

Abstract
The importance of climate in determining biodiversity patterns has been well documented. However, the relationship between climate and rates of genetic evolution remains controversial. Latitude and elevation have been associated with rates of change in genetic markers such as cytochrome b. What is not known, however, is the strength of such associations and whether patterns found among these genes apply across entire genomes. Here, using bumblebee genetic data from seven subgenera of Bombus, we demonstrate that all species occupying warmer elevations have undergone faster genome-wide evolution than those in the same subgenera occupying cooler elevations. Our findings point to a critical biogeographic role in the relative rates of whole species evolution, potentially influencing global biodiversity patterns.
Keywords: Bombus, diversity, genetic evolution, integrated evolutionary speed, genome, mitochondrial, nuclear
Species diversity patterns are correlated with environmental variables such as temperature, primary productivity, and biome area (McBride et al. 2014; Gillman et al. 2015; Jonathan and Walter 2015), and declining species richness among bees with increasing elevation has been associated with temperature (Alice et al. 2015). Rensch (1959) suggested that high diversity may derive from greater evolutionary speed in warmer climates, possibly due to shorter generation times and/or higher mutation rates (Rohde 1992). The strength and importance of relationships among environmental and geographical variables and rates of genetic evolution is, however, controversial (Gillman et al. 2011; Weir and Schluter 2011; Rolland et al. 2016). Faster rates of genetic evolution have been found in warmer climates, such as at lower elevations, for a range of taxa including endotherms and ectotherms (Gillman and Wright 2014; Dugo-Cota et al. 2015), and greater intraspecific divergence has been reported from populations occupying warmer environments (Oppold et al. 2016). Life-history traits, including generation time, longevity, metabolic rate, and body mass have also been related to rates of substitution (Nabholz et al. 2008; Santos 2012; Lehtonen and Lanfear 2014; Bromham et al. 2015) and may underpin the geographic and climatic associations with rates of evolution. However, these studies have all relied on relatively small genetic markers from mitochondria and, less often, nuclear DNA, to characterize species-level rates of evolution, and it is unknown whether the results are representative of genome-wide evolution. Therefore, the importance of environment in mediating the rate of evolution remains unclear. Here, we address this issue by comparing rates of genetic evolution across whole nuclear and mitochondrial genomes for seven pairs of bumblebee species that occupy contrasting elevational distributions.
The genus Bombus (Hymenoptera: Apidae) includes ∼250 species within 15 subgenera (Cameron et al. 2007; Williams et al. 2008). Elevational segregation of closely related species enabled us to select seven pairs of species, within seven subgenera, with elevations differing between species within each pair by an average of 1,867 m (two-tailed paired t-test, P = 9.5 × 10−4) (supplementary table S1, Supplementary Material online). We sequenced 12 of the 14 species (SRA accession No. PRJNA508540; supplementary table S2, Supplementary Material online) and obtained published genome sequences (Sadd et al. 2015) for the remaining two (B. impatiens and B. terrestris). Nuclear genome sequences (NUC) and mitochondrial genome sequences (MIT) were each concatenated for phylogenetic analysis. A total of 7,738 nucleotide genes with 4,641,249 bp and 13 mitochondrial genes with 10,842 bp were used in our analyses.
Maximum likelihood with CODON models was used for phylogenetic analysis. The best-fit model for NUC and MIT was GY+F+R4 and GY+F+R5, respectively. The topologies of the two phylogenetic trees (fig. 1) were consistent with each other, and with previous studies (Williams et al. 2008). Branch lengths for all lowland species were longer than their high-elevation counterparts for both NUC and MIT (fig. 1, table 1). Consequently, mean log transformed branch lengths of lowland species were longer than those for high-elevation species (table 2). We also calculated the tip age (total branch length from each tip to the root) of each species (supplementary table S2, Supplementary Material online) and performed correlation analyses between log transformed tip ages and their corresponding mean elevations. Both nucleotide (r = −0.653, P = 0.011, n = 14) and mitochondrial (r = −0.736, P = 0.003, n = 14) tip ages were negatively correlated with mean elevations.
Fig. 1.
