

ABSTRACT
Protein arginine methyltransferases (PRMTs) have emerged as important regulators of skeletal muscle metabolism and regeneration. However, the direct roles of the various PRMTs during skeletal muscle remodeling remain unclear. Using skeletal muscle-specific prmt1 knockout mice, we examined the function and downstream targets of PRMT1 in muscle homeostasis. We found that muscle-specific PRMT1 deficiency led to muscle atrophy. PRMT1-deficient muscles exhibited enhanced expression of a macroautophagic/autophagic marker LC3-II, FOXO3 and muscle-specific ubiquitin ligases, TRIM63/MURF-1 and FBXO32, likely contributing to muscle atrophy. The mechanistic study reveals that PRMT1 regulates FOXO3 through PRMT6 modulation. In the absence of PRMT1, increased PRMT6 specifically methylates FOXO3 at arginine 188 and 249, leading to its activation. Finally, we demonstrate that PRMT1 deficiency triggers FOXO3 hyperactivation, which is abrogated by PRMT6 depletion. Taken together, PRMT1 is a key regulator for the PRMT6-FOXO3 axis in the control of autophagy and protein degradation underlying muscle maintenance.
Abbreviations: Ad-RNAi: adenovirus-delivered small interfering RNA; AKT: thymoma viral proto-oncogene; AMPK: AMP-activated protein kinase; Baf A1: bafilomycin A1; CSA: cross-sectional area; EDL: extensor digitorum longus; FBXO32: F-box protein 32; FOXO: forkhead box O; GAS: gatrocnemieus; HDAC: histone deacetylase; IGF: insulin-like growth factor; LAMP: lysosomal-associated membrane protein; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; mKO: Mice with skeletal muscle-specific deletion of Prmt1; MTOR: mechanistic target of rapamycin kinase; MYH: myosin heavy chain; MYL1/MLC1f: myosin, light polypeptide 1; PRMT: protein arginine N-methyltransferase; sgRNA: single guide RNA; SQSTM1: sequestosome 1; SOL: soleus; TA: tibialis anterior; TRIM63/MURF-1: tripartite motif-containing 63; YY1: YY1 transcription factor
KEYWORDS: Arginine methylation, FOXO3, muscle atrophy, PRMT1, PRMT6
Introduction
Skeletal muscle dynamically responds to environmental cues via multiple pathways involving protein turnover or synthesis [1–5]. Muscle atrophy is caused by excessive protein degradation, and it is detrimental for metabolic health and aggravates other chronic diseases, leading to increased morbidity and mortality [6,7]. The key mediators for muscle atrophy are the FOXO (forkhead box O) family transcription factors and the growth promoting IGF-AKT pathway that induces a shift towards protein synthesis by inhibiting FOXOs and protein degradation [8,9]. In atrophying muscles, FOXO transcription factors are activated, which in turn regulate multiple components of the ubiquitin-proteasome pathway, such as the muscle-specific ubiquitin ligases TRIM63/MURF-1 and FBXO32/ATROGIN1, leading to accelerated proteolysis and atrophy [10–12]. The importance for the protein degradation pathway in muscle atrophy has been underlined by several studies with knockout models for genes encoding FOXOs or FBXO32 and TRIM63 that show a resistance to muscle loss or force decline in response to fasting or denervation [13,14]. FOXOs are involved in autophagy, which plays a pivotal role in tissue homeostasis in response to various stimuli, such as cellular stress, nutrient deprivation, amino acid starvation and cytokines [15–17]. The autophagy-proteasomal degradation system is tightly controlled for the removal of aged or dysfunctional organelles to ensure the maintenance of muscle function [4]. The perturbed autophagic flux, either impaired or excessive autophagy, has been implicated in muscle dystrophy [18,19]. Duchenne muscular dystrophy (DMD) patients exhibit impaired autophagy with accumulation of damaged organelles [20]. Conversely, the AKT-MTOR pathway inhibits autophagy. Previous studies with distinct dystrophic mouse models or patients showed the importance of increased autophagy accompanied by AKT inactivation or FOXO hyperactivation [18,19]. Despite identification of several key regulators, mechanisms controlling muscle atrophy are still incompletely understood. In particular, the mechanism by which FOXO transcription factors are regulated in skeletal muscle appears to be a key issue that needs to be investigated to better understand muscle homeostasis and dysfunction.
Arginine methylation is one of the post-translational modifications that regulate histone or nonhistone substrates in various cellular responses. Among nine identified protein arginine methyltransferases (PRMTs), PRMT1 is the predominant form that is responsible for at least ~85% of all arginine methylation in human cells [21–23]. Consistently, the deletion of PRMT1 function causes early embryonic lethality occurring before embryonic day 6.5 [22,24]. PRMT1 has been implicated in diverse biological processes, including gene transcription, cell proliferation and death, DNA damage responses or hepatic glucose metabolism [22,25–27]. Investigations in non-muscle cell types including hepatocytes discovered that PRMT1 regulates several nonhistone substrates such as PPARGC1A/PGC1α, ESR (estrogen receptor), and FOXO1 that regulate muscle phenotype determination and remodeling [25,28–30]. PRMT1 is also expressed in skeletal muscle and muscle progenitors [31,32]. A recent report has shown a critical role for PRMT1 in muscle regeneration by using a satellite cell-specific prmt1 deletion mouse model [33]. PRMT1 is required for myogenic differentiation of muscle progenitors during muscle regeneration. In the current study, we attempted to answer the question of whether and how PRMT1 regulates skeletal muscle function by using a MYL1/MLC1f (myosin, light polypeptide 1)-Cre driven Prmt1 deletion mouse model. Our study reveals that PRMT1 is critical for the maintenance of muscle mass and function. PRMT1 deficiency leads to muscle atrophy. Furthermore, PRMT1 deficiency causes abnormal expression of FOXO3, TRIM63 and FBXO32, likely contributing to muscle loss. PRMT1 regulates FOXO3 through suppression of PRMT6, which methylates and activates FOXO3. Thus, PRMT1 deficiency results in excessive autophagy through perturbed nutrient- and energy-sensing pathways with an elevated PRMT6-FOXO3 axis.
Results
Skeletal muscle-specific ablation of PRMT1 leads to muscle loss
Skeletal muscle-specific knockout mice for Prmt1 were generated by crossing Prmt1flox/flox (f/f) mice heterozygous for the floxed allele that also expressed Cre recombinase under the control of the Myl1 promoter (Myl1-Cre) which is mainly active in the postmitotic type II myofibers (Figure S1(a)). Immunoblot analysis for PRMT1 in various tissues confirmed the specific ablation of PRMT1 in skeletal muscles, but not other tissues including heart or liver (Figure S1(b)). Skeletal muscle-specific deletion of Prmt1 (hereafter designated as mKO) mice were indistinguishable in gross appearance from Prmt1f/f littermates (hereafter designated as f/f). The body weight of 6-months-old mKO mice did not show any difference relative to the wild type, whereas 12-months-old mKO mice had reduced body weights (Figure 1(a)). The analysis of the body composition by using an NMR analyzer revealed that the lean mass was decreased in mKO mice while the fat mass was increased, compared to the control mice (Figure S2(a,b)). Consistently, the relative weight of visceral and subcutaneous white adipose tissue in mKO mice was significantly increased whereas the relative weights of heart and liver were not significantly different (Figure S2(c)). In contrast, the analysis of individual low hindlimb muscles revealed that the weights of tibialis anterior (TA), gastrocnemieus (GAS) and extensor digitorum longus (EDL) muscles were significantly decreased in mKO mice at both age groups (Figure 1(b)). In addition, the relative weight of soleus (SOL) muscle containing largely type I myofibers was not significantly altered in 6-months-old mKO mice; however, they were decreased in 12-months-old mKO mice. These data suggest that PRMT1 ablation causes muscle loss.
Figure 1.
