

ABSTRACT
As a master regulator of the macroautophagy/autophagy-lysosomal pathway, TFEB (transcription factor EB) plays a prominent role in regulating neurodegenerative diseases and cancer. The transcription activity of TFEB is tightly controlled by phosphorylation and dephosphorylation. Phosphorylated S211 (p-S211) of TFEB can be recognized by YWHA/14-3-3 proteins for TFEB cytoplasmic localization. Here, we characterized the interactions between phosphorylated TFEB and YWHA/14-3-3 proteins and determined the structures of YWHA/14-3-3 proteins in complex with a TFEB p-S211-peptide. Although the critical arginine for YWHA/14-3-3 recognition is missing in the N terminus of the TFEB p-S211-peptide, the C-terminal additional hydrophobic residues of the peptide unexpectedly occupy nearly half of the target-binding groove of YWHA/14-3-3 proteins, which compensates for the N-terminal defect and is distinct from the canonical YWHA/14-3-3-binding mode. Mutations of essential residues in the interaction interface between TFEB and YWHA/14-3-3 proteins disrupted their interactions and severely impaired the cytoplasmic localization of TFEB, which altered the expression of TFEB target genes and affected autophagy. Thus, YWHA/14-3-3 proteins recognize phosphorylated TFEB by a noncanonical mode for controlling TFEB cytoplasmic localization and its activity.
Abbreviation: ACTB: actin beta; ALP: autophagy-lysosomal pathway; ATP6V1H: ATPase H+ transporting V1 subunit H; bHLH: basic helix-loop-helix; CLEAR: coordinated lysosomal expression and regulation; Co-IP: co-immunoprecipitation; CTSB: cathepsin B; CTSD: cathepsin D; LAMP1: lysosomal associated membrane protein 1; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MITF: melanocyte inducing transcription factor; NLS: nuclear localization signal; TFEB: transcription factor EB; YWHA/14-3-3: tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein.
KEYWORDS: Autophagy, autophagy-lysosomal pathway, protein interaction, structural biology, transcription factor, X-ray crystallography
Introduction
The autophagy-lysosomal pathway (ALP) is critical for eliminating misfolded proteins, invasive pathogens and dysfunctional organelles [1]. Defects in ALP result in abnormal protein aggregation, and the accumulation of damaged organelles and other toxic components. Given that nondividing neuron cells are particularly sensitive to these hazards, it is widely accepted that dysfunctions of ALP are strongly correlated with neurodegenerative disease, including Alzheimer disease, Parkinson disease and Huntington disease [2–9]. Moreover, it is thought that ALP is also intricately involved in the development and progression of cancer; i.e., it acts as either a tumor promoter or a tumor suppressor in a context- and type-dependent manner [10]. Given the above intimate correlation between ALP and numerous diseases, a series of regulators of ALP have been characterized to be potential therapeutic targets [1].
TFEB (transcription factor EB), a member of the conserved microphthalmia-associated transcription factor/transcription factor E (MiT/TFE) family, is a master regulator of ALP and is also a promising therapeutic target [1,11,12]. As a key transcription factor, TFEB contains an N-terminal glutamine-rich domain, a transcriptional activation domain (AD) followed by a basic helix-loop-helix (bHLH) and a leucine zipper, and a C-terminal proline-rich domain (Figure 1(a)). The bHLH domain of TFEB is capable of binding to a 10-base pair palindromic sequence motif ‘GTCACGTGAC’ in the promoter region of the coordinated lysosomal expression and regulation (CLEAR) network that regulates the expression of a large subset of lysosomal and autophagic genes [13–15]. The transcriptional activity of TFEB predominantly depends on its cytoplasmic or nuclear localization and is precisely controlled by post-translational modifications and protein-protein interactions [11]. Under nutrient-rich conditions, TFEB can be phosphorylated directly by MTORC1 [14,16,17], and phosphorylated TFEB is then retained in the cytoplasm by interacting with the regulatory YWHA/14-3-3 proteins [11]. Under starvation conditions, MTORC1 is inactivated and PPP3/calcineurin can dephosphorylate TFEB, which leads to dissociation of TFEB from YWHA/14-3-3 proteins. TFEB will then be translocated to the nucleus for activation [18]. During this dynamic process, YWHA/14-3-3 proteins are the critical regulatory factors for controlling the switch of TFEB between cytoplasmic and nuclear localization.
Figure 1.
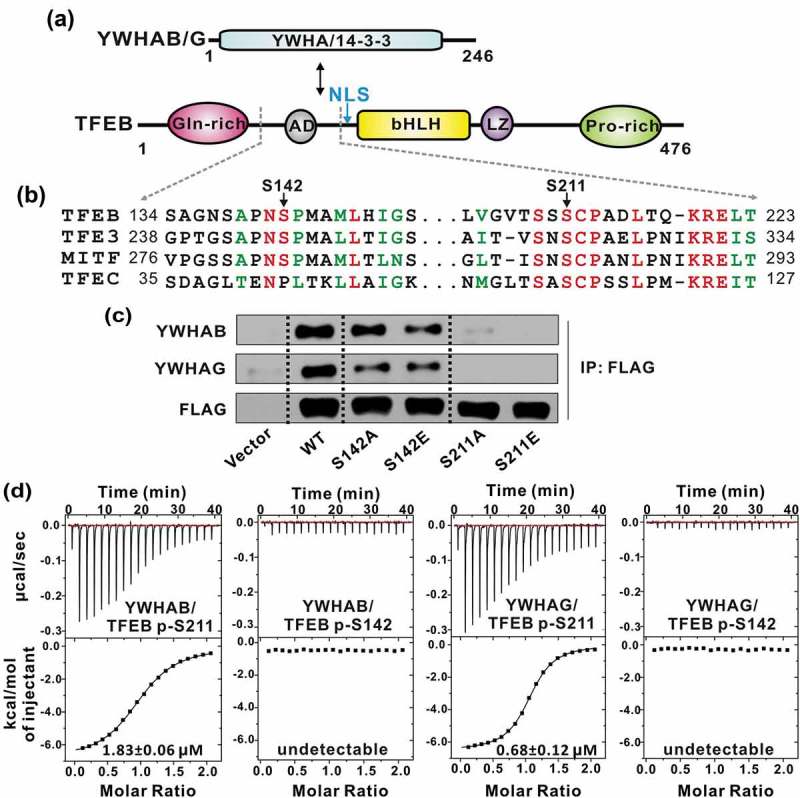
TFEB S211 is responsible for phosphorylation-dependent interaction with YWHA/14-3-3 proteins. (a) Domain organizations of YWHA/14-3-3 proteins and TFEB. TFEB contains an N-terminal glutamine-rich domain, a transcriptional activation domain (AD) followed by a bHLH and a leucine zipper (LZ), and a C-terminal proline-rich domain. (b) Sequence alignment of human TFEB, TFE3, MITF and TFEC from the MiT/TFE family. The identical residues are colored in red and the highly conserved residues are colored in green. Notably, the extremely conserved S211 in TFEB is very close to the NLS. (c) Co-IP assay of the interactions between YWHA/14-3-3 proteins and TFEB. Both YWHAB and YWHAG can co-immunoprecipitate with wild-type TFEB. Mutations at S211 abolished the interactions but the same type of mutations at S142 did not significantly impair the binding. (d) The binding affinities between TFEB phosphorylated peptides and YWHA/14-3-3 proteins determined by ITC experiments. YWHAB and YWHAG both interact with the p-S211-peptide but not the p-S142-peptide. The binding affinities are indicated in each panel.
YWHA/14-3-3 proteins are a large family of phospho-regulatory proteins that exist primarily as homo- and heterodimers (i.e., there at least 7 isoforms in mammals: YWHAB, YWHAG, YWHAE, YWHAH, SFN, YWHAZ and YWHAQ) [19]. In general, YWHA/14-3-3 proteins can recognize phosphorylated serine/threonine with a conserved sequence motif, i.e., ‘RSXpS/pTXP’ and ‘RXXXpS/pTXP’ (where pS/pT is phosphorylated serine/threonine and X is any residue) [19,20]. In the previous studies, YWHA/14-3-3 proteins were reported to interact with TFEB in a phosphorylation-dependent manner to regulate TFEB cytoplasmic localization [11]. Among the multiple phosphorylation sites of TFEB, phosphorylated S211 (p-S211) has been indicated to be the critical site for interacting with YWHA/14-3-3 proteins. However, the primary sequence analysis demonstrates that the p-S211-containing sequence of TFEB is unable to fit well into the canonical YWHA/14-3-3-binding motif; i.e., the critical arginine is missing in the N terminus of the p-S211 site of TFEB (Figure S1), suggesting that YWHA/14-3-3 proteins may bind to phosphorylated TFEB in a different manner. Nevertheless, the mechanisms underlying the recognition of TFEB by YWHA/14-3-3 proteins and the subsequent regulation of TFEB subcellular localization are poorly understood.