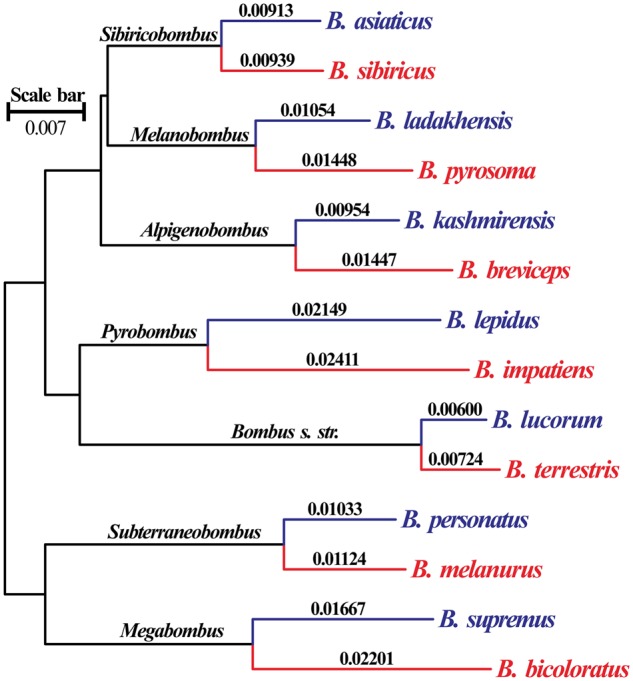
Phylogenetic tree of the 14 bumblebee species based on the concatenated nuclear sequences. The subgenus is given above the branch; the species branch length is given above each terminal branch; the species colored blue occur at high elevations and the species colored red occur at low elevations.
Table 1.
Branch Lengths, Nonsynonymous (dN), Synonymous (dS), and dN/dS Ratios for High- and Low-Elevation Bombus Species.
| Group | Species | NUC |
MIT |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Length | dN | dS | dN/dS | Length | dN | dS | dN/dS | ||
| High elevation | B. asiaticus | 0.00913 | 0.00110 | 0.00786 | 0.14035 | 2.40887 | 0.02487 | 1.19214 | 0.02086 |
| B. ladakhensis | 0.01054 | 0.00130 | 0.00900 | 0.14430 | 1.79839 | 0.01922 | 0.88297 | 0.02177 | |
| B. kashmirensis | 0.00954 | 0.00114 | 0.00824 | 0.13814 | 3.37945 | 0.03769 | 1.64224 | 0.02295 | |
| B. lepidus | 0.02149 | 0.00283 | 0.01790 | 0.15794 | 2.61395 | 0.02799 | 1.28283 | 0.02182 | |
| B. lucorum | 0.00600 | 0.00076 | 0.00507 | 0.14960 | 1.24106 | 0.01370 | 0.60463 | 0.02266 | |
| B. personatus | 0.01033 | 0.00123 | 0.00895 | 0.13685 | 1.53831 | 0.01768 | 0.74190 | 0.02383 | |
| B. supremus | 0.01667 | 0.00205 | 0.01426 | 0.14346 | 1.97232 | 0.02328 | 0.94470 | 0.02464 | |
| Low elevation | B. sibiricus | 0.00939 | 0.00115 | 0.00803 | 0.14352 | 2.63253 | 0.02838 | 1.28988 | 0.02200 |
| B. pyrosoma | 0.01448 | 0.00184 | 0.01221 | 0.15101 | 2.94224 | 0.03046 | 1.45514 | 0.02093 | |
| B. breviceps | 0.01447 | 0.00177 | 0.01240 | 0.14247 | 3.51815 | 0.03937 | 1.70821 | 0.02305 | |
| B. impatiens | 0.02411 | 0.00319 | 0.02003 | 0.15933 | 4.30636 | 0.04482 | 2.12729 | 0.02107 | |
| B. terrestris | 0.00724 | 0.00090 | 0.00617 | 0.14554 | 1.71056 | 0.01957 | 0.82593 | 0.02370 | |
| B. melanurus | 0.01124 | 0.00132 | 0.00978 | 0.13454 | 2.13841 | 0.02271 | 1.05144 | 0.02160 | |
| B. bicoloratus | 0.02201 | 0.00289 | 0.01833 | 0.15785 | 4.58913 | 0.04984 | 2.24455 | 0.02221 | |
Note.—NUC, concatenated nuclear sequence; MIT, concatenated mitochondrial sequences.
