

Abstract
Intravascular administration of plasminogen activators is a clinically important thrombolytic strategy to treat occlusive vascular conditions. A major issue with this strategy is the systemic off-target drug action, which affects hemostatic capabilities and causes substantial hemorrhagic risks. This issue can be potentially resolved by designing technologies that allow thrombus-targeted delivery and site-specific action of thrombolytic drugs. To this end, leveraging a liposomal platform, we have developed platelet microparticle (PMP)-inspired nanovesicles (PMINs), that can protect encapsulated thrombolytic drugs in circulation to prevent off-target uptake and action, anchor actively onto thrombus via PMP-relevant molecular mechanisms and allow drug release via thrombus-relevant enzymatic trigger. Specifically, the PMINs can anchor onto thrombus via heteromultivalent ligand-mediated binding to active platelet integrin GPIIb-IIIa and P-selectin, and release the thrombolytic payload due to vesicle destabilization triggered by clot-relevant enzyme phospholipase-A2. Here we report on the evaluation of clot-targeting efficacy, lipase-triggered drug release and resultant thrombolytic capability of the PMINs in vitro, and subsequently demonstrate that intravenous delivery of thrombolytic-loaded PMINs can render targeted fibrinolysis without affecting systemic hemostasis, in vivo, in a carotid artery thrombosis model in mice. Our studies establish significant promise of the PMIN technology for safe and site-targeted nanomedicine therapies in the vascular compartment.
Keywords: Targeted thrombolysis, Nanomedicine, Platelet-derived microparticle, Lipid vesicle, Drug delivery, Enzyme-triggered release
1. Introduction
Vascular pathologies like myocardial infarction, stroke and peripheral arterial disease are major causes of morbidities and mortalities on a global scale [1]. A common clinical presentation in such disease pathologies is the formation of occlusive clots (thrombi) in blood vessels, that restrict blood flow to critical organs [2]. Therefore, rapid removal of occlusive thrombi to restore blood flow is a critical component of treating these conditions. One established clinical strategy for clot removal is the intravascular administration of thrombolytic (fibrinolytic) drugs [3]. These drugs, e.g. streptokinase (SK), urokinase-type plasminogen activators (uPA) and tissue plasminogen activators (tPA), act by facilitating conversion of plasminogen to plasmin, which in turn can break down the fibrin in the clot. While this fibrinolytic action is essential for therapeutic activity at the clot site, systemic off-target action of these drugs to convert circulating plasminogen to plasmin is harmful because this circulating plasmin can then break down circulating fibrinogen (systemic fibrinogenolysis), which affects normal hemostatic capabilities and leads to hemorrhagic side-effects [4,5]. According to the American Academy of Emergency Medicine (AAEM) and National Institute of Neurological Disorders and Stroke (NINDS), about 6% of stroke patients undergoing tPA-based thrombolytic therapy suffer from intracranial hemorrhage with about 45% fatality risk [6]. Another issue with the direct intravascular administration of thrombolytic agents is their rapid deactivation by plasma components (e.g. by plasminogen activator inhibitors) and resultant short circulation life, that in turn reduces their availability at the clot site [7]. All these issues can be potentially mitigated by designing technologies that can (i) encapsulate and protect the drug in circulation, (ii) anchor actively onto clot site under hemodynamic flow environment and (iii) allow triggered release of the drug specifically at the clot site to minimize off-target effects.
In consideration of these design criteria, we have developed a vascular nanomedicine technology inspired by platelet-derived microparticles (PMPs). PMPs, originally reported by Wolf in the 1960s as ‘platelet dust’, are membrane fragments shed from activated platelets [8]. These PMPs are characteristically known to be lipid bilayer vesicles 100nm-1μ in diameter, with a high surface-presentation of pro-coagulant anionic phosphatidylserine (PS) lipid, active integrin GPIIb-IIIa, P-selectin, GPIb-type receptors, thrombospondin, C-X-C type chemokine receptors and thrombin receptors [9,10]. Fig. 1A shows representative cartoon of a PMP, with (A1) showing fluorescence microscopy image of red fluorescent active platelets (shown with blue arrows) shedding PMPs (shown with yellow arrows) and (A2) showing representative scanning electron microscopy (SEM) images of the same at high resolution, demonstrating that the PMPs are sub-micron size vesicular structures. The PS-rich surface of PMPs facilitate intrinsic coagulation mechanisms, while the presence of active integrin GPIIb-IIIa and P-selectin moieties facilitate binding interactions with fibrinogen (Fg), fibrin, stimulated GPIIb-IIIa motifs on active platelets via fibrinogen-mediated interactions and P-selectin Glycoprotein Ligand-1 (PSGL-1) on leukocytes and active platelets (schematic shown in Supplementary Figure S1). Also, PMPs secrete several chemokines, cytokines, pro-thrombotic and pro-inflammatory molecules in the clot milieu [9–12]. Therefore, PMPs essentially represent (i) lipid vesicle nano-containers loaded with bioactive molecules, that can (ii) actively bind to thrombus-associated cellular phenotypes (e.g. platelets and leukocytes) via heterotypic ligand-receptor interactions, and (iii) secrete their payload locally to influence the thrombus pathology. Drawing inspiration from these structural and mechanistic aspects of PMPs (but not their pro-thrombotic functional aspect), we have chosen to construct (i) liposomal nanovesicles with non-coagulant lipid membrane, that can (ii) undergo active anchorage to platelet-rich thrombi via PMP-inspired heteromultivalent ligand-receptor interactions, and (iii) upon active anchorage to thrombus, can release encapsulated thrombolytic drug to render site-specific thrombolytic action. We rationalized that these PMP-inspired nanovesicles (PMIN) will enhance therapeutic availability at the clot site, while protecting the encapsulated drug from plasma and minimizing systemic off-target side effects.
Fig. 1.
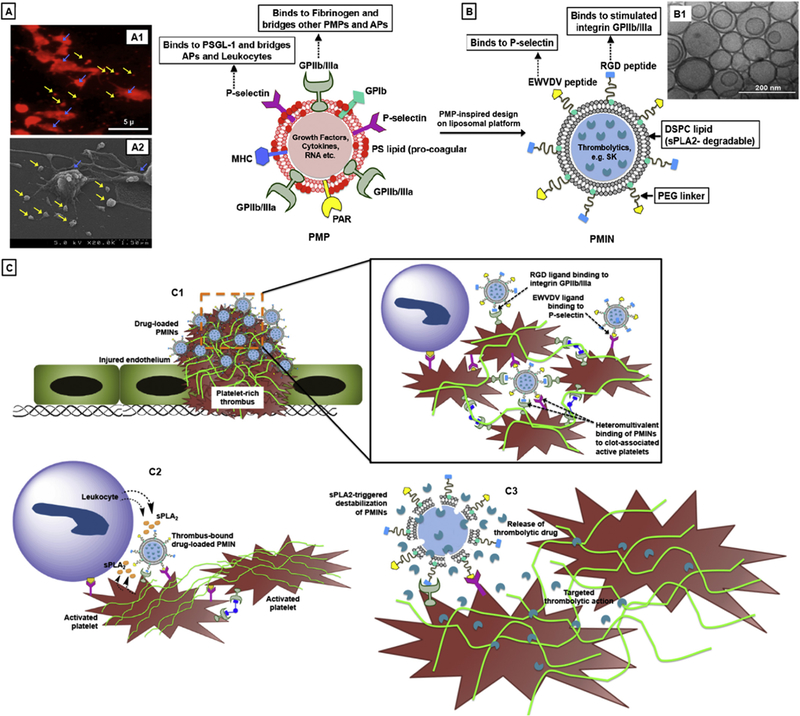
[A] Schematic representation of platelet-derived microparticles (PMP) showing characteristic surface entities, with (A1) showing representative red fluorescence image of PE-anti-CD62P stained active platelets (stained for P-selectin, shown with blue arrows) shedding PMPs (shown with yellow arrows, and (A2) showing representative high resolution SEM image of active platelet shedding microparticle, with PMPs (shown with yellow arrows) visible as sub-micron vesicular structures; [B] Schematic representation of PMP-inspired nanovesicle (PMIN), with (B1) showing representative cryo-TEM image of PMINs developed for the studies; [C] Envisioned mechanism of targeted thrombolytic action using PMINs, where (C1) PMINs can actively anchor onto platelet-rich thrombi by virtue of heteromultivalent binding to integrin GPIIb-IIIa and P-selectin on active platelets, (C2) clot-bound PMINs get acted upon by sPLA2 enzymes secreted from leukocytes and active platelets in the thrombus milieu and (C3) drug released from degraded PMINs renders site-specific fibrinolysis.
