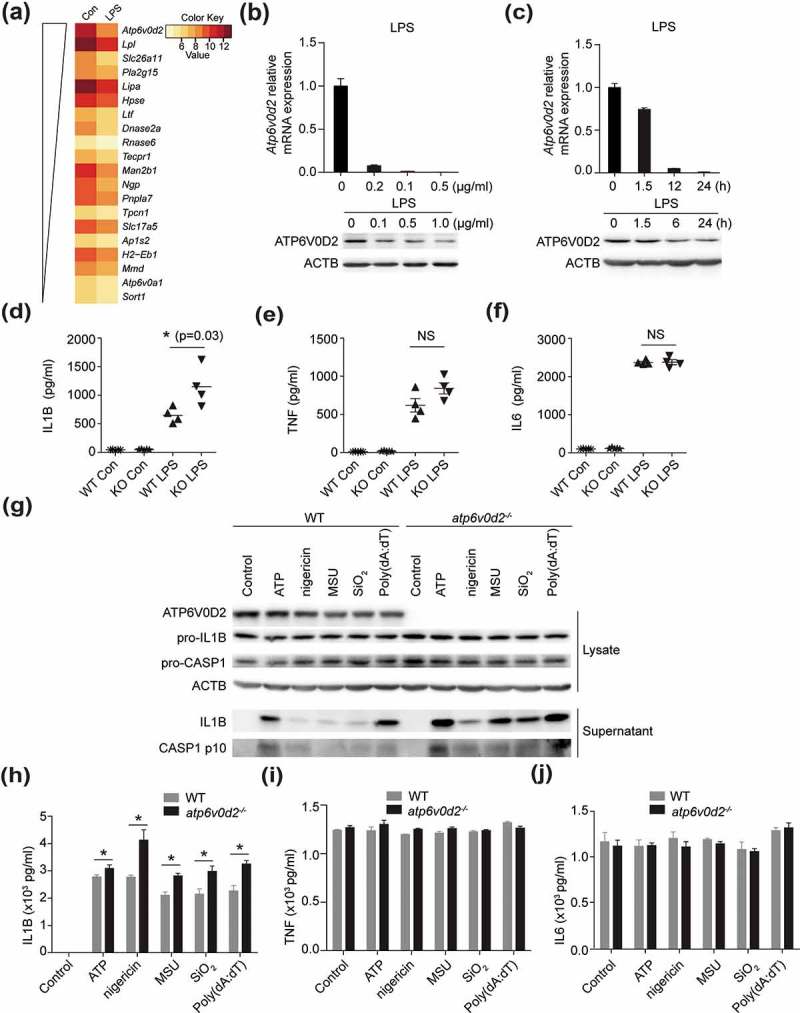
ATP6V0D2 inhibits inflammasome activation both in vitro and in vivo. (a) Heatmap showing relative expression of lysosome genes suppressed by LPS, data taken from GSE53986. (b and c) qRT-PCR analysis of Atp6v0d2 mRNA expression and immunoblot analysis of ATP6V0D2 in BMDMs stimulated with 20, 100, or 500 ng/ml LPS for 4 h (b), and 20 ng/ml LPS for 1.5, 6, or 24 h (c). (d–f) ELISA of IL1B, TNF and IL6 in serum of WT or atp6v0d2−/- mice intraperitoneally injected with either PBS (Con) or LPS (20 mg/kg) for 4 h. (g–j) Immunoblot measurement of IL1B and CASP1 (g) together with ELISA measurement of IL1B (h), TNF (i) or IL6 (j) in cultured supernatants of LPS-primed WT or atp6v0d2−/- BMDMs (LPS 100 ng/ml) for 4 h followed by ATP (5 mM), nigericin (5 μM), MSU (400 μg/ml), SiO2 (500 μg/ml) or 1.5 μg/ml poly (dA:dT) transfected with Lipofectamine 2000. Data are representative of at least 3 independent experiments (c–j). Histograms depict mean values ± s.e.m. *P < 0.05.
To elucidate the role of ATP6V0D2 in the immune system, we generated mice lacking Atp6v0d2 by targeting exon 1. Consistent with a previous report [21], atp6v0d2−/- mice were born at expected Mendelian frequencies with no sex bias and did not manifest any gross developmental defects (data not shown). To explore the role of ATP6V0D2 in inflammation in vivo, we used a model of endotoxic shock by intraperitoneal (i.p.) injection of E. coli LPS into WT and atp6v0d2−/- mice. atp6v0d2−/- mice exhibited significantly elevated levels of circulating IL1B after LPS treatment (Figure 1(d)). By contrast, the serum levels of inflammasome-independent cytokines, TNF and IL6, were comparable (Figure 1(e,f)).
Next, we assessed the function of ATP6V0D2 on inflammasome activation as this is known to regulate secretion of IL1B [22,23]. Stimulation of WT and atp6v0d2−/- BMDMs with various NLRP3 inflammasome agonists led to significantly greater IL1B secretion in atp6v0d2−/- macrophages than WT macrophages (Figure 1(g,h)). By contrast, TNF and IL6 secretion was not affected (Figure 1(i,j)). Together, these data demonstrate that ATP6V0D2 constrains inflammasome activation both in vitro in isolated macrophages and in vivo.
Macrophage intrinsic ATP6V0D2 restricts inflammation in DSS-induced colitis
Elevated IL1B production can exacerbate gut pathology, particularly the DSS model of colitis [24]. Using this model, atp6v0d2−/- animals demonstrated significantly increased weight loss, shortening of colon length, and increased pathology compared with littermate controls after administration of DSS (Figures 2(a–e) and S2(a)).
Figure 2.
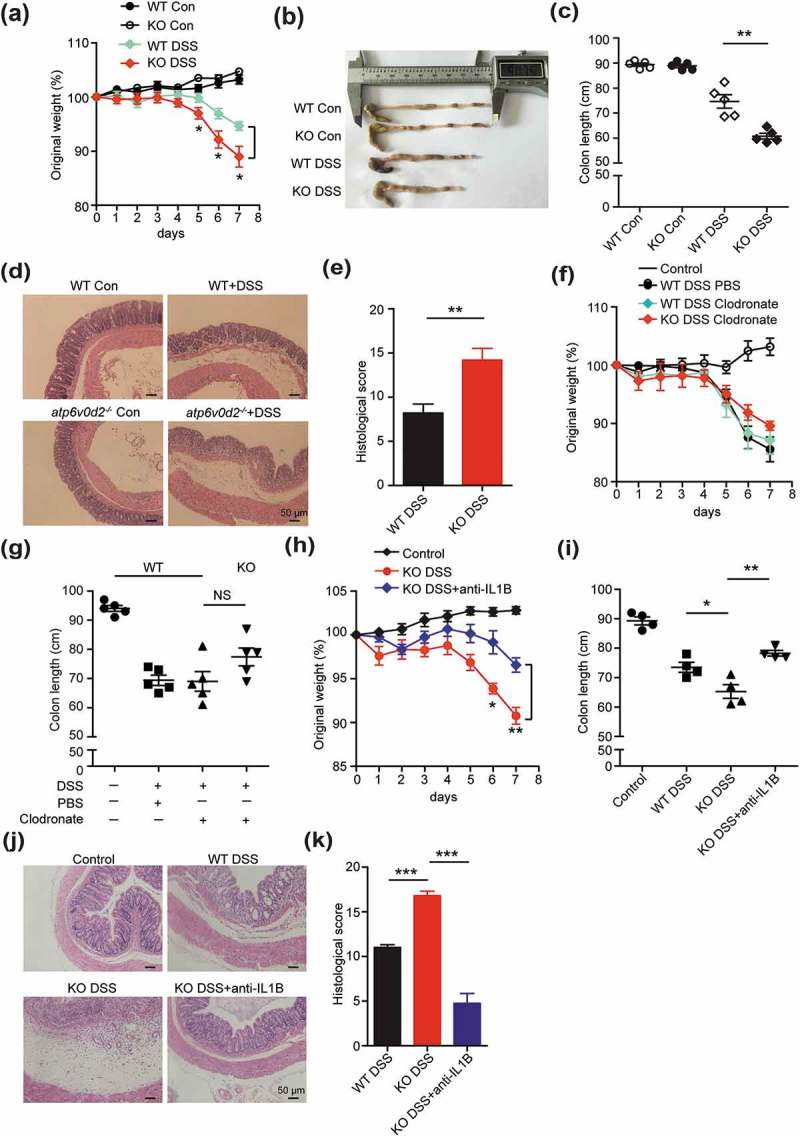
ATP6V0D2 inhibits inflammasome-dependent colitis. (a–e) Acute colitis was induced in WT and atp6v0d2−/- mice by adding 2.5% DSS to drinking water ad libitum for 7 consecutive days. Comparison of body weight (a), representative examples (b) and mean values (c) of colon length, histology 20X (d) and histology score (e) between WT and atp6v0d2−/- mice after 7 days. Data are shown from 1 experiment (n = 5) that is representative of 3 separate experiments. (f and g) DSS colitis was induced in WT and atp6v0d2−/- mice upon depletion of macrophages. Comparison of body weight (f) and mean values (g) of colon length between WT and atp6v0d2−/- mice after 7 days (n = 5). (h–k) DSS colitis was repeated in WT and atp6v0d2−/- mice in the presence or absence of anti-IL1B (300 μg/mouse) injected subcutaneously at day 1, 3, and 5. Comparison of mean weight loss (h), mean values (i) of colon length, histology (j) and histological scores (k). Data shown are from 1 experiment (n = 4–5) that is representative of 3 (a–e) or 2 (f–k) separate experiments. Histograms depict mean values ± s.e.m.*P < 0.05; **P < 0.01;***P < 0.001.