Table 2.
Mean Branch Lengths, dN, dS, and dN/dS ratios of High- and Low-Elevation Species.
| Sequence | Index | High Elevation | Low Elevation | Statistics (df = 6)a |
|---|---|---|---|---|
| NUC | Length | 0.01196 ± 0.00528 | 0.01471 ± 0.00630 | t = −3.864, P = 0.008 |
| dN | 0.00149 ± 0.00071 | 0.00187 ± 0.00087 | t = −3.732, P = 0.010 | |
| dS | 0.01018 ± 0.00436 | 0.01242 ± 0.00513 | t = −3.885, P = 0.008 | |
| dN/dS | 0.14438 ± 0.00735 | 0.14775 ± 0.00887 | t = −1.454, P = 0.196 | |
| MIT | Length | 2.13605 ± 0.72434 | 3.11963 ± 1.07576 | t = −3.615, P = 0.011 |
| dN | 0.02349 ± 0.00787 | 0.03359 ± 0.01137 | t = −3.889, P = 0.008 | |
| dS | 1.04163 ± 0.35518 | 1.52892 ± 0.53026 | t = −3.548, P = 0.012 | |
| dN/dS | 0.02265 ± 0.0013 | 0.02208 ± 0.00101 | t = 1.044, P = 0.337 |
Note.—NUC, concatenated nuclear sequence; MIT, concatenated mitochondrial sequences. Values are given as mean ± SD.
Two-tailed paired-sample t-tests for log transformed mean differences.
Thus, all comparisons conformed to the pattern of greater genome evolution in lower-elevation species. Our results confirm a pattern previously found among both ectotherms and endotherms using limited genetic data from marker genes (Gillman and Wright 2014; Dugo-Cota et al. 2015). However, the results from previous studies were less consistent, with approximately one-third of comparisons in each of these studies producing a result contrary to the overall pattern. Here, we show a stronger, indeed universal pattern across our data set derived from full genomes.
Faster rates of genetic evolution at lower elevations may be due to a population size effect. Nearly neutral theory predicts elevated rates of substitution in small populations due to relaxed purifying selection of slightly deleterious mutations (Ohta 1972) thereby elevating nonsynonymous substitutions (dN) relative to synonymous substitutions (dS). We therefore examined dN and dS. Lower-elevation species in all cases exhibited higher dN and dS in both mitochondrial and nuclear DNA than the contrasted higher-elevation species (table 1). The log transformed means for dN and dS of both NUC and MIT were all significantly larger in low-elevation species (table 2). By contrast, there was no indication of a mean difference in dN/dS between high- and low-elevation species for either NUC or MIT (paired-samples t-test for log transformed values, P = 0.196 and 0.337, respectively) (table 2). Relaxed selection due to small populations cannot therefore explain our results.
The opposite population effect, whereby total mutations increase with population size and thus increase rates of substitution, has been previously modeled (Kimura 1979), and the integrated evolutionary speed hypothesis predicts elevated rates of genetic evolution in larger populations (Gillman and Wright 2014). Longer branch lengths have been reported for mainland bird species with larger populations relative to island species (Wright et al. 2009). The low-elevation species in our study occur in higher densities and are more widely distributed than the high-elevation species (An et al. 2014) and therefore this mechanism provides a potential explanation for our results.
Life-history traits such as body size, metabolic rate, longevity, and generation time have been negatively associated with temperature and with substitution rates (Martin and Palumbi 1993; Welch et al. 2008; Bromham et al. 2015). Life history therefore provides plausible explanations for lower rates in cooler environments. However, in many cases life-history traits, such as longevity and body size, have failed to explain latitudinal or elevational relationships (Gillman et al. 2012; Gillman and Wright 2013; Lourenço et al. 2013). Neither generation time nor longevity is a tenable explanation for the divergent substitution rates we measured because all of the study species have an annual life cycle (Goulson 2010). Likewise, larger bodies at higher elevations are an unlikely explanation for slower substitution rates because the high-elevation species in our study are generally smaller, not larger, than their low-elevation counterparts (Williams et al. 2009).
