

ABSTRACT
Cancer stem-like cells (CSLCs) reside as a small population within tumors, which mostly contain a larger population of differentiated cells. With their unique self-renewing abilities, CSLCs remain refractory to various therapeutic interventions, which otherwise kill differentiated cancer cells, and thus are a major culprit behind cancer treatment failures and cancer relapse. Recently, the process of macroautophagy/autophagy has emerged as a potential therapeutic target for eliminating CSLCs, as autophagic homeostasis has been discovered to play an important role in the growth of cancer and normal stem cells, and is required for the maintenance of the non-differentiated state of CSLCs. Our current work now shows that the so-called ‘tumor suppressor’ TP73/p73 plays an unconventional role in CSLC biology, and positively regulates the growth and stemness of CSLCs through the modulation of autophagy. Our data show that TP73/p73 deficiency, promotes autophagy in CSLCs by activating the autophagy machinery involving AMPK-TSC-MTOR signaling. Mechanistically, TP73/p73 deficiency-induced autophagy occurs as a result of reduced ATP levels resulting from the metabolic perturbations within the proline regulatory axis. Collectively, these findings unveil novel therapeutically-relevant implications for autophagy in the TP73/p73-dependent regulation of stemness within CSLCs.
KEYWORDS: Autophagy, brain tumor-initiating cells, cancer stem cells, glutamine, metabolism, TP73/p73, proline-regulatory axis, tumor suppressors
Tumor suppressors, can ‘suppress’ the growth of cancerous cells, and thus are pursued therapeutically to formulate antitumor strategies. From these tumor suppressors, TP53/p53, the most well-studied member, is known for its role in maintaining genomic integrity and preventing the uncontrolled proliferation of transformed cells. Unfortunately, the tumor suppressing activities of TP53/p53 often remain unexploitable as it is lost or mutated in most cancers. TP73/p73 is a protein that shares close structural homology to TP53/p53. Interestingly, TP73/p73 shares similar tumor-suppressing characteristics as that of TP53/p53; however, unlike TP53/p53, TP73/p73 is rarely mutated in cancers. Hence, TP73/p73 has emerged as a potential antitumor therapeutic target.
Many aggressive tumors consist of heterogeneous populations wherein the bulk of the tumor is made up of differentiated cancer cells, while the rest is composed of less differentiated stem-like cancer cells (CSLCs). The latest advances now demonstrate that CSLCs are biologically different than their differentiated counterparts and thus often bear differential susceptibility to the therapeutic interventions. In this scenario, CSLCs often survive the therapeutic assault, self renew and cause cancer relapse. In this context, the effect of potential anticancer therapeutic modalities on CSLCs must be understood. Whereas TP73/p73 has been demonstrated to suppress the growth of differentiated cancer cells, the efficacy of TP73/p73 in targeting CSLCs remained unknown and was studied in our recently published work in Clinical Cancer Research [1].
During this work, we found that the addition of exogenous TP73/p73, in an anticipation to harvest its ‘suppressive’ actions, does not affect the viability or stemness of CSLCs. In contrast, we found that the silencing of TP73/p73 drastically suppresses the viability and stemness characteristics of CSLCs. We found that TP73/p73 knockdown (KD) abrogates the self-renewal potential of CSLCs. Moreover, TP73/p73 deficiency decreases the expression of genes (POU5F1/OCT4, NANOG and SOX2) associated with the embryonic stem cell (ESC)-like signature. This TP73/p73 deficiency-induced loss of self-renewal and ESC-like signature is accompanied by an increase in the expression of differentiation markers from several distinct lineages. Next, restoring the expression of the ESC-like transcription factor POU5F1/OCT4 rescues the growth and suppresses the upregulation of differentiation in TP73/p73-deficient CSLCs. These findings are also evident in vivo where tumors derived from xenografted TP73/p73-deficient CSLCs harbor significantly lower levels of POU5F1/OCT4, and this corresponds with drastically decreased tumor burden, as compared to the control tumors generated with wild-type CSLCs. Altogether, these findings indicate that the loss of TP73/p73 suppresses stem-like features in CSLCs and shifts the phenotype of these cells towards a more differentiated, less aggressive state. At this juncture, it should be noted that such therapy-induced differentiation within CLSCs is highly desired, as it makes CSLCs amenable to therapeutic interventions.