For the PMIN design (Fig. 1B), the clot-specific active anchorage was rendered by heteromultivalently surface-decorating glycer-ophospholipid-based liposomal vesicles with peptide ligands that can specifically bind to stimulated integrin GPIIb/IIIa and P-selectin on activated platelets. Activated platelets are an ideal cellular target for PMIN binding to thrombi, since aggregation of activated platelets and platelet-mediated promotion of coagulation mechanisms are hallmark events in thrombosis [13]. Specifically, the fibrinogen-derived peptide sequence GSSSGRGDSPA was used for active GPIIb-IIIa-binding and the sequence DAEWVDVS was used for P-selectin binding [14,15]. For thrombolytic drug encapsulation, we selected streptokinase (SK) as a model fibrinolytic drug, especially since its direct systemic use is known to cause significant off-target hemorrhagic side-effects (and therefore will be a good control to compare). For clot-relevant stimulus, we selected secreted phospholipase A2 (sPLA2, group II) as a candidate, since it is reported to be produced from activated platelets and inflammatory cells in athero-thrombotic milieu and can cleave the sn-2 ester bonds in glycerophospholipids, thereby destabilizing lipid vesicles for payload release [16–18]. In fact, PLA2 action can result in release of arachidonic acid from PMPs to augment transactivation and aggregation of platelets in thrombosis [9]. Building on these design components, our central hypothesis was that intravenously administered SK-loaded PMINs can actively anchor onto thrombi (Fig. 1C, C1), where thrombus-associated sPLA2 activity can destabilize the vesicles to render clot site-selective SK release for targeted fibrinolytic action (Fig. 1C, C2 and C3).
2. Materials and methods
2.1. Materials
Phosphate Buffered Saline (PBS), 3.8% w/v sodium citrate, Bovine Serum Albumin (BSA), chloroform, methanol, ethanol, AlexaFluor 488-conjugated fibrinogen (AF488-Fg) and 1,2-Bis-BODIPY®FL-C11-sn-Glycero-3-Phosphocholine (bis-BODIPY®FL C11-PC) were obtained from Thermo Fisher Scientific (Pittsburgh, PA, USA). Cholesterol, secreted phospholipase A2 (sPLA2), calcium chloride and collagen were obtained from Sigma Aldrich (St. Louis, MO, USA). Adenosine Diphosphate (ADP) was purchased from Bio/ Data Corporation (Horsham, PA, USA). Cysteine-terminated peptides CGSSSGRGDSPA and CDAEWVDVS were custom-synthesized and purchased from Genscript (Piscataway, NJ, USA). Distearyl phosphatidyl choline (DSPC), maleimide-terminated polyethylene glycol-conjugated distearyl phosphatidyl ethanolamine (DSPE-PEG2000-Mal) and Rhodamine-B-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (DHPE-RhB) were obtained from Avanti Polar Lipids (Alabaster, AL, USA). Polycarbonate membrane filters with 200 nm pores for vesicle extrusion were obtained from Whatman (Kent, UK). For streptokinase analysis the chromogenic assay Chromogemix S-2251 was purchased from Diapharma (West Cjester, OH, USA). The Parallel Plate Flow Chamber (PPFC) system was purchased from Glycotech (Gaithersburg, MD, USA). Aggregometry studies were done on a ChronoLog aggregometer. All inverted fluorescence microscope studies were carried out using a Zeiss Axio Observer D1 microscope fitted with a CCD camera. For in vivo studies on mice, anesthesia agents ketamine was obtained from Fort Dodge Animal Health, IA, USA and xylazine was obtained from Hospira, IL, USA. Rhodamine 6G for in vivo mouse platelet labeling and ferric chloride (FeCl3) for carotid artery thrombus induction were obtained from Sigma. All intravital microscopy studies were carried out using a Leica DMLFS fluorescent microscope with a Gibraltar Platform (EXFO, Quebec, Canada).
2.2. Preparation of heteromultivalently ligand-decorated PMINs
The peptides CGSSSGRGDSPA and CDAEWVDVS were conjugated via thioether reaction to DSPE-PEG2000-Mal through the cysteine termini to form DSPE-PEG2000-GSSSGRGDSPA and DSPE-PEG2000-DAEWVDVS conjugates. The peptides and resulting DSPE-PEG-peptide conjugates were characterized by mass spectrometry. For PPFC-based in vitro platelet-rich clot-targeting studies DSPC, DhPE-RhB, cholesterol and DSPE-PEG-peptide conjugates were dissolved in 1:1 chloroform: methanol, and this lipid mixture was subjected to standard reverse phase evaporation and lipid hydration technique followed by extrusion through 200 nm pore size polycarbonate membrane in a pneumatically controlled lipid extruder (Northern Lipids). Resultant vesicles were characterized for their size distribution using dynamic light scattering (DLS) and cryo transmission electron microscopy (cryoTEM) and the size was found to be approximately 150–170 nm in diameter. For the various vesicle formation, DSPC content was always maintained at 50 mol% and DHPE-RhB content was always maintained at 1 mol% of total lipid. For homomultivalently decorated vesicles (single peptide decoration), the corresponding DSPE-PEG-peptide content was maintained at 2.5, 5, or 10 mol % of total lipid, while for hetero-multivalently decorated vesicles (bearing both peptides) the total DSPE-PEG-peptide was kept at 5 mol%, with each lipid-peptide conjugate at 2.5 mol%. The remainder of the lipid membrane content was cholesterol, since it is known to interdigitate with lipids to enhance membrane stability of bilayer vesicles [19]. It is to be noted that the heteromultivalent composition was used as a test condition for the current thrombolytic studies, but if needed the heteromultivalent composition can be modulated to vary relative peptide ratios as well as the total peptide content if needed. For analysis of sPLA2-induced vesicle degradation, vesicles were synthesized with adjusting the DSPC component to have DSPC: bis-BODIPY®FL C11-PC at 20:1 ratio in mole% (as per ThermoFisher specification), keeping everything else constant. For SK encapsulation and release studies, 25kU/ml of SK was dissolved in the PBS used for lipid film hydration during the vesicle preparation and after extrusion the SK-loaded vesicles were purified from unencapsulated SK using Amicon ultracentrifugal filters on an ultracentrifuge.
2.3. In vitro studies of vesicle binding and retention on active platelet-rich clots
Parallel plate flow chamber (PPFC) studies were done by adapting the methods reported by Kruchten et al. [20], where platelet-rich thrombi was formed by incubating activated platelet-rich plasma (PRP) over Type III collagen-coated circular region on glass slides. For this, acid washed glass microscope slides were incubated for 1 h with suspensions of BSA and type III collagen in two adjacent circular areas respectively delineated by 8 mm diameter O-rings (schematic shown in Fig. 3A). The suspensions were gently washed off with PBS and the coated areas were incubated with 200 μl of platelet-rich plasma (PRP) containing ADP and Ca++. The active platelets in PRP adhered spontaneously to the collagen-coated region and simulated a platelet-rich clot but not to albumin-coated region (confirmed by SEM shown in Fig. 3A). Please note that since the incubations are done with PRP, the platelet-rich collagen-coated surface shows dense presence of central bodies of adjacent activated platelets along with fibrin strands and individual ‘spread platelet’ pseudopodal morphology is not clearly visible. Similar incubation done with ‘washed platelets’ only (instead of PRP) allows confirmation of activated ‘spread platelet’ pseudopodal morphologies as shown in Supplementary Figure S2. The PRP was gently washed off with PBS, the slides were fixed with 4% paraformaldehyde, vacuum sealed within a parallel plate flow chamber system placed under an inverted fluorescence microscope (Zeiss AxioObserver.D1) and were exposed to the flow of the RhB-labeled vesicles (homomultivalently decorated, heteromultivalently decorated, or unmodified) at various flow rates producing wall shear rates of300–4000 sec–1 in a recirculating loop for 30 min. This was followed by flow of PBS only (at the same flow rates and hence shear values) for an additional 15 min in an open loop to remove any loosely bound vesicles. Thus, imaging up to 30 min time point allowed analysis of ‘binding’ and following that at the 45min time point allowed analysis of ‘retention’ of vesicles on platelet-rich versus platelet-poor surfaces. Images at various time points were analyzed for surface-averaged RhB fluorescence intensity (as a quantitative measure of vesicle binding and retention), using the Zeiss AxioVision software. For the current studies, experiments and analyses were done for homomultivalent decorations of 2.5 mol%, 5 mol% and 10 mol% peptide compositions compared with heteromultivalent peptide composition of 5 mol% (2.5 mol% of each peptide). Additional PPFC experiments were also done by varying the relative ratios of peptides while keeping the total heteromultivalent peptide composition at 5 mol%.
Fig. 3.