Lower basal metabolic rates among the high-elevation species are another potential explanation for our results. However, empirical testing with large data sets has previously failed to find significant relationships between basal metabolic rate and substitution rate (Bromham et al. 1996; Lanfear et al. 2007). Active, rather than basal, metabolic rates have, by contrast, been positively associated with rates of genetic evolution (Santos 2012; Gillman and Wright 2013) and may be suppressed at higher elevations due to colder temperatures. A low substitution rate in two thermophiles is thought to result from internal control mechanisms that counterbalance potentially deleterious effects of elevated mutations at extreme high temperatures (Drake 2009; Swami 2009). Similarly, a conserved genetic structure might result due to hypoxia stress among bumblebees occupying cold environments (Cai et al. 2013; Zhang et al. 2013). As potential explanations for variable rates of evolution, these mechanisms deserve further empirical investigation.
We clearly demonstrate faster molecular evolution among low-elevation bumblebee species than among high-elevation species. Further work is needed to identify the mechanisms producing this pattern. Our genomic data also provide fertile ground for investigation into functional implications of genetic divergence in contrasting environments. The unequivocal pattern we reveal among our study species suggests a critical biogeographic role in the relative rates of whole species evolution. Tests for an association between substitution rate, diversification rate, and species richness have produced variable results (Lanfear et al. 2010; Goldie et al. 2011; Bromham et al. 2015). Nonetheless, we suggest that differential rates of species evolution have the potential to influence global patterns of biodiversity.
Materials and Methods
Bumblebees (adult workers) were live-trapped using sweep nets and stored in a refrigerator (supplementary table S3, Supplementary Material online). DNA was extracted from each bumblebee and a 350-bp library constructed. The paired-end library was sequenced (Illumina HiSeq2000) with both directions of 150-bp reads representing ∼100× coverage of the genome. Using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/; last accessed March 14, 2019) the sequenced data were filtered by removal of adaptors, low-quality reads, and ambiguous reads. The clean reads were used for de novo assembly with IDBA-UD (Peng et al. 2012). The 200-bp longer contigs were used for further analyses. In addition to the 12 species sequenced by us, previously published genomes (Sadd et al. 2015) of B. terrestris (RefSeq assembly accession: GCF_000214255.1) and B. impatiens (RefSeq assembly accession: GCF_000188095.2) were used.
Mean elevation data for all species we sequenced were obtained by An et al. (2014). For B. terrestris and B. impatiens, distributions were obtained from the GBIF database (https://www.gbif.org/; last accessed March 14, 2019), and average elevations were determined using the ArcGIS platform and the SRTM30 digital elevation model (https://dds.cr.usgs.gov/srtm/version2_1/SRTM30/; last accessed March 14, 2019). Seven sister pairs of bumblebee species, belonging to seven subgenera, were compared with respect to rates of molecular evolution and elevation.
First, we edited the gff file of B. terrestris (GCF_000214255.1_Bter_1.0_genomic.gff) downloaded from the National Centre for Biotechnology Information (NCBI) RefSeq assembly database, only keeping the coding sequence (CDS) of the longest exon of each gene. We then extracted the CDSs (CDS-L) with the gffread program from the Cufflinks package (Trapnell et al. 2010) using the gff file and the genomic assembly (GCF_000214255.1_Bter_1.0_genomic.fna). Second, we translated CDS-L into peptide sequences (PEP-L) using MEGA (Kumar et al. 2016). With PEP-L as query sequences, we used TBlastN from the BLAST program package (Altschul et al. 1997) and seqtk (https://github.com/lh3/seqtk; last accessed March 14, 2019) to determine and extract potential homologous sequences (plus 1,000 bp of upstream/downstream regions). Third, we extracted the CDS region of the potential homologous sequences using the Exonerate program (Slater and Birney 2005). Finally, we extracted CDS-L and PEP-L of each species and identified and aligned the orthologous genes successively using the programs InParanoid (Remm et al. 2001), MultiParanoid (Alexeyenko et al. 2006), MACSE (Ranwez et al. 2011), and PRANK (Löytynoja and Goldman 2005), as previous described (Lin et al. 2014).