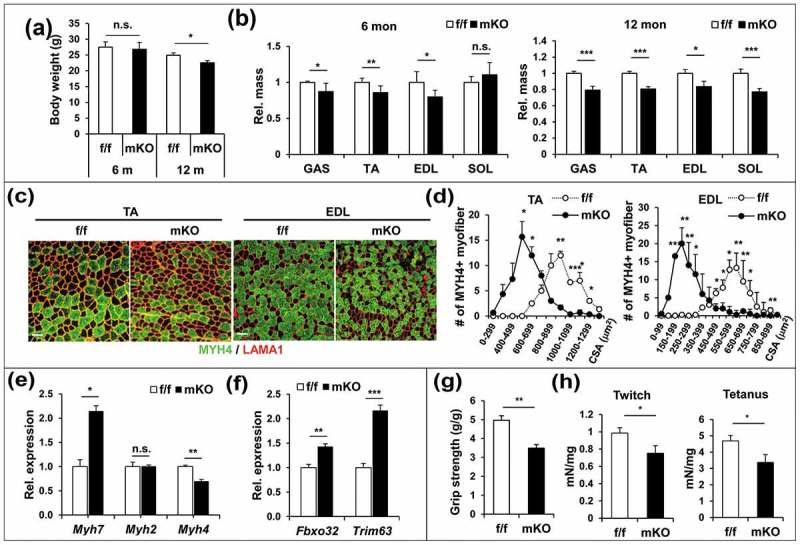
PRMT1 deficiency causes muscle loss and weakness. (a) Body weights from 6 months and 12 months-old control and prmt1 mKO mice (n = 8–10). Values represent the mean ± SD. *P < 0.05. (b) The relative mass of four muscle types divided by the body weight of control and prmt1 mKO mice. Values of control muscles were set to 1.0 (n = 8–10). (c) Immunostaining for MYH4 (Myh type IIb) and LAMA1 (laminin) in TA and EDL muscles from 6 months-old control and prmt1 mKO mice. Scale bar: 50 μm. (d) Quantification of the cross-sectional area of MYH4-positive myofibers in TA and EDL muscles of 6 months old mice. (n = 4). Data represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. (e and f) qRT-PCR analysis of TA muscles from 6-months-old control and prmt1 mKO mice. Data represent mean ±SD. *P < 0.05, **P < 0.01, ***P < 0.001 (n = 4). (g) Measurement of grip strength from 6-months-old mice (n = 8). Data represent mean ± SD. **P < 0.01. (h) Amplitude of electrical-triggered force by pacing designed to induce single twitch and tetanus (n = 4). Values represent the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
The 6-months-old muscles were subjected to detailed analysis for the muscle phenotype of PRMT1 deficiency. TA and EDL muscles of control and mKO mice were immunostained for MYH (myosin heavy chain) isoform types. The result revealed that mKO muscles exhibited significantly reduced type II myofiber size, whereas type I myofibers were increased in number and size, relative to control muscles (Figures 1(c,d) and S3). Consistently, the mRNA expression of Myh7 (Myh type I) and Myh4 (Mhy type IIb) was increased or decreased in mKO muscles, respectively (Figure 1(e)). These data suggest that PRMT1 deficiency causes alterations in the myofiber composition with decreased type IIb myofiber size. We then examined the expression of atrophy-associated ubiquitin ligases, FBXO32 and TRIM63 in f/f and mKO TA muscles. In agreement with the muscle atrophy phenotype, PRMT1-deficient muscles displayed enhanced expression of Fbxo32 and Trim63, compared to f/f muscles (Figure 1(f)).
In order to determine the consequence of this alteration in muscle function, control and mKO mice were subjected to grip strength assessment (Figure 1(g)). mKO mice exhibited declined grip strength, which is largely dependent on the glycolytic muscles composed of type II myofibers, compared to f/f mice. We further assessed the contractile function of EDL muscles from 6-months-old f/f and mKO mice by measuring the isometric force and force-frequency analysis. The glycolytic EDL muscles had significantly reduced isometric twitch and tetanic force (Figure 1(h)). The decreased force generation might reflect the alterations in contractile proteins as shown above. These data collectively suggest that the muscle-specific ablation of PRMT1 evokes premature muscle loss and weakness.
PRMT1 deficiency causes perturbed nutrient and energy-sensing pathway in skeletal muscles
To examine the mechanism of muscle atrophy caused by PRMT1 depletion, we next assessed the expression of PRMT1 in different energy states. TA muscles harvested from 3 different conditions, normally fed, fasted for 16 h and refed for 6 h, were examined for the expression of PRMT1, p-AMPK, AMPK and LC3-II (also known as LC3B). The phosphorylated form of AMPK (p-AMPK), a key energy sensor, was greatly increased in fasted muscles and decreased in refed muscles without changes in total AMPK levels (Figure 2(a,b)). The LC3-II level was greatly elevated in fasted muscles, whereas it was diminished in refed muscles, indicating the autophagic response to energy states. Interestingly, the level of PRMT1 protein was decreased in fasted muscles, whereas it was increased in the refed condition. These data suggest a potential role for PRMT1 in the control of energy sensing pathways.
Figure 2.
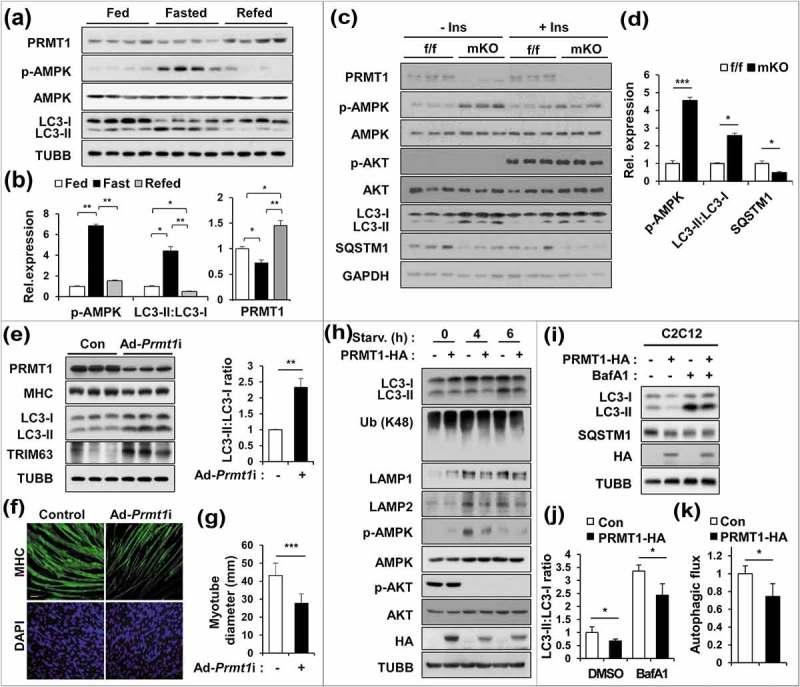
PRMT1-deficient muscles exhibit elevated catabolic pathways with increased autophagy. (a) Immunoblot analysis of fed, fast (16 h) or re-fed (6 h) TA muscles for expression of various regulators. (b) Quantification of the relative protein levels of p-AMPK, LC3-II and PRMT1. The intensity of p-AMPK, LC3-II and PRMT1 were normalized to total AMPK, LC3-I and TUBB/β-tubulin, respectively. The value of the fed state was set to 1.0 (n = 4). Data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. (c) Immunoblot analysis of control and prmt1 mKO muscles treated with control or insulin (0.1 U/mouse) for 10 min (n = 3). (d) Quantification of the relative protein levels of p-AMPK, LC3-II and SQSTM1 from control and prmt1 mKO muscles without insulin treatment. The intensity of p-AMPK, LC3-II and SQSTM1 were normalized to total AMPK, LC3-I and TUBB, respectively. The value of control muscles was set to 1.0 (n = 3). Data represent means ± SD. *P < 0.05, ***P < 0.001. (e) Immunoblot analysis of C2C12 cells infected with adenoviruses for control RNAi (ad-control) or Prmt1 RNAi (ad-Prmt1i) at D3 for the expression of MHC, LC3 and TRIM63. Quantification of the relative protein levels of LC3-II. The intensities of LC3-II were normalized to LC3-I. Data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. (f) Immunostaining for MHC in C2C12/ad-control or C2C12/ad-Prmt1i cells at D3. Scale bar: 50 μm. (g) Quantification of myotube diameter in C2C12/ad-control or C2C12/ad-Prmt1i cells at D3. Data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. (h) C2C12/pcDNA-HA and C2C12/PRMT1-HA cells were starved in DMEM containing 2% HS for the indicated hours followed by immunoblot analysis for the indicated markers. (i–k) Immunoblot analysis and quantifications of LC3-II levels from C2C12/pcDNA-HA and C2C12/PRMT1-HA cells treated with or without 10 nM bafilomycin A1 for 3 h. (n = 3). Data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
To gain insight into the effect of PRMT1 deficiency on energy sensing pathways, control f/f and mKO mice were fasted for 6 h and treated with insulin (0.1 U/mouse) for 10 min. Without insulin treatment, mKO muscles exhibited greatly enhanced levels of p-AMPK and LC3-II, and decreased levels of SQSTM1/p62, compared to the f/f muscles (Figure 2(c,d)). As expected, the insulin treatment augmented AKT activation in control and mKO muscles, suggesting that PRMT1 is dispensable for the AKT activation in response to insulin. The level of p-AMPK was partially decreased in insulin-treated mKO muscles, but it was still higher than the insulin-treated control muscles. Interestingly, LC3-II levels stayed higher in insulin-treated mKO muscles, compared to the control muscles. In addition, the level of SQSTM1 was lower than the control muscles. These data suggest that PRMT1 deficiency enhances energy deprivation signals in skeletal muscles.
To further define this mechanism, near confluent C2C12 myoblasts were induced to differentiate for 24 h, followed by infection with either adenovirus expressing control RNAi or Prmt1 RNAi. Three days later, the effect of PRMT1 depletion was examined (Figure 2(e–g)). Similar to PRMT1-depleted muscles, the relative levels of LC3-II and TRIM63 were greatly increased in PRMT1-depleted myotubes, compared to the control. PRMT1-depleted cells exhibited a reduction in myosin heavy chain expression and formed thinner myotubes. In a converse experiment, control or PRMT1-overexpressing C2C12 cells were serum-deprived for the indicated hours and the autophagic response and AMPK activation were assessed (Figure 2(h–k)). At 4 h after serum deprivation, p-AMPK levels were greatly enhanced in both cultures; however, the induction level of p-AMPK was blunted in PRMT1-overexpressing cells. Consistently, PRMT1 overexpression resulted in decreased LC3-II levels compared to control cells. Furthermore, the induction of LAMP1 and LAMP2 as well as the level of ubiquitinated proteins measured by antibody against ubiquitinated K48 were reduced in PRMT1-overexpressing cells. To assess the autophagic flux, cells were treated with the lysosome inhibitor bafilomycin A1 (Baf A1). PRMT1-overexpressing cells had less LC3-II accumulation in the presence of Baf A1 and showed decreased autophagic flux, compared to the control cells. These data suggest that PRMT1 deficiency results in perturbed regulation of the energy deprivation signal and autophagy.
