

ABSTRACT
The Parkinson disease‐associated proteins PINK1 and PRKN coordinate the ubiquitination of mitochondrial outer membrane proteins to tag them either for degradation or for autophagic clearance of the mitochondrion. The proteins include the mitochondrial trafficking proteins RHOT1 and RHOT2, the removal of which may be required for immobilization of mitochondria prior to mitophagy. Here, we demonstrate that RHOT1 and RHOT2 are not only substrates for PINK1-PRKN‐dependent degradation but that they also play an active role in the process of mitophagy. RHOT1, and likely also RHOT2, may act as a docking site for inactive PRKN prior to mitochondrial damage, thus keeping PRKN in close proximity to its potential substrates and thereby facilitating mitophagy. We also show that RHOT1 functions as a calcium-sensing docking site for PRKN, and we suggest that calcium binding to RHOT is a key step in the calcium‐dependent activation of mitophagy machinery.
KEYWORDS: Cytosolic calcium, mitochondrial trafficking, mitophagy, neuron, PRKN translocation
Mutations in PRKN (parkin RBR E3 ubiquitin protein ligase) and PINK1 (PTEN induced kinase 1) are the most prevalent causes of autosomal recessive early-onset Parkinson disease. PRKN and PINK1 are involved in the selective degradation of damaged mitochondria by autophagy (mitophagy). PINK1 accumulates on the outer membrane of depolarized mitochondria, where it phosphorylates ubiquitin and PRKN at their S65 residues. The latter then activates PRKN to catalyze the ubiquitination of mitochondrial outer membrane proteins, which are then either degraded or serve as a signal for autophagic clearance of the mitochondrion. The mitochondrial trafficking proteins RHOT1 and RHOT2 are among the first proteins to be ubiquitinated and removed after activation of the PINK1-PRKN pathway. RHOTs are small GTPases with calcium-sensing EF-hands that function as adaptors between the mitochondrion and its molecular motors. It has been suggested that removal of RHOT1 is required for immobilization of mitochondria prior to mitophagy.
We have recently shown that RHOT proteins are not only substrates for PINK1-PRKN‐dependent degradation but that they also play an active role in the process of mitophagy [1]. Our data demonstrated that RHOT1 and RHOT2 themselves may recruit a low level of PRKN to normal (undamaged) mitochondria. In RHOT1- and RHOT2-overexpressing cells, fluorescently labelled PRKN translocates to rod-shaped mitochondria in a number of different cell types. This ‘translocation’ did not show the same pattern typically observed in the case of damaged mitochondria: here, these mitochondria remain polarized and retain their shape, distribution and ability to move. These data suggest that a pool of PRKN interacts with RHOT proteins prior to mitochondrial damage. Importantly, RHOT1 overexpression also induces PRKN translocation in the presence of PINK1 shRNA or in PINK1-deficient mouse embryonic fibroblasts, showing that the process is independent of PINK1 accumulation on the mitochondrial outer membrane. RHOT1 overexpression also attracts ligase-dead mutants of PRKN to mitochondria, suggesting that the RHOT1-associated pool of mitochondrial PRKN is inactive. Overexpression of the RHOT1 mutant K572R, which cannot be efficiently ubiquitinated by PRKN, nonetheless induces PRKN translocation to mitochondria, confirming that initial PRKN translocation does not involve ubiquitination of RHOT1. With respect to depolarized mitochondria, we also demonstrated that RHOT1 and RHOT2 are required for mitochondrial damage-induced PRKN translocation to mitochondria and subsequent mitophagy. Removal of RHOT1 or RHOT2 by specific shRNAs slow down PRKN translocation to mitochondria after depolarization or PINK1 overexpression. These data suggest that the observed RHOT-PRKN interaction is required for downstream events in the mitophagy cascade.
Thus, our first major conclusion is that RHOT1, and probably also RHOT2, act as docking stations for PRKN. The small pool of PRKN attached to RHOT1 remains inactive and unable to ubiquitinate RHOT1, and it does not affect basal mitochondrial function. However, when the mitochondrion depolarizes and PINK1 accumulates, PRKN then becomes activated and ubiquitinates mitochondrial targets including RHOT1 and RHOT2. This in turn leads to swift cessation of mitochondrial movement, mitochondrial fragmentation and other events favoring autophagosomal engulfment of dysfunctional mitochondria. We speculate that the close proximity of the small pool of inactive PRKN to its future substrates in the mitochondrial outer membrane would speed up the recruitment of cytosolic PRKN after mitochondrial damage and thus facilitate the removal of damaged mitochondria.
