

ABSTRACT
A plethora of studies over several decades has demonstrated the importance of autophagy in aging and age-related neurodegenerative disease. The role of autophagy in damage clearance and cell survival is well established, and supports a prevailing view that increasing autophagic activity can be broadly beneficial, and could form the basis of anti-aging interventions. However, macroautophagy/autophagy also promotes some elements of senescence. For example, in C. elegans hermaphrodites it facilitates conversion of intestinal biomass into yolk, leading to sex-specific gut atrophy and senescent steatosis.
KEYWORDS: Aging, autophagy, atg-13, C. elegans, IIS, intestine, lipoprotein, pathology, reproduction, sexual dimorphism, vitellogenin, yolk
A key objective of research on aging (senescence) is to discover its initial causes (or etiologies). Although a tractable model for research on aging, the causes of senescence in C. elegans remain frustratingly elusive. After much testing, a primary role for stochastic molecular damage appears unlikely, and although protein aggregation occurs during C. elegans aging, its causes and impact on aging are unclear. Taking a different approach, we have shifted our focus from lifespan to senescent pathologies (i.e., diseases of aging) and their development as a metric of aging [1].
Developmental and correlation analyses of visible worm aging pathologies revealed that most of them originate surprisingly early in adulthood (from day 4 out of an ~20 day lifespan). Moreover, individual pathologies develop in relative synchrony, suggesting a syndrome of multimorbidity. Two visceral pathologies show a particularly close correlation with one another and with lifespan: intestinal atrophy and accumulation of vitellogenins (yolk proteins) and lipids into extracellular, pseudocoelomic lipoprotein pools (PLPs) – a form of senescent steatosis. The latter results from apparently futile run-on of yolk production in later life, leading to up to 7-fold increases in vitellogenin and 8-fold increases in triglyceride. Moreover, as worms age, neutral lipid stores translocate from the gut to other organs, and to the PLPs.
The close correlation of gut atrophy and PLP accumulation suggests a common etiology. Yolk is synthesized by the C. elegans intestine, and this synthesis continues during aging despite an age decline in feeding rate. This led us to hypothesize that the worm gut consumes its own biomass to maintain yolk production, and that run-on of gut-to-yolk biomass conversion leads to gut atrophy. Consistent with this, inhibiting vitellogenin synthesis by RNA-mediated interference (RNAi) inhibits both gut atrophy and PLP accumulation. Moreover, neither pathology occurs in C. elegans males, which do not synthesize yolk; however, induction of ectopic yolk production in males by mutation of mab-3 induces both pathologies, and shortens lifespan.
Consumption of intestinal biomass for yolk synthesis suggests a possible role of autophagy, whose intestinal levels increase at sexual maturity. We therefore asked: does inhibiting autophagy (by mutation or RNAi of atg genes) suppress gut atrophy and PLP accumulation? The answer is that it does. For example, both pathologies are inhibited by mutation or adult-specific and intestine-limited RNAi of atg-13, whereas the effect of atg-13 mutation on intestinal atrophy is reversed by gut-limited expression of atg-13-coding constructs. atg-13 inhibition also rescues visceral pathologies in yolk-producing mab-3 males, and leads to a more youthful lipidomic profile in older hermaphrodites. These results suggest that autophagy does indeed promote worm visceral aging.
Attenuation of insulin/IGF-1 signaling (IIS) by mutation of the IIS receptor daf-2 increases lifespan, and this effect requires the daf-16/FOXO transcription factor. daf-2 also suppresses visceral pathologies in a daf-16-dependent manner. Moreover, atg gene knockdown rescues visceral pathologies in daf-16 mutants, suggesting that autophagy acts either in parallel to or downstream of IIS. A further implication here is that levels of intestinal autophagy are reduced in daf-2 mutants. This is consistent with their observed reduction in protein turnover rate, though not the suppression of daf-2 longevity by inactivation of some atg genes. Our new findings imply that autophagy-dependent gut-to-yolk biomass conversion is a primary etiology of C. elegans senescence promoted by wild-type IIS.