Mechanistically, the hampering of stemness within CSLCs following loss of TP73/p73 is accompanied by metabolic perturbations within the proline-regulatory axis (PRA) that interconnects proline, glutamine and urea metabolism (Figure 1). We found that TP73/p73 KD decreases the mRNA levels of PYCR1 (pyrroline-5-carboxylate reductase 1; an enzyme that catalyzes proline synthesis from pyrroline-5-carboxylate [P5C]) and GLS (glutaminase; converts glutamine to glutamate), and increases the expression of OAT (ornithine aminotransferase; reversible conversion of P5C into ornithine) with no effect on the other enzymes in the PRA. Owing to these effects on the PRA enzymes, the metabolite levels of proline and glutamate are decreased, whereas metabolites of the urea cycle are increased, following TP73/p73 KD. Moreover, we also found that loss of TP73/p73 inhibits glutamine uptake by decreasing the expression of the transcription factor MYC, which in turn suppresses the mRNA expression of the glutamine uptake receptor SLC1A5. Thus, as summarized in Figure 1, TP73/p73 KD causes perturbations within the PRA and shifts the balance between proline synthesis and urea cycle metabolism by altering glutamine uptake. In line with these metabolic changes, we found that TP73/p73 KD affects the growth and viability of CSLCs via autophagy. We found that the metabolic distress caused by the PRA perturbations following silencing of TP73/p73 is accompanied by decreased levels of ATP and activation of AMPK. This activated AMPK promotes the activation of the tuberous sclerosis complex via phosphorylation of TSC2, leading to the inhibition and downregulation of phosphorylated MTOR, a negative regulator of autophagy, ultimately causing enhanced autophagy flux (Figure 2). Finally, supporting the role of TP73/p73 KD-induced metabolic perturbations in autophagy regulation, the replenishment of the metabolite glutamine hampers the upregulation of autophagy and rescues the growth and stemness of TP73/p73-deficient CSLCs.
Figure 1.

Loss of TP73/p73 in CSLCs causes metabolic perturbations within the proline regulatory axis (PRA). Downregulation of TP73/p73 inhibits the expression of MYC, which decreases the transcription of the glutamine uptake receptor SLC1A5. Loss of TP73/p73 also affects the PRA by decreasing the expression of the enzymes GLS and PYCR1 and increasing the expression of OAT. Collectively, these TP73/p73 deficiency in CSLCs decrease proline and glutamate synthesis (as highlighted in red) and increase urea cycle metabolism (as highlighted in green) within the PRA. PRODH; proline dehydrogenase1.
Figure 2.