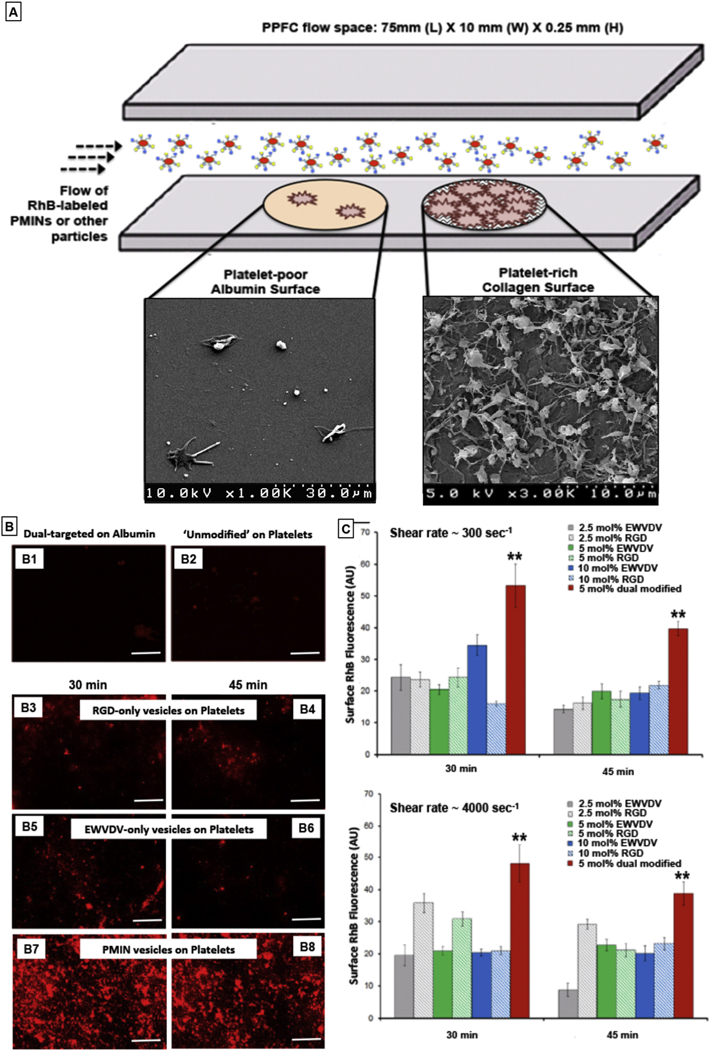
[A] Experimental set-up schematic of parallel plate flow chamber (PPFC) where platelet-poor albumin-coated regions (control surface) and active platelet-rich thrombus region (on collagen-coated area) were created and unmodified RhB-labeled (red fluorescent) unmodified vesicles or singly modified vesicles (bearing RGD decorations or EWVDV decorations only) or dual modified PMINs (bearing both peptide decorations) were flowed over these surfaces at various flow rates (low-to-high shear rates); [B] Representative fluorescent images of particle binding shows that (B1) dual-targeted particles (i.e. PMINs) have minimal binding on albumin-coated surface and (B2) unmodified particles have minimal binding on the platelet-rich thrombus surface; (B3, B4) RGD-decorated vesicles and (B5, B6) EWVDV-decorated vesicles have reasonable extent of binding and retention on the platelet-rich thrombus surface, but the level of binding and retention levels are significantly enhanced for (B7, B8) dual modified PMINs; [C] Quantitative analysis of vesicle binding and retention (based on surface-averaged RhB fluorescence intensity) from multiple batches of experiments show that irrespective of flow conditions (low or high shear rate), dual modified vesicles (PMINs) have significantly higher binding and retention capabilities compared to singly modified vesicles even when the mol% composition of single peptide modification is to twice (10 mol%) that of dual peptide modification (5 mol%).
2.4. Characterization of sPLA2-induced vesicle degradation and payload (SK) release
The ability of sPLA2 to degrade and destabilize the PMINs for payload release was characterized in vitro by incorporating fluorescently labeled (BODIPY-labeled) lipid within the vesicle membrane and monitoring the release of BODIPY over time under lipase action. The bulk of the lipid in the vesicle membrane was DSPC, which is a known substrate for sPLA2 [21]. An analogous BODIPY-labeled phosphatidylcholine, bis-BODIPY®FL C11-PC, (Excitation: 488 nm, Emission: 530 nm) was incorporated at 5 mol% along with DSPC in the vesicle fabrication. In this assay, the BODIPY is self-quenched initially in the intact membrane, but cleavage of the BODIPY-labeled membrane lipid by sPLA2 results in release of BODIPY and corresponding increase in emitted fluorescence signal. Bis-BODIPY® FL C11-PC-labeled PMINs thus prepared were incubated in the presence of sPLA2 (final concentration 1 μg/ml) in 450 μl assay volume per well in 96-well plate and resultant fluorescence signal from released BODIPY (due to membrane degradation by sPLA2) was monitored over time using a fluorescence platelet reader. Next, to determine whether such lipase-induced vesicle degradation leads to payload release, model thrombolytic drug streptokinase (SK) was encapsulated within similarly prepared PMINs and incubated in absence versus in presence of sPLA2. For this, SK was loaded within the vesicles using the RPEE technique with 25kU/ml SK in PBS to reconstitute the lipid film and free SK was removed using ultracentrifugation. Encapsulation efficiency was evaluated by disrupting the vesicles with Triton-X and exhaustively releasing the encapsulated SK, which was analyzed spectrometrically (on a UV–Vis platelet reader) using Chromogenix S-2251 assay. For this, the SK-loaded vesicles were suspended in 5 mM Tris-HCl + 1 mM CaCl2 buffer in microcentrifuge tubes in the presence or absence of 2.5 ng/ml sPLA2 for 0–5 h on a gyratory shaker (60 rpm) at 37 °C, and 100 μl of the suspension was aliquoted at various time points to measure the released SK by the S-2251 assay. In this assay, released SK converts the plasminogen reagent to plasmin, which in turn acts on the chromogenic substrate reagent to liberate p-nitroaniline (pNA) that is measured spectrophotometrically at 405 nm. This signal is in linear calibration with respect to active SK concentration, and hence the absorbance signal represents released SK concentration.
2.5. Evaluation of thrombolytic capacity of vesicles in vitro
SK-loaded Rh-B-labeled (red fluorescent) heteromultivalently decorated (2.5 mol% of each peptide at total 5 mol%) PMINs were prepared as described previously. Glass microscope slides were incubated with collagen suspension in a circular region for 30 min (similar to slide preparation for PPFC-based binding studies) and then gently washed with 1 × PBS. PRP with AlexaFluor 488-conjugated fibrinogen (6% v/v of 1.5 mg/mL, green fluorescent) was briefly mixed with α-thrombin and 0.5 M CaCl2, to final concentrations of 5 nM and 25 μM respectively, to initiate clot formation and the mixture was pipetted (100 μl volume) onto the collagen-coated surface region. The clot was allowed to incubate for 1 h and then gently washed with 1 × PBS to remove loosely bound components. The slide thus formed with platelet-rich green fluorescent fibrin clot on its surface, was sealed into the parallel plate flow chamber (PPFC) system, to be imaged under the inverted fluorescence microscope. The objective was focused on an area at the edge of the clot such that the initial field of view enabled observation of the green fluorescence of the fibrin clot throughout the whole area of view, and the clot lysis could then be observed as the loss of this fluorescence. SK-loaded RhB-labeled hetero-multivalently platelet-targeted or untargeted PMINs in platelet-poor plasma (final concentration of 2.1 × 1011 particles/mL) were flowed along with sPLA2 (2.5 ng/ml) over the clot at 300 sec–1 shear rate and the clot (stability or lysis) was imaged (one image per minute) over a period of 60 min with a 10 × objective. In this experimental set-up, sPLA2-triggered degradation of clot-bound PMIN vesicles and resultant SK release was expected to convert the plasminogen to plasmin and thereby lyse the fibrin of the clot. This mechanism would result in reduction of red fluorescence (degradation of clot-bound PMINs) and green fluorescence (lysis of fibrin in clot), as depicted in Fig. 5A, imaged over time under inverted fluorescence microscope.
Fig. 5.
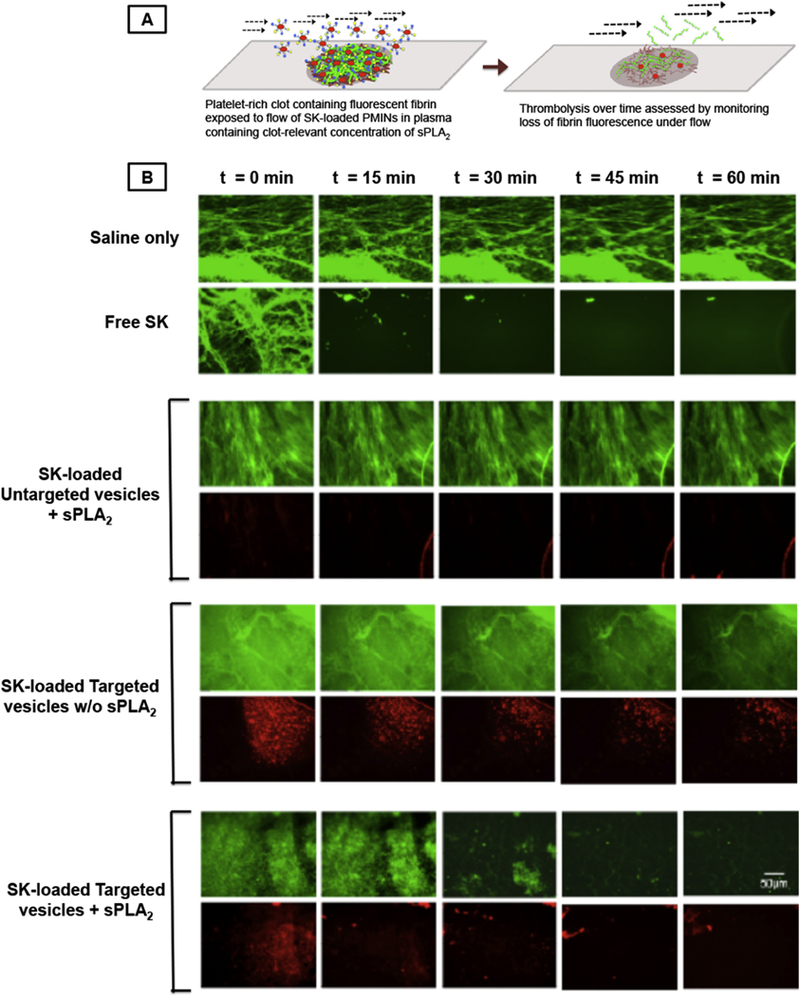
[A] In vitro experimental set-up where SK-loaded RhB-labeled (red fluorescent) clot-targeted PMINs (or untargeted vesicles) were flowed over platelet-rich clots containing green fluorescent fibrin, in presence (or absence) of clot-relevant concentration of sPLA2, and clot lysis was monitored by imaging fate of vesicle (red) and clot fluorescence (green) over time; [B] representative images where ‘saline only’ was unable to affect clot fluorescence while free SK caused substantial loss of clot fluorescence (hence thrombolysis); SK-loaded targeted PMINs in presence of sPLA2 showed similar clot lysis along with vesicle degradation, compared to the control conditions (SK-loaded targeted vesicles w/o sPLA2 or untargeted vesicles with sPLA2).