The 13 coding genes in the mitochondrial genome were extracted and aligned manually. We downloaded a mitochondrial genome from GenBank (accession number: KT368150.1) and extracted the 13 coding genes. Using each coding gene as the query sequence, we identified and extracted homologous sequences from the other 13 species with the TBlastN program. We then aligned each of the 13 genes manually in MEGA.
All the aligned nuclear and mitochondrial sequences were concatenated into two alignment sets (supplementary tables S4 and S5, Supplementary Material online). The substitution saturation of each of NUC and MIT was tested manually using DAMBE (Xia 2013). We used maximum likelihood with CODON models for phylogenetic tree reconstruction because they are thought to be biologically more realistic than other substitution models for protein-coding sequence evolution (Gil et al. 2013; Galinskaya et al. 2014). IQ-TREE (Nguyen et al. 2015) was used to reconstruct trees with 1,000 bootstrap replicates. The ModelFinder (Darriba et al. 2012; Kalyaanamoorthy et al. 2017) program was automatically invoked by IQ-TREE to select the best-fitting substitution model for each alignment according to the Bayesian information criterion. The root setting with outgroup taxa does not influence the topology of ingroup taxa. In order to avoid loss of information in the processes of orthologous gene identification between the ingroup and outgroup, we did not use outgroup taxa in the reconstruction. Finally, the branch length as well as the tip age for each species was directly read from the phylogenetic trees. The “several ω ratio” branch model (model = 2) in CODEML in the PAML package (version 4.9h) was used to calculate dN/dS (Yang et al. 2000). The 14 external branches corresponding to the species were viewed as different foregrounds, whereas all the internal branches were viewed as a common background (supplementary table S6, Supplementary Material online). The prior branch lengths generated by IQ-TREE were used with the fix_blength = 3 (proportional) set.
Statistical analyses were performed using SPSS. Several data sets deviated from normal (one-sample Kolmogorov–Smirnov test) and therefore, in order to be consistent, all branch lengths, tip ages, dN, dS, and dN/dS were natural logarithmic (Ln) transformed. The two-tailed paired-samples t-test was used to compare means of each index between the high- and low-elevation species. The Spearman correlation test was performed to test the relationship between the tip ages and their corresponding mean elevations.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Acknowledgments
This study was supported by Youth Innovation Promotion Association of Chinese Academy of Sciences (No. 2015352), Science and Technology Foundation of Jiangxi Provincial Department of Education (No. GJJ170655), and Construction Fund for Qinghai Key Laboratories (2017-ZJ-Y23).
Author Contributions
G.L., L.W., T.Z., and Z.C. carried out bumblebee sampling. G.L., Z.H., L.N.G., and F.Z. wrote the paper. L.N.G. and F.Z. managed the project and the team. All authors read and approved the final manuscript.