In this study, we confirmed that the p-S211 site of TFEB mediates the interactions with YWHA/14-3-3 proteins and solved the structures of YWHA/14-3-3 proteins in complex with the TFEB p-S211-peptide. In addition to p-S211-mediated electrostatic interactions, the TFEB p-S211-peptide contains additional hydrophobic residues at the C terminus that occupy nearly half of the target-binding groove of YWHA/14-3-3 proteins, which compensates for the lack of the critical arginine in TFEB. This recognition mechanism of YWHA/14-3-3 proteins is distinct from the canonical YWHA/14-3-3-binding mode. Mutations of the essential residues in the interaction interface between YWHA/14-3-3 proteins and TFEB disrupted their interactions and interfered with the subcellular localization of TFEB, which further affected the expression of TFEB target genes and autophagy. Thus, YWHA/14-3-3 proteins recognize the TFEB p-S211-peptide by a noncanonical mode for controlling TFEB cytoplasmic localization and its cellular functions.
Results
S211 in TFEB is responsible for phosphorylation-dependent interaction with YWHA/14-3-3 proteins
It has been reported that phosphorylation of TFEB at S142 and/or S211 is essential for controlling TFEB subcellular localization and affecting its transcriptional activity possibly by interacting with YWHA/14-3-3 proteins (Figure 1(a–b)) [21]. To further check which site is responsible for binding to YWHA/14-3-3 proteins, we initiated this study by performing the Co-IP assay with 2 isoforms of YWHA/14-3-3 proteins, YWHAB and YWHAG. As expected, TFEB could associate with both YWHAB and YWHAG in vivo (Figure 1(c)). The mutation of S211 to an alanine abolished the interaction but the same type of mutation at S142 did not significantly impair the binding (Figure 1(c)), indicating that S211 is the key phosphorylation site of TFEB responsible for binding to YWHA/14-3-3 proteins. In addition, the phospho-mimic S211E mutant could not interact with YWHA/14-3-3 proteins (Figure 1(c)), suggesting that the interactions between TFEB and YWHA/14-3-3 proteins are phospho-serine specific. Consistently, the same mutation at S142 had no significant impact on the binding between TFEB and YWHA/14-3-3 proteins.
To further characterize the interactions between TFEB and YWHA/14-3-3 proteins, we synthesized the TFEB peptides with phosphorylated S211 (p-S211) or S142 (p-S142) and measured the binding affinities between the peptides and YWHA/14-3-3 proteins by an isothermal titration calorimetry (ITC) assay. Consistent with the Co-IP experiments, the TFEB p-S211-peptide (with the sequence of ‘LVGVTSSpSCPADLTQKRELT’) bound to YWHAB and YWHAG with a relatively high affinity (1.83 ± 0.06 μM and 0.68 ± 0.12 μM for YWHAB and YWHAG, respectively) (Figure 1(d)). In contrast, the binding affinities between the TFEB p-S142-peptide (with the sequence of ‘SAGNSAPNpSPMAMLHIGS’) and YWHAB and YWHAG were undetectable (Figure 1(d)). Taken together, all the data demonstrated that, in the 2 essential phosphorylation sites of TFEB, S211 is the critical site responsible for phosphorylation-dependent interaction with YWHA/14-3-3 proteins. Moreover, given the conservation of S211 in the MiT/TFE family (Figure 1(b)), it is possible that other MiT/TFE family members such as TFE3, TFEC and MITF may also bind to YWHA/14-3-3 proteins in a similar manner.
Overall structure of YWHA/14-3-3 proteins in complex with the TFEB p-S211-peptide
TFEB p-S211 is the key site for binding to YWHA/14-3-3 proteins, but the fragment including p-S211 is unable to be well aligned with the canonical YWHA/14-3-3-binding motifs (such as ‘RSXpS/pTXP’ and ‘RXXXpS/pTXP’) due to the absence of the N-terminal signature arginine (Figure S1), which prompted us to further investigate the mechanism underlying the recognition of the TFEB p-S211-peptide by YWHA/14-3-3 proteins. We next performed co-crystallization of YWHAB and YWHAG with the TFEB p-S211-peptide. After extensive trials with crystal screening and optimization, high quality crystals of both YWHAB and YWHAG in complex with the p-S211-peptide (with the sequence of ‘204LVGVTSSpSCPADLTQ218’) were obtained. The structures of the YWHAB-p-S211-peptide and YWHAG-p-S211-peptide complexes were determined using the molecular replacement method and were further refined to 2.0 Å and 2.1 Å, respectively (Figure 2(a–b) and Table S1).
In the structures of the 2 complexes, YWHAB and YWHAG both adopted a canonical YWHA/14-3-3 fold with 9 antiparallel α-helices (from αA to αI). With the aid of the first 4 helices (αA to αD), YWHAB and YWHAG formed a horseshoe-shaped dimer and the 2 subunits of the dimer were related by a 2-fold symmetry (Figure 2(a–b)), consistent with the previous structural studies of YWHA/14-3-3 proteins [22]. In each subunit of the dimer, the 4 helices αC, αE, αG and αI constituted an amphipathic target-binding groove that could recognize the phosphorylated peptide (Figure 2(a–b)). The overall structures of the YWHAB-p-S211-peptide and YWHAG-p-S211-peptide complexes resembled each other with a root-mean-square deviation of ~0.59 Å for all atoms (Figure 2(c)). The electron density maps of the p-S211-peptide were clearly resolved in both complex structures and the p-S211-peptide was found to fully occupy the target-binding grooves of YWHAB and YWHAG (Figure 2(d–e)).
Figure 2.
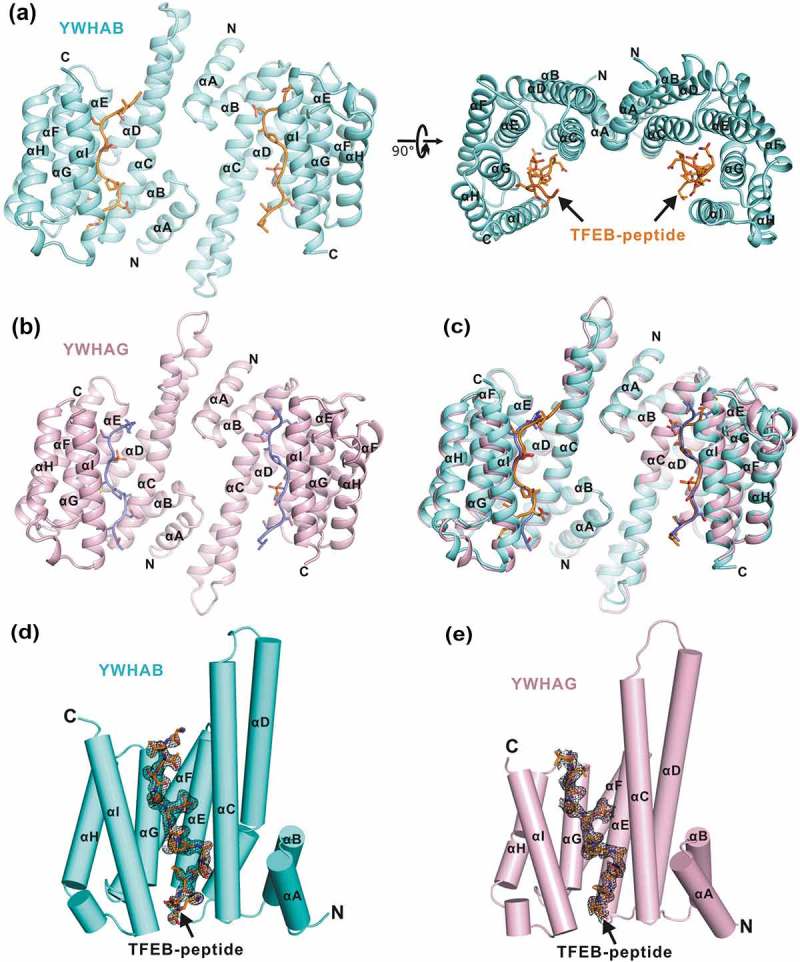
The overall structures of YWHA/14-3-3 proteins in complex with the TFEB p-S211-peptide. (a-b) Ribbon diagrams of the YWHAB-p-S211-peptide (a) and YWHAG-p-S211-peptide (b) complexes. YWHAB and YWHAG are colored in cyan and pink, respectively. The sidechains of the residues in the p-S211-peptide are shown as sticks. (c) Superimposition of the structures of the YWHAB-p-S211-peptide and YWHAG-p-S211-peptide complexes. (d-e) The resolved electron density maps of the p-S211-peptide in the structures of the YWHAB-p-S211-peptide (d) and YWHAG-p-S211-peptide (e) complexes. In this drawing, YWHA/14-3-3 proteins are shown as cylinders and colored as those in panel A and B. The omit electron density maps of the p-S211-peptide in the 2 structures are shown and contoured at the 1.5 σ level.