PRMT1 depletion causes elevated levels of total and nuclear FOXO3 proteins
FOXO transcription factors, especially FOXO3 are involved in autophagy and induction of the ubiquitin ligases TRIM63 and FBXO32 linked with protein degradation and muscle atrophy [10–12]. Thus, in combination with the above experimental data, it can be speculated that PRMT1 inhibits the action of FOXO in skeletal muscle. To test this hypothesis, control and PRMT1-deficient muscles were subjected to immunoblotting and qRT-PCR analysis for FOXO1 and FOXO3 levels. The protein levels of FOXO1 were not altered, whereas FOXO3 protein levels were dramatically increased in PRMT1-deficient muscles (Figure 3(a)). In addition, qRT-PCR analysis of TA muscles from control and mKO mice revealed that mKO muscles exhibited a 2-fold increase in Foxo3 transcripts whereas the increase of Foxo1 mRNA expression was minimal (Figure 3(b)). In the immunostaining analysis, mKO muscles had more myonuclei positive for FOXO3, including a less distributed centralized nucleus of myofiber (Figure 3(c)). Furthermore, control or PRMT1-depleted C2C12 cells were examined for FOXO1 and FOXO3 levels by cell fractionation (Figure 3(d,e)). PRMT1 depletion resulted in elevated levels of total and nuclear FOXO1 and FOXO3 proteins, suggesting enhanced FOXO activation in PRMT1-depleted cells, which is consistent with increased expression of Trim63 and Fbxo32 (Figure 3(f)). Taken together, these data suggest that PRMT1 negatively regulates the expression and activity of FOXO3.
Figure 3.
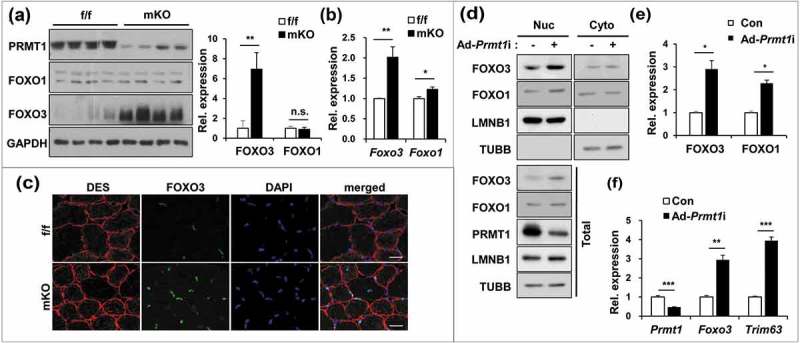
PRMT1 deficiency enhances the expression and activity of FOXO3 in skeletal muscle. (a) Immunoblot analysis of 6-months-old control and prmt1 mKO TA muscles for FOXO1 and FOXO3. Quantification of relative protein levels of FOXO3 and FOXO1. The intensities were normalized to GAPDH. Data represent means ± SD. **P < 0.01, n.s. = not significant. (b) qRT-PCR analysis of Foxo1 and Foxo3 levels in 6-months-old control and prmt1 mKO TA muscles (n = 4). (c) Representative images of immunostaining for FOXO3 (green) and DES (red) with TA sections from 6-months-old control and prmt1 mKO mice. Scale bar: 20 μm. (d) Nuclear fractionation of C2C12/ad-control and C2C12/ad-Prmt1i cells. (e) Quantification of the relative amounts of nuclear FOXO3 and FOXO1 protein levels. The intensities were normalized to LMNB1. Data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. (f) qRT-PCR analysis of Prmt1, Foxo3 and Trim63 levels in C2C12/ad-control and C2C12/ad-Prmt1i cells at D3 (n = 4). Data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
PRMT1 represses PRMT6 expression through YY1 and HDAC2/3
Previous studies have shown that arginine methylation of FOXO1 by PRMT1 promotes its activity in hepatic glucose metabolism [25]. Thus, we have examined whether PRMT1 inhibits FOXO3 through methylation. PRMT1 interacted with FOXO3 but failed to methylate it (Figure S4). Hereto we have assessed the expression of other PRMTs in control f/f and mKO muscles (Figure 4(a)). Although the expression of several PRMTs was altered in mKO muscles, Prmt6 mRNA was upregulated with the highest level in PRMT1-deficient muscles, which was further confirmed at the protein level (Figure 4(b,c)). Furthermore, the immunostaining for PRMT6 revealed increased immunopositivity in mKO muscles, especially in myonuclei, whereas control muscles showed weak staining in myonuclei (Figure 4(d)). In agreement with the muscle analysis, PRMT1 depletion in C2C12 cells enhanced PRMT6 approximately 3-fold, along with TRIM63 proteins (Figure 4(e,f)), whereas PRMT1 overexpression reduced Prmt6 transcript levels (Figure 4(g)). These data suggest that PRMT1 represses PRMT6 expression.
Figure 4.
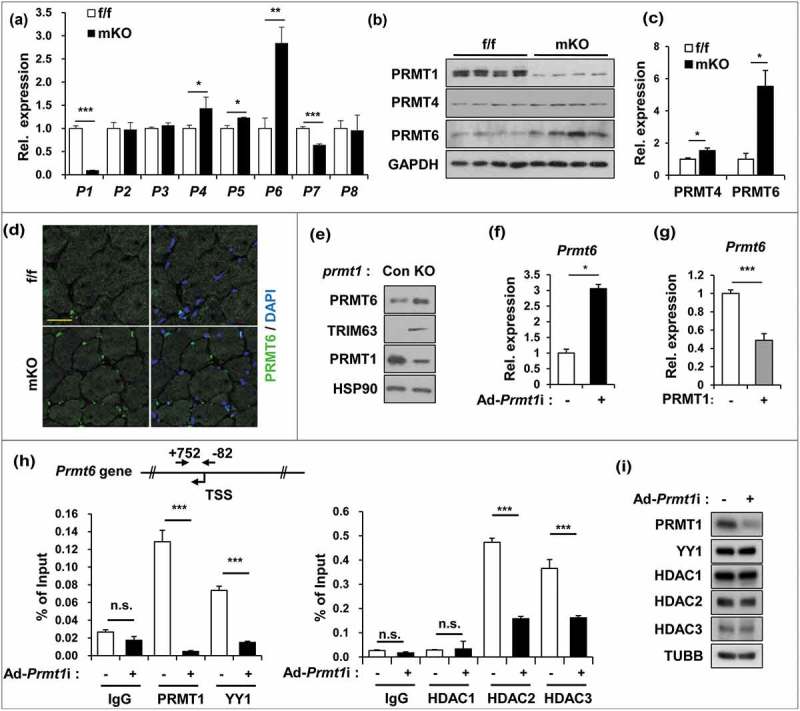
PRMT1 suppresses PRMT6 in skeletal muscle and myoblasts. (a) qRT-PCR analysis of PRMTs levels from 2-months-old control and prmt1 mKO TA muscles. (n = 4) (b) Immunoblot analysis for PRMT1 and PRMT6 protein expression in control and prmt1 mKO TA muscles. (c) Quantification of relative protein levels of PRMT4 and PRMT6. The intensities were normalized to GAPDH (n = 4). Data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. (d) Immunostaining for PRMT6 (green) and DAPI (blue) from 6-months-old TA muscles. Scale bar: 20 μm. (e) Immunoblot analysis of PRMT1, PRMT6 and TRIM63 expression in control and prmt1 knockout C2C12 cells at D3. (f) qRT-PCR analysis of Prmt6 levels in C2C12/ad-control or C2C12/ad-Prmt1i cells at D3. Data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. (g) qRT-PCR analysis of Prmt6 levels in C2C12/pcDNA-HA or C2C12/PRMT1-HA cells at D3. Data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. (h) Chromatin-immunoprecipitation (ChIP) assay of Prmt6 promoter region from +752 to −82 in C2C12/ad-control and C2C12/ad-Prmt1i cells at D3. Values are means of triplicate determinants ± SD. *P< 0.05, **<0.01, ***<0.001. (i) Immunoblot analysis for indicated markers in C2C12/ad-control and C2C12/ad-Prmt1i cells at D3.