2.6. Vesicle binding to mouse carotid artery thrombus in vivo
The mouse model experiments were carried out in accordance to Cleveland Clinic Foundation IACUC-approved protocols, using ferric chloride (FeCl3)-induced vascular injury and thrombosis model in mouse [22]. For this, 8–12 week old male C57BL6 mice were anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg) via intraperitoneal injection, and anesthesia was confirmed by toe clipping. The right common carotid artery (CA) was exposed. A 1 × 2 mm piece of Whatman #1 filter paper was saturated with 7.5% FeCl3 solution and placed directly on the CA for ~1 min (Fig. 6A). In our experience, this method produces a thrombus that occludes the vessel ~50% in 5 min and almost full occlusion around 11–12 min after injury. Representative SEM images of substantial platelet-rich clot formation on the luminal side of the artery upon such vascular injury is shown supplementary Figure S6 where progressive magnification scales show high accumulation of platelets upon vessel injury (S.6. B and C) and fibrin generation on individual platelets (S.6.D). RhB-labeled (red fluorescent) non-targeted (unmodified) vesicles or platelet-targeted (heteromultivalently decorated) PMINs in saline were then injected at 30 mg/kg in the right jugular vein and vesicle interaction with thrombus was imaged in real time in the anesthetized mice using a Leica intravital microscope. In parallel studies, similarly treated mice were euthanized with an overdose of anesthesia cocktail (IACUC approved protocol), the thrombosed artery was excised, sectioned longitudinally, fixed in PFA, placed on glass microscope slides and imaged with the luminal side facing the objective to observe RhB fluorescence in the thrombus. In additional experiments, FeCl3-induced thrombi were created in the carotid of mice (n = 3) as before, 100 μl RhB-labeled targeted PMIN was injected into the mouse circulation system via the jugular vein catheter, the circulation was maintained for another 10 min in a 37 °C warming chamber and then mice were euthanized. Lung, liver, kidney and spleen as well as the thrombosed carotid artery (right, ~ 2–3 mm length section) and non-thrombosed carotid artery (left, ~ 2–3 mm length section) were harvested, snap-frozen in liquid nitrogen and dried in a lyophilizer. The dry weight of the organs and tissues was recorded. The organs and tissues were then homogenized at 4000 rpm for 2 cycles of 25 s with a 5 s delay using a BeadBug Microtube Homogenizer (Benchmark Scientific, Edison, NJ) with 3.0 mm high impact zirconium beads. Samples were shaken overnight at 750 rpm at 37 °C in a 1:1 solution of methanol/ chloroform to extract RhB-conjugated conjugated lipids. These samples were centrifuged at 12,000 g for 20 min and the supernatant, containing RhB-labeled lipids, was collected. For the thrombosed versus non-thrombosed carotid samples, the overall fluorescence intensities of the processed samples were recorded using a platelet-reader. Assuming that the RhB fluorescence in these samples originate from the anchorage and localization of the PMINs in the carotid, the fluorescence intensities (hence PMIN localization) within thrombosed versus non-thrombosed carotid of the same animal were recorded. For the samples from clearance organs, the RhB-labeled lipids were resolved using Waters Acquity UPLC system (column: Waters BEH C8 1.7 μm) and analyzed with a fluorescence detector (Waters Corporation, Milford, MA, USA) using excitation wavelength 560 nm and emission wavelength of 580 nm. Accordingly, the uptake of PMINs in the clearance organs was determined by calculating the percent (%) injected dose using an RhB-fluorescence based calibration curve for PMINs.
Fig. 6.
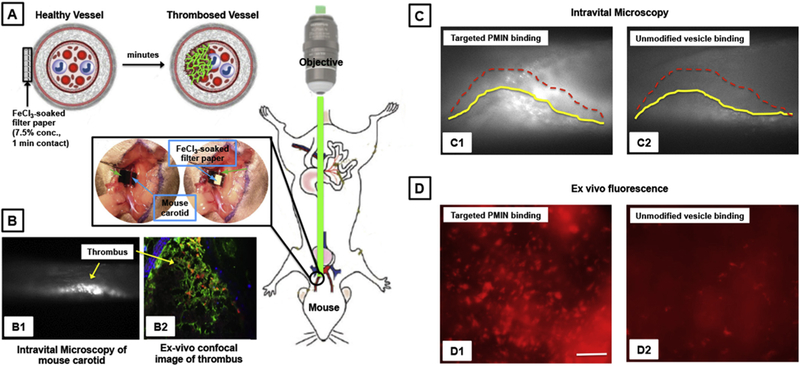
[A] Schematic of experimental set-up and resultant clot formation in the carotid artery of mouse upon application of FeCl3-soaked filter paper on the adventitial side and real-time observation of clot (thrombus) by intra-vital microscopy; [B] Representative image of thrombosed carotid as observed by (B1) intravital microscopy and (B2) ex vivo immunofluorescence; [C] Representative intravital microscopy images of vesicle binding to carotid thrombus upon intravenous (through jugular) administration shows that (C1) active platelet-targeted PMINs can bind to the thrombi at substantially high levels compared to (C2) unmodified vesicles (thrombus surface profile shown by yellow and dotted red lines); [D] Representative ex vivo fluorescence microscopy images of excised arteries show that (D1) targeted PMINs have high level of binding to thrombosed arteries, compared to binding of (D2) unmodified vesicles.
2.7. Targeted thrombolysis in vivo using sPLA2-responsive PMINs
For targeted thrombolytic capacity assessment, Rhodamine 6G solution was first injected into the right jugular vein of the mouse to render direct fluorescent labeling of circulating platelets and the FeCl3-induced thrombus was created in the carotid as before, such that the formation of the thrombus and extent of vessel occlusion can be observed directly by monitoring the fluorescent platelets in the artery with intra-vital microscopy [23]. For this, the carotid artery was thrombosed as before and 100 μl of Rhodamine 6G solution was injected into the right jugular vein that results in direct fluorescent labeling of circulating platelets. This allows for imaging platelet-rich thrombus formation (and lysis) in real time. Free SK, or, non-fluorescent SK-loaded enzyme-responsive non-targeted (unmodified) vesicles, or, non-fluorescent SK-loaded enzyme-responsive PMINs, or non-fluorescent ‘blank’ (i.e. no SK loading) enzyme-responsive PMINs were injected via the jugular catheter, ~5 min after thrombus induction. Free SK suspension, SK-loaded unmodified vesicles, as well as, SK-loaded targeted PMIN vesicles, all contained ~400 IU of SK and suspended in 200 μL saline. The effect of these various treatments on delaying the thrombus growth over time was imaged by intravital microscopy, with images recorded for 10 s every minute for the first 10 min and then 10 s every other minute until the end of the experiment. In this experimental framework the thrombolytic drug is expected to constantly lyse the forming thrombus and hence delay vessel occlusion, when compared to no drug treatment. The end points of the experiment were: 1) when blood flow had ceased for >30 s due to vessel occlusion, or 2) if occlusion was not observed in 30 min after SK (free or vesicle-encapsulated) administration. In our experience with this model, at ~5 min after FeCl3-induced endothelial damage the thrombus occludes about 30–50% of the blood vessel and upon further growth of the occlusive thrombus, numerous larger cells (leukocytes) start to roll on the vessel wall at the proximal site of the thrombus and the blood flow usually stops within 2–3 min of the appearance of these large cells [22]. The thrombolytic effect was quantitatively expressed as ‘time to delay’ for vessel occlusion (blood flow cessation) if vessel was fully occluded before 30 min post-injury or the 30 min data point if vessel occlusion did not occur by 30 min.