In steady state, in contrast with macrophages, colon epithelial cells and colon tissue did not express ATP6V0D2 (Figure S2(b,c)), indicating a macrophage-specific role of ATP6V0D2 in colitis. To confirm that the enhanced pathology in atp6v0d2−/- animals was due to a macrophage-intrinsic effect, we used clodronate to deplete macrophages (Figure S2(d)) and compared the disease severity. Depletion of macrophages completely reversed the susceptibility of atp6v0d2−/- mice to DSS, compared with wild-type animals (Figures 2(f,g) and S2(e-f)). To confirm that these findings were dependent on elevated IL1B expression in atp6v0d2−/- animals, the experiment was repeated with the addition of anti-IL1B, which again reversed the differences seen in weight loss, colon length, and histological score (Figures 2(h-k) and S2(g)).
Enhanced accumulation of damaged mitochondria in atp6v0d2−/- macrophages
Next we investigated whether the enhanced inflammasome activation in the atp6v0d2−/- macrophages was due to the augmented mitochondrial damage. We explored the effect of ATP6V0D2 on mitochondrial membrane potential by flow cytometry. The proportion of depolarized mitochondria, indicated by a loss of MitoTracker Deep Red stain, was increased in the Atp6v0d2-deficient macrophages upon stimulation with various NLRP3 inflammasome agonists (Figure 3(a,b). To confirm our flow cytometric findings, we directly assessed mitochondrial integrity in WT and atp6v0d2−/- BMDMs using electron microscopy (EM). Whereas LPS plus ATP induced mitochondria swelling in WT cells, this effect was strongly exaggerated in atp6v0d2−/- BMDMs, which were characterized by morphological alteration, the loss of cristae, and the presence of electron-dense particles inside the mitochondria (Figure 3(c)).
Figure 3.
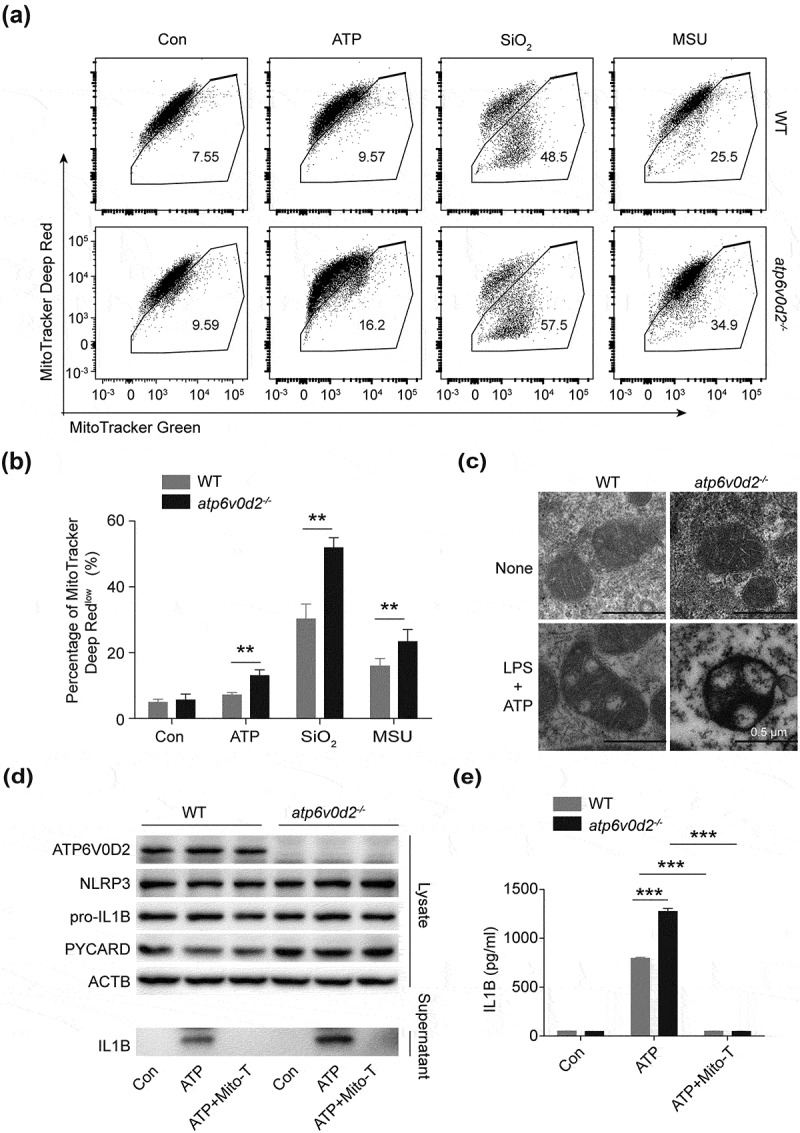
ATP6V0D2 limits accumulation of damaged mitochondria after inflammasome activation. (a and b) Mitochondria membrane integrity, determined with MitoTracker Green and MitoTrack Deep Red staining followed by flow cytometry in WT and atp6v0d2−/- BMDMs left unstimulated or LPS-primed (100 ng/ml, 4 h) then stimulated with ATP (2 mM), SiO2 (250 μg/ml) and MSU (200 μg/ml). Representative flow cytometry (a) and a histogram plot of percentage of Mitotracker Deep Red low (b) is shown. (c) Images of mitochondrial structure determined by transmission electron microscopy in WT and atp6v0d2−/- BMDMs left unstimulated or LPS-primed (20 ng/ml, 4 h) then stimulated with ATP (2 mM) for 1 h. (d and e) Immunoblot analysis of NLRP3, PYCARD/ASC and pro-IL1B in the cell lysate; IL1B in the supernatant of cultured WT and atp6v0d2−/- BMDMs. Cells were primed with LPS (100 ng/ml, 4 h) alone, or stimulated with ATP (5 mM) for 30 min after priming with (LPS+ATP+TEMPO) or without Mito-TEMPO (500 µM) (LPS+ATP) (d). IL1B concentration determined by ELISA in the supernatant of cultured WT and atp6v0d2−/- BMDMs (e). The data are representative of 3 experiments, histograms depict mean values ± s.e.m.**P < 0.01; ***P < 0.001.
We next asked whether the enhanced inflammasome activation in atp6v0d2−/- BMDMs was due to the accumulation of damaged mitochondria. Inhibition of the mitochondria ROS and intracellular ROS by Mito-TEMPO and N-acetylcysteine (NAC), respectively, resulted in reduced IL1B secretion in atp6v0d2−/- BMDMs, which was comparable to the WT BMDMs (Figures 3(d,e) and S3(a)). Conversely, overexpression of ATP6V0D2 using retroviral infection, resulted in reduced mitochondrial ROS production (Figure S3(b,c)), IL1B secretion, and CASP1 (caspase 1) activation upon inflammasome stimulation (Figure S3(d,e)). Together, these data suggest that ATP6V0D2 limits inflammasome activation in macrophages through its ability to clear damaged mitochondria.
ATP6V0D2 is required for the macrophage-specific completion of autophagy
Because autophagy plays a central role in the removal of damaged mitochondria, we speculated that ATP6V0D2 is required for autophagy in general. Stimulation of ATP led to a significant increase of autophagosomes inside atp6v0d2−/- BMDMs compared with WT cells (Figure 4(a,b)), suggesting either an increased autophagy flux or a defect in the completion of autophagy. To test this, we transduced a lentiviral plasmid encoding GFP-RFP-LC3 into WT and knockout (KO) BMDMs. Under steady state conditions, KO cells had accumulated GFP-RFP puncta that were colocalized; after autophagy induction with rapamycin, the GFP was degraded in the WT cells but the majority of LC3 dots in KO cells were both RFP and GFP positive, indicating a defect of lysosomal delivery (which would result in quenching of the GFP signal) in KO cells (Figure 4(c,d) upper and middle panels). Addition of bafilomycin A1 blocked degradation in both WT and KO cells (Figure 4(c,d) lower panel). To further substantiate this, WT and atp6v0d2−/- BMDMs were treated with rapamycin and the levels of SQSTM1/p62 and LC3-II were measured. With time, SQSTM1/p62, PYCARD/ASC and LC3-II, were degraded in WT but not atp6v0d2−/- BMDMs (although the LC3 level was slightly reduced), suggesting that ATP6V0D2 was required for degradation of autophagosomes (Figure 4(e)). However, cultured T cells and B cells from WT and atp6v0d2−/- mice showed no difference in SQSTM1/p62 and LC3-II levels upon rapamycin treatment, indicating the regulatory effect of ATP6V0D2 on autophagy was macrophage specific (Figure S4(a,b)).