For co-crystallization of the 2 complexes, the p-S211-peptide we used was 15 residues. In the structure of the YWHAB-p-S211-peptide complex, nearly all of the residues of the p-S211-peptide could be traced except for the 2 residues at the N terminus (Figure 2(d)). However, in the structure of the YWHAG-p-S211-peptide complex, the N-terminal 3 residues and the C-terminal 1 residue were untraceable in the electron density maps (Figure 2(e)). Given the high similarity of the 2 complex structures and the structural completeness of the p-S211-peptide in the structure of the YWHAB-p-S211-peptide complex, this complex structure was chosen as the representative one for further structural analysis in detail.
Interaction interface between YWHA/14-3-3 proteins and the TFEB p-S211-peptide
In the structure of the representative YWHAB-p-S211-peptide complex, the target-binding groove of YWHA/14-3-3 proteins was amphipathic and the interaction interface between YWHA/14-3-3 proteins and the TFEB p-S211-peptide was mediated by hydrophobic, electrostatic and hydrogen-bonding interactions (Figure 3(a)). In the upper part of the interaction interface (from the N to C terminus of the p-S211-peptide and divided at p-S211), the first 3 residues (G206, V207 and T208) protruded from the target-binding groove and made little contact with YWHA/14-3-3 proteins (Figure 3(b)); i.e., only some hydrophobic contacts were found between V207 from the peptide and L229 from αI. The major contacts of the upper part were contributed by a hydrogen-bonding network formed by the side-chains of S209 from the peptide, W230 from αI and E182 from αG (Figure 3(b)). In addition, another hydrogen bond was formed between the backbone of S210 from the peptide and the side-chain of N226 from αI (Figure 3(b)). Consistent with the functional importance of S211, p-S211 sat deeply in the core of the target-binding groove and made extensive electrostatic and hydrogen-bonding contacts with YWHA/14-3-3 proteins. The negatively charged side-chain of p-S211 formed electrostatic interactions with the side-chains of R58, K51 from αC and R219 from αE (Figure 3(c)). Moreover, a hydrogen bond was also formed between the side-chains of p-S211 and Y130 from αE (Figure 3(c)).
Figure 3.
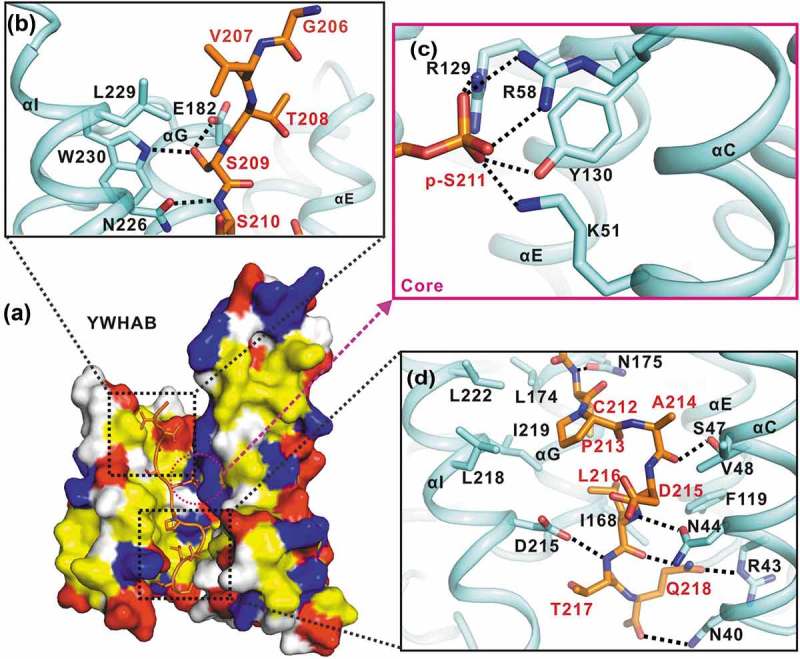
The interaction interface between YWHAB and the p-S211-peptide. (a) A combined surface and stick representation showing the interaction interface between YWHAB and the p-S211-peptide. The p-S211-peptide is in the stick representation (colored in orange) and YWHAB is in the surface representation. In this surface drawing, the hydrophobic, positively charged, negatively charged residues and the rest of the residues are colored in yellow, blue, red and white, respectively. The upper and lower interaction sites are highlighted by dashed boxes. (b-d) A combined ribbon-and-stick model illustrates the interaction interface between YWHAB and the p-S211-peptide. In this drawing, YWHAB and the p-S211-peptide are colored following the color patterns of Figure 1(b) and the sidechains of the residues involved in the interaction interface are shown as sticks. Hydrogen bonds and salt bridges are indicated by dashed lines.
The lower part of the interaction interface is predominantly contributed by hydrophobic interactions. Specifically, in YWHA/14-3-3 proteins, I168 and L174 from αG, and L218, I219, L222 from αI, together with F119 from αE and V48 from αC, formed a large hydrophobic pocket (Figure 3(d)). The hydrophobic residues C212, P213, A214 and L216 from the peptide concomitantly pack into this hydrophobic pocket to further stabilize the interaction interface. In addition to these hydrophobic contacts, several hydrogen bonds were also formed by the backbones of C212, A214, L216 and T217 from the peptide and the side-chains of N175 from αG, S47 and N44 from αC and D215 from αI, respectively (Figure 3(d)). At the bottom of the interaction interface, another hydrogen-bonding network was constituted by the backbone and side-chain of Q218 from the peptide and N40 and R43 from αC (Figure 3(d)). Notably, from the overall packing track of the p-S211-peptide in the target-binding groove of YWHA/14-3-3 proteins, the upper part of the peptide was regularly straight but the lower part was irregularly curved (Figure 3(d)), which was likely caused by P213 that would generate some kinks in the peptide.
Based on the sequence alignment of the different isoforms of YWHA/14-3-3 proteins, the target-binding grooves of YWHA/14-3-3 proteins were highly conserved (Figure S2A). Consistently, the structural comparison of the YWHAB/p-S211-peptide and YWHAG/p-S211-peptide complexes demonstrated that the p-S211-peptide bound to YWHAB and YWHAG in a similar manner (Figure S2B-C). However, after superimposition of the 2 complex structures, some minor structural differences were observed at both the N- and C-termini of the p-S211-peptide (Figure S2D-E), possibly due to the intrinsic dynamic flexibilities at the 2 termini. Notably, in comparison to the YWHAB-p-S211-peptide complex, L216 from the peptide in the YWHAG-p-S211-peptide complex moved downward but was still embedded in the hydrophobic pocket (Figure S2D-E), further suggesting that the hydrophobic packing in the lower part of the target-binding groove was somewhat nonspecific and dynamic.
YWHA/14-3-3 proteins recognize the TFEB p-S211-peptide by a noncanonical mode
It has been well established that YWHA/14-3-3 proteins can recognize phosphorylated serine/threonine in a consensus sequence of ‘RSXpS/pTXP’ (mode I) and ‘RXXXpS/pTXP’ (mode II) [23]. The peptide library screening and binding experiments also suggest a high selectivity for a positively charged residue at (−3) or (−4) positions of the phosphorylated peptide [24]. The structures of YWHA/14-3-3 proteins in complex with mode I/II phospho-peptides have revealed that the signature arginine at the N terminus of the peptide formed the electrostatic interactions with the upper part of the target-binding groove (Figure 4(a–b)). Moreover, in the complex structures, P(+2) induced a sharp turn in the peptide, which would likely direct the following residues to be out of the target-binding groove. In addition to canonical mode I/II, there was another mode for YWHA/14-3-3 proteins to recognize the carboxyl tail peptide (mode III), in which the N-terminal arginine was absent but the carboxyl tail of the peptide formed additional electrostatic interactions with the target-binding groove to enhance the binding (Figure 4(c)).