These findings suggest that high wild-type levels of autophagy in the hermaphrodite gut promote visceral aging. Why would natural selection favor this? We found that combined knockdown of vitellogenesis and autophagy greatly reduces fertility, whereas mating – which increases egg production – accelerates gut atrophy. Thus, autophagy-dependent gut-to-yolk biomass may mediate a trade-off between hermaphrodite reproduction and aging; here, in principle, natural selection could favor basal gut autophagy levels that exceed cell maintenance requirements.
This study has a number of broad implications concerning autophagy. First, by providing a new example of how increased autophagy can promote rather than suppress senescent pathology, it emphasizes the Janus-faced nature of autophagy’s role in aging. Second, here autophagy promotes aging via a mechanism apparently unrelated to stochastic damage accumulation. If later gut-to-yolk biomass conversion is indeed futile, this would involve autophagy in a ‘quasi-program’ (the destructive run-on of a biological function that promotes fitness earlier in life), as proposed by Blagosklonny. Third, these findings provide an example of how sex differences in autophagic action can specify sexual dimorphism in senescent pathology (Figure 1).
Figure 1.
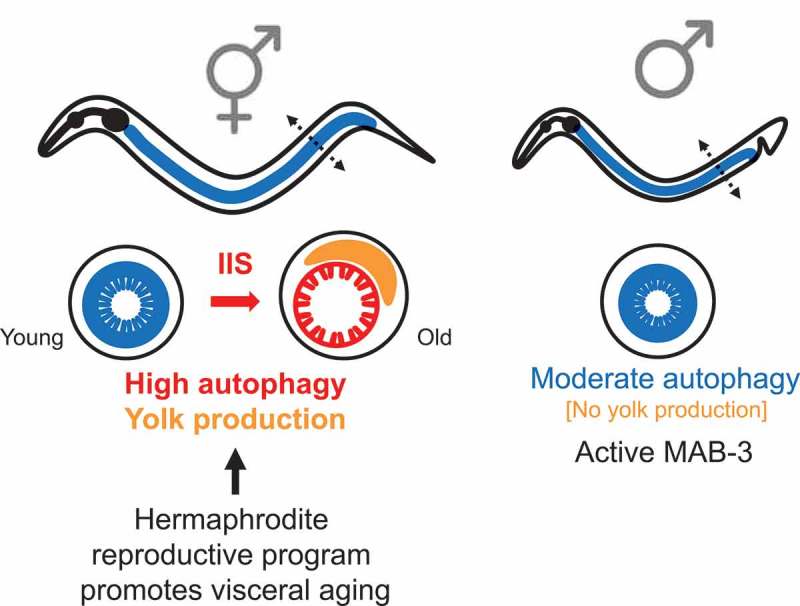
Autophagy promotes aging in a gender-specific manner in C. elegans. High autophagy in the gut (blue) allows adult C. elegans hermaphrodites (left) to produce more yolk and maximize reproductive output. However, it leads to senescent gut atrophy (red) and yolk steatosis (orange). This is driven by an IIS-activated reproductive program that is repressed in males (right) by mab-3 activity.
What then of anti-aging therapies based on enhancing autophagic capacity? These findings confirm the importance of autophagy in the biology of aging, but oppose a simple model where autophagy protects against damage and, thereby, organismal senescence. Instead, this work underscores how delaying different facets of aging may require either enhancing or suppressing autophagy. Wild-type autophagy levels may in principle be set too low or too high relative to what is optimal for healthy aging, depending on the tissue, sex, genotype, species or environment. Understanding such context dependency is key to the development of autophagy-targeted interventions to improve late-life health.
Funding Statement
This work was supported by the Lancaster University [Startup Grant]; Wellcome Trust [098565/Z/12/Z].
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Ezcurra M, Benedetto A, Sornda T, et al. C. elegans eats its own intestine to make yolk leading to multiple senescent pathologies. Curr Biol. 2018;28:2544–2556. PubMed PMID: 30100339. [DOI] [PMC free article] [PubMed] [Google Scholar]