Figure 5.
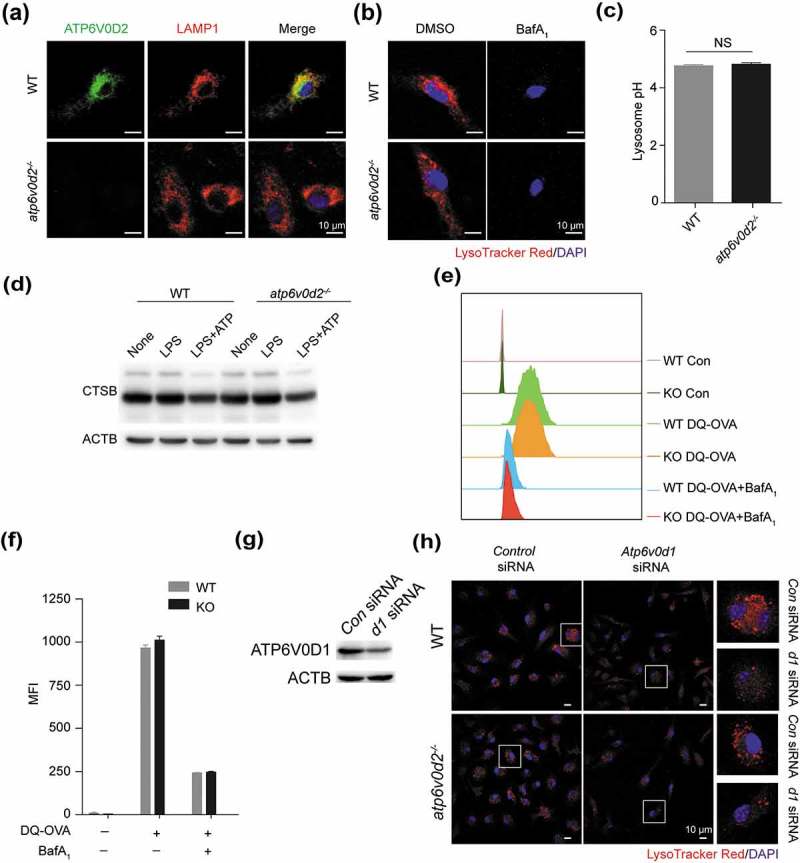
ATP6V0D1 but not ATP6V0D2 is required for lysosome acidification and maturation. (a and b) Intracellular distribution of ATP6V0D2 and colocalization with LAMP1 (a) or LysoTracker Red staining (b) in WT and atp6v0d2−/- BMDMs treated with DMSO or bafliomycin A1 for 30 min and examined by confocal microscopy. (c) The pH of lysosomes of WT and atp6v0d2−/- BMDMs was measured with LysoSensor staining. (d) Immunoblot of CTSB analysis of 100 ng/ml LPS-primed WT and atp6v0d2−/- BMDMs with or without ATP (5 nM) for 30 min. (e and f) WT and atp6v0d2−/- BMDMs were incubated with FITC-labeled DQ-OVA for 1 h. Intracellular FITC fluorescence intensity measured by flow cytometry of a representative sample is shown (e) together with a histogram of mean values (f). (g) Immunoblot analysis of ATP6V0D1 24 h after transfection of control siRNA (Con) or Atp6v0d1 siRNA (d1) in WT BMDMs. (h) LysoTracker Red staining of WT and atp6v0d2−/- BMDMs after transfection of control siRNA or Atp6v0d1 siRNA in WT and atp6v0d2−/- BMDMs. The data shown are representative of 3 (a–f) or 2 (g-h) independent experiments; histograms depict mean values ± s.e.m. MFI, mean fluorescence intensity.
Figure 4.
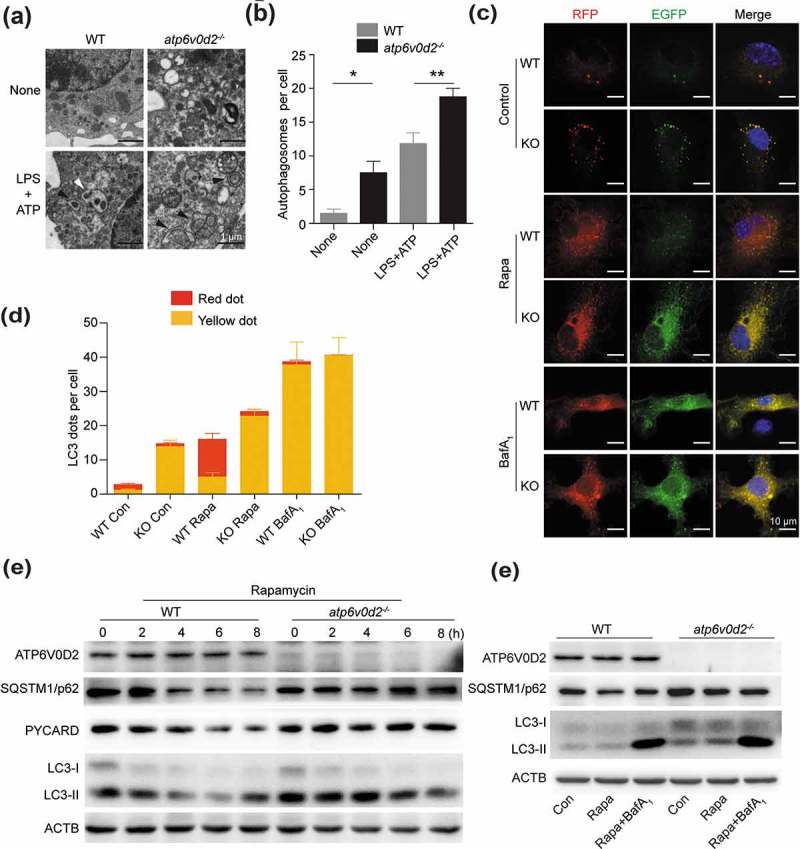
ATP6V0D2 is essential for the degradation of autophagic components. (a and b) Autophagosome analysis by transmission electron microscopy of WT and atp6v0d2−/- BMDMs that were either unstimulated, or primed with LPS (20 ng/ml) for 4 h followed by 2 mM ATP stimulation for 1 h. Representative images are shown (a) together with mean values of numbers of autophagosomes per cell (b). The average number of autophagosome per cell (10 cells) were quantified manually. (c and d) WT and atp6v0d2−/- BMDMs that were transduced with RFP-GFP-LC3-expressing lentivirus. Cells were untreated, or treated with rapamycin (Rapa) or bafilomycin A1 (BafA1) for 6 h. Fluorescence were examined by confocal microscopy (c). The mean number of RFP and GFP dots were determined manually (n = 10 cells) (d). (e) Immunoblot analysis of ATP6V0D2, SQSTM1/p62, PYCARD/ASC, and LC3 in WT and atp6v0d2−/- BMDMs that were either untreated or treated with rapamycin (100 ng/ml) for 2–8 h. (f) Immunoblot analysis of ATP6V0D2, SQSTM1/p62, and LC3 in WT and atp6v0d2−/- BMDMs that were either untreated, treated with rapamycin alone (100 ng/ml), or rapamycin (100 ng/ml) in the presence of bafilomycin A1 (100 nM) for 3 h. The data are representative of 3 independent experiments, histograms depict mean values ± s.e.m.*P < 0.05; **P < 0.01.
Bafilomycin A1 enhanced SQSTM1/p62 and LC3-II in both WT and KO macrophages, however, after bafilomycin A1 treatment, the levels of SQSTM1/p62 and LC3-II were comparable between WT and KO macrophages (Figure 4(f)). Furthermore, addition of 3-MA enhanced IL1B secretion into the supernatant, whereas rapamycin inhibited secretion in WT BMDMs, but to a lesser extent in KO macrophages (Figure S4(c)). More importantly, the secreted levels of IL1B were comparable between WT and KO macrophages (Figure S4(c)). Taken together, these data suggest that the effect of ATP6V0D2 deficiency on inflammasome activation was related to its effect on autophagosome degradation.
ATP6V0D1 but not ATP6V0D2 is required for lysosome acidification and maturation in macrophages
Using immunofluorescence costaining of ATP6V0D2 and LAMP1, a lysosome membrane marker, we found that the majority of ATP6V0D2 was localized in the lysosome (Figure 5(a)). Next, we explored if ATP6V0D2 is required for acidification and maturation of lysosomes in macrophages. Surprisingly, deletion of ATP6V0D2 did not alter lysosome acidification as measured by fluorescence of pH-sensitive dyes (Figure 5(b,c)).