Figure 4.
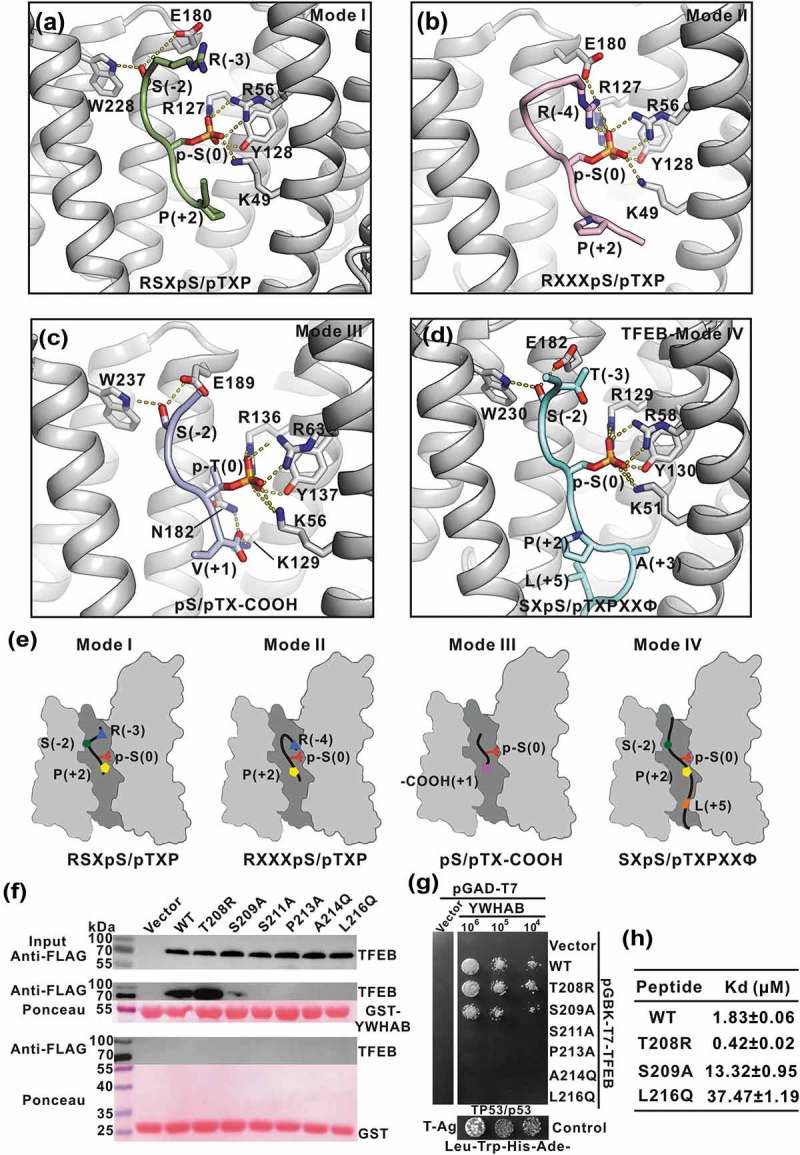
YWHA/14-3-3 proteins recognize TFEB p-S211-peptide by a non-canonical mode. (a-c) A combined ribbon-and-stick representation of the ‘mode I’ (PDB: 1QJB) (a), ‘mode II’ (PDB: 1QJA) (b) and ‘mode III’ (PDB: 1Q9D) (c) binding motifs for YWHA/14-3-3 proteins. The consensus binding motifs for each mode are indicated in each panel. Besides the phosphorylated-serine-mediated electrostatic interactions, R(−3) and S(−2) in mode I, R(−4) in mode II and the carboxyl tail of V(+1) in mode III also contribute to the binding. (d) TFEB binds to YWHA/14-3-3 proteins via a noncanonical mode. Compared with the canonical mode I and II binding motifs, the TFEB p-S211-peptide lacks the N-terminal arginine but contains C-terminal hydrophobic residues that bind to the lower part of the target-binding groove, which can be defined as mode IV. (e) The schematic models of the different YWHA/14-3-3-binding modes (mode I-IV). In this drawing, YWHA/14-3-3 proteins are colored in gray and their central target-binding grooves are highlighted in dark gray. The phosphorylated peptides are drawn as black lines and the key residues for binding to YWHA/14-3-3 proteins in different modes are depicted as green dots (S[−2]), blue triangles (R[−3] or R[−4]), red forks (p-S[0]), yellow pentagons (P[+2]), pink squares (V[+1]-COOH) and orange parallelogram (L[+5]). (f-g) GST affinity-isolation analysis (f) and yeast two-hybrid assay (g) of the interactions between the TFEB mutants and YWHAB. As compared to the wild-type protein, the point mutations in TFEB affected its binding to YWHAB. The interaction between TP53/p53 and T-Ag were used as the positive control for yeast two-hybrid assay. T-Ag, SV40 T antigen. (h) The binding affinities between TFEB p-S211-peptide (with mutations) and YWHAB determined by ITC experiments. The T208R mutation can enhance the binding affinity but the S209A and L216Q mutations both reduce it.
In contrast to the canonical YWHA/14-3-3-binding modes, the critical arginine was also missing in the N terminus of the TFEB p-S211-peptide (with the sequence of ‘GVTSSpSCPADLTQ’), and instead, the (−3) and (−4) positions of the peptide were occupied by T(−3) and V(−4), respectively. However, the binding affinities between YWHA/14-3-3 proteins and the TFEB p-S211-peptide were not compromised despite the lack of the N-terminal positively charged arginine (Figure 1(d)). In the YWHA-p-S211-peptide complex structure, P(+2) induced a sharp turn of the peptide and concomitantly allowed the resides L216, T217 and Q218 to intimately contact with the target-binding groove of YWHA/14-3-3 proteins (Figures 3(d) and 4(d)). These extensive hydrophobic and hydrogen-bonding interactions between the C-terminal half of the peptide and the target-binding groove of YWHA/14-3-3 proteins (with a surface area of ~895 Å2) compensated for the absence of the N-terminal arginine. Thus, the recognition mode of YWHA/14-3-3 proteins for the TFEB p-S211-peptide was significantly distinct from canonical mode I/II (Figure 4(e)). Moreover, this noncanonical mode of YWHA/14-3-3 proteins for the TFEB p-S211-peptide was also different from mode III for the carboxyl tail peptide and could be referred to as a new target recognition mode of YWHA/14-3-3 proteins (mode IV) (Figure 4(e)).
In comparison to previously identified mode I/II/III, the obvious difference in the new mode IV is the additional extensive hydrophobic and hydrogen-bonding interactions between the peptide and the lower part the target-binding groove (Figure 4(a–d)), indicating that the target-binding groove of YWHA/14-3-3 proteins is intrinsically capable of binding to the hydrophobic peptide. Consistently, YWHA/14-3-3 proteins can also recognize the nonphosphorylated hydrophobic peptide [25]. The structure of the YWHA-nonphosphorylated peptide complex demonstrated that the peptide indeed formed extensive hydrophobic and hydrogen-bonding interactions with the lower part of the target-binding groove of YWHA/14-3-3 proteins (Figure S3), resembling the binding between the C-terminal half of the TFEB p-S211-peptide and the target-binding groove. Thus, the lower hydrophobic part of the target-binding groove may endow the plasticity and versatility of YWHA/14-3-3 proteins for recognizing a diverse range of different targets.