Supplementary Material
References
- Alexeyenko A, Tamas I, Liu G, Sonnhammer E.. 2006. Automatic clustering of orthologs and inparalogs shared by multiple proteomes. Bioinformatics 2214: e9–e15. [DOI] [PubMed] [Google Scholar]
- Alice C, PM K, KW J, Tim A, EC D, GM W, Andreas H, Thomas N, Ingolf S-D.. 2015. Temperature versus resource constraints: which factors determine bee diversity on Mount Kilimanjaro, Tanzania? Glob Ecol Biogeogr. 24:642–652. [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ.. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 2517: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Huang J, Shao Y, Zhang S, Wang B, Liu X, Wu J, Williams PH.. 2014. The bumblebees of North China (Apidae, Bombus Latreille). Zootaxa 38301: 1.. [DOI] [PubMed] [Google Scholar]
- Bromham L, Hua X, Lanfear R, Cowman PF.. 2015. Exploring the relationships between mutation rates, life history, genome size, environment, and species richness in flowering plants. Am Nat. 1854: 507–524. [DOI] [PubMed] [Google Scholar]
- Bromham L, Rambaut A, Harvey PH.. 1996. Determinants of rate variation in mammalian DNA sequence evolution. J Mol Evol. 436: 610–621. [DOI] [PubMed] [Google Scholar]
- Cai Q, Qian X, Lang Y, Luo Y, Xu J, Pan S, Hui Y, Gou C, Cai Y, Hao M, et al. 2013. Genome sequence of ground tit Pseudopodoces humilis and its adaptation to high altitude. Genome Biol. 143: R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron SA, Hines HM, Williams PH.. 2007. A comprehensive phylogeny of the bumble bees (Bombus). Biol J Linn Soc. 911: 161–188. [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D.. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 98: 772.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW. 2009. Avoiding dangerous missense: thermophiles display especially low mutation rates. PLoS Genet. 5:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugo-Cota Á, Castroviejo-Fisher S, Vilà C, Gonzalez-Voyer A.. 2015. A test of the integrated evolutionary speed hypothesis in a Neotropical amphibian radiation. Glob Ecol Biogeogr. 247: 804–813. [Google Scholar]
- Galinskaya TV, Suvorov A, Okun MV, Shatalkin AI.. 2014. DNA barcoding of Palaearctic Ulidiidae (Diptera: Tephritoidea): morphology, DNA evolution, and Markov codon models. Zool Stud. 53:51. [Google Scholar]
- Gil M, Zanetti MS, Zoller S, Anisimova M.. 2013. CodonPhyML: fast maximum likelihood phylogeny estimation under codon substitution models. Mol Biol Evol. 306: 1270–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillman LN, McBride P, Keeling DJ, Ross HA, Wright SD.. 2011. Are rates of molecular evolution in mammals substantially accelerated in warmer environments? Reply. Proc R Soc B. 2781710: 1294–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillman LN, McCowan LS, Wright SD.. 2012. The tempo of genetic evolution in birds: body mass and climate effects. J Biogeogr. 399: 1567–1572. [Google Scholar]
- Gillman LN, Wright SD.. 2013. Patterns of evolutionary speed: in search of a causal mechanism. Diversity 54: 811–823. [Google Scholar]
- Gillman LN, Wright SD.. 2014. Species richness and evolutionary speed: the influence of temperature, water and area. J Biogeogr. 411: 39–51. [Google Scholar]
- Gillman LN, Wright SD, Cusens J, McBride PD, Malhi Y, Whittaker RJ.. 2015. Latitude, productivity and species richness. Glob Ecol Biogeogr. 241: 107–117. [Google Scholar]
- Goldie X, Lanfear R, Bromham L.. 2011. Diversification and the rate of molecular evolution: no evidence of a link in mammals. BMC Evol Biol. 11:1471–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulson D. 2010. Bumblebees: behaviour, ecology, and conservation. New York: Oxford University Press. [Google Scholar]
- Jonathan B, Walter J.. 2015. Relative roles of ecological and energetic constraints, diversification rates and region history on global species richness gradients. Ecol Lett. 18:563–571. [DOI] [PubMed] [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS.. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 146: 587.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1979. Model of effectively neutral mutations in which selective constraint is incorporated. Proc Natl Acad Sci U S A. 767: 3440–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K.. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 337: 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R, Ho SYW, Love D, Bromham L.. 2010. Mutation rate is linked to diversification in birds. Proc Natl Acad Sci U S A. 10747: 20423–20428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R, Thomas JA, Welch JJ, Brey T, Bromham L.. 2007. Metabolic rate does not calibrate the molecular clock. Proc Natl Acad Sci U S A. 10439: 15388–15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen J, Lanfear R.. 2014. Generation time, life history and the substitution rate of neutral mutations. Biol Lett. 1011: 20140801.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G-H, Wang K, Deng X-G, Nevo E, Zhao F, Su J-P, Guo S-C, Zhang T-Z, Zhao H.. 2014. Transcriptome sequencing and phylogenomic resolution within Spalacidae (Rodentia). BMC Genomics 15:32.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenço JM, Glémin S, Chiari Y, Galtier N.. 2013. The determinants of the molecular substitution process in turtles. J Evol Biol. 261: 38–50. [DOI] [PubMed] [Google Scholar]