CTSB (cathepsin B) is a major lysosome hydrolase, and its maturation requires lysosomal acidification. We compared CTSB maturation in WT and atp6v0d2−/- BMDMs. Deficiency of ATP6V0D2 did not affect the processing and maturation of CTSB (Figure 5(d)). We measured the ability of WT and atp6v0d2−/- BMDMs to degrade DQ-OVA and found no difference. The addition of the lysosomal acidification inhibitor bafilomycin A1 had a similar effect in inhibiting DQ-OVA in both groups (Figure 5(e,f)). By contrast, knockdown of Atp6v0d1, significantly disrupted the acidification of lysosomes (Figure 5(g,h)), indicating that ATP6V0D1 and ATP6V0D2 have nonredundant roles in regulation of lysosomal function.
ATP6V0D2 promotes autophagosome-lysosome membrane fusion
Completion of autophagy is dependent on both lysosome function and autophagosome-lysosome fusion. As the former was intact in the absence of ATP6V0D2, we investigated the latter. To test this, we compared the colocalization of LC3-II with LAMP1, in WT and atp6v0d2−/- cells upon inflammasome induction. In the absence of ATP6V0D2, the colocalization of LC3-II and LAMP1 was significantly reduced upon either nigericin or ATP stimulation, and the difference was abolished by the addition of bafilomycin A1 (Figure 6(a,b)). We sought to confirm this result by assessing colocalization of a mitochondrial marker, TOMM20, and the autophagosome protein STX17, with LAMP1. After stimulation with NLRP3 agonists, both TOMM20 and STX17 formed punctate structures (Figure S5(a)). These TOMM20 and STX17 puncta were colocalized with LAMP1 in the WT BMDMs but the colocalization was significantly reduced in atp6v0d2−/- BMDMs cells (Figure S5(b)).
Figure 6.
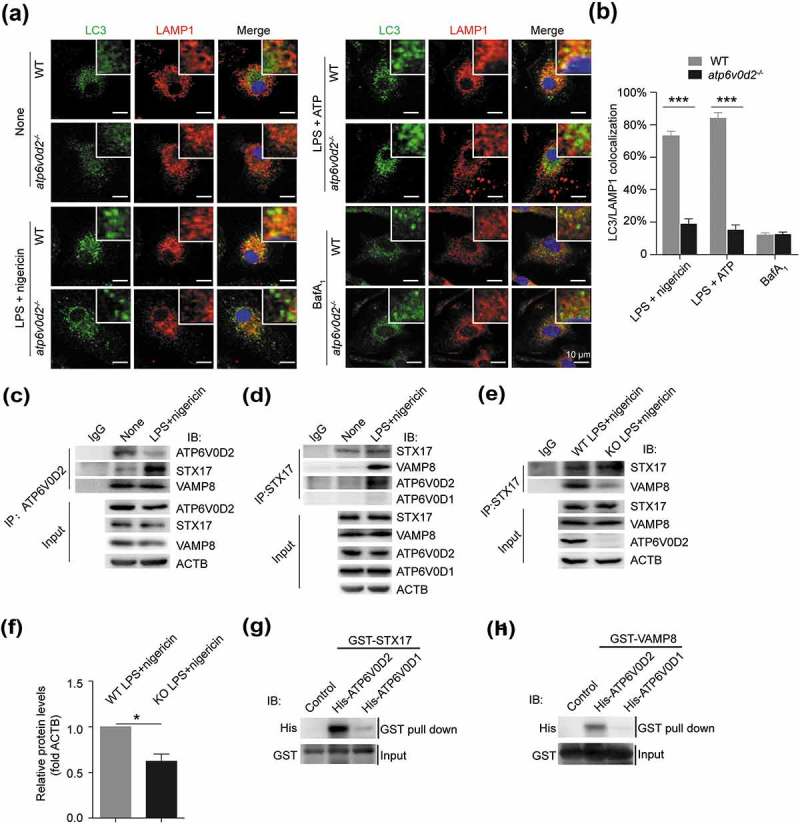
ATP6V0D2 promotes the fusion between autophagosomes and lysosomes. (a and b) Colocalization of LC3 with LAMP1 examined by confocal microscopy in WT and atp6v0d2−/- BMDMs unstimulated or stimulated with either LPS plus nigericin, LPS plus ATP or in the presence of bafilomycin A1 (a). Histograms showing colocalization of LC3 with LAMP1 that was determined manually (b). Bar charts show mean values ± s.e.m. of colocalized puncta in each field (n = 3) ***P < 0.001. (c and d) WT BMDMs were either unstimulated or stimulated with LPS+nigericin. Cell lysates were immunoprecipitated either with anti-ATP6V0D2 (c) or STX17 (d). The immune complex was subjected to western blotting and probed with antibodies as indicated. (e and f) WT and atp6v0d2−/- BMDMs were primed with LPS for 4 h, followed with stimulation of nigericin for 1 h. Cells lysates were immunoprecipitated with anti-STX17, subjected to western blotting and probed with either anti-STX17 antibody or anti-VAMP8 (e). Histograms depict mean values of band intensity (f) ± s.e.m.*P < 0.05. (g and h) Interaction between purified recombinant His-tagged ATP6V0D2 and ATP6V0D1 with GST-STX17 (g) or GST-VAMP8 (h) using in vitro GST affinity-isolation assay followed by immunoblot.
Next, we asked if ATP6V0D2 interacts with SNARE proteins during autophagosome-lysosome fusion. In transfected HEK293T cells, FLAG-tagged STX17, SNAP29 and VAMP8 were found to associate with intact HA-tagged ATP6V0D2 but not HA-tagged ATP6V0D1 (Figure S6(a)). To determine which part of ATP6V0D2 interacts with the SNARE complex, we generated 3 truncated forms of ATP6V0D2 and found none of those interacted with the SNARE complex (Figure S6(a)).
In primary macrophage cell lysates, which were immunoprecipitated with anti-ATP6V0D2, western blotted and probed for STX17 and VAMP8, we found inflammasome stimulation-dependent association of the former, but constitutive association of the latter, despite reduced expression of ATP6V0D2 upon stimulation (Figure 6(c)). We repeated this experiment, immunoprecipitating cell lysates with anti-STX17. Again, VAMP8 and ATP6V0D2 could only be found in the immunoprecipitated lysates of cell after stimulation with LPS+nigericin whereas ATP6V0D1 could never be detected (Figure 6(d)). The experiment was repeated using LPS+nigericin-stimulated wild-type and Atp6v0d2-deficient macrophage cell lysates. We found a significant reduction in the association of STX17 with VAMP8 in the absence of ATP6V0D2 (Figure 6(e,f)). A GST affinity isolation assay revealed that both STX17 and VAMP8 were associated with ATP6V0D2 but not ATP6V0D1, indicating a direct interaction between STX17 or VAMP8 with ATP6V0D2 (Figure 6(g,h)). Thus, ATP6V0D2, but not ATP6V0D1, forms a complex with VAMP8 and STX17, and is required for the optimal stabilization of this complex in activated macrophages.
Loss of ATP6V0D2 in macrophages impairs clearance of ingested bacteria
Next, we asked if this defect in autophagosome-lysosome fusion would affect other macrophage functions that are dependent on this process. The same machinery that enables autophagy to play a major role in the ability of macrophages to clear phagocytosed bacteria including S. typhimurium. To investigate this, we infected WT and atp6v0d2−/- BMDM cells with S. typhimurium and measured the ability of infected macrophages to clear ingested bacteria using a gentamicin protection assay. After incubation of macrophages with Salmonella bacteria, we found significantly higher numbers of bacteria in the lysate of atp6v0d2−/- BMDM cells (Figure 7(a)), indicating that ATP6V0D2 is necessary for the clearance of phagocytosed intracellular bacteria. To better characterize the clearance defect of atp6v0d2−/- macrophages, we next compared the ability of WT and atp6v0d2−/- BMDM cells to phagocytose GFP-labeled bacteria. When incubated with GFP-labeled S. typhimurium, there was enhanced GFP accumulation in the Atp6v0d2-deficient macrophages with time (Figure 7(b)). However, when macrophages were incubated with FITC-dextran, there was no difference in the degree of FITC stain between WT and atp6v0d2−/- cells (Figure 7(c)), indicating that ATP6V0D2 does not affect phagocytosis.
Figure 7.