Evaluation of the key residues in TFEB for binding to YWHA/14-3-3 proteins
With the structures of YWHA/14-3-3 proteins in complex with the TFEB p-S211-peptide, we next evaluated the key residues in TFEB for binding to YWHA/14-3-3 proteins. Based on the above structural analysis, we made a series of point mutations in full-length TFEB to disrupt the interaction interface and checked the binding between these TFEB mutants and YWHA/14-3-3 proteins by a GST affinity-isolation assay and yeast two-hybrid experiment (Figure 4(f–g)). Compared with the wild-type full-length TFEB protein, mutation of the core residue S211 (S211A) in the interaction interface abolished the binding between TFEB and YWHA/14-3-3 proteins. In addition, mutations of the hydrophobic residues in the lower part of the interaction interface (P213A, A214Q and L216Q) also disrupted the binding (Figure 4(f–g)), supporting the critical roles of the additional hydrophobic interactions in the new mode IV for binding to YWHA/14-3-3 proteins. In contrast, mutation of the residue in the upper part of the interaction interface (S209A) decreased (but did not abolish) the binding (Figure 4(f–g). Given the absence of the critical arginine in TFEB (Figure S1B), we further introduced arginine back into TFEB (T208R) and found that this mutation could enhance the binding between TFEB and YWHA/14-3-3 proteins (Figure 4(f–g)), consistent with canonical mode I/II. To further characterize the binding between YWHA/14-3-3 proteins and different TFEB mutants, we performed the ITC assay by using the synthesized p-S211-peptide. Consistently, the T208R mutation could enhance the binding affinity (0.42 ± 0.02 μM), whereas the S209A and L216Q mutations both reduced it (13.32 ± 0.95 μM and 37.47 ± 1.19 μM, respectively) in comparison to the wild-type p-S211-peptide (1.83 ± 0.06 μM) (Figure 4(h) and S4). Taken together, all the mutational data demonstrated that the residues of TFEB involved in the interaction interface packing are essential for binding to YWHA/14-3-3 proteins.
The interactions between YWHA/14-3-3 proteins and TFEB are essential for TFEB subcellular localization
After the biochemical characterization, we checked the subcellular localization of TFEB and evaluated the effects of the point mutations (that disrupt the interaction interface) on TFEB at the cellular level. Under nutrient-rich conditions, wild-type TFEB was largely distributed in the cytoplasm (Figure 5(a–b)), consistent with previous studies. As expected, the TFEB mutants with the point mutations to disrupt the TFEB-YWHA interaction (S209A, S211A, P213A, A214Q and L216Q) showed significantly increased nuclear localization under nutrient-rich conditions, whereas the TFEB mutant with the point mutation to enhance the TFEB-YWHA interaction (T208R) did not (Figure 5(a–b)), indicating the essential role of the TFEB-YWHA binding for TFEB cytoplasmic localization. In contrast, under starvation conditions, wild-type TFEB and the TFEB mutants (S209A, S211A, P213A, A214Q and L216Q) were mainly localized in the nucleus, but in comparison to wild-type TFEB, the TFEB mutant (T208R) showed decreased nuclear localization (Figure 5(a–b)), possibly due to the enhanced binding of YWHA/14-3-3 proteins (Figure 5(c) and see discussion below). Taken together, all the cellular data demonstrated that the interactions between YWHA/14-3-3 proteins and TFEB are essential for controlling TFEB subcellular localization.
Figure 5.
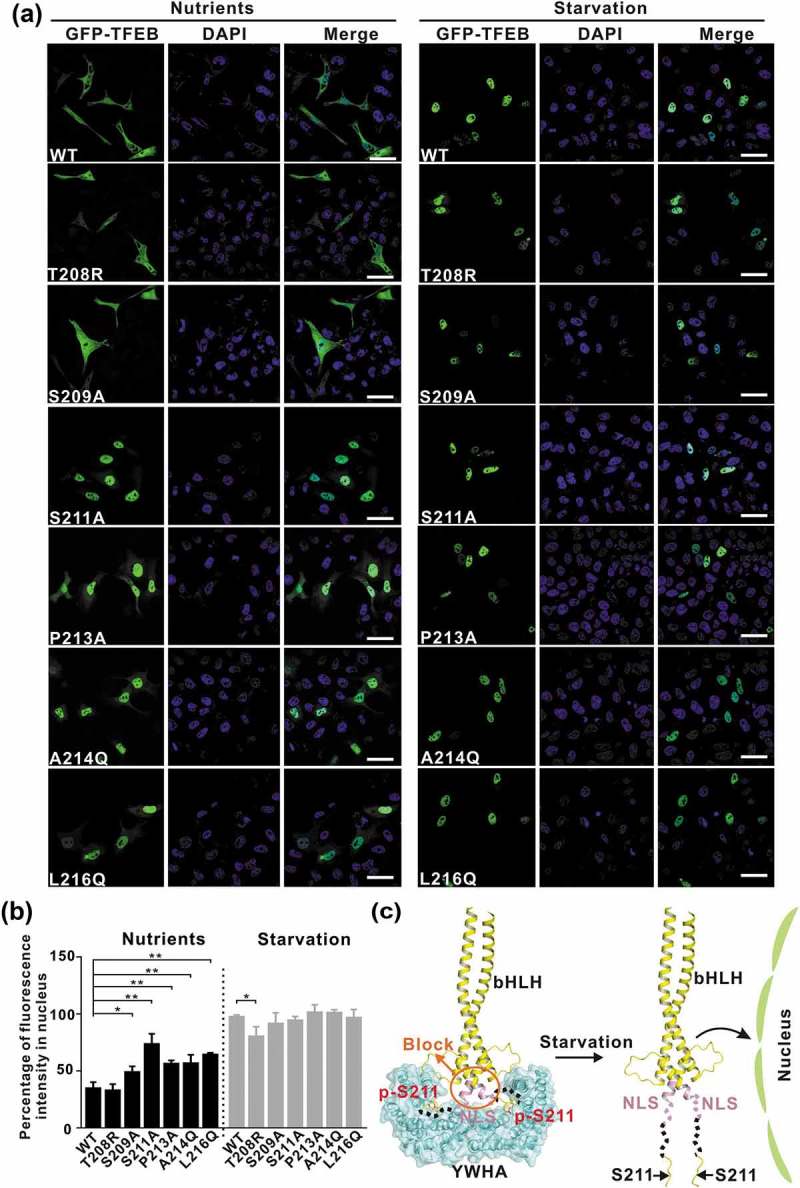
The interactions between YWHA/14-3-3 proteins and TFEB are essential for TFEB subcellular localization. (a) Subcellular distribution of TFEB and its mutants under nutrient-rich and starvation conditions. Under nutrient-rich conditions, wild-type TFEB is largely distributed in the cytoplasm. The TFEB mutants (S209A, S211A, P213A, A214Q and L216Q) showed increased nuclear localization, whereas the TFEBT208R mutant did not. Upon starvation, wild-type TFEB and the TFEB mutants are mainly localized in the nucleus. Scale bar: 50 μm. (b) Quantification of the subcellular distribution data shown in panel A. The percentage of the fluorescence intensity of each construct in the nucleus was quantified (average of 4 experiments, n > 50 cells for each experiment). Each bar represents the mean ±SD, **p < 0.01, *p < 0.05. (c) A schematic model illustrating the YWHA/14-3-3-mediated regulation of TFEB subcellular localization. Briefly, the binding of YWHA/14-3-3 proteins could induce the conformational changes of the p-S211-site and the region between the NLS and the p-S211-site, which would potentially interfere with the NLS. The structural model was built based on the structures of the bHLH domain of MITF (PDB: 4ATH) and the YWHA-p-S211-peptide complex.
Modulation of the binding of TFEB to YWHA/14-3-3 proteins affects TFEB cellular functions
Finally, we evaluated the effects of the mutations of TFEB on its cellular functions by checking autophagy and the expression of TFEB target genes. To probe the autophagic flux, we checked the autophagy marker LC3-II in the presence of bafilomycin A1 (an inhibitor of autophagosome-lysosome fusion). As expected, the T208R mutation in TFEB that enhances the binding to YWHA/14-3-3 proteins impaired the autophagic flux under both nutrient-rich and starvation conditions as indicated by a decreased LC3-II level (Figure 6(a) and see discussion below). Consistently, the expression levels of TFEB target genes (such as ATP6V1H, CTSB, CTSD and LAMP1) were also decreased by the T208R mutation in TFEB (Figure 6(b)). In contrast, all the other mutations (S209A, S211A and L216Q) in TFEB that disrupt the binding to YWHA/14-3-3 proteins could somewhat promote the autophagic flux under both nutrient-rich and starvation conditions as indicated by the increased LC3-II level (Figure 6(a) and see discussion below), and the expression levels of TFEB target genes were also increased by these mutations in TFEB (Figure 6(b)). Taken together, modulations of the binding of TFEB to YWHA/14-3-3 proteins can have an impact on both the cellular autophagy activity and the expression of TFEB target genes.
Figure 6.