Previously, PRMT1 has been implicated in YY1-mediated gene regulation, and YY1 can modulate gene expression through interaction with various histone modifiers, like histone acetyltransferases (HATs) or histone deacetylases (HDACs) [34,35]. The sequence analysis of the Prmt6 promoter region predicted 2 potential YY1 binding sites around transcription initiation sites, thus we have assessed whether PRMT1, YY1 and HDACs (1–3) were recruited to the Prmt6 promoter by chromatin immunoprecipitation assays (Figure 4(h)). PRMT1 and YY1 were recruited to the Prmt6 promoter region in control cells, whereas PRMT1 depletion abrogated the YY1 recruitment. Furthermore, HDAC2 and HDAC3 were enriched at this region in control cells while PRMT1 depletion blunted this recruitment. However, this alteration in the recruitment to the Prmt6 promoter region was not due to altered levels of these proteins in PRMT1-depleted cells (Figure 4(i)). Taken together, these data suggest that PRMT1 suppresses Prmt6 transcription through binding to its promoter region together with YY1 and HDAC2/3.
PRMT6 upregulates FOXO3 and autophagy in PRMT1-depleted cells
To further define the relationship between PRMT1 and PRMT6 on FOXO3 regulation, control or prmt1 knockout C2C12 cells incubated for 3 days in 2% horse serum were subjected to cellular fractionation assays. PRMT1 depletion greatly elevated the level of FOXO3 and PRMT6 proteins in the nucleus and total lysates, which correlated well with elevated TRIM63 expression (Figure 5(a)). To examine the effect of these PRMTs on FOXO3 activity, C2C12 cells were transfected with a plasmid encoding FOXO3 in combination with either PRMT1 or PRMT6 and subjected to a FOXO3-luciferase reporter assay. PRMT6 greatly enhanced the FOXO3-mediated luciferase activity, whereas PRMT1 expression had no effect on it (Figure 5(b)). In line with this, PRMT6 overexpression in C2C12 myoblasts greatly elevated total and nuclear FOXO3 levels (Figure 5(c)). To examine the effect of PRMT6 on autophagy, C2C12 cells overexpressing PRMT6 were switched to low-serum medium for 12 and 24 h (Figure 5(d)). PRMT6 overexpression further elevated the level of LC3-II, compared to the control cells. Conversely, PRMT6-depleted C2C12 cells had less LC3-II in both control and Baf A1 treated samples, and exhibited decreased autophagic flux, compared to the control cells (Figure 5(e–g)), suggesting a positive role of PRMT6 in autophagy. Taken together, these data suggest that PRMT1 depletion-induced FOXO3 activation and autophagy are likely mediated by PRMT6.
Figure 5.
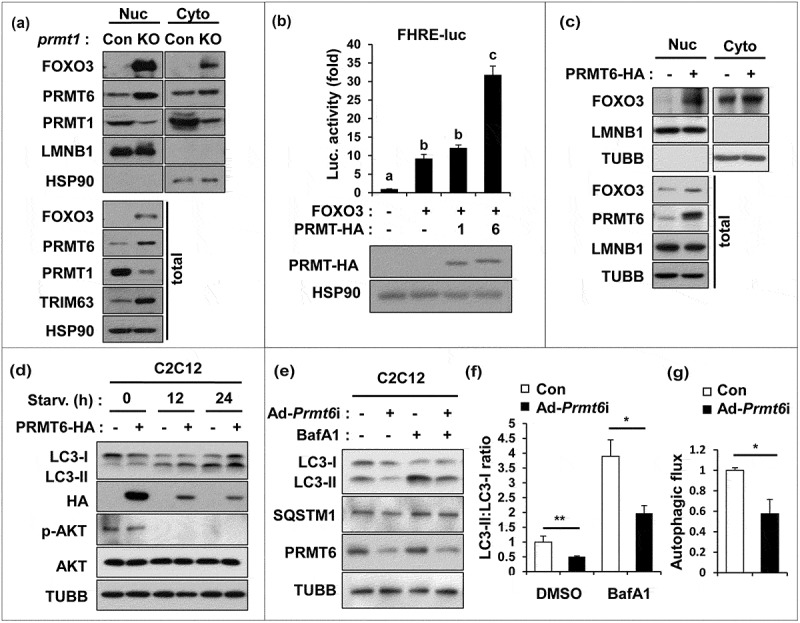
PRMT1 inhibits FOXO3 through PRMT6 suppression. (a) Cellular fractionation of control or prmt1-knockout C2C12 cells followed by immunoblot analysis with the indicated markers. (b) Luciferase assay of C2C12 cells cotransfected with FOXO3-reporter, control or FOXO3 and control, PRMT1 or PRMT6 expression vectors. Data represent means ± SD. Letters indicate statistically distinct group (one-way ANOVA<0.01). (c) Cellular fractionation of C2C12 cells at D1 transfected with pcDNA-HA or a plasmid encoding PRMT6-HA followed by immunoblot analysis for the indicated markers. (d) C2C12/pcDNA-HA and C2C12/PRMT6-HA cells were incubated in DMEM containing 2% HS for the indicated hours followed by immunoblot analysis for the indicated markers. (e–g) Immunoblot analysis and quantifications of LC3-ll levels from C2C12/ad-control and C2C12/ad-Prmt6i cells treated with or without 10 nM Baf A1 for 3 h. (n = 3). Data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
PRMT6 activates FOXO3 via methylation of arginine residues 188 and 249
To examine whether PRMT6 activates FOXO3 by methylation, 10T1/2 cells were switched to low-serum-containing medium for 6 or 18 h and the asymmetric methylation of FOXO3 was assessed by immunoprecipitation and immunoblotting with asymmetric dimethylarginine-specific D6A8 antibodies. In line with the data obtained in skeletal muscles, PRMT1 protein level was decreased in response to serum starvation, whereas PRMT6 levels were greatly enhanced with the concomitant FOXO3 methylation (Figure 6(a)). To further define this molecular mechanism, the physical interaction between FOXO3 and PRMT6 was examined. 293T cells were transfected with vectors for HA-tagged PRMT1 or PRMT6 along with Flag-tagged FOXO3. In agreement with the previous data, PRMT1 proteins were less efficiently coprecipitated with FOXO3 and failed to methylate it. In contrast, PRMT6 efficiently coprecipitated with FOXO3 and asymmetrically dimethylated it as recognized by D6A8 antibodies that are specific for asymmetrically dimethylated arginines (Figure 6(b)). Next, wild-type PRMT6 or a methyltransferase activity-deficient PRMT6 (V86K D88A) were cotransfected with a plasmid encoding FOXO3 and the methylation and activity of FOXO3 was assessed (Figure 6(c,d)). Wild-type PRMT6 robustly enhanced asymmetrically methylated FOXO3 levels while the V86K D88A mutant abrogated it. Consistently, the FOXO3 reporter activity was significantly elevated by wild-type PRMT6, but not by the V86K D88A mutant. These data suggest that the methylation of FOXO3 by PRMT6 promotes FOXO3 activation. Consistently, the expression of Fbxo32 and Trim63 was enhanced by wild-type PRMT6, whereas mutant PRMT6 had no or a minimal effect (Figure 6(e)). These data suggest that PRMT6 promotes FOXO3 activation via its methylation.
Figure 6.