2.8. Effect of free SK versus PMIN-encapsulated SK on systemic hemostasis in mice
‘Tail bleeding time’ is a standard method for assessing hemostatic disorders in mouse models and therefore this was used to assess the systemic effect on hemostasis of free SK versus nanovesicle-encapsulated SK administration in mice [24]. For this, 8–12 weeks old C57Bl/6 mice were anesthetized as before with ketamine/xylazine cocktail, and free SK or PMIN-encapsulated SK (400 IU/mouse) in 200 μL saline suspension was injected into the mouse via right jugular vein as before. The mice were kept in 37 °C warming chamber for 30 min. For tail bleeding analysis, 1 cm length of tail from the tail-tip was surgically removed with a sharp scalpel, the transected tail was immediately immersed into warm saline (37 °C) and time for bleeding to stop (designated as ‘bleeding time’) was recorded. In our experience, the normal bleeding time for tail bleeding in this strain of mice is 100–120 s as shown in our revised Fig. 6C. Therefore any drastic variation of this bleeding time range was considered to be a systemic effect of SK on hemostasis (e.g. circulating plasminogen activation and systemic fibrinogenolysis). At the end of experiments, mice were euthanized by overdose of anesthesia.
2.9. Statistical analysis
Statistical analysis of the PPFC-based in vitro binding and retention studies of heteromultivalent versus homomultivalent targeting of nanovesicles on thrombus-relevant platelet-rich surfaces was done using one-way ANOVA with the Tukey Method. For these studies, the quantitative analysis of surface-averaged fluorescence intensity (from platelet-bound particles on the test and control surfaces) was analyzed for multiple images (n = 10 per test condition). While Fig. 3 shows histogram data of these studies, we have added our Mean ± SD data tables in the supplementary information Figure S4. The sPLA2-triggered drug release data was derived from n = 3 studies, and statistical analysis of this was done using a paired student’s T-test between the groups. For the in vivo thrombotic occlusion time delay analysis, since we set up 30 min as one of the end points, the data do not have normal distribution. Therefore, we chose to report these results analyzed around the estimated mean (SE), and not by how much the sample differs from the sample mean (SD). For the tail-bleeding assay we used Mann-Whitney U test as this data is also not in a normal distribution. In all analyses, significance was considered to be p < 0.05.
3. Results
3.1. In vitro evaluation of thrombus-targeting capabilities ofPMINs
The peptides CGSSSGRGDSPA and CDAEWVDVS and the corresponding DSPE-PEG-peptide conjugates were characterized by mass spectrometry as shown in Fig. 2. Representative cryo-TEM image of PMINs vesicles formed by combining DSPE-PEG-peptide molecules with distearyl phosphatidylcholine (DSPC) and cholesterol via self-assembly using the reverse phase evaporation and extrusion (RPEE) technique, is shown in Fig. 1, B1. Glass slides with adjacent circular areas of collagen-coated platelet-rich surface (simulating platelet-rich thrombi) and albumin-coated platelet-poor surface (control surface) were placed within the parallel plate chamber, and Rhodamine B (RhB)-labeled (red fluorescence) un-modified vesicles (no peptide decoration), or PMINs bearing both peptides (heteromultivalent at 1:1 ratio of 2.5 mol% each with respect to total lipid), or either peptide (homomultivalent at 2.5, 5 or 10 mol% with respect to total lipid) were flowed over the slides (schematic in Fig. 3A). Vesicle binding (RhB fluorescence) and retention was imaged under an inverted fluorescence microscope as described in the methods section. Fig. 3B shows representative fluorescence microscopy images of particle binding and retention on platelet-rich (or control) surfaces at high shear rate (4000 sec–1) conditions, while Fig. 3C shows quantitative results of homomultivalent versus heteromultivalent particle binding on platelet-rich surfaces at low (300 sec–1) and high (4000 sec–1) shear flow conditions. Additional results in Supplementary Figure S3 show representative fluorescence images of vesicle binding (30 min time point) and retention (45 min time point) at low (300 sec–1), medium (2000 sec–1) and high (4000 sec–1) shear rate conditions, for vesicles decorated homomultivalently (2.5 mol% or 5 mol% or 10 mol% single peptide) compared with PMINs bearing 5 mol% heteromultivalent decoration, along with the quantitative data from these studies for the mid-range (2000 sec–1) shear rate condition. The corresponding Mean ± SD data sets based on which such histograms of Fig. 3B and Figure S3 were prepared, are shown in Supplementary Figure S4. As evident from these results, homo-multivalently decorated (singly modified) vesicles (bearing RGD-peptide only or EWVDV-peptide only) were able to substantially bind to activated platelet-rich surface and stay retained under flow conditions (B3: binding and B4: retention for RGD-decorated vesicles, B5: binding and B6: retention for EWVDV-decorated vesicles), but this capability was tremendously enhanced when the peptides decorations were combined on dual modified PMINs, i.e. heteromultivalent decoration (B7: binding, B8: retention). Supplementary Figure S5 shows representative images of particle fluorescence and corresponding platelet-rich surface brightfield images in the same field of view, for single targeted (homomultivalent) versus dual targeted (heteromultivalent) binding, confirming that the fluorescence indeed is from platelet-bound particles and that dual targeted particles have a significantly higher binding/retention capability on active platelets under a flow environment. This enhanced platelet-binding capability of heteromultivalently decorated PMINs was evident both at low (300 sec–1) and high (4000 sec–1) shear conditions even when the homomultivalent peptide decoration was increased to twice (10 mol%) that of heteromultivalent (5 mol% total) (quantitative histogram data in Fig. 3C and Mean ± SD intensity analysis data in Supplementary Figure S4.). Related studies also indicated that within fixed total heteromultivalent modification (e.g. 5 mol% total peptide incorporation), modulating relative peptide ratios (80:20, 60:40, 50:50, 40:60, 20:80) may have some effect on binding, depending on the shear rate (representative fluorescence images and quantitative data shown in Supplementary Figure S6.), but all vesicles with various ratios of heteromultivalent decoration compositions are capable of binding and retention on active platelet-coated surfaces substantially more than their homomultivalent (100:0 or 0:100) counterpart. As for the controls, unmodified vesicles showed minimal non-specific binding to platelet-rich collagen surface, as did the heteromultivalently decorated PMINs on platelet-poor albumin surface (Fig. 3B, B1 and B2). These results further confirm that combining PMP-inspired heteromultivalent platelet-binding mechanisms significantly enhances clot-binding specificity and clot-specific retention of PMINs under low-to-high shear flow environment.
Fig. 2.
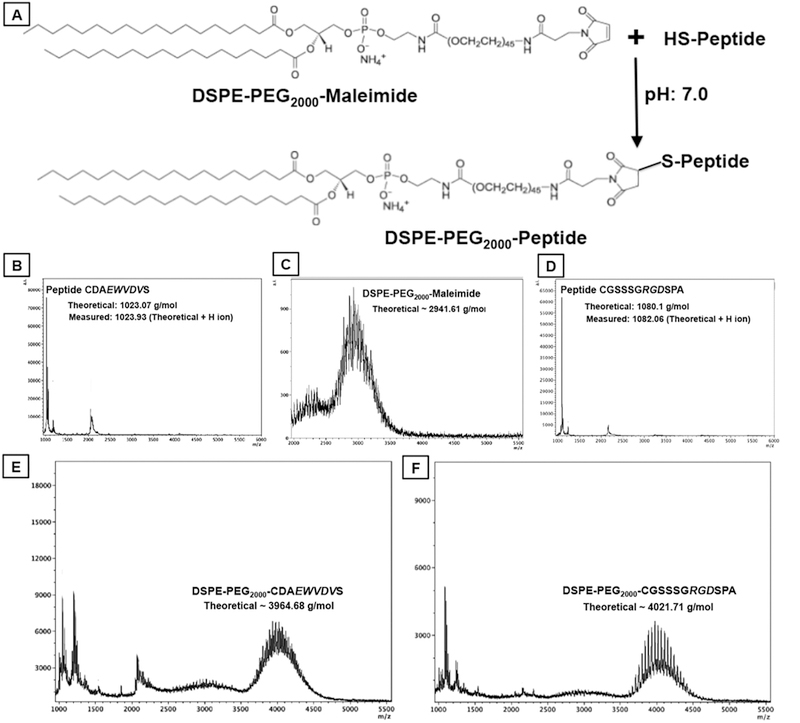
[A] shows reaction schematic for conjugating Cysteine-terminated peptides to DSPE-PEG2000-Maleimide via thioether linkage to form DSPE-PEG2000-peptide molecules; [B]-to-[F] shows representative mass spectrometry data for peptides, DSPE-PEG2000-Maleimide and final DSPE-PEG-peptide conjugates.