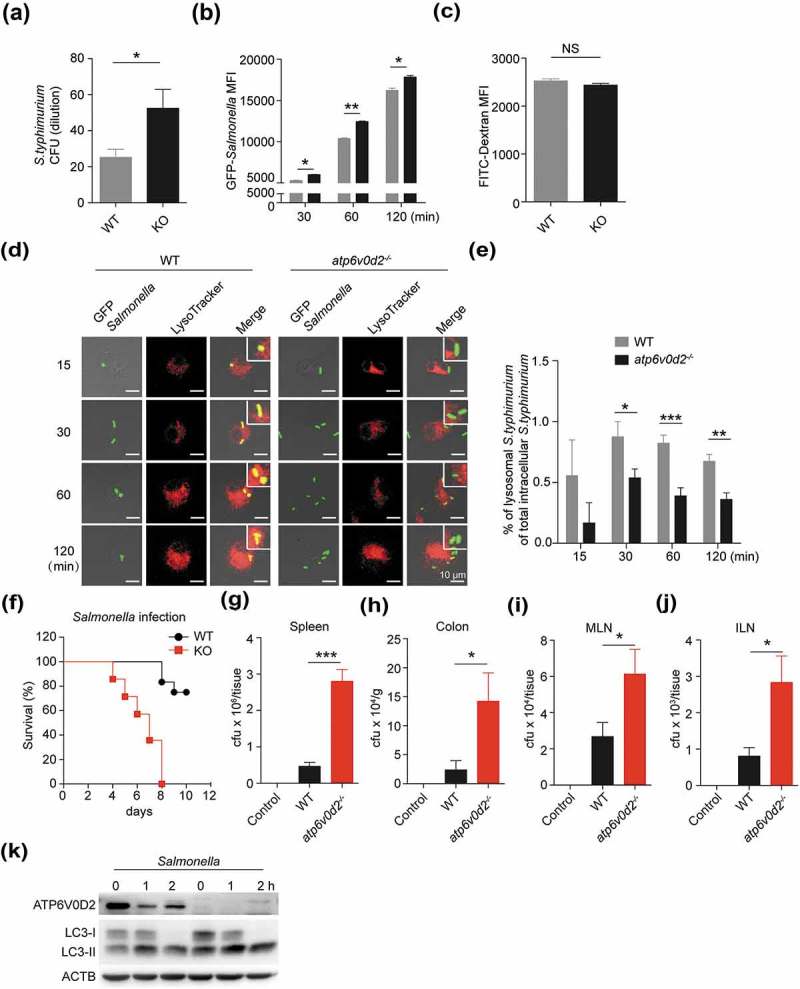
ATP6V0D2 is required for the clearance of Salmonella typhimurium. (a) WT and atp6v0d2−/- BMDMs were infected with S. typhimurium for 1 h. After washing and incubation with gentamicin-containing medium for 1 h, cells were lysed and serial dilutions of cell lysate were plated onto agar plates for 24 h at 37ºC and resulting bacterial colonies are shown. (b) WT and atp6v0d2−/- BMDMs were infected with GFP-S. typhimurium for 30, 60, and 120 min. Mean fluorescence intensities (MFI) of intracellular FITC/GFP measured with flow cytometry are shown. (c) WT and atp6v0d2−/- BMDMs were incubated with FITC-Dextran for 60 min. Mean fluorescence intensities measured with flow cytometry are shown. (d and e) WT and atp6v0d2−/- BMDMs were infected with GFP-S. typhimurium for 15, 30, 60, and 120 min. Colocalization of LysoTracker with GFP, analyzed by confocal microscopy (d). Quantification of mean percentage of colocalized lysosome staining with GFP-labelled bacteria is shown (e). (f–j) WT and atp6v0d2−/- mice were infected with 1 × 108 Salmonella by oral gavage. Kaplan-Meier survival curve of WT and atp6v0d2−/- mice (f) (n = 14). 5 days later, CFUs from spleen (g), colon (h), mesenteric lymph node (MLN) (i), and inguinal lymph node (ILN) (j) were determined. (n = 5). (k) Immunoblot analysis of ATP6V0D2, LC3 in WT and atp6v0d2−/- BMDMs upon infection with GFP-S. typhimurium for the indicated times. Data are representative of either 3 independent experiments (a–c, k) or 2 independent experiments (d–j) Histograms depict mean values ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001. CFU, colony-forming units.
Upon costaining the GFP-labeled bacteria with a lysosome dye, we found a significant reduction in the proportion of bacteria colocalized with lysosomes in the atp6v0d2−/- BMDM cells, confirming that there was a defect in the fusion of the phagosome and lysosome (Figure 7(d,e)). Moreover, atp6v0d2−/- mice were more susceptible to Salmonella infection-induced death and developed enhanced splenomegaly and pathology (Figures 7(f) and S7(a–d)), together with a significant increase in the numbers of bacteria cultured from the spleen, colon and lymph nodes, compared with infected wild-type mice (Figure 7(g–j)). Moreover, infection of Salmonella led to reduced ATP6V0D2 expression, consistent with inflammation-induced downregulation of ATP6V0D2, and atp6v0d2−/- BMDM cells showed enhanced levels of LC3-II (Figure 7(k)), indicating an autophagy defect in the absence of ATP6V0D2 in infection. Taken together, these data suggest ATP6V0D2 is necessary for bacterial clearance via promotion of autophagosome-lysosome fusion.
Discussion
In response to nutrient deprivation or cell stress signals, autophagy is initiated to maintain cellular homeostasis [8,25,26]. Cells of the innate immune system take advantage of this mechanism to degrade intracellular pathogens and constrain inflammation [27]. In this study, we determined that ATP6V0D2, a highly expressed isoform of one of the subunits of the V-ATPase in macrophage cells, was required for efficient autophagosome-lysosome fusion through facilitating the association of STX17 with VAMP8. Deletion of Atp6v0d2 in mice leads to enhanced inflammasome activation, impaired clearance of ingested bacteria in vitro and DSS-induced colitis. Therefore, ATP6V0D2 acts as an important component of the molecular circuitry to prevent hyperactivation of inflammation with potential implications in chronic infection and autoimmunity.
The notion that the V-ATPase regulates membrane fusion has been articulated previously in multiple organisms, although it remains controversial [15,28]. A single subunit of V0 promotes fusion of vacuolar membranes in yeast [29,30]. Similarly, Peri et al. found that ATP6V0A1/subunit a1, a specific isoform expressed in microglia, is essential for autophagosome-lysosome fusion and clearance of engulfed apoptotic neural cells in Zebrafish, but not for acidification of microglial lysosomes [31] whereas ATP6V0A1 is generally involved in vesicle acidification [32–34]. However, depletion of individual V-ATPase subunits in Drosophila fat body [35] or disruption of the whole complex of V-ATPase by deletion of the gene encoding the V-ATPase accessory protein ATP6AP2 in mouse macrophages [36] is dispensable for phagosome-lysosome fusion. Our data, together with these findings, suggest that specific isoforms of the V0 subunit regulate fusion in a species- and cell-type specific fashion independent of its pump activity. ATG14 directly binds to the STX17-SNAP29 complex on autophagosomes and promotes STX17-SNAP29-VAMP8-mediated autophagosome-lysosome fusion [14], similar to our findings with ATP6V0D2. Given that disruption of the V-ATPase complex by deletion of Atp6ap2 does not affect phagosome-lysosome fusion in murine macrophage cells [36], ATP6V0D2 may form a protein complex with SNARE proteins, distinct from the classically described V-ATPase complex, to mediate autophagosome-lysosome fusion. Furthermore, V0 subunits are directly involved in membrane and vesicle fusion, which is unrelated to their proton transport activity [37]. It is possible that ATP6V0D2 plays a role in membrane and vesicle fusion in addition to autophagosome-lysosome fusion.
The inflammasome and autophagy pathways are closely linked and mutually regulated [8,38,39]. A variety of inflammasome stimuli activate both inflammasome and autophagy in macrophages simultaneously. Yet activation of autophagy limits inflammasome activity via both degradation of the inflammasome adaptor PYCARD/ASC [7] and removal of mitochondrial ROS [5,6]. We found that deletion of Atp6v0d2 led to enhanced production of mitochondrial ROS and the accumulation of damaged mitochondria, suggesting a defect of autophagy. Consistent with this, deficiency of SQSTM1/p62 and PRKN/PARK2/Parkin, 2 factors necessary for mitophagy, results in pronounced accumulation of damaged mitochondria and excessive IL1B-dependent inflammation [40].
In addition to its role in regulating activation of the inflammasome, autophagy is essential for eliminating phagocytosed bacteria, including Salmonella enterica, serovar typhimurium [41], Mycobacterium tuberculosis [42], Legionella pneumophila [43], and Listeria monocytogenes [44]. For example, mice expressing mutant ATG16L1T300A have increased sensibility to CASP3 (caspase 3) cleavage on ATG16L1, heightened IL1B production and decreased antibacterial defense [45,46]. Chronic granulomatous disease (CGD) is due to mutations of genes encoding members of the NADPH oxidase complex, traditionally regarded as essential to generate superoxide necessary to kill phagocytosed bacteria and fungi. The complex is additionally required to recruit LC3 to the phagosome, and macrophages from patients with CGD have enhanced IL1B production [47]. It is of note that the use of IL1B inhibitors in these patients ameliorates disease severity [48], suggesting a close relationship between heightened inflammation and defective bacterial clearance.
If ATP6V0D2 is essential for macrophage clearance of ingested bacteria, it is counterintuitive that the presence of LPS would limit its expression. However, this finding is consistent with the notion that a subsequent status of immunosuppression exists following septic shock, including an inability to clear bacteria, despite heightened inflammation [49]. From the perspective of the bacteria, the advantages are obvious. Apart from allowing survival of the organisms within macrophages; the enhanced inflammatory stimulus results in the recruitment of more phagocytes that become potential hosts for more bacteria [50]. It is important to note that although expression of Atp6v0d2 was inhibited by LPS, wild-type BMDM still express enough residual protein to degrade bacteria to a significantly greater extent than atp6v0d2−/- macrophages both in vitro and in vivo. Thus, an uneasy stalemate exists in the macrophages of wild-type animals.