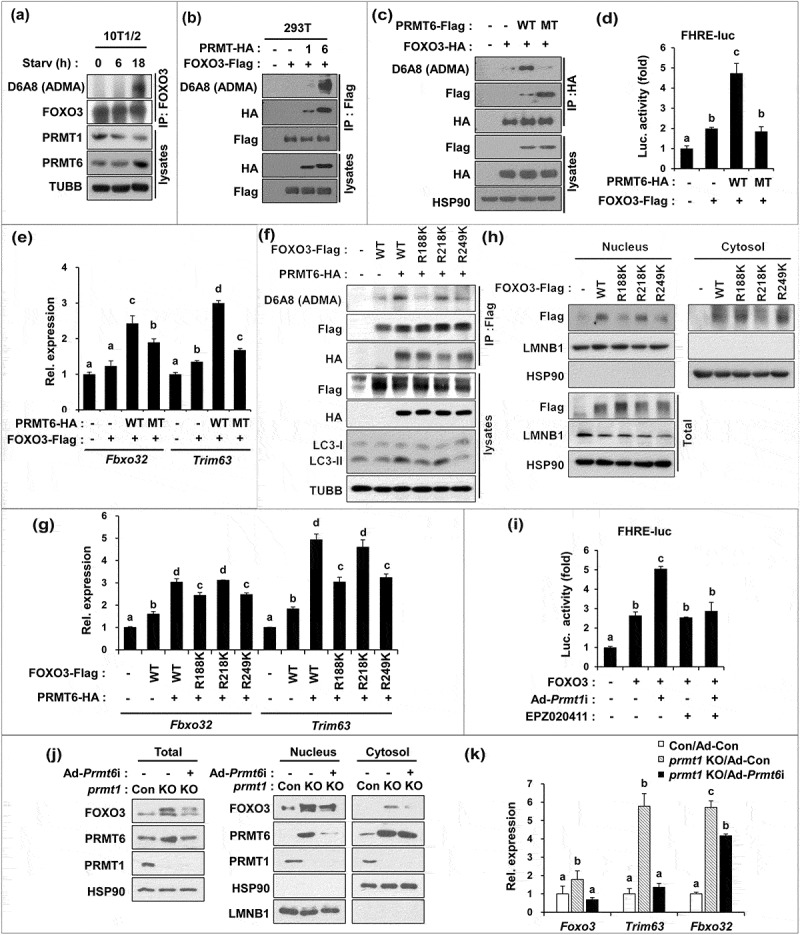
PRMT6 methylates FOXO3 on arginine 188 and 249 critical for its activity. (a) Lysates of 10T1/2 mouse embryonic fibroblasts incubated with DMEM containing 0.1% FBS for the indicated hours were subjected to immunoprecipitation with FOXO3 antibodies and immunoblot analysis with indicated antibodies. (b) Co-immunoprecipitation of control, PRMT1-HA or PRMT6-HA and control or FOXO3-flag transfected 293T cells with flag antibodies and immunoblot analysis with indicated antibodies. (c) Coimmunoprecipitation from 293T cells transiently expressing control, PRMT6 WT or PRMT6V86K,D88A mutant and control or FOXO3-HA proteins. Anti-HA immunoprecipitates were analyzed by immunoblotting analysis via indicated antibodies. (d) Luciferase assay of C2C12 cells cotransfected with FOXO3-reporter, FOXO3 and PRMT6 WT or PRMT6V86K/D88A mutant. Data represent means ± SD. Letter indicate statistically distinct group (One way ANOVA, P < 0.01). (e) qRT-PCR analysis for the expression of Fbxo32 and Trim63 in FOXO3 and PRMT6 WT or PRMT6V86K,D88A transfected C2C12 cells at D3. Data represent means ± SD. Letters indicate statistically distinct groups (one-way ANOVA, P < 0.01). (f) Coimmunoprecipitation of PRMT6-HA and flag-tagged FOXO3 WT, R188K, R218K or R249K transiently transfected 293T cells with flag antibodies and immunoblot analysis with the indicated antibodies. (g) qRT-PCR analysis for the expression of Fbxo32 and Trim63 in FOXO3 WT or R188K, R218K or R249K transfected C2C12 cells at D3. Data represent means ± SD. Letter indicate statistically distinct group (One way ANOVA, P < 0.01). (h) Cellular fractionation of C2C12 cells at D3 transfected with flag-tagged pcDNA, FOXO3 WT, R188K, R218K or R249K followed by immunoblot analysis for the indicated markers. (i) Luciferase assay with C2C12/ad-control or C2C12/ad-Prmt1i cells cotransfected with FOXO3-reporter, control or FOXO3 and treated with vehicle or 10 μM EPZ020411 for 24 h. Data represent means ± SD. Letters indicate statistically distinct groups (One way ANOVA, P < 0.01). (j) Control or prmt1-knockout C2C12 cells were transduced by ad-control or ad-Prmt6i and cultured for 3 days, followed by cellular fractionation and immunoblotting. (k) qRT-PCR analysis for the expression of Foxo3, Trim63 and Fbxo32 in C2C12 cells at D3. Data represent means ± SD. Letter indicate statistically distinct group (One way ANOVA, P < 0.01).
To determine the potential methylation sites in FOXO3, mass spectrometry analysis was performed and 3 potential methylation sites (R188, 218, and 249) in FOXO3 were identified (Figure S5). R188 and R218 are localized in the forkhead domain that are not conserved in FOXO1, whereas R249 is located in the nuclear localization signal and conserved in FOXO1. There are 3 reported AKT phosphorylation sites [8,36–38] and R249 is located in the vicinity of the serine 252 that can be phosphorylated and inhibited by AKT [37,39], suggesting a potential crosstalk between arginine methylation and AKT-mediated inhibition of FOXO3. To further assess the functional significance of these modifications, we generated arginine-to-lysine mutants in FOXO3 for these sites and analyzed the effect on methylation and the subsequent transcriptional activity. The methylation of wid-type and R218K were equally enhanced in response to ectopic PRMT6, compared to that of wild-type FOXO3 without ectopic PRMT6 (Figure 6(f)). In contrast, R188K and R249K mutants failed to show this increased methylation by PRMT6, suggesting that R188 and R249 are target sites for PRMT6. In addition, the expression of wild-type FOXO3 alone did not greatly alter LC3-II levels. The expression of wild-type or the R218K mutant with PRMT6 substantially enhanced the level of LC3-II, which was abrogated in cells expressing R188K or R249K with PRMT6. To assess the functional consequence, PRMT6 and FOXO3 proteins were coexpressed in C2C12 cells and the expression level of TRIM63 and FBXO32 was determined (Figure 6(g)). Compared to the control cells, the expression of wild-type FOXO3 alone increased Trim63 and Fbxo32 levels and they were further enhanced by PRMT6 co-expression. Like wild-type, R218K had a similar response to PRMT6, whereas R188K and R249K exhibited a reduced response to PRMT6. Next, we examined whether arginine methylation of FOXO3 regulates its nuclear function. C2C12 cells were transfected with plasmids encoding FOXO3 proteins and their nuclear localization was examined by nuclear fractionation (Figure 6(h)). The wild-type and R218K mutant proteins were relatively higher in the nuclear fraction, whereas the nuclear levels of the R188K and R249K mutants were lower. Taken together, these data suggest that PRMT6 methylates the R188 and R249 residues of FOXO3.
Finally, we examined whether the PRMT6 inhibition can prevent the elevated FOXO3 reporter activity elicited by PRMT1-depletion (Figure 6(i)). The FOXO3 reporter activity was increased by PRMT1 depletion, which was blunted by PRMT6 inhibition with a specific inhibitor, EPZ020411. In addition, control or prmt1 knockout C2C12 cells were infected with adenovirus expressing control or shPrmt6 followed by a cellular fractionation assay. In agreement with the previous data, prmt1 knockout C2C12 cells exhibited a dramatic increase in FOXO3 and PRMT6 levels in total lysates as well as in the nuclear fraction which was blunted by PRMT6 depletion (Figure 6(j)). Furthermore, PRMT6 depletion in prmt1 knockout C2C12 cells rescued the level of Foxo3 and Trim63 almost to the level of control cells, with a small but significant impact on Fbxo32 expression as well (Figure 6(k)). Our mechanistic study suggests that PRMT1 deficiency causes upregulation of PRMT6, which directly interacts with and methylates FOXO3 leading to FOXO3 hyperactivation. In conclusion, PRMT1 deficiency-induced muscle atrophy is largely due to the upregulation of PRMT6 and FOXO3, augmenting the autophagic response and protein degradation pathways in skeletal muscles.
Discussion
Our results highlight the role of PRMT1 as a key regulator to orchestrate a complex regulatory network for muscle maintenance. PRMT1 deficiency in glycolytic fibers causes muscle atrophy. PRMT1-deficient muscles exhibit a reduced exercise capacity, and the decreased contractile force likely is attributed to this alteration in fiber types. The fiber type conversion toward type I fibers and the force decline in PRMT1-deficient muscles resembles age-related alterations in skeletal muscle associated with muscle weakness and atrophy [40,41]. Together with results from the previous study showing the critical function of PRMT1 in muscle regeneration [33], our current study underlines the importance of PRMT1 in skeletal muscle maintenance.
Muscle atrophy is a complex and multi-factorial process, which is incompletely understood. Regardless of the causes, muscle atrophy is characterized by a reduction in protein content, fiber diameter and force production. Muscle mass is maintained via a delicate balance between anabolic and catabolic factors, and an imbalance between protein synthesis and breakdown is the major underlying mechanism for muscle loss [1–5]. In catabolic pathways, FOXO transcription factors play a key role through induction of autophagy and the ubiquitin-proteasome system [15–17]. In various pathological conditions, FOXOs are dysregulated, causing metabolic disorders and atrophy [10–12]. Our data suggest that PRMT1 deficiency-induced muscle loss and atrophy is likely due to deregulation of energy sensing pathways rather than defects in anabolic pathways. In line with this, PRMT1-deficient muscles showed normal AKT activation in response to insulin suggesting that PRMT1 deficiency does not cause defects in AKT activation. It is conceivable that PRMT1 suppresses energy-deprivation signaling. Consistently, PRMT1 is decreased in fasted muscles and subsequently elevated in refed muscles. Furthermore, PRMT1-deficient muscles and myoblasts exhibit elevated AMPK activation and FOXO3 levels, accompanied by induction of the proteolytic regulators TRIM63 and FBXO32. Conversely, PRMT1 overexpression reduced the level of p-AMPK, LAMP1, LAMP2, LC3-II and the protein ubiquitination, induced by serum starvation.