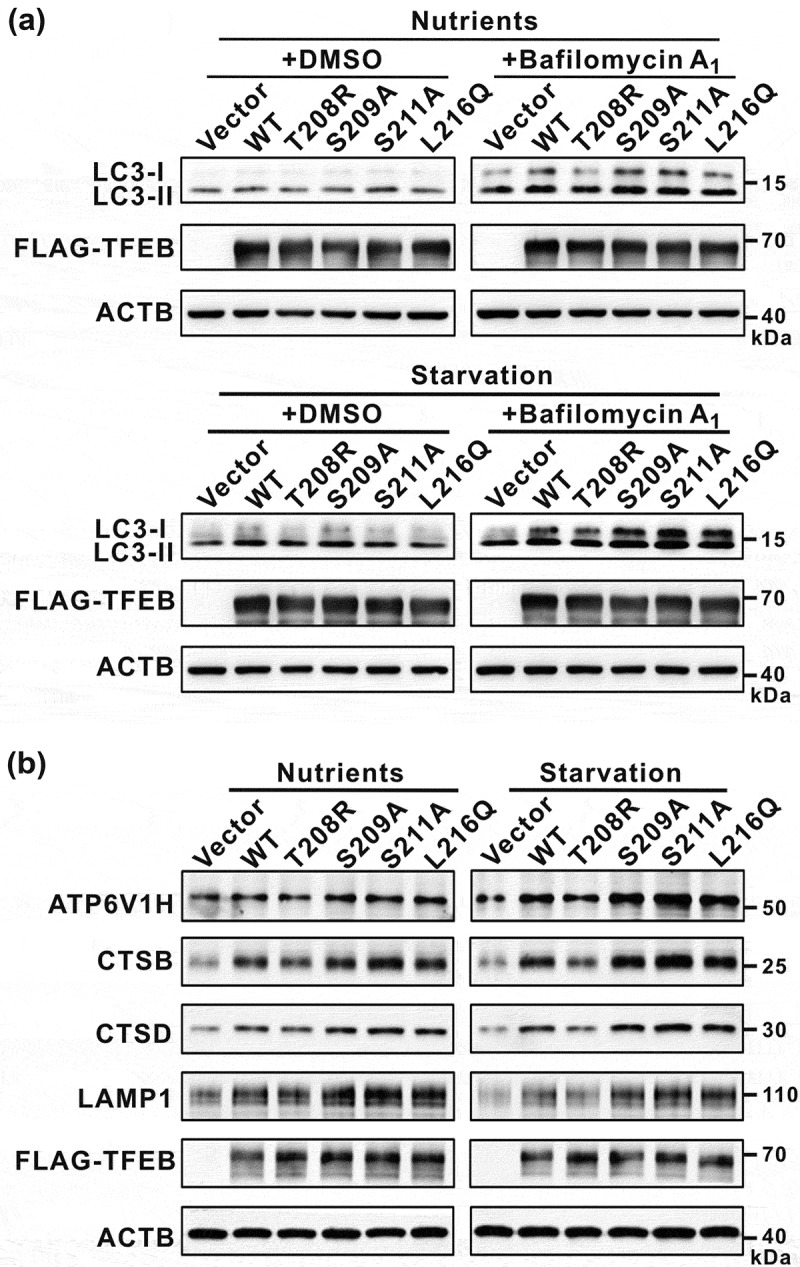
Modulations of the binding of TFEB to YWHA/14-3-3 proteins affect TFEB cellular functions. (a) Immunoblotting analysis of LC3 in TFEB-overexpressing cells (with wild-type TFEB and its various mutants) in the absence and presence of bafilomycin A1 under nutrient-rich and starvation conditions. (b) Western-blot analysis of the expression of TFEB target genes (ATP6V1H, CTSB, CTSD and LAMP1) after transfection with wild-type TFEB and its various mutants. ACTB was used as the loading control.
Discussion
A new binding motif in TFEB for YWHA/14-3-3 proteins
YWHA/14-3-3 proteins are phospho-regulatory proteins that can specifically recognize phosphorylated serine/threonine with a well-established sequence motif ‘RSXpS/pTXP’ (mode I) and ‘RXXXpS/pTXP’ (mode II) (Figure 4) [23]. In addition to the canonical mode I/II, YWHA/14-3-3 proteins can also bind to the phosphorylated carboxyl tail peptide ‘pS/pTX-COOH’ (mode III) (Figure 4) [26]. Based on structural studies of the YWHA-TFEB complex, we here found a new binding motif embedded in TFEB ‘SXpS/pTXPXXφ’ (mode IV) for YWHA/14-3-3 proteins. In this new noncanonical mode, the N-terminal critical arginine for binding to YWHA/14-3-3 proteins is absent, but instead, the C terminus contains additional hydrophobic residues that can intimately bind to the lower part of the target-binding groove of YWHA/14-3-3 proteins (Figure 3), which would compensate for the N-terminal defect and significantly enhance the binding. Mutations of these hydrophobic residues severely disrupted the binding between TFEB and YWHA/14-3-3 proteins (Figure 4), supporting the critical roles of the hydrophobic interactions in the binding interface for mode IV. Given that the sequence including the binding mode IV is extremely conserved in the MiT/TFE family (Figure 1(b)), other members of the MiT/TFE family such as TFE3, MITF and TFEC may bind to YWHA/14-3-3 proteins by employing a similar binding mode. Moreover, the new binding motif in TFEB can be further found in other known YWHA/14-3-3-binding partners that may also use this mode IV to mediate the interactions with YWHA/14-3-3 proteins (Table S2).
Target recognition versatility of YWHA/14-3-3 proteins
Based on the structural analysis of YWHA/14-3-3 proteins in complex with various phosphorylated peptides, the target-binding groove of YWHA/14-3-3 proteins are highly conserved in different isoforms and can be further divided into the upper and lower parts (Figure 3 and S2). In previously identified mode I/II/III, the upper part of the target-binding groove is chiefly involved in target recognition. In addition to binding to phosphorylated serine/threonine, the upper part of the target-binding groove can also interact with the N-terminal arginine of the peptides in canonical mode I and II (Figure 4). In contrast, the lower part of the target-binding groove is largely hydrophobic and can further recognize the hydrophobic residues from the peptide. In newly identified mode IV, the C-terminal hydrophobic residues of the peptide are deeply buried in the lower part of the target-binding groove (Figure 3). More intriguingly, the lower part of the target-binding groove is also directly involved in mediating the interactions between YWHA/14-3-3 proteins and nonphosphorylated peptides (that form an amphipathic helix to bind to this site) (Figure S3). Thus, YWHA/14-3-3 proteins can alternately use the 2 parts of the target-binding groove or combine both of them to recognize a diverse range of phosphorylated peptides with different binding modes (Figure 4). The 2-part-designed target-binding groove is intrinsically dedicated for accommodating different target proteins and endows the target-binding versatility of YWHA/14-3-3 proteins, consistent with the prevalent regulatory role of YWHA/14-3-3 proteins in controlling different cellular processes.
Potential mechanism for YWHA/14-3-3-mediated regulation of TFEB cytoplasmic localization and its cellular functions
Previous studies demonstrated that phosphorylation of S211 in TFEB by MTORC1 under nutrient-rich conditions is critical for binding to YWHA/14-3-3 proteins to keep TFEB in the cytoplasm [17]. We confirmed the binding between TFEB and YWHA/14-3-3 proteins and determined the structure of YWHA/14-3-3 proteins in complex with the TFEB p-S211-peptide (Figures 1 and 2). In the complex structure, YWHA/14-3-3 proteins form a dimeric structure that can simultaneously capture 2 TFEB p-S211-peptides. We further found that purified YWHA/14-3-3 proteins and full-length TFEB both exist in a dimeric state (Figure S5). Based on the previous structural studies of TFEB, the dimerization of full-length TFEB is most likely caused by the bHLH domain that can form a coiled-coil dimer [27]. Thus, dimeric full-length TFEB would expose 2 p-S211-sites that would tend to simultaneously bind to dimeric YWHA/14-3-3 proteins (Figure 5(c)), although the possibility of forming a high-order oligomeric complex could not be excluded at the current stage. In this dimeric model, the nuclear localization signal (NLS) of TFEB (from residues 241 to 252, responsible for TFEB nuclear localization) is close to the p-S211-site (i.e., ~20 residues from the YWHA/14-3-3-binding p-S211-site). The binding of YWHA/14-3-3 proteins would induce conformational changes of the p-S211-site and the region between the NLS and the p-S211-site, which would somewhat interfere with the NLS and block its nuclear localization function (Figure 5(c)). Consistent with this hypothesis, mutations of the interaction interface between YWHA/14-3-3 proteins and TFEB significantly affected the subcellular localization of TFEB (Figure 5(a–b)). In contrast, upon starvation and dephosphorylation of S211, YWHA/14-3-3 proteins would dissociate from TFEB, and the NLS could be exposed for mediating the nuclear localization of TFEB (Figure 5(c)). Nevertheless, the structure of YWHA/14-3-3 proteins in complex with full-length TFEB warrants further investigations.