In summary, we showed that ATP6V0D2 facilitates fusion of autophagosomes with lysosomes, a critical but often overlooked step of autophagy in macrophage cells. Our findings indicate that cell-type specific autophagy machinery exists in macrophage cells, that it is a critical component in the inflammatory response against stresses and infections, and that it is necessary for the elimination of damaged organelles and restriction of over-exuberant inflammation. Manipulation of this fusion step of autophagy may lead to novel therapies in the treatment of a variety of inflammatory and autoimmune diseases.
Materials and methods
Animals
All experiments were performed according to the guidelines of the Institutional Animal Care and Use Committee of Tongji Medical School, Huazhong University of Science and Technology (HUST, Wuhan).
Reagents
Pam3CSK4 (tlrl-pms), Poly(I:C) (tlrl-pic), LPS-B5 Ultrapure (tlrl-pb5lps), ATP (tlrl-atpl), nigericin (tlrl-nig), SiO2 (tlrl-sio), monosodium urate crystal (MSU; tlrl-msu) and poly(dA:dT; tlrl-patn) were from Invivogen. LPS (L2630) for the in vivo experiment was from Sigma Aldrich. MitoSOX (M36008), LysoTracker Red DND-99 (L7528) and LysoSensor Yellow/Blue dextran (L22460) were from ThermoFisher Scientific. Anti-IL1B (BE0246) for the in vivo experiment was from BioXcell. Antibodies used for immunoblotting analysis were: anti-ATP6V0D2 (Sigma Aldrich, SAB2103221), anti-ATP6V0D1 (Abcam, ab202889), anti-mouse IL1B (Cell Signaling Technology, 12507), anti-mouse NLRP3 (Adipogen, AG-20B-0014C100), anti-mouse PYCARD/ASC (Adipogen, AG-25b-0006-C100), anti-mouse CASP1 p10 (Santa Cruz Biotechnology, sc-514), anti-SQSTM1/p62 (MBL, PM045), anti-LC3B (Cell Signaling Technology, 12741), anti-CTSB (Cell Signaling Technology, 31718), anti-GST (Cell Signaling Technology, 2625), anti-His (Cell Signaling Technology, 12698) and anti-ACTB (Cell Signaling Technology, 4970). Mito-TEMPO (sc-221945) was from Santa Cruz Biotechnology. PerCP/Cy5.5 anti-mouse ADGRE1/F4/80 (123127) and FITC anti-mouse ITGAM/CD11b (101205) were from BioLegend. 3-MA (s2767) was from Selleck. Bafilomycin A1 (ab120497) and rapamycin (ab1202224) were from Abcam. Constructs encoding flag-tagged full-length human STX17 (CH863186), VAMP8 (CH855804) and SNAP29 (CH800417), and HA-tagged human ATP6V0D2 (custom service) were from Vigene Bioscience. Truncated forms of HA-ATP6V0D2 were cloned into pcDNA3.1 (ThermoFisher Scientific, V79020) with BamHI and XhoI. Full-length mouse Atp6v0d2 was cloned into pMYs-IRES-GFP (Cell Biolabs, RTV-021) with BamHI and NotI for generation of retrovirus.
Generation of Atp6v0d2-deficient mice
atp6v0d2−/− mice were generated with Transcription activator-like effector nucleases (TALEN) technology. A TALEN binding pair was chosen from the Atp6v0d2 coding sequence in the first exon. The genomic recognition sequences of TALEN left and right arms are CGAGGATGCAAAGCCAGCC (L) and GCACTAGGTTGACATA (R), spaced by 16 base pairs and anchored by a preceding T base at the −1 position to meet the optimal criteria for natural TAL proteins. TALEN vectors of left and right arms – -TALEN-atp6v0d2-L and TALEN-atp6v0d2-R- were obtained by one-step ligation using the Fast TALETM TALEN Assembly Kit (SIDANSAI Biotechnology, 2801) according to the manufacturer’s instructions. mRNA (400 ng/μl, 10 pl) was injected into cytoplasm of 180 1-cell embryos. After incubation for 24 h, the selected 2-cell embryos were transferred into the oviduct of 7 pseudopregnant C57BL/6 mice. We confirmed the genotype of F0 mice by DNA sequencing.
Cell culture and stimulation
Primary BMDMs were generated by culturing mouse bone marrow cells in the presence of 50 ng/ml CSF1/M-CSF (PeproTech, 315-02) conditional medium for 7 days. BMDMs were seeded in 6-well, 12-well or 48-well plates overnight. On the second day, after pretreatment with ultrapure LPS (100 ng/ml) for 4 h, BMDMs (1x106 cells/ml) were further stimulated with ATP (5 mM) or nigericin (5 μM) for 30 min unless otherwise indicated or silica (500 μg/ml) and MSU (400 μg/ml) for 6 h. For AIM2 inflammasome activation, macrophages were primed with LPS (100 ng/ml) followed by transfection of poly (dA:dT; Invivogen, tlrl-patn) using Lipofectamine 2000 (ThermoFisher Scientific, 11668019) according to the manufacturer’s protocol. Culture supernatants were collected 6 h post infection, and IL1B release was measured by ELISA. Supernatants and cell lysates were collected for ELISA and immunoblot analyses.
RNA isolation and real-time PCR
RNA was isolated from BMDMs and was reverse transcribed, and real-time PCR was performed. Primer sequences are as follows.
Atp6v0d2-F: 5ʹ-TGCGGCAGGCTCTATCCAGAGG-3ʹ;
Atp6v0d2-R:5ʹ-CCACTGCCACCGACAGCGTC-3ʹ;
Actb/β-actin-F: 5ʹ-CCTTCCAGCAGATGTGGATCA-3ʹ;
Actb/β-actin-R: 5ʹ-AACGCAGCTCAGTAACAGTCC-3ʹ.
SiRNA transfection
Control siRNA and Atp6v0d1 siRNA (GenePharma, Mus-1015) were transfected into WT and atp6v0d2−/- BMDMs using Lipofectamine RNA iMAX (ThermoFisher Scientific, 13778500) according to the manufacturer’s instructions. The sequences are as follows: Atp6v0d1 siRNA forward: (CAUCGAGAUAAUCCGAAAUTTT); Atp6v0d1 siRNA reverse: (AUUUCGGAUUAUCUCGAUGTT); Control siRNA forward: (UUCUCCGAACGUGUCACGUTT); Control siRNA reverse: (ACGUCACACGUUCGGAGAATT).
ELISA
Paired (capture and detection) antibodies and standard recombinant mouse IL1B (R&D Systems, MLB00C), TNF (R&D Systems, MTA00B), and IL6 (R&D Systems, M6000B) were used to determine cytokine concentrations in cell culture supernatants and mouse sera according to the manufacturer’s instructions.
Western blot and immunoprecipitation assay
Cells were lysed in RIPA buffer (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% NP-40 (Sigma Aldrich, NP40S), 1% sodium deoxycholate (Sigma Aldrich, D6750), 0.1% SDS (Sigma Aldrich, 74255)) containing protease inhibitor cocktail (Roche, 11836153001) and phosphatase inhibitor cocktail (Sigma Aldrich, P5726). Protein concentrations were quantified using a BCA Protein Assay Kit (Pierce, 23225). Equal amounts of protein were loaded onto SDS-PAGE gels and transferred onto nitrocellulose membranes. The membranes were then incubated with antibodies against SQSTM1/p62, PYCARD/ASC, NLRP3, ACTB, CTSB, and IL1B (as described above), followed by appropriate secondary HRP-conjugated goat anti-rabbit IgG (Proteintech, SA00001-2) or HRP-conjugated goat anti-mouse IgG (Proteintech, SA00001-1), then developed with ECL (GE healthcare, RPN2232). For immunoprecipitation, cell lysates were precipitated with 2 μg anti-HA (Abcam, Ab9110) overnight and the precipitated immunocomplex were separated by SDS-PAGE and detected with anti-Flag (Sigma Aldrich, F1804) antibodies. The band intensities in the linear range (0–255) were quantified with ImageJ.
Mitochondrial and intracellular ROS detection
Mitochondrial ROS was measured using MitoSOX (ThermoFisher Scientific, M36008) following the manufacturer’s instructions. In brief, macrophages were first primed with LPS (100 ng/ml) followed by treatment with ATP (2 mM; Invivogen, tlrl-atpl) for 30 min or nigericin (5 µM; Invivogen, tlrl-nig) for 1 h, after which the cells were washed twice with PBS (Sigma Aldrich, P5368), loaded with 4 µM of MitoSOX for 10 min and washed twice with PBS. The presence of ROS was detected by flow cytometry using a FACS Verse (BD Bioscience).