- Löytynoja A, Goldman N.. 2005. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 10230: 10557–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AP, Palumbi SR.. 1993. Body size, metabolic rate, generation time, and the molecular clock. Proc Natl Acad Sci U S A. 909: 4087–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride PD, Cusens J, Gillman LN.. 2014. Revisiting spatial scale in the productivity–species richness relationship: fundamental issues and global change implications. AoB Plants 6:plu057.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabholz B, Glémin S, Galtier N.. 2008. Strong variations of mitochondrial mutation rate across mammals—the longevity hypothesis. Mol Biol Evol. 251: 120–130. [DOI] [PubMed] [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1): 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T. 1972. Population size and the rate of evolution. J Mol Evol. 14: 305–314. [DOI] [PubMed] [Google Scholar]
- Oppold A-M, Pedrosa JAM, Bálint M, Diogo JB, Ilkova J, Pestana JLT, Pfenninger M.. 2016. Support for the evolutionary speed hypothesis from intraspecific population genetic data in the non-biting midge Chironomus riparius. Proc R Soc B. 2831825: 20152413.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Leung HCM, Yiu SM, Chin F.. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2811: 1420–1428. [DOI] [PubMed] [Google Scholar]
- Ranwez V, Harispe S, Delsuc F, Douzery E.. 2011. MACSE: multiple alignment of coding sequences accounting for frameshifts and stop codons. PLoS One 69: e22594.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remm M, Storm CEV, Sonnhammer E.. 2001. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol. 3145: 1041–1052. [DOI] [PubMed] [Google Scholar]
- Rensch B. 1959. Evolution above the species level. London: Methuen. [Google Scholar]
- Rohde K. 1992. Latitudinal gradients in species diversity: the search for the primary cause. Oikos 653: 514–527. [Google Scholar]
- Rolland J, Loiseau O, Romiguier J, Salamin N.. 2016. Molecular evolutionary rates are not correlated with temperature and latitude in Squamata: an exception to the metabolic theory of ecology? BMC Evol Biol. 16:95.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadd BM, Barribeau SM, Bloch G, de Graaf DC, Dearden P, Elsik CG, Gadau J, Grimmelikhuijzen CJ, Hasselmann M, Lozier JD, et al. 2015. The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol. 16:76.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JC. 2012. Fast molecular evolution associated with high active metabolic rates in poison frogs. Mol Biol Evol. 298: 2001–2018. [DOI] [PubMed] [Google Scholar]
- Slater GSC, Birney E.. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 61: 31.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swami M. 2009. Turning up the heat. Nat Rev Genet. 108: 512–513. [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L.. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 285: 511.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir JT, Schluter D.. 2011. Are rates of molecular evolution in mammals substantially accelerated in warmer environments? Proc R Soc B. 2781710: 1291–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch J, Bininda-Emonds O, Bromham L.. 2008. Correlates of substitution rate variation in mammalian protein-coding sequences. BMC Evol Biol. 8:53.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams P, Tang Y, Yao J, Cameron S.. 2009. The bumblebees of Sichuan (Hymenoptera: Apidae, Bombini). Syst Biodivers. 72: 101–189. [Google Scholar]
- Williams PH, Cameron SA, Hines HM, Cederberg B, Rasmont P.. 2008. A simplified subgeneric classification of the bumblebees (genus Bombus). Apidologie 391: 46–74. [Google Scholar]
- Wright SD, Gillman LN, Ross HA, Keeling DJ.. 2009. Slower tempo of microevolution in island birds: implications for conservation biology. Evolution 639: 2275–2287. [DOI] [PubMed] [Google Scholar]
- Xia X. 2013. DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol. 307: 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen A-M.. 2000. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Lin G, Nevo E, Yang C, Su J.. 2013. Cytochrome b gene selection of subterranean rodent Gansu zokor Eospalax cansus (Rodentia, Spalacidae). Zool Anz. 2521: 118–122. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.