The muscle atrophy observed in PRMT1-deficient mice resembled the atrophic phenotype observed in mice transgenic for FOXO1 or FOXO3. Skeletal muscle-specific FOXO1 transgenic overexpression induces a reduction in body size and muscle mass associated with enhanced expression of FBXO32 and TRIM63 [42]. Furthermore, the genetic activation of FOXO3 results in atrophy associated with FBXO32 upregulation, whereas knockdown of FOXO3 by siRNA prevents muscle atrophy [43]. Because PRMT1 has been previously reported to activate FOXO1 in the control of hepatic gluconeogenesis [25], our current results showing FOXO3 upregulation in PRMT1-deficient muscles are rather surprising and further suggest a unique regulatory mechanism for FOXO3 by PRMTs in skeletal muscle that is distinct from that in hepatic gluconeogenesis. We propose that the underlying mechanism of tissue-specific regulation is the suppressive effect of PRMT1 on PRMT6 in skeletal muscle. It appears that PRMT1 depletion upregulates PRMT6, which in turn activates FOXO3 and the ubiquitin-proteasome degradation pathway in skeletal muscle. Interestingly, both PRMT1 and PRMT6 can interact with FOXO3, but only PRMT6 induces asymmetric arginine methylation on R188 and R249 residues. Our present data and a previous report showing that PRMT6 interacts with FOXO1 but does not methylate it suggest the target selectivity of PRMTs [28]. The basis of this selectivity or the exact mechanism by which the arginine methylation of FOXO3 by PRMT6 regulates its activity is unclear. Because R188 is located in the forkhead domain, its methylation could promote its activity through DNA binding ability, whereas the methylation of R249 might interfere with the inhibitory phosphorylation by AKT. This needs to be addressed in the future to elucidate the exact mechanism of FOXO3 regulation by PRMT6.
PRMT1 indirectly regulates FOXO3 through PRMT6 suppression in skeletal muscle. PRMT1 is co-recruited to the promoter region of Prmt6 with YY1 and HDAC2/3, and PRMT1 depletion resulted in no enrichment of these transcription regulators, which likely is relieving the repression of the Prmt6 gene. Arginine methylation of histone H4 arginine 3 by PRMT1 is generally linked to transcriptional activation, whereas arginine methylation of non-histone transcriptional regulators by PRMT1 has been associated with transcriptional activation or repression [44,45]. Our current data suggest that PRMT1 is required for the co-recruitment of YY1 and HDAC2/3 to the Prmt6 promoter region to repress its expression. Considering that PRMT6 depletion abrogates FOXO3 activity caused by PRMT1 depletion, PRMT1 might largely regulate atrophic signaling through the PRMT6-FOXO3 pathway. The in vivo function of PRMT6 is currently less well characterized. Similar to PRMT1, PRMT6 can methylate glycine- and arginine-rich sequences in proteins and has been implicated in biological processes, including transcription, DNA repair, replication and signal transduction [46,47]. Unlike PRMT1, PRMT6 is not essential for the survival of mice, because mice lacking PRMT6 are viable [48]. However, PRMT6-deficient embryonic fibroblasts undergo rapid senescence, implying a role for PRMT6 in cell proliferation [49]. Thus, it will be of great interest to investigate the muscle phenotype of prmt6 knockout mice in response to energy deprivation or atrophy signaling. Taken together, our data demonstrate that the loss of PRMT1 in skeletal muscle results in muscle atrophy. Our study uncovers a specific role of PRMT1 in suppression of catabolic pathways through the PRMT6-FOXO3 axis. Although the importance of PRMT1 in early embryonic development is reported with the whole body knockout mouse model, our findings in the current study suggest that PRMT1 acts as a gatekeeper of the activity of the master regulator of autophagy, FOXO3, preventing excessive autophagy during nutrient and energy deprivation. Therefore, PRMT1 is a potential target for maintaining muscle integrity and mass, providing new avenues for the prevention and intervention of muscle atrophy associated with aging or pathological conditions.
Materials and methods
Animal studies
All procedures were performed according to approved protocols by the Korea University Institutional Animal Care and Use Committee. Mice bearing the Prmt1-floxed allele (Prmt1f/f, obtained from EUCOMM) were crossed with transgenic mice expressing Cre under the control of a Myl1 promoter (Myl1/MLC1f-Cre, provided by Chul-Ho Lee from KRIBB). The primers used for genotyping are as following: Prmt1 primers: forward 5ʹ-GTGCTTGCCATACAAGAGATCC-3ʹ; backward 5ʹ-ACAGCCGAGTAGCAAGGAGG-3ʹ, Cre primers: forward 5ʹ-GCGGTCTGGCAGTAA AAACTATC-3ʹ; backward 5ʹ-GTGAAACAGCATTGCTGTCACTT-3ʹ. For the assessment of muscle exercise capacity, the grip strength of the forelimb was measured with a grip strength meter (Bioseb, Bio-GS3) from 6-months-old control and mKO mice. All grip strength readings (measured in grams) were normalized to body weight. Fat and lean masses were analyzed by using an NMR analyzer Bruker Minispec LF50 (Bruker Optics Inc.) at the Korea Mouse Phenotyping Center (KMPC), Seoul National University.
Muscle physiology
The muscle contractile properties of extensor digitorum longus (EDL) muscles isolated from 6-months-old control and mKO mice were examined. The isolated muscles were transferred to a chamber containing Krebs-Ringer buffer (118 mM NaCl, 4.75 mM KCl, 24.8 mM NaHCO3, 1.18 mM KH2PO4, 2.5 mM CaCl2 · 2 H2O, 1.18 mM MgSO4, 10 mM glucose, pH 7.4) and oxygenated with 95% O2 and 5% CO2, thermostatically maintained at 25°C. The proximal tendon was fixed using a clamp, and the distal tendon was fixed to a force transducer (Grass Instruments, FT03D). The optimum muscle length was determined as the length producing the highest twitch force (mN) with a supramaximal pulse of 0.2 ms duration. Isometric tetanic contractions (mN) were determined using an electrical stimulator (Grass Instruments, S48) at a stimulation frequency of 100 Hz for the EDL muscle, a stimulation current of 100 V, and a duration of 200 ms. The muscle contration was normalized to EDL muscle mass.
Cryosections, immunostaining analysis
Freshly isolated TA muscles were snap frozen in optimal cutting temperature (Sakura finetek, 4583) and sectioned with 7-μm thickness on a cryostat microtome (Leica, Wetzlar, Germany). For immunostaining, muscle sections were fixed, permeabilized, and processed for incubation with primary antibodies against MYH7/Myh type I (Developmental Studies Hybridoma Bank, BA-D5), MYH4/Myh type IIb (DSHB, BF-F3), MYH2/Myh type IIa (DSHB, BF-32), MYH1/Myh type IIx (DSHB, 6H1), FOXO3 (Cell Signaling Technology, 12829), PRMT6 (Cell Signaling Technology, 14641), LAMA1/Laminin (abcam, 11575) and DES/desmin (Santa Cruz Biotechnology, sc23879) and secondary antibodies (Life technologies, A-11001 and A-11012). Images were captured under Nikon ECLIPS TE-2000U and NIS-Elements F software (Nikon, Tokyo, Japan). Myofibers were traced, and their area was measured using NIS-Elements F software (Nikon).
Cell cultures, luciferase assay
293T cells and C2C12 myoblasts were cultured as previously described [25,50]. Recombinant adenoviruses expressing control RNAi (5ʹ-GGCATTACAGTATCGATCAG-3ʹ), Prmt1 RNAi (5ʹ-GCAACTCCATGTTTCACAATC-3ʹ) [25], and Prmt6 RNAi (5ʹ-ACAAGATACGGACATTTCC-3ʹ) were generated using the AdEasy system [51]. Adenoviruses were purified using a cesium chloride density gradient method for infection into C2C12 cells. To induce differentiation of C2C12 myoblasts, cells at near confluence were changed from DMEM containing 15% fetal bovine serum to DMEM containing 2% horse serum (Gibco, 26050–088) and were harvested at the indicated days. Luciferase assays were performed as previously described [52]. All experiments were carried out as triplicates and repeated at least 3 times. Constructs used in this study are as follows: FHRE-luc (Addgene, 1789; deposited by Dr. Michael Greenberg), pcDNA3-PRMT1-HA [53,54], pcDNA3-PRMT6-HA [54], pcDNA3-FOXO3-flag. To construct the flag-tagged FOXO3 expression vectors, the entire coding region of FOXO3 was amplified by polymerase chain reaction (PCR) and the PCR products were cloned into flag-tagged pcDNA3 (Addgene, 20011; deposited by Dr. Stephen Smale). To generate the point mutants for the arginine-to-lysine replacement at R188, R218 and R249 of mouse FOXO3, the site directed mutageneses were performed by using QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene, 200518).
Generation of a PRMT1 knockout C2C12 cell line
PRMT1 guide RNA was produced using the gRNA design tool (Genscript). The pSpCas9 plasmid was purchased from Addgene (62988; deposited by Dr. Feng Zhang) [55]. PRMT1 sgRNA and the pSpCas9 plasmid were transiently transfected into the C2C12 cell line using TransIT-LT1 transfection reagent (Mirus, MIR5400). Targeted sequence of clones was confirmed by sequencing. Guide RNA sequences used in this study are as following: PRMT1 gRNA: forward 5ʹ-CACCGCTCACCATGGTCTAACTTGT-3ʹ, backward 5ʹ-AAACACAAGTTAGACCATGGTGAGC-3ʹ.