The subcellular localization of TFEB has been proposed to be immediately coupled with its cellular functions. Consistent with this hypothesis, modulations of the binding of TFEB to YWHA/14-3-3 proteins were demonstrated to affect both the expression of TFEB target genes and the cellular autophagy activity (Figure 6). However, the effects of the mutations of TFEB on its cellular functions were shown to be somewhat different from that on its subcellular localization under different conditions, i.e., all the mutations affected TFEB cellular functions under both nutrient-rich and starvation conditions but significantly affected its subcellular localization only under either nutrient-rich or starvation condition (Figures 5 and 6). The possible explanation for this discrepancy is that slight differences in TFEB subcellular localization caused by the mutations might be further amplified to significant changes in its cellular functions. Nevertheless, the mutations in TFEB that either enhance or disrupt the binding to YWHA/14-3-3 proteins can modulate its cellular functions, supporting the idea that the interactions between YWHA/14-3-3 proteins and TFEB are of functional importance.
Materials and methods
Protein expression and purification
DNA sequences of human YWHAB (residues 1-247) and YWHAG (residues 1-246) were each cloned into a modified pET32a (Novogen, 69337-3) vector. Recombinant proteins were expressed in Escherichia coli BL21 (DE3) host cells at 16°C. The TRX-His6-tagged fusion proteins were purified using Ni2+-NTA affinity chromatography (GE Healthcare Life Sciences, 17517201) followed by size-exclusion chromatography (Superdex 200 10/300 GL column; GE Healthcare Life Sciences, 17517501). After cleavage of the tag, the resulting proteins were further purified by another step of size-exclusion chromatography with buffer containing 20 mM HEPES, pH 7.0, 150 mM NaCl, 1 mM DTT, 5% (v:v) glycerol (Amresco, 0854). Full-length TFEB was cloned into a modified pFastBac1 vector (Invitrogen, 10359-016). Recombinant protein was expressed in a baculovirus system in Sf9 (Invitrogen, 12552014) insect cells and was purified by Ni2+-NTA and Strep-Tactin (IBA, 2-1201-010) affinity chromatography followed by size-exclusion chromatography (Superdex 200).
Co-IP assay
For Co-IP assay, DNA sequences encoding full-length TFEB and its mutants were cloned into the pcDNA3.1-3× FLAG vector (Genechem, GV141). HeLa cells were cultured at 37°C with 5% CO2 in DMEM supplemented with 10% (v:v) fetal bovine serum (FBS; WISENT, 087–150), 100 U/ml penicillin and 100 mg/ml streptomycin (ThermoFisher Scientific, 15140122). Upon 70% confluency, the cells were transfected with a plasmid encoding FLAG-tagged wild-type TFEB and its mutants and were further incubated for 48 h. The cells were then washed with phosphate buffered saline (PBS; ThermoFisher Scientific, 10010023), lysed with lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% [v:v] Triton X-100 [Sigma-Aldrich, X100], protease [Amresco, M250] and phosphatase [ThermoFisher Scientific, A32957] inhibitors) and were centrifuged for 15 min at 14,000 g to remove insoluble debris. The supernatants were transferred to new tubes and were incubated with anti-FLAG M2 affinity resin beads (Sigma-Aldrich, A2220) for 4–5 h at 4°C. After being resuspended and washed 3 times in TBS (20 mM Tris-HCl, pH 8.0, 150 mM NaCl), the resin beads were mixed with 2 × SDS-loading buffer, boiled at 95°C for 10 min and centrifuged for 10 min at 14,000 g. The supernatants were then resolved by SDS-PAGE and the proteins were detected by immunoblotting using the anti-YWHAB (Cell Signaling Technology, 9636), anti-YWHAG (Proteintech, 12381-1-AP) and anti-FLAG antibodies (Proteintech, 66008-2-Ig).
Crystallization, data collection and structural determination
Purified YWHA/14-3-3 proteins (20 mg/ml in 20 mM HEPES, pH 7.0, 150 mM NaCl, 1 mM DTT, and 5% [v:v] glycerol) were mixed with commercially synthesized TFEB p-S211-peptide with a molar ratio of 1:5 (protein to peptide). Crystals of the YWHAB-p-S211-peptide complex were grown in 0.1 M sodium malonate (Sigma-Aldrich, 63409), pH 4.0, 12% (w:v) PEG 3350 (Sigma-Aldrich, P3640). Crystals of the YWHAG-p-S211-peptide complex were grown in 0.1 M Tris-HCl, pH 7.3, 0.2 M calcium acetate (Sigma-Aldrich, 21056), 20% (w:v) PEG 3350. All the crystals were obtained using the vapor diffusion method (sitting drop) at 16°C. Before being flash-frozen in liquid nitrogen, crystals were cryo-protected in their mother liquor supplemented with 15% (v:v) ethylene glycol. Diffraction data were collected at the beamline BL17U at the Shanghai Synchrotron Radiation Facility (SSRF) using a wavelength of 0.979 Å at 100 K [28], and were processed and scaled using HKL2000 [29]. Both structures were determined by the molecular replacement method with the structure of YWHAB (PDB code: 2BQ0) and YWHAG (PDB code: 3UZD) as searching models using PHASER [30]. The p-S211-peptide in each complex was manually modeled into the structure according to the 2Fo-Fc and Fo-Fc electron density maps using COOT [31]. The structures were further fitted and refined with PHENIX [32] and the overall quality of the final structural models was assessed using PROCHECK [33]. The structure figures were prepared with the program PyMOL (http://www.pymol.org). The statistics for data collection and structural refinement were summarized in Table S1.
Isothermal titration calorimetry (ITC) assay
The ITC assay was performed using a MicroCalorimeter ITC200 (GE Healthcare Life Sciences, USA) at 25°C. Prior to the experiment, the protein samples were dialyzed in a buffer containing 20 mM Tris-HCl, pH 7.5, 200 mM NaCl. YWHA/14-3-3 proteins were in the sample cell and the synthetic phosphorylated peptides of TFEB were in the syringe of the instrument. In each experiment, the peptide (~200 μM) was sequentially injected into the stirred calorimeter cell initially containing the YWHA/14-3-3 protein sample (~40 μM) with the injection sequence of 20 × 2 µl at 2 min intervals. The heat of dilution obtained by the titration of the peptide into the buffer was subtracted. The integrated, corrected, and concentration-normalized peak areas of the raw data were finally fitted with a model of one binding site using ORIGIN 7.0 (OriginLab).
Yeast two-hybrid experiment
The inserts containing DNA sequences encoding YWHAB and TFEB were cloned into the vectors pGAD-T7 (Clontech, 630442) and pGBK-T7 (Clontech, 631604) respectively. Plasmids for transient expression experiments were extracted with an EndoFree Plasmid MiDi Kit (CWBIO, CW2105). The yeast strain AH109 (Clontech, 630444) was transformed with pGADT7 and pGBKT7 as a negative control, and was also transformed with T-Ag and TP53/p53 as a positive control. The Gal4 AD-YWHAB fusion plasmids and Gal4 BD-TFEB (wild-type and the T208R, S209A, S211A, P213A, A214Q and L216Q mutants) fusion plasmids were co-transformed into yeast strain AH109 following a lithium acetate protocol [34], and the transformants were grown on synthetically defined SD/-Trp-Leu dropout medium plates for 2–3 days. The yeast cells with co-transformation of the 2 fusion plasmids were further dropped on SD/-Trp-Leu-His-Ade dropout medium plates with a series of 10-fold dilutions (from 106 to 104) for checking the interactions.