3.2. Characterization of lipase-triggered vesicle degradation and payload release from PMINs
The degradation mechanism of bis-BODIPY®FL C11-PC lipid by sPAL2 is the same as that for DSPC lipid as shown in Fig. 4A and therefore the increase in BODIPY fluorescence emission is reflective of PMIN vesicle membrane degradation via sn-2 ester cleavage of DSPC by sPLA2. As shown in Fig. 4B, incubation of BODIPY-labeled PMINs with sPLA2 resulted in substantial enhancement of BOD-IPY fluorescence emission within 30 min period, indicating sub-stantial membrane degradation of the PMINs by the enzyme. Streptokinase (SK) was encapsulated in these PMINs as described in methods, the vesicles were purified from free (un-encapsulated) SK by using Amicon ultracentrifugal filters and the isolated vesicles were treated with Triton X to exhaustively release the loaded SK. The SK thus released was quantified using an SK-specific chromo-genic assay (S-2251, see Methods and Supplementary Figure S7.) and encapsulation efficiency (EE) of SK for various batches of PMIN preparation was thus assessed. As shown in Fig. 4C, the EE for SK in the PMINs was found to be approximately 40% for the various batches. Following this, temporal release of SK from the PMINs with or without sPLA2 exposure was monitored over 10 h. Exposure of SK-loaded PMINs to sPLA2 resulted in significant enhancement of the SK release levels, compared to that without sPLA2 exposure (Fig. 4D). Within the first 2 h, the percent (%) release of SK from the PMINs was about 4 times more with sPLA2 exposure than without the enzyme exposure. Altogether, these results establish the capability of the PMINs to be amenable to clot-relevant enzyme-responsive membrane destabilization and resultant release of encapsulated drug payload.
Fig. 4.
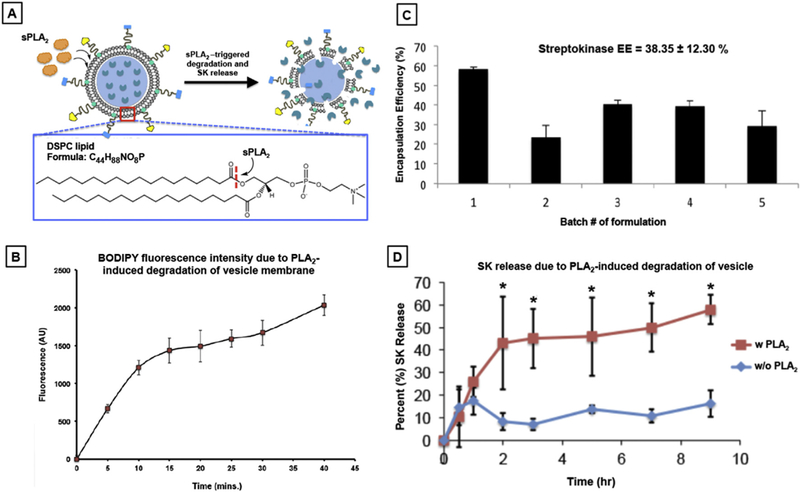
[A] Schematic mechanism of sPLA2-induced membrane degradation due to cleavage of sn-2 acyl group of the phosphatidyl choline lipids; [B] Vesicle membrane degradation kinetics as monitored by measuring the increase in BODIPY fluorescence over time due to cleavage of the BODIPY moiety from the bis-BODIPY®FL C11-PC due to sPLA2 action; [C] Encapsulation efficiency (EE) assessment for streptokinase (SK) encapsulation various batches of PMINs show an average EE of ~38%; [D] Release kinetics assessment of SK from PMINs with or without sPLA2 incubation shows that upon sPLA2 exposure the percent (%) release of SK from PMINs is significantly enhanced (~4 fold) compared passive release (principally via diffusion) without sPLA2 exposure.
3.3. In vitro evaluation of targeted thrombolytic capacity of PMINs
The synergistic validation of platelet-rich thrombus anchorage capability and the lipase-triggered thrombolytic drug release capability of the PMINs, led to the integration of these capabilities to create SK-loaded clot-targeted enzyme-responsive PMINs and their evaluation for targeted thrombolytic (fibrinolytic) capability in vitro. For this, platelet-rich thrombi was created on collagen-coated circular regions on glass microscope slides as before but with PRP containing AlexaFluor 488-labeled (green fluorescent) fibrinogen (6% v/v of 1.5 mg/mL), such that the platelet-rich fibrin clot on the collagen-coated region could be visualized for green fluorescence under an inverted fluorescence microscope (see Methods for details). SK-loaded, platelet-targeted (or untargeted) RhB-labeled (red fluorescent) PMINs (or control vesicles) were flowed over these green fluorescent clots in the PPFC system in a closed loop along with sPLA2, and the vesicles and clots were imaged under an inverted fluorescence microscope. Fig. 5A shows the schematic of this set-up where the fibrinolytic effect was assessed by imaging the green fluorescent clot with red fluorescent vesicles binding to it and monitoring the loss of green fluorescence (indicating fibrinolysis) and red fluorescence (indicating vesicle degradation) in the field of view. Fig. 5B shows representative fluorescent images from these studies captured over 60 min flow period for the groups ‘Saline only’ treatment, ‘Free SK’ treatment, ‘SK-loaded untargeted vesicles + sPLA2’, ‘SK-loaded thrombus-targeted vesicles (PMINs) without sPLA2’, and ‘SK-loaded thrombus-targeted PMINs with sPLA2’. Please note that for the first two groups, representative images have only clot fluorescence (green) since no red fluorescent particles are involved in these two groups of studies. For the next three groups, since untargeted or targeted vesicles are involved in the studies, the top row shows the clot (green) fluorescence while the bottom row shows the particle (red) fluorescence in the same field of view. Also please note that the fibrin morphology (shown by green fluorescence) has some variability between the various groups compared, but the starting condition in all groups shows a green fluorescent dense fibrin-covered surface. As evident from the images, free saline is unable to cause any clot lysis (no change in green clot fluorescence) while free SK effectively lyses clots within 15 min of flow (significant loss of green fluorescence). For the vesicle treated groups, SK-loaded untargeted vesicles are unable to bind to the clot and therefore unable to release any SK in presence of sPLA2, as evident from minimal loss of green clot fluorescence and minimal presence of bound red vesicles over the various time points spanning 60 min. For SK-loaded targeted PMINs, the vesicles are able to substantially bind to the clot as shown by the initial red fluorescence of the bound vesicles on the green clots in corresponding fields of view. However, without the presence of sPLA2, these vesicles do not degrade and release adequate amount of SK for clot lysis, as reflected by minimal loss of green fluorescence of clots (and red fluorescence of vesicles) over time. Some reduction in fluorescence is seen at longer time points (at 45 min and beyond), possibly because of low levels of vesicle destabilization and release of SK. In contrast, in presence of sPLA2, the SK-loaded thrombus-targeted PMINs degrade and release SK to render substantial thrombolysis, as evident from progressive decrease in green clot fluorescence (reflecting fibrin lysis) and red vesicle fluorescence (reflecting vesicle degradation). Corresponding representative videos for these experiments are shown in Supplementary Info Movies S1-S5. These studies confirm that the SK-loaded thrombus-targeted PMINs are capable of targeted anchorage to platelet-rich clots and release SK triggered by sPLA2-induced vesicle degradation for substantial clot lysis to levels comparable to free SK action.
3.4. In vivo evaluation of clot-targeted binding capacity of PMINs
Before carrying out binding studies in vivo, the effect of the peptides on quiescent mouse platelets were evaluated by adding them at concentrations equivalent to that used for PMINs to murine washed platelets with or without platelet agonist (ADP or collagen) and assessing the effect of the peptides on resting versus activated platelets via turbidimetric measurement on an aggregometer. As shown in supplementary Figure S8, the peptides were found to have no effect on quiescent platelets without agonist and the platelet aggregation was found to occur only when agonist was added. This indicated that the peptides (and hence peptide-decorated vesicles) themselves were not capable of activating and aggregating quiescent platelets, which can otherwise lead to systemic pro-thrombotic risks. The ferric chloride (FeCl3)-induced carotid artery thrombosis model, viewed under intravital microscopy, was used to evaluate the binding of PMINs in vivo (setup shown in Fig. 6A). This model results in oxidative damage of arterial wall, leading to rapid development of platelet-rich occlusive thrombus, as confirmed by intra-vital microscopy (Fig. 6B, Bl), as well as, by immunofluorescence-based histology of excised thrombosed artery (Fig. 6B, B2) and by scanning electron microscopy (supplementary Figure S9). In this model, action of fibrinolytic drug can inhibit clot formation and thereby delay the vessel occlusion, which is considered as a measure of thrombolytic efficacy. First, the ability of the PMINs to bind to the carotid artery thrombus was assessed by creating the thrombi in mouse carotid with non-fluorescent platelets, injecting RhB-labeled (red fluorescent) PMINs or unmodified vesicles intravenously (through the jugular) ~5 min after thrombus induction on the carotid, and imaging the binding of the PMINs (versus control unmodified vesicles) onto the platelet-rich thrombi. The studies confirmed that intravenously administered PMINs could actively bind and accumulate on the thrombus at significantly higher levels (Fig. 6C, Cl) compared to unmodified vesicles (Fig. 6C, C2). This was further confirmed by excising the thrombosed artery, sectioning it longitudinally and imaging the luminal surface for vesicle (RhB) fluorescence. Ex vivo images of thrombi exposed to targeted PMINs showed significantly high levels of red fluorescent clusters (Fig. 6D, Dl), compared to that exposed to unmodified vesicles (Fig. 6D, D2), indicating enhanced binding of the RhB-labeled PMINs to thrombus-associated active platelets. In additional experiments, after thrombus induction followed by targeted PMIN injection in the mice, the thrombi was observed for ~10 min under intravital microscopy, the mice were then euthanized, and the thrombosed carotid, non-thrombosed carotid, as well as, clearance organs (liver, lung, spleen and kidney) were excised, processed and analyzed for PMIN fluorescence to analyze percent localization (see Methods for experiment details). As shown in Supplementary Figure S10A, the targeted PMINs showed a significantly high level of localization (higher fluorescence intensity) in the thrombosed carotid compared to the non-thrombosed carotid. Also, during the 15 min circulation period of the experimental time window, only ~20% of the total injected dose were cleared cumulatively in the various clearance organs, with liver and spleen being the major organs of clearance (Figure S10B). This is not surprising considering the fact that the PMINs are built upon a PEG-ylated liposome platform known to have low clearance and long circulation periods [25]. These results therefore suggest that much of the injected PMINs might still be in the vascular compartment of the injected mice.