RNA, protein analysis
Quantitative real-time PCR analysis was measured as previously described [56]. Total RNA was isolated from cultured cells or muscle tissues using an RNeasy plus mini Kit (Qiagen, 4106). cDNA was obtained using a GoScript Reverse Transcription System (Promega, A5001) and analyzed by quantitative real-time PCR using SYBR Green (TAKARA, RR420A) or SensiFAST SYBR No-ROX kit (Bioline, BIO-98050). The data were normalized to expression of ribosomal gene RPL32 or GAPDH. The primer sequences are shown in Table S1. Western blot analysis was performed as previously described [57]. Briefly, cells were lysed in cell extraction buffer (10 mmol/L Tris-HCl, pH 8.0, 150 mmol/L NaCl, 1 mmol/L EDTA, 1% Triton X-100 (Sigma-Aldrich, T8787) containing complete protease inhibitor cocktail (Roche, 04693132001), followed by SDS-PAGE and incubated with primary and secondary antibodies. Tissue extracts were prepared by cryo-pulverization with liquid nitrogen and lysed in cell extraction buffer. Primary antibodies used were PRMT1 (Upstate, 07–404), PRMT4 (Cell Signaling Technology, 4438), PRMT6 (Cell Signaling Technology, 14641), asymmetric dimethylarginine (Cell Signaling Technology, D6A8; custom order [58]), p-AMPK (Cell Signaling Technology, 2535), AMPK (Cell Signaling Technology, 2532), p-AKT (Cell Signaling Technology, 9271), AKT (Cell Signaling Technology, 9272), SQSTM1 (Cell Signaling Technology, 5114), LC3A/B (Cell Signaling Technology, 4108), FOXO1 (Cell Signaling Technology, 9454), FOXO3 (Cell Signaling Technology, 12829), HSP90 (Santa Cruz Biotechnology, sc7947), GAPDH (Santa Cruz Biotechnology, sc25778), HA (Santa Cruz Biotechnology, sc7392), TRIM63/MURF-1 (Gene Tex, 110475), LMNB1/LaminB1 (Abcam, 16048), LAMP1 (Abcam, 24170), LAMP2 (Abcam, 25631), HDAC1 (Abcam, 7028), HDAC2 (Abcam, 2540), HDAC3 (Abcam, 7030), YY1 (Abcam, 109237), Flag (Sigma-aldrich, F3165), TUBB/β-tubulin (Sigma-aldrich, 5293), MHC (DSHB, MF20) and K48-ubiquitin (Millipore, 05–1307). A cellular fractionation assay was performed as previously described [59]. Briefly, C2C12 cells were resuspended in 100 µl of cytosolic fractionation buffer containing 40 mM Tris–HCl, pH 7.4, 10 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol (DTT) and protease inhibitors and incubated on ice for 10 min. After the addition of 7.5 µl of 10% NP-40 (Sigma-Aldrich, 74385), cells were briefly vortexed and centrifuged at 13,000 g at 4°C for 10 min. After transferring the cytoplasmic supernatant, nuclear pellets were dispersed in 50 µl of nuclear fractionation buffer containing 40 mM Tris-HCl, pH 7.4, 420 mM NaCl, 10% glycerol, 1 mM EDTA, 1 mM DTT and proteinase inhibitors followed by incubation for 15 min on ice and centrifugation at 13,000 g for 10 min. The supernatant containing the nuclear fraction was transferred into a new tube, and the fractions were analyzed by immunoblotting. For controls, we have utilized TUBB or HSP90 as a cytosolic marker and LMNB1 as a nuclear marker.
Immunoprecipitation and chromatin immunoprecipitation analysis
Immunoprecipitation analysis was examined as previously described [57,60]. HEK293T cells were transfected with plasmids using TransIT-LT1 transfection reagent (Mirus, MIR5400). The cells were lysed in lysis buffer containing 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100 (Sigma-Aldrich, T8787), 2.5 mM Sodium pyrophosphate, 50 mM NaF, 5 mM β-glycerophosphate, 1 mM Na3VO4, proteinase inhibitor cocktail (Roche, 04693132001). The lysates were incubated with anti-flag M2 affinity gel (Sigma-aldrich, A2220) at 4°C. The immunoprecipitates were resolved by SDS-PAGE and immunoblotted. Chromatin immunoprecipitation analysis was performed as previously described [52]. Antibodies used were rabbit IgG (abcam, ab37415), PRMT1 (abcam, ab3768), YY1 (abcam, ab109237), HDAC1 (abcam, ab7028), HDAC2 (abcam, ab7029) and HDAC3 (abcam, ab7030). The primers used to amplify the Prmt6 promoter region between −82 to +752 are listed in Table S1.
Statistical analysis
Values are expressed as means ±SD or ±SEM, as indicated in the figure legends. Statistical significance was calculated using paired or unpaired two-tailed Student’s t test. For comparison between multiple groups, statistical significance was tested by one-way ANOVA test using SPSS (version 12.0; SPSS, Chicago, IL). Differences were considered statistically significant at or under values of P < 0.05.
Funding Statement
This research was supported by the National Research Foundation of Korea Grant funded by the Korean Government (MSIP) (NRF-2016R1A2B2007179, NRF-2017M3A9D8048710 and NRF-2016R1A5A2945889 to J.S.K., and NRF-2018R1A2B3001540, NRF-2015R1A5A1009024 and NRF-2017M3A9D5A01052447 to S.H.K.).
Acknowledgments
We thank Drs Hana Cho and Ruth Simon for critical reading of the manuscript. Authors are grateful to Hye Young Lee for the technical assistance with mouse work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Egerman MA, Glass DJ.. Signaling pathways controlling skeletal muscle mass. Crit Rev Biochem Mol Biol. 2014. Jan-Feb;49(1):59–68. PubMed PMID: 24237131; PubMed Central PMCID: PMC3913083. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cohen S, Nathan JA, Goldberg AL.. Muscle wasting in disease: molecular mechanisms and promising therapies [Review]. Nat Rev Drug Discov. 2015;14(1):58–74. [DOI] [PubMed] [Google Scholar]
- [3].Sartorelli V, Fulco M. Molecular and cellular determinants of skeletal muscle atrophy and hypertrophy. Sci STKE. 2004. July 27;2004(244):re11 PubMed PMID: 15292521; eng. [DOI] [PubMed] [Google Scholar]
- [4].Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. 2013. January;6(1):25–39. PubMed PMID: 23268536; PubMed Central PMCID: PMC3529336. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Baldwin KM, Haddad F, Pandorf CE, et al. Alterations in muscle mass and contractile phenotype in response to unloading models: role of transcriptional/pretranslational mechanisms. Front Physiol. 2013. October 11;4:284 PubMed PMID: 24130531; PubMed Central PMCID: PMC3795307. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dutta C. Significance of sarcopenia in the elderly. J Nutr. 1997. May;127(5 Suppl):992S–993S. PubMed PMID: 9164281; eng. [DOI] [PubMed] [Google Scholar]
- [7].Vinciguerra M, Musaro A, Rosenthal N. Regulation of muscle atrophy in aging and disease. Adv Exp Med Biol. 2010;694:211–233. PubMed PMID: 20886766; eng. [DOI] [PubMed] [Google Scholar]
- [8].Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999. March 19;96(6):857–868. PubMed PMID: 10102273; eng. [DOI] [PubMed] [Google Scholar]
- [9].Stitt TN, Drujan D, Clarke BA, et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004. May 7;14(3):395–403. PubMed PMID: 15125842; eng. [DOI] [PubMed] [Google Scholar]
- [10].Lecker SH, Jagoe RT, Gilbert A, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004. January;18(1):39–51. PubMed PMID: 14718385; eng. [DOI] [PubMed] [Google Scholar]
- [11].Senf SM, Dodd SL, Judge AR. FOXO signaling is required for disuse muscle atrophy and is directly regulated by Hsp70. Am J Physiol Cell Physiol. 2010. January;298(1):C38–C45. PubMed PMID: 19864323; PubMed Central PMCID: PMC2806148. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sandri M, Sandri C, Gilbert A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004. April 30;117(3):399–412. PubMed PMID: 15109499; PubMed Central PMCID: PMC3619734. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Milan G, Romanello V, Pescatore F, et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun. 2015. April 10;6:6670 PubMed PMID: 25858807; PubMed Central PMCID: PMC4403316. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bodine SC, Latres E, Baumhueter S, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001. November 23;294(5547):1704–1708. PubMed PMID: 11679633; eng. [DOI] [PubMed] [Google Scholar]
- [15].Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013. February;14(2):83–97. PubMed PMID: 23325358; eng. [DOI] [PubMed] [Google Scholar]
- [16].Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014. April;39(4):159–169. PubMed PMID: 24630600; PubMed Central PMCID: PMC4021867. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008. April;20(2):126–136. PubMed PMID: 18394876; PubMed Central PMCID: PMC2387118. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Neel BA, Lin Y, Pessin JE. Skeletal muscle autophagy: a new metabolic regulator. Trends Endocrinol Metab. 2013. December;24(12):635–643. PubMed PMID: 24182456; PubMed Central PMCID: PMC3849822. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sandri M, Coletto L, Grumati P, et al. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J Cell Sci. 2013. December 01;126(Pt 23):5325–5333. PubMed PMID: 24293330; eng. [DOI] [PubMed] [Google Scholar]