GST affinity-isolation assay
The GST affinity-isolation assay was performed using immobilized GST or GST-tagged YWHAB fusion protein incubated with the total cell lysate of HeLa cells transfected with various FLAG-tagged TFEB proteins (wild-type and the T208R, S209A, S211A, P213A, A214Q and L216Q mutants). Briefly, the cell suspensions were prepared in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 10 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 0.5% sodium deoxycholate [Sigma-Aldrich, D6750], 1% Triton X-100) containing protease inhibitors. After centrifugation, the supernatants were first incubated with 50 μl of glutathione Sepharose 4B Fast Flow (GE Healthcare Life Sciences, 17075604) 50% slurry beads in RIPA buffer for 30 min. The cleared supernatants were then transferred to a new batch of 50 μl 50% slurry beads, mixed with 200 µg GST or GST-tagged YWHAB for 3 h at 4°C. After washing the beads 3 times with RIPA buffer, the proteins captured by the beads were eluted by boiling with the SDS-PAGE sample buffer. GST and GST-tagged YWHAB were detected by ponceau S staining and FLAG-tagged TFEB proteins were analyzed by western blot using the anti-FLAG antibody.
Western blot analysis and antibodies
Cells were transfected with a plasmid encoding wild-type TFEB, its various mutants (T208R, S209A, S211A and L216Q) and control vector and were incubated for 36 h. For starvation treatment, cells were further incubated in Earle’s balanced salt solution (ThermoFisher Scientific, 14155063) for 4 h. Cells were then rinsed with PBS and lysed with RIPA buffer supplemented with protease and phosphatase inhibitors for 20 min at 4°C. Lysates were cleared by centrifugation at 16,000 g for 20 min at 4°C. Loading buffer (5×) was added and samples were boiled for 10 min. Each sample equivalent of 20 μg total protein was separated by SDS-PAGE followed by electrophoretic transfer onto polyvinylidenedifluoride membrane. Membranes were saturated with 5% (w:v) defatted milk in TBST (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% Tween-20 [Amresco, 0777]) for 2 h at room temperature and were incubated overnight at 4°C with the following antibodies: LC3 (a generous gift from Prof. Hong Zhang, Institute of Biophysics, CAS), FLAG, CTSB (R&D Systems, AF953), CTSD (Abcam, ab72915), LAMP1 (Cell Signaling Technology, 3243), ATP6V1H (Proteintech, 26683-1-AP) and ACTB (Proteintech, 20536-1-AP). After being washed in TBST 3 times, membranes were probed with corresponding secondary antibodies (Sangon Biotech, D110058/D110087/D110117) for 1 h at 25°C. Membranes were then washed in TBST and bands were visualized using the ECL detection reagent (Tanon, 180-5001).
Assay of the autophagic flux
To check the autophagic flux, cells transfected with a plasmid encoding wild-type TFEB and various mutants were treated with bafilomycin A1 (MedChemExpress, HY-100558), an inhibitor that blocks the fusion of autophagosomes with lysosomes, at the concentration of 100 nM for 24 h, and then were followed by starvation treatment. The cellular autophagic flux was estimated by the western blot analysis of LC3.
Cell culture, imaging and data analysis
DNA sequences encoding full-length TFEB and its various mutants were cloned into the pEGFP-N1 (Clontech, 6085-1) vector. HeLa cells were cultured in DMEM (Dulbecco’s modified Eagle medium; GE Healthcare Life Sciences, SH30021) supplemented with 10% (v:v) FBS and were grown at 37°C in a humidified atmosphere containing 5% (v:v) CO2. The cells were transfected with wild-type TFEB and various mutants with Lipofectamine 2000 (ThermoFisher Scientific, 11668019) according to the manufacturer’s instructions. For starvation experiments, the cells were washed with PBS twice at room temperature and then were switched to Earle’s balanced salt solution for an additional 4 h. Florescence images were obtained on a FV1000 laser scanning confocal microscope (Olympus, Japan) equipped with a 60× oil-immersion objective lens (NA = 1.42). Confocal settings used for image capture were held constant in comparison experiments. All the fluorescence images were processed and analyzed using ImageJ (National Institutes of Health). The final quantification graphs were generated by EXCEL (Microsoft).
Statistical analysis
Data were plotted as means ± SD and statistical significance was determined using a two-tailed student’s t-test, where * p < 0.05, ** p < 0.01.
Accession numbers
The atomic coordinates of the YWHAB-p-S211-peptide complex and the YWHAG-p-S211-peptide complex have been deposited in the Protein Data Bank with the accession numbers 6A5Q and 6A5S, respectively.
Funding Statement
This work was supported by the National Key R&D Program of China under grant 2017YFA0503501 and 2017YFA0205501; the National Major Basic Research Program of China under grant 2014CB910202, and the National Natural Science Foundation of China under grants 31470746, 31770786, 31600608, 31600622 and 31470814.
Acknowledgments
We thank the beam-line BL17U of the Shanghai Synchrotron Radiation Facility for the beam time and Yongwei Wang for technical help.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary materials
Supplemental data for this article can be accessed here.
References
- [1].Martini-Stoica H, Xu Y, Ballabio A, et al. The Autophagy-lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 2016;39:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tsunemi T, Ashe TD, Morrison BE, et al. PGC-1alpha rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Thompson LM, Aiken CT, Kaltenbach LS, et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009;187:1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rui YN, Xu Z, Patel B, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. 2015;17:262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ross CA, Tabrizi SJ.. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. [DOI] [PubMed] [Google Scholar]
- [6].Qi L, Zhang XD, Wu JC, et al. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS One. 2012;7:e46834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ochaba J, Lukacsovich T, Csikos G, et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci USA. 2014;111:16889–16894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Martinez-Vicente M, Talloczy Z, Wong E, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13:567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Menzies FM, Fleming A, Rubinsztein DC.. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–357. [DOI] [PubMed] [Google Scholar]
- [10].Gozuacik D, Akkoc Y, Ozturk DG, et al. Autophagy-regulating microRNAs and cancer. Front Oncol. 2017;7:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129:2475–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rehli M, Den Elzen N, Cassady AI, et al. Cloning and characterization of the murine genes for bHLH-ZIP transcription factors TFEC and TFEB reveal a common gene organization for all MiT subfamily members. Genomics. 1999;56:111–120. [DOI] [PubMed] [Google Scholar]
- [13].Settembre C, Di MC, Va P, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links MTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Palmieri M, Impey S, Kang H, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–3866. [DOI] [PubMed] [Google Scholar]
- [16].Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, et al. Regulation of TFEB and V-ATPases by MTORC1. Embo J. 2011;30:3242–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wilker EW, Grant RA, Artim SC, et al. A structural basis for 14-3-3sigma functional specificity. J Biol Chem. 2005;280:18891–18898. [DOI] [PubMed] [Google Scholar]
- [20].Tzivion G, Avruch J. 14-3-3 proteins: active cofactors in cellular regulation by serine/threonine phosphorylation. J Biol Chem. 2002;277:3061–3064. [DOI] [PubMed] [Google Scholar]
- [21].Tong Y, Song F. Intracellular calcium signaling regulates autophagy via calcineurin-mediated TFEB dephosphorylation. Autophagy. 2015;11:1192–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rittinger K, Budman J, Xu J, et al. Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol Cell. 1999;4:153–166. [DOI] [PubMed] [Google Scholar]
- [23].Muslin AJ, Tanner JW, Allen PM, et al. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. [DOI] [PubMed] [Google Scholar]
- [24].Yaffe MB, Rittinger K, Volinia S, et al. The structural basis for 14-3-3: phosphopeptidebinding specificity. Cell. 1997;91:961–971. [DOI] [PubMed] [Google Scholar]
- [25].Ottmann C, Yasmin L, Weyand M, et al. Phosphorylation-independent interaction between 14-3-3 and exoenzyme S: from structure to pathogenesis. Embo J. 2007;26:902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Choe JY, Nelson SW, Arienti KL, et al. Inhibition of fructose-1,6-bisphosphatase by a new class of allosteric effectors. J Biol Chem. 2003;278:51176–51183. [DOI] [PubMed] [Google Scholar]
- [27].Pogenberg V, Ogmundsdottir MH, Bergsteinsdottir K, et al. Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF. Genes Dev. 2012;26:2647–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang QS, Yu F, Huang S, et al. The macromolecular crystallography beamline of SSRF. Nucl Sci Tech. 2015;26:12–17. [Google Scholar]
- [29].Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. [DOI] [PubMed] [Google Scholar]
- [30].McCoy AJ. Solving structures of protein complexes by molecular replacement with phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. [DOI] [PubMed] [Google Scholar]
- [32].Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Laskowski RA, Macarthur MW, Moss DS, et al. Procheck - a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- [34].Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2:31–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.