3.5. In vivo evaluation of targeted thrombolytic capacity of PMINs
For targeted thrombolytic capacity assessment, Rhodamine 6G solution was first injected into the right jugular vein of the mouse to render direct fluorescent labeling of circulating platelets and the FeCl3-induced thrombus was created in the carotid as before, such that the formation of the thrombus and extent of vessel occlusion can be observed directly by monitoring the fluorescent platelets in the artery with intra-vital microscopy. Blank (i.e. without SK-loading) non-fluorescent PMINs or SK-loaded non-fluorescent PMINs or unmodified vesicles or free SK or saline only, was administered via the jugular vein (5 min after FeCl3-induced thrombus induction) to observe thrombolytic effect (delay in thrombus growth and vessel occlusion) at the carotid site, in real time. Fig. 7A shows representative 2 min and 10 min post-injection time-point images for various treatment conditions, while Fig. 7B shows overall quantitative data (4 animals per group) from the various condition studies. Series of images from 2 min to 12 min time points for ‘free SK’ treatment group, ‘SK-loaded targeted PMIN’ treatment group and ‘SK-loaded untargeted PMIN’ treatment group are shown in supplementary Figure S11. As evident from these results, administration of SK-loaded clot-targeted PMINs resulted in significant delay in vessel occlusion, at levels similar to that rendered by free SK administration. In comparison, administration of SK-loaded untargeted vesicles failed to render such delay in vessel occlusion and the vessel appeared majorly occluded by 12 min. These results indicate that the enhanced anchorage of SK-loaded PMINs at the carotid thrombus site enabled enzyme-triggered release of the encapsulated SK to cause targeted thrombolytic action and delay the vessel occlusion, while this was not the case for unmodified (untargeted) SK-loaded vesicles. Furthermore, targeted ‘blank’ PMINs (i.e. without SK loading) did not seem to have any thromboprotective effect by themselves. This is probably because of the fact that the multivalent decoration of platelet-interactive ligands on the PMIN surface possibly allows them to act as ‘bridging’ particles between active platelets, thereby supplementing the thrombotic aggregation. This effect is potentially offset when thrombolytic drug (e.g. SK) is released from the PMINs and hence the delay in vessel occlusion is essentially an effect of targeted delivery and release of PMIN-encapsulated SK. In separate experiments, free SK or PMIN-encapsulated SK was intravenously administered in mice and 30 min post-administration the effect of SK on systemic hemostatic capability (i.e. off-target systemic effect) was assessed by tail-vein bleeding time measurement. Free SK administration resulted in significant prolongation of tail bleeding time (indicating systemic off-target fibrinogenolysis) compared to normal (no treatment) mice, while administration of PMIN-encapsulated SK had minimal effect on bleeding time (Fig. 7C). Altogether these results establish that the SK-loaded enzyme-responsive PMINs can render thrombolysis almost as effectively as free SK but in a targeted fashion, while minimizing off-target effects on systemic hemostatic capabilities.
Fig. 7.
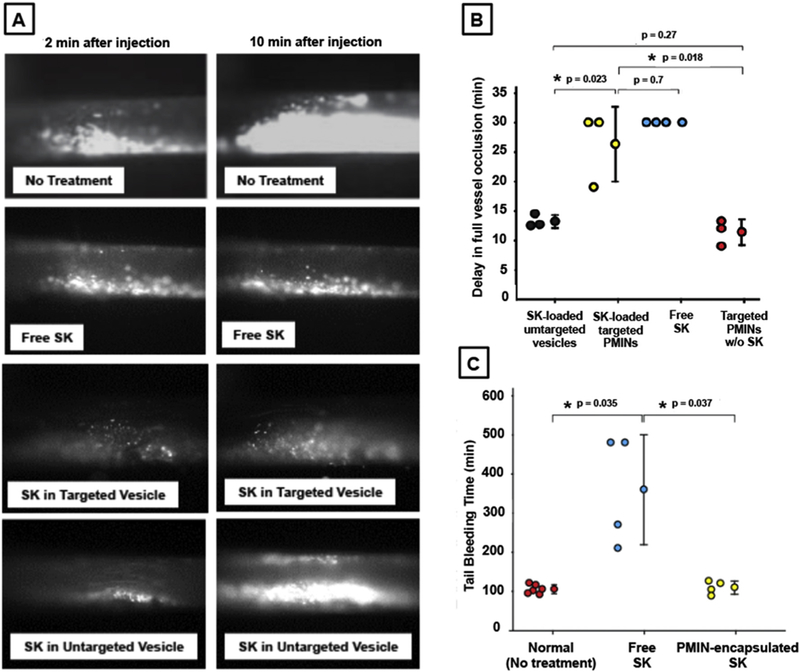
[A] Representative intravital microscopy images (2 min and 10 min time point post treatment injection) of Rhodamine G-labeled platelet-rich thrombi (FeCl3-induced) within mouse carotid after intravenous (through jugular) administration of free SK or SK-loaded targeted PMIN vesicles or SK-loaded unmodified (untargeted) vesicles or no treatment, shows that without any treatment the artery is significantly occluded within 10 min, free SK treatment keeps the artery significantly open due to persistent thrombolysis, treatment with SK-loaded targeted PMINs shows similar thrombolytic effect as free SK, and treatment with SK-loaded untargeted vesicles have much reduced thrombolytic effect; [B] Quantitative results of delay in vessel occlusion emphasizes the fact that SK-loaded targeted PMINs can delay vessel occlusion with an efficacy close to that of free SK, targeted PMINs without SK cannot effectively delay vessel occlusion, and SK-loaded untargeted vesicles are unable to delay vessel occlusion to the level of SK-loaded targeted PMINs; [C] Tail bleeding studies on mouse injected with free SK versus SK-loaded PMINs show that free SK have systemic off-target effect on hemostatic capability (tail bleeding time increased significantly compared to normal ‘no treatment’ mice) while PMIN-encapsulated SK does not have such drastic effect.
4. Discussion
Fibrinolytic therapy using plasminogen-activating agents remains a clinical mainstay in therapeutic intervention of occlusive vascular pathologies [26]. A persistent issue with the direct intravascular administration of plasminogen-activating drugs is their off-target action to cause systemic fibrinogenolysis and thereby severely affect body’s natural hemostatic capabilities to put patients at hemorrhagic risks. In fact, this issue was solely responsible for the abandonment of SK as the thrombolytic drug of choice in the US and UK, and development (and clinical approval) of recombinant tPA as the thrombolytic drug of choice instead, since tPA is known to have relatively higher affinity to clot-associated plasminogen than circulating plasminogen (hence relatively more clot-specific action). Interestingly, the data from the GUSTO clinical trial that compared tPA (under the name Alteplase®) versus Streptokinase (SK) in thrombolytic treatment of acute MI shows that the 30 day mortality benefit for tPA is only about a percentage better than that for SK (6.3% for tPA versus 7.4% for SK) and also showed that there is a significant excess of hemorrhagic strokes for accelerated tPA treatment compared with streptokinase treatment [27]. In addition, the GISSI-2 trial and the ISIS-3 trial did not show any significant benefit of tPA over SK. In these studies, overall mortality was comparable for SK and tPA, with re-infarction rate slightly lower with tPA (2.93%) compared to SK (3.47%), while strokes occurred slightly higher with tPA (1.39%), compared to SK (1.04%). Furthermore, SK still remains a clinically relevant thrombolytic drug in many developing countries, since it is significantly less expensive than the recombinant tPA agents [28]. A recent Cochrane review has also indicated that symptomatic hemorrhagic transformation rates are not significantly higher in SK patients, relative to tPA [29]. These reports have revived clinical interest in SK for treating acute myocardial infarction and stroke, especially in developing countries. To this end, we believe that a nanomedicine platform that allows clot site-selective ‘targeted delivery and release’ can significantly benefit SK-based therapy. Therefore in the current studies, we chose to carry out the design and experiments using SK as a model drug. The comparable clinical performance of tPA and SK in several trials suggests that such a nanomedicine platform can also further improve the therapeutic benefit of tPA-based therapies. Therefore, future studies with this PMIN approach will also be directed towards adapting the system for targeted delivery of tPA since tPA is currently the clinically approved thrombolytic of choice in the US and UK.