Mitochondrial membrane integrity detection
Mitochondrial membrane integrity was detected with MitoTracker Deep Red FM (ThermoFisher Scientific, M22426), which is membrane potential dependent and MitoTracker Green FM (ThermoFisher Scientific, M7514), which is membrane potential independent. In brief, LPS-primed BMDMs were stimulated with various NLRP3 agonists for the indicated times, then cocultured with 50 nM MitoTracker Deep Red FM and 50 nM MitoTracker Green FM for 20 min. Next, the BMDMs were washed twice with PBS, treated with trypsin-EDTA (ThermoFisher Scientific, 25200056) and finally resuspended with 1% BSA (Sigma Aldrich, B2064) in PBS. Stained mitochondria were identified by a change in mean fluorescence intensity by flow cytometry using a FACS Verse.
GST affinity isolation assay
His-tagged ATP6V0D1 and ATP6V0D2 were constructed in the pET28a vector (Addgene, 26094). Recombinant His tag proteins were expressed in E. coli strain Rosetta (DE3) and purified with an Ni-NTA agarose column (Qiagen, 30230). STX17 and VAMP8 were expressed individually as N-terminal GST-fused proteins from pGEX-4T-3 (BioVector NTCC Inc, pGEX-4T-3) in DE3 E. coli, and bacterial lysates were bound to Glutathione HiCap resin (Qiagen, 30900) for 4 h at 4°C. Purified His-ATP6V0D1 or His-ATP6V0D2 was added into the resin and incubated overnight at 4°C. After washing with PBS, the resin-bound proteins were eluted and then analyzed by western blot.
Phagocytosis of DQ-OVA and FITC-dexan
WT and atp6v0d2−/- BMDMs were plated in 48-well plates, FITC-DQ-OVA (ThermoFisher Scientific, D12053) or FITC-Dextran (Sigma Aldrich, FD4) was added to each well, then incubated for 1 h at 37ºC. Subsequently, cells were washed 3 times with PBS, and the fluorescence intensity was measured by flow cytometry (FACS Verse, BD Bioscience).
Salmonella culture, gentamicin protection assay and infection of Salmonella in BMDMs
Bacteria was cultured in plates containing ampicillin (Sigma Aldrich, A9518) for 24 h at 37ºC. Single clones of bacteria were harvested to culture in 5 ml LB medium for 4–5 h to reach an OD600 of 0.5–0.6. The total number of bacteria was calculated according to instrument Precision determination.
Mature BMDMs were plated in 24-well plates and infected with Salmonella at a desired multiplicity of infection (MOI) of 2. Cells were incubated with bacteria for 60 min. After bacterial incubation, BMDMs were washed 3 times with PBS, and resuspended in fresh medium containing gentamicin (Sigma Aldrich, E003632) at a final concentration of 300 μg/ml for 1 h at 37ºC, followed by a second incubation in culture medium containing 100 μg/ml gentamicin. BMDMs were again washed 3 time in DPBS, then lysed by the addition of 0.02% Triton X-100 (Sigma Aldrich, T8787) and serial dilutions of lysate were placed on blood agar (Sigma Aldrich, 70133). Bacterial colonies were counted after incubation for 24 h at 37ºC.
For quantification of bacterial fluorescence upon infection, WT and atp6v0d2−/- BMDMs were infected with GFP-Salmonella at an MOI of 50 on ice for 30 min, and then incubated for 15, 30, 60, or 120 min at 37ºC. After extracellular fluorescence was quenched by trypan blue (Sigma Aldrich, T6146), cells were washed twice with PBS and the fluorescence intensity was measured by flow cytometry. For colocalizations of LysoTracker Red and GFP-bacteria, Salmonella (10 MOI)-infected WT and atp6v0d2−/- BMDMs were stained with LysoTracker Red for 15 min prior to cell harvest. Subsequently, extracellular fluorescence was quenched with trypan blue. These cells were fixed with 4% paraformaldehyde (Sigma Aldrich, 158127) and imaged with a laser scanning confocal microscope (Olympus, Japan).
Infection of GFP-Salmonella in vivo
WT and atp6v0d2−/- mice were treated with 20 mg streptomycin (Sigma Aldrich, V900929) by oral gavage. On the next day, mice were infected orally with 1 × 108 ampicillin-resistant Salmonella diluted in PBS in a 0.2-ml volume. Five days later, mice were sacrificed, tissues were homogenized, and bacterial burdens were enumerated by serial dilution on ampicillin-containing LB agar plates. Haematoxylin (Sigma Aldrich, H9627) and eosin (Sigma Aldrich, E4009)-stained formaldehyde (Sigma Aldrich, F8775)-fixed sections of intestines were examined.
Immunofluorescent staining, confocal microscopy and transmission electron microscopy
BMDMs were seeded at 1 × 105 cells per well on glass slides, and rested overnight for proper attachment. Then the cells were primed with LPS (20 ng/ml) for 4 h, and treated with ATP (2 mM) or nigericin (5 µM) for 1 h. After the treatment, cells were washed twice with sterile PBS and fixed with 4% paraformaldehyde (PFA), permeabilized with 0.05% Triton X-100 and blocked in 5% BSA. Anti-LC3B (Cell Signaling Technology, 12741), anti-TOMM20 (Abcam, ab186735), anti-STX17 (Sigma Aldrich, HPA001204), and anti-LAMP1 (BD Bioscience, 555798) were incubated overnight at 4°C. Secondary fluorescent antibodies (FITC (111–095-045), Cy3 (115–165-003) from Jackson Laboratories) were added for 1 h and DAPI was used for nuclear counterstaining. For experiments that used LysoTracker Red, cells were stained with LysoTracker Red DND-99 prior to PFA fixation. Samples were imaged through a laser scanning confocal microscope (Olympus, Japan).
After LPS priming and NLRP3 agonist stimulation, macrophages were fixed in 2.5% glutaraldehyde (Sigma Aldrich, G5882) in 0.1 M phosphate buffer (pH 7.4, pre-warmed at 37°C for 5 min) at room temperature followed by 2 h on ice. After fixation, cells were rinsed 5 times for 1 min each in 0.1 M phosphate buffer, then post-fixed in 2% OsO4 in 0.1 M phosphate buffer for 2 h. After 2 washes for 1 min each in 0.1 M phosphate buffer and 3 washes for 1 min each in double-distilled water, cells were en bloc stained with 2% aqueous uranyl acetate overnight at 4°C. After 5 rinses for 1 min each in double-distilled water, cells were dehydrated in a cold graded ethanol series (50%, 70%, 80%, 90%, 100%) for 15 min each on ice, then rinsed once at room temperature with 100% ethanol, and embedded using the SPI-PonTMKit (SPI Supplies, 90529-77-4). Sections were cut with a diamond knife (Daitome Ultra 45) at a thickness of 60–80 nm and visualized using a HT-7700 electron microscope (Hitachi HT-7700) at an accelerating voltage of 150 kV. Images were recorded on film at either 4000x or 10,000x magnification.
LPS-induced systemic inflammation
Septic shock was induced by i.p. injecting 8- to 12-weeks old sex-matched WT and atp6v0d2−/- mice with 20 mg/kg body weight LPS. Mouse sera were collected 4 h post LPS injection. Serum IL1B, TNF and IL6 concentrations were measured by ELISA.
DSS-induced colitis
Acute colitis was induced in WT and atp6v0d2−/- mice by oral gavage with 200 μl of 2.5% DSS (MP Biomedicals, 02160110; MW 36,000–50,000) to minimize variation, followed by adding 2.5% DSS to drinking water ad libitum for 7 consecutive days. On day 8, the animals were sacrificed and tissues analyzed by flow cytometry and histology on haematoxylin and eosin-stained formaldehyde-fixed tissue sections. For macrophage depletion, WT and atp6V0d2−/- mice were intraperitoneally injected with 0.2 ml clodronate liposomes 4 days prior to DSS treatment and on day 0, 3, and 5 during DSS colitis induction. In a subset of experiments, mice received anti-IL1B 300 µg per mouse i.p. at day 1, 3, and 5. Histology scores were determined by 2 blinded pathologists as previously described [51].
Statistics
All data are shown as means ± s.d. or means ± s.e.m. as indicated. Statistical analysis was performed using a two-tailed Student’s t-test. For all tests, p-values lower than 0.05 were considered significant.
Funding Statement
This work was supported by the National Scientific Foundation of China [31470851, 81671539, and 31870892].
Acknowledgments
We thank Drs. Xiaopeng Qi, Huiming Peng, Yueguang Rong for the gifts of bacteria and technical assistance and discussions. This work was supported by grants from the National Scientific Foundation of China to X.P.Y (#31470851, #81671539, 31870892), 973 Program to F.G. (2013CB530505) and Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College, HUST to X.P.Y. A.L. is supported by the Crohn’s and colitis foundation of America.
Disclosure statement
No potential conflict of interest was reported by the authors.
Author contributions
YX, JJO, JJC, AL, and XPY designed experiments, analyzed data, and wrote the manuscript; YX, NL, XX, HB, LL, GB, JZ, and YY performed experiments; XD and KJ preformed analysis; ZT, BY, JW, ZL, CY, XC and FG provided reagents and funding; ZL and XPY supervised the project.