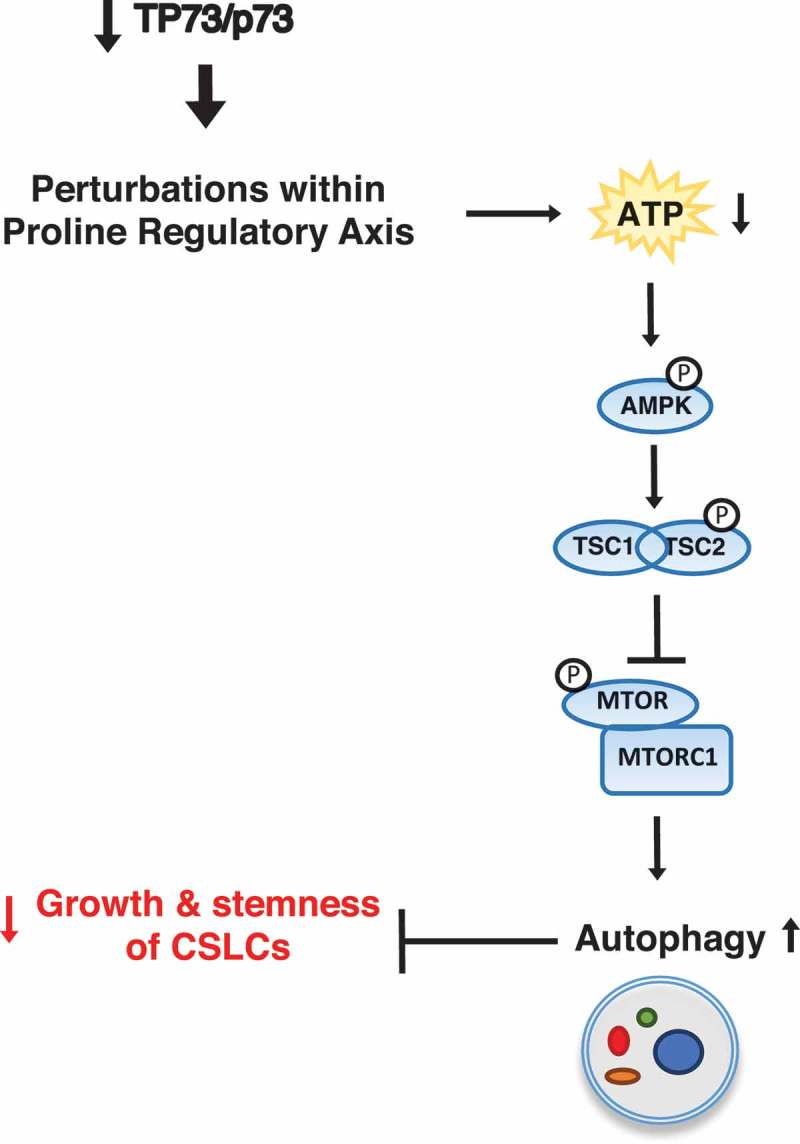
Metabolic perturbations following loss of TP73/p73 in CSLCs activate autophagy via regulation of the AMPK-TSC-MTOR signaling nexus. Knockdown of TP73/p73 affects the PRA, which is accompanied by the decreased levels of ATP and activation of p-AMPK. This p-AMPK activates the tuberous sclerosis complex via phosphorylation of TSC2, which subsequently inhibits phosphorylation of MTOR (p-MTOR), leading to the activation of autophagy. These TP73/p73 deficiency-mediated effects on autophagic homeostasis negatively affect the growth and stemness of CSLCs.
Importantly, the clinical implications for TP73/p73 are also evident in PROM1/CD133High patient-derived brain-tumor initiating cells (BTICs). We found that silencing of TP73/p73 suppresses self-renewal and stemness features in PROM1/CD133High BTICs, derived from 3 different patients. Moreover, we also found that TP73/p73 KD modifies metabolism and promotes autophagy, as measured by activation of AMPK and inhibition of MTOR, which corresponds with an upregulation of autophagosome-associated protein MAP1LC3/LC3 isoforms A and B. This role of TP73/p73 in metabolic regulation of autophagy appears to be unique in CSLCs, as non-stem-like PROM1/CD133Low patient-derived glioblastoma (GBM) cells are relatively unaffected by the loss of TP73/p73. Altogether, these findings unveil a novel and therapeutically relevant role for TP73/p73 in the regulation of the PRA-autophagy nexus in CSLCs.
Funding Statement
This work was supported by the Canadian Institute for Health Research [MOP-142289].
Acknowledgments
This work was supported through the Canadian Institutes of Health Research, Beatrice Hunter Cancer Research Institute, Dalhousie Medical Research Foundation, Tier 1 Canada Research Chair in Human Brain Cancer Stem Cell Biology, and Terry Fox Research Institutes’ New Frontiers Program Project Grant.
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Sharif T, Dai C, Martell E, et al. TAP73 modifies metabolism and positively regulates growth of cancer stem-like cells in a redox-sensitive manner. Clin Cancer Res. 2018. December 28 DOI: 10.1158/1078-0432.CCR-17-3177 PubMed PMID: 30593514. [DOI] [PubMed] [Google Scholar]