- [20].De Palma C, Morisi F, Cheli S, et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 2012. November 15;3:e418 PubMed PMID: 23152054; PubMed Central PMCID: PMC3542595. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009. January 16;33(1):1–13. PubMed PMID: 19150423; PubMed Central PMCID: PMC3372459. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yu Z, Chen T, Hebert J, et al. A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol Cell Biol. 2009. June;29(11):2982–2996. PubMed PMID: 19289494; PubMed Central PMCID: PMC2681996. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tang J, Kao PN, Herschman HR. Protein-arginine methyltransferase I, the predominant protein-arginine methyltransferase in cells, interacts with and is regulated by interleukin enhancer-binding factor 3. J Biol Chem. 2000. June 30;275(26):19866–19876. PubMed PMID: 10749851; eng. [DOI] [PubMed] [Google Scholar]
- [24].Pawlak MR, Scherer CA, Chen J, et al. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol Cell Biol. 2000. July;20(13):4859–4869. PubMed PMID: 10848611; PubMed Central PMCID: PMC85937. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Choi D, Oh KJ, Han HS, et al. Protein arginine methyltransferase 1 regulates hepatic glucose production in a FoxO1-dependent manner. Hepatology. 2012. October;56(4):1546–1556. PubMed PMID: 22532369; eng. [DOI] [PubMed] [Google Scholar]
- [26].Auclair Y, Richard S. The role of arginine methylation in the DNA damage response. DNA Repair (Amst). 2013. July;12(7):459–465. PubMed PMID: 23684798; eng. [DOI] [PubMed] [Google Scholar]
- [27].Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol Cell. 2005. April 29;18(3):263–272. PubMed PMID: 15866169; eng. [DOI] [PubMed] [Google Scholar]
- [28].Yamagata K, Daitoku H, Takahashi Y, et al. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell. 2008. October 24;32(2):221–231. PubMed PMID: 18951090; eng. [DOI] [PubMed] [Google Scholar]
- [29].Le Romancer M, Treilleux I, Leconte N, et al. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell. 2008. July 25;31(2):212–221. PubMed PMID: 18657504; eng. [DOI] [PubMed] [Google Scholar]
- [30].Teyssier C, Ma H, Emter R, et al. Activation of nuclear receptor coactivator PGC-1alpha by arginine methylation. Genes Dev. 2005. June 15;19(12):1466–1473. PubMed PMID: 15964996; PubMed Central PMCID: PMC1151663. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Iwasaki H, Yada T. Protein arginine methylation regulates insulin signaling in L6 skeletal muscle cells. Biochem Biophys Res Commun. 2007. December 28;364(4):1015–1021. PubMed PMID: 17971302; eng. [DOI] [PubMed] [Google Scholar]
- [32].Kim SJ, Yoo BC, Uhm CS, et al. Posttranslational arginine methylation of lamin A/C during myoblast fusion. Biochim Biophys Acta. 2011. February;1814(2):308–317. PubMed PMID: 21111849; eng. [DOI] [PubMed] [Google Scholar]
- [33].Blanc RS, Vogel G, Li X, et al. Arginine methylation by PRMT1 regulates muscle stem cell fate. Mol Cell Biol. 2017. February 01;37(3):e00457–16. PubMed PMID: 27849571; PubMed Central PMCID: PMC5247616. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baumeister P, Luo S, Skarnes WC, et al. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol Cell Biol. 2005. June;25(11):4529–4540. PubMed PMID: 15899857; PubMed Central PMCID: PMC1140640. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rezai-Zadeh N, Zhang X, Namour F, et al. Targeted recruitment of a histone H4-specific methyltransferase by the transcription factor YY1. Genes Dev. 2003. April 15;17(8):1019–1029. PubMed PMID: 12704081; PubMed Central PMCID: PMC196041. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dobson M, Ramakrishnan G, Ma S, et al. Bimodal regulation of FoxO3 by AKT and 14-3-3. Biochim Biophys Acta. 2011. August;1813(8):1453–1464. PubMed PMID: 21621563; PubMed Central PMCID: PMC3237389. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Brunet A, Park J, Tran H, et al. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol. 2001. February;21(3):952–965. PubMed PMID: 11154281; PubMed Central PMCID: PMC86685. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta. 2011. November;1813(11):1938–1945. PubMed PMID: 21708191; eng. [DOI] [PubMed] [Google Scholar]
- [39].Brunet A, Kanai F, Stehn J, et al. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol. 2002. March 4;156(5):817–828. PubMed PMID: 11864996; PubMed Central PMCID: PMC2173313. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chan S, Head SI. Age- and gender-related changes in contractile properties of non-atrophied EDL muscle. PLoS One. 2010. August 23;5(8):e12345 PubMed PMID: 20808812; PubMed Central PMCID: PMC2925956. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Moran AL, Warren GL, Lowe DA. Soleus and EDL muscle contractility across the lifespan of female C57BL/6 mice. Exp Gerontol. 2005. December;40(12):966–975. PubMed PMID: 16243468; eng. [DOI] [PubMed] [Google Scholar]
- [42].Kamei Y, Miura S, Suzuki M, et al. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated Type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J Biol Chem. 2004. September 24;279(39):41114–41123. PubMed PMID: 15272020; eng. [DOI] [PubMed] [Google Scholar]
- [43].Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007. December;6(6):458–471. PubMed PMID: 18054315; eng. [DOI] [PubMed] [Google Scholar]
- [44].Wang H, Huang ZQ, Xia L, et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001. August 03;293(5531):853–857. PubMed PMID: 11387442; eng. [DOI] [PubMed] [Google Scholar]
- [45].Strahl BD, Briggs SD, Brame CJ, et al. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr Biol. 2001. June 26;11(12):996–1000. PubMed PMID: 11448779; eng. [DOI] [PubMed] [Google Scholar]
- [46].Waldmann T, Izzo A, Kamieniarz K, et al. Methylation of H2AR29 is a novel repressive PRMT6 target. Epigenetics Chromatin. 2011. July 20;4:11 PubMed PMID: 21774791; PubMed Central PMCID: PMC3164600. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hyllus D, Stein C, Schnabel K, et al. PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev. 2007. December 15;21(24):3369–3380. PubMed PMID: 18079182; PubMed Central PMCID: PMC2113036. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013. January;13(1):37–50. PubMed PMID: 23235912; eng. [DOI] [PubMed] [Google Scholar]
- [49].Neault M, Mallette FA, Vogel G, et al. Ablation of PRMT6 reveals a role as a negative transcriptional regulator of the p53 tumor suppressor. Nucleic Acids Res. 2012. October;40(19):9513–9521. PubMed PMID: 22904064; PubMed Central PMCID: PMC3479207. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Leem YE, Jeong HJ, Kim HJ, et al. Cdo Regulates Surface Expression of Kir2.1 K+ Channel in Myoblast Differentiation. PLoS One. 2016;11(7):e0158707 PubMed PMID: 27380411; PubMed Central PMCID: PMC4933383. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Luo J, Deng ZL, Luo X, et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc. 2007;2(5):1236–1247. PubMed PMID: 17546019; eng. [DOI] [PubMed] [Google Scholar]
- [52].Jeong HJ, Lee HJ, Vuong TA, et al. Prmt7 deficiency causes reduced skeletal muscle oxidative metabolism and age-related obesity. Diabetes. 2016. July;65(7):1868–1882. PubMed PMID: 27207521. [DOI] [PubMed] [Google Scholar]
- [53].Kim HJ, Jeong MH, Kim KR, et al. Protein arginine methylation facilitates KCNQ channel-PIP2 interaction leading to seizure suppression. Elife. 2016. July 28;5:e17159 PubMed PMID: 27466704; PubMed Central PMCID: PMC4996652. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Han HS, Jung CY, Yoon YS, et al. Arginine methylation of CRTC2 is critical in the transcriptional control of hepatic glucose metabolism. Sci Signal. 2014. February 25;7(314):ra19 PubMed PMID: 24570487; eng. [DOI] [PubMed] [Google Scholar]
- [55].Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013. November;8(11):2281–2308. PubMed PMID: 24157548; PubMed Central PMCID: PMC3969860. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jeong MH, Leem YE, Kim HJ, et al. A Shh coreceptor Cdo is required for efficient cardiomyogenesis of pluripotent stem cells. J Mol Cell Cardiol. 2016. April;93:57–66. PubMed PMID: 26906632; eng. [DOI] [PubMed] [Google Scholar]
- [57].Jeong MH, Kim HJ, Pyun JH, et al. Cdon deficiency causes cardiac remodeling through hyperactivation of WNT/beta-catenin signaling. Proc Natl Acad Sci U S A. 2017. February 21;114(8):E1345–E1354. PubMed PMID: 28154134; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Guo A, Gu H, Zhou J, et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol Cell Proteomics. 2014. January;13(1):372–387. PubMed PMID: 24129315; PubMed Central PMCID: PMC3879628. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jeong MH, Ho SM, Vuong TA, et al. Cdo suppresses canonical Wnt signalling via interaction with Lrp6 thereby promoting neuronal differentiation. Nat Commun. 2014. November 19;5:5455 PubMed PMID: 25406935; PubMed Central PMCID: PMCPMC4412020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Vuong TA, Leem YE, Kim BG, et al. A Sonic hedgehog coreceptor, BOC regulates neuronal differentiation and neurite outgrowth via interaction with ABL and JNK activation. Cell Signal. 2017. January;30:30–40. PubMed PMID: 27871935; eng. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.