Supplementary Material
Supplementary material for this article can be accessed here.
References
- [1].Levine B, Deretic V.. Unveiling the roles of autophagy in innate and adaptive immunity [Review Article]. Nat Rev Immunol. 2007. 10/01/online;7:767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion [Review Article]. Nature. 2008. 02/28/online;451:1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Huang J, Brumell JH.. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol. 2014. February;12(2):101–114. PubMed PMID: 24384599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008. November 13;456(7219):264–268. PubMed PMID: 18849965. [DOI] [PubMed] [Google Scholar]
- [5].Zhou R, Yazdi AS, Menu P, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011. January 13;469(7329):221–225. PubMed PMID: 21124315. [DOI] [PubMed] [Google Scholar]
- [6].Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011. March;12(3):222–230. PubMed PMID: 21151103; PubMed Central PMCID: PMC3079381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012. January 29;13(3):255–263. PubMed PMID: 22286270; PubMed Central PMCID: PMC4116819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013. October;13(10):722–737. PubMed PMID: 24064518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011. January 20;469(7330):323–335. PubMed PMID: 21248839; PubMed Central PMCID: PMC3131688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010. December 23;22(2):124–131. PubMed PMID: PMC2854249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009. December 1;43(1):67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. PubMed PMID: 21801009. [DOI] [PubMed] [Google Scholar]
- [13].Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012. December 7;151(6):1256–1269. PubMed PMID: 23217709. [DOI] [PubMed] [Google Scholar]
- [14].Diao J, Liu R, Rong Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015. April 23;520(7548):563–566. PubMed PMID: 25686604; PubMed Central PMCID: PMC4442024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cotter K, Stransky L, McGuire C, et al. Recent insights into the structure, regulation, and function of the V-ATPases. Trends Biochem Sci. 2015. October;40(10):611–622. PubMed PMID: 26410601; PubMed Central PMCID: PMC4589219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nishi T, Kawasaki-Nishi S, Forgac M. Expression and function of the mouse V-ATPase d subunit isoforms. J Biol Chem. 2003. November 21;278(47):46396–46402. PubMed PMID: 12963731. [DOI] [PubMed] [Google Scholar]
- [17].Smith AN, Francis RW, Sorrell SL, et al. The d subunit plays a central role in human vacuolar H(+)-ATPases. J Bioenerg Biomembr. 2008. August;40(4):371–380. PubMed PMID: 18752060; PubMed Central PMCID: PMC2782108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Toei M, Saum R, Forgac M. Regulation and isoform function of the V-ATPases. Biochemistry. 2010. June 15;49(23):4715–4723. PubMed PMID: 20450191; PubMed Central PMCID: PMC2907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wu H, Xu G, Li YP. Atp6v0d2 is an essential component of the osteoclast-specific proton pump that mediates extracellular acidification in bone resorption. J Bone Miner Res. 2009. May;24(5):871–885. PubMed PMID: 19113919; PubMed Central PMCID: PMC2672205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Noubade R, Wong K, Ota N, et al. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature. 2014. May 08;509(7499):235–239. PubMed PMID: 24739962. [DOI] [PubMed] [Google Scholar]
- [21].Lee SH, Rho J, Jeong D, et al. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat Med. 2006. December;12(12):1403–1409. PubMed PMID: 17128270. [DOI] [PubMed] [Google Scholar]
- [22].Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013. June;13(6):397–411. PubMed PMID: 23702978; PubMed Central PMCID: PMC3807999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity. 2013. September 19;39(3):432–441. PubMed PMID: 24054327; PubMed Central PMCID: PMC3835203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ravindran R, Loebbermann J, Nakaya HI, et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature. 2016. March 24;531(7595):523–527. PubMed PMID: 26982722; PubMed Central PMCID: PMC4854628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014. March 27;157(1):65–75. PubMed PMID: 24679527; PubMed Central PMCID: PMC4020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005. January 28;120(2):159–162. PubMed PMID: 15680321. [DOI] [PubMed] [Google Scholar]
- [27].Shahnazari S, Brumell JH. Mechanisms and consequences of bacterial targeting by the autophagy pathway. Curr Opin Microbiol. 2011. February;14(1):68–75. PubMed PMID: 21112809. [DOI] [PubMed] [Google Scholar]
- [28].Maxson ME, Grinstein S. The vacuolar-type H(+)-ATPase at a glance - more than a proton pump. J Cell Sci. 2014. December 01;127(Pt 23):4987–4993. PubMed PMID: 25453113. [DOI] [PubMed] [Google Scholar]
- [29].Baars TL, Petri S, Peters C, et al. Role of the V-ATPase in regulation of the vacuolar fission-fusion equilibrium. Mol Biol Cell. 2007. October;18(10):3873–3882. PubMed PMID: 17652457; PubMed Central PMCID: PMC1995711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Takeda K, Cabrera M, Rohde J, et al. The vacuolar V1/V0-ATPase is involved in the release of the HOPS subunit Vps41 from vacuoles, vacuole fragmentation and fusion. FEBS Lett. 2008. April 30;582(10):1558–1563. PubMed PMID: 18405665. [DOI] [PubMed] [Google Scholar]
- [31].Peri F, Nusslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 2008. May 30;133(5):916–927. PubMed PMID: 18510934. [DOI] [PubMed] [Google Scholar]
- [32].Saw NMN, Kang S-YA, Parsaud L, et al. Vacuolar H+-ATPase subunits Voa1 and Voa2 cooperatively regulate secretory vesicle acidification, transmitter uptake, and storage. Mol Biol Cell. 2011. September 15;22(18):3394–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bagh MB, Peng S, Chandra G, et al. Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model [Article]. Nat Commun. 2017. 03/07/online;8:14612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lee J-H, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by alzheimer-related PS1 mutations. Cell. 2010. June 25;141(7):1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mauvezin C, Nagy P, Juhasz G, et al. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun. 2015. May 11;6:7007 PubMed PMID: 25959678; PubMed Central PMCID: PMC4428688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kissing S, Hermsen C, Repnik U, et al. Vacuolar ATPase in phagosome-lysosome fusion. J Biol Chem. 2015. May 29;290(22):14166–14180. PubMed PMID: 25903133; PubMed Central PMCID: PMC4447986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].El Far O, Seagar M. A role for V‐ATPase subunits in synaptic vesicle fusion? J Neurochem. 2011. May 1;117(4):603–612. [DOI] [PubMed] [Google Scholar]
- [38].Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016. November;16(11):661–675. PubMed PMID: 27694913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rodgers MA, Bowman JW, Liang Q, et al. Regulation where autophagy intersects the inflammasome. Antioxid Redox Signal. 2014. January 20;20(3):495–506. PubMed PMID: 23642014; PubMed Central PMCID: PMC3894701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016. February 25;164(5):896–910. PubMed PMID: 26919428; PubMed Central PMCID: PMC4769378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jia K, Thomas C, Akbar M, et al. Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc Natl Acad Sci USA. 2009. August 25;106(34):14564–14569. PubMed PMID: 19667176; PubMed Central PMCID: PMC2731839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kim JJ, Lee HM, Shin DM, et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe. 2012. May 17;11(5):457–468. PubMed PMID: 22607799. [DOI] [PubMed] [Google Scholar]
- [43].Tung SM, Unal C, Ley A, et al. Loss of dictyostelium ATG9 results in a pleiotropic phenotype affecting growth, development, phagocytosis and clearance and replication of legionella pneumophila. Cell Microbiol. 2010. June;12(6):765–780. PubMed PMID: 20070309. [DOI] [PubMed] [Google Scholar]
- [44].Yano T, Mita S, Ohmori H, et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat Immunol. 2008. August;9(8):908–916. PubMed PMID: 18604211; PubMed Central PMCID: PMC2562576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lassen KG, Kuballa P, Conway KL, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci USA. 2014. May 27;111(21):7741–7746. PubMed PMID: 24821797; PubMed Central PMCID: PMC4040621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Murthy A, Li Y, Peng I, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014. February 27;506(7489):456–462. PubMed PMID: 24553140. [DOI] [PubMed] [Google Scholar]
- [47].Huang J, Canadien V, Lam GY, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci USA. 2009. April 14;106(15):6226–6231. PubMed PMID: 19339495; PubMed Central PMCID: PMC2664152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].de Luca A, Smeekens SP, Casagrande A, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci USA. 2014. March 4;111(9):3526–3531. PubMed PMID: 24550444; PubMed Central PMCID: PMC3948220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013;13(3):260–268. PubMed PMID: PMC3798159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gordon S. Phagocytosis: an immunobiologic process. Immunity. 2016. March 15;44(3):463–475. PubMed PMID: 26982354. [DOI] [PubMed] [Google Scholar]
- [51].Elson CO, Beagley KW, Sharmanov AT, et al. Hapten-induced model of murine inflammatory bowel disease: mucosa immune responses and protection by tolerance. J Immunol. 1996;157(5):2174–2185. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.