As described previously, RHOT proteins carry calcium-sensing EF-hands, which, when bound by calcium, decrease affinity of RHOT1 for its mitochondrial trafficking-related binding partners. We investigated whether calcium binding to RHOT1 EF-hands also affected its ubiquitination by PRKN. We found that a RHOT1 EF-hands mutant, which is unable to bind calcium, cannot be polyubiquitinated by PRKN after mitochondrial damage. Moreover, PRKN translocation to mitochondria is suppressed when the RHOT1 EF‐hands mutant is overexpressed, suggesting that conformational changes in the EF-hands may make the RHOT1 ubiquitination site(s) less accessible for PRKN.
We hypothesized that RHOT1 may function as a calcium-sensing docking site for PRKN, and we tested whether calcium levels affect PRKN translocation to mitochondria. We followed PRKN translocation in calcium-free conditions or in the presence of a calcium-channel opener in a dopaminergic PC6 cell line. In neurons, we applied glutamate to open NMDA receptor channels, thereby stimulating calcium influx. Our data showed that excessive cytosolic calcium facilitates, whereas calcium chelation inhibits, PRKN translocation to mitochondria. RHOT1 overexpression increases glutamate-induced PRKN translocation, and the number of mitochondria in autophagosomes, and decreases further the neuronal levels of ATP and neuronal viability. In contrast, the RHOT1 EF-hands mutant inhibits mitophagy, restores neuronal ATP levels and partially rescues glutamate toxicity. Thus, the second major conclusion from our study is that RHOT1 functions as a calcium-sensing docking site for PRKN and that calcium binding to RHOT is a major step in the calcium‐dependent activation of mitophagy machinery.
Taken together, our data show that RHOT proteins maintain a pool of PRKN on mitochondria that becomes activated upon mitochondrial damage (Figure 1). We suggest that, upon calcium binding to RHOT1 EF‐hands, PRKN can access its preferred lysine(s) and/or PINK1 can phosphorylate ubiquitins on RHOT1. These events amplify the PRKN activation cycle and ensure faster PRKN translocation to mitochondria. Without calcium, RHOT1 remains protected and cannot be ubiquitinated by PRKN. This could be an additional safety mechanism for mitochondria still attached to microtubules to prevent their removal and recycling by active PRKN.
Figure 1.
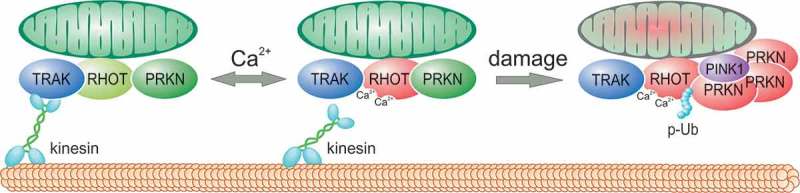
RHOT proteins act as calcium-sensitive docking sites for PRKN. A small pool of PRKN is normally attached to RHOT proteins at mitochondria. This pool remains inactive and mitochondrial function remains unaffected. In this state, RHOT proteins remain in complex with the transport machinery (TRAK1/2 and kinesins). Calcium binding to RHOT1 EF-hands has 2 consequences. First, it reversibly derails the mitochondrion from the microtubule and, second, it exposes RHOT1 lysines to PRKN and/or PINK1. However, in the absence of PINK1 accumulation, PRKN cannot be activated. If the mitochondrion is severely damaged and depolarized, PINK1 accumulates and phosphorylates ubiquitin (p-Ub) and PRKN, thereby triggering the PRKN activation cycle. PRKN then polyubiquitinates RHOT1 and facilitates the removal of the mitochondrion (mitophagy). Together, our results show that RHOT proteins act as a docking station for PRKN and mediate the effects of calcium signaling to coordinate mitochondrial trafficking and mitochondrial degradation.
Funding Statement
This work was supported by the Estonian Research Council [IUT2‐5, PUT513, ETF8810, PRG400], European Union Horizon 2020 Framework Program [692202] and the European Regional Development Fund [2014‐2020.4.01.15‐0012].
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Safiulina D, Kuum M, Choubey V, et al. Miro proteins prime mitochondria for Parkin translocation and mitophagy. Embo J. 2019;38(2):e99384. [DOI] [PMC free article] [PubMed] [Google Scholar]