In the context of ‘targeted thrombolysis’ approaches, several recent research reports have shown the ability of increasing the systemic circulation safety and lifetime of SK by encapsulating it in microparticles and nanoparticles (including liposomes), as well as, facilitating clot site-specific targeted action of SK, as well as, other thrombolytic drugs like urokinase and tPA, by directly modifying the drug (or site-activable prodrug) molecules with clot-selective (e.g. platelet-specific or fibrin-specific) antibodies and ligands [30–35]. While encapsulation of thrombolytic agents within micro and nanoparticles may increase their circulatory safety and residence time, that may not ensure their preferential delivery and release at the clot site. On the other hand, directly modifying drug molecules with ligands and antibodies may affect drug activity and may not prevent rapid plasma-induced deactivation or clearance. In this framework, several experts in the field have recently demonstrated and emphasized that advanced drug delivery systems can provide unique solutions regarding encapsulation, site-selective delivery and targeted action of thrombolytics [36–39]. Advanced drug delivery systems should be capable of carrying a therapeutic payload safely through the circulation without non-specific delivery or plasma-induced deactivation, deliver the payload preferentially at the target site via response to site-specific stimuli and biodegrade in the body for safe metabolic elimination [40]. Application of these design criteria in thrombolytic therapies is emerging as an exciting area of research, as evident from recent reports of encapsulating thrombolytic agents in fibrin-targeted perfluorocarbon nanoparticles or thrombin-responsive platelet-targeted polyoxazoline capsules, conjugating the agent on surface-engineered RBCs, loading the agent on magnetic nanoparticles for magnetically guided thrombus-specific localization, encapsulating the agents within peptide (e.g. RGD) or antibody decorated echoliposomes and microbubbles for targeted ultrasound-triggered sonothrombolysis, etc. [41–47]. In this metric, we have previously demonstrated the design feasibility of creating nano-and microparticle systems (liposomal, polymeric etc.) that can utilize targeting of active platelet-relevant receptor moieties to specifically bind to clots and this approach has also been utilized recently by other research groups to demonstrate clot-directed delivery of therapeutic agents [48–51]. Building on this framework, we have developed a PMP-inspired nanovesicular (PMIN) delivery platform using self-assembly of heterotypic lipid-peptide conjugates such that, the vesicles can (i) encapsulate therapeutically relevant doses of a thrombolytic drug to protect it in circulation, (ii) prevent non-specific distribution, off-target side-effects and plasma-induced deactivation of the drug, (iii) enhance clot-specific availability of the drug by virtue of active anchorage of the vesicles to clot-associated active platelets, and (iv) allow release of the drug upon vesicle destabilization by clot-relevant enzyme trigger for site-selective thrombolytic action.
For the heteromultivalent decoration of our PMIN vesicles, we have utilized a fibrinogen-relevant RGD-containing peptide (CGSSSGRGDSPA) for binding to active platelet GPIIb-IIIa and a PSGL-1-relevant EWVDV-containing peptide (CDAEWVDVS) for binding to active platelet P-selectin. In the past we have characterized the binding behavior of liposomes decorated with these peptides, using flow cytometry, to show that the peptide-decorated liposomes cannot bind to platelets significantly unless the platelets are agonist-activated [48,49]. Per our aggregometry studies reported in the Supplementary section of this manuscript, these peptides do not activate and aggregate quiescent platelets. Also, in our in vivo studies to analyze systemic off-target effects of SK-loaded PMIN vesicles in mice (i.e. tail bleeding studies), we did not observe any signs of systemic discomfort or respiratory distress in the mice once the peptide-decorated vesicles were injected and allowed to circulate for 30 min before tail clip. Therefore, we rationalize that the PMINs decorated with these peptides will have minimal interaction with circulating ‘resting’ platelets and thereby pose minimal systemic thrombotic risk.
It is to be noted here that the RGDS amino acid sequence itself is ubiquitous for several integrin binding, including platelet GPIIb/IIIa (αIIbbβ), endothelial αVβ3 and α5β1 [52,53]. However, the flanking amino acids in an RGDS-containing sequence can have major influence on its affinity and selectivity [54]. The RGD-containing linear sequence that we have used here was adapted from past reports of AFM-based studies of mechanistic investigation on ligand binding to active platelet GPIIb-IIIa [55,56]. We rationalized that this sequence is reasonable to be utilized for targeted binding of PMIN vesicles to the ‘active’ form of GPIIb-IIIa on platelets as a ‘proof-of-concept’ for PMIN targeting to active platelet-rich thrombus. Previously we have also explored utilization of conformationally constrained cyclic RGD sequences to enhance affinity and selectivity of liposomal particles towards active platelet GPIIb/ IIIa [57]. Based upon our past and current studies we believe that very high affinity RGD ligands pose the risk of actually amplifying the aggregation of active platelets due to high extent of platelet clustering around the ligand-decorated particles, while low-to-moderate affinity RGD ligands avoid this risk but may not have sufficient extent of binding and retention under a flow environment. Therefore in our current studies we decided to explore the benefits of combining a moderate affinity GPIIb/IIIa-binding RGD sequence with another active platelet-binding moiety, specifically, P-selectin binding EWVDVS sequence, to cooperatively enhance binding and retention of PMINs on platelet-rich thrombi under flow environment. While this ‘proof-of-concept’ design has demonstrated promising capabilities of targeted binding and site-selective thrombolysis as reported in the current manuscript, we believe that the ideal ‘heteromultivalent’ decoration should involve ligands with moderate affinity (kinetic parameter) but high specificity (thermodynamic parameter). To this end, our ongoing and future studies are focused on further exploring a library of peptides involving RGD, AGD and KGD sequences that can bind with moderate affinity but high specificity to GPIIb/IIIa compared to αVβ3 and α5β1 integrins.
It is also to be noted that the peptide sequences were conjugated by reacting the sulfhydryl (–SH) of the Cysteine termini to mal-eimide (Mal) of lipid-PEG-Mal to form the ‘thioether’ (C-S-C) linkage, which is a common bioconjugate technique for chemically modifying many proteins and peptides in the pharmaceutical and vaccine industry. Although this thioether conjugate is generally considered irreversible and stable, several recent studies have suggested the possibility of this linkage being reversible in presence of other thiols in plasma [58,59]. The extent of this possibility for our PMIN vesicle systems will be heterogenous depending on the equilibrium constant for the reaction of lipid-PEG-Mal to cysteine-terminated peptide compared to that for the reaction of lipid-PEG-Mal to other available thiol groups in plasma molecules, and we have not studied this aspect specifically in our current studies. Our in vivo studies indicated that a considerable extent of PMIN vesicles were capable of binding to the platelet-rich thrombi and hence we proceeded to load these PMIN vesicles with the thrombolytic drug and investigate their targeted fibrinolytic capability compared to free SK and SK-loaded untargeted vesicles. In the context of thioether instability possibilities in vivo, our future studies will explore alternative strategies of bioconjugation, especially using copper-free click chemistry. In the context of drug loading efficacy, liposomal encapsulation of streptokinase (SK) has been reported previously by a few groups, with values ranging in 10–35% EE [60–63]. In our studies we have observed a range of ~20–45% EE for SK in liposomes, based on processing parameters [64]. Based on these data, we believe that the ~38% EE observed in our current studies is comparable to reported ranges. Dosing of the animals was calculated based on this EE such that free SK suspension, SK-loaded unmodified vesicles and SK-loaded targeted PMIN vesicles all contained ~400 IU of SK and suspended in 200 μL saline, as stated in the methods section.
The platelet-inspired heteromultivalent ligand-decoration strategy can be also adapted to other drug delivery platforms (e.g. micelles, biodegradable polymeric particles etc.) to allow encapsulation of a variety of payloads and also modulate encapsulation efficiency and release kinetics. Interestingly, several research groups have recently reported on utilizing isolated platelet membrane for particle surface-coating to enhance target-specific drug delivery [65–67]. While these reports emphasize the advantage of leveraging platelet-inspired heterotypic surface-interactions via bio-derived surface modifications, clinical translation of platelet membrane-based systems may face logistical challenges of ensuring sufficient platelet availability, minimizing contamination risks and maintaining reasonably long storage life, and ensuring batch-to-batch biofunctional reproducibility, which are existing issues in platelet-based transfusion products. To that end, capturing bio-inspired heterotypic interactions on fully synthetic customizable delivery platforms, such as the PMIN system, may represent an alternative strategy with substantial advantages. Our future studies will also focus on utilizing fluorescently or radioactively labeled drug so as to be able to determine the levels of drug released and residual particles (and drugs) after targeted thrombolytic action in vivo, as well as, characterize the risk of embolic residues, in real time post thrombolysis.
5. Conclusion
The current work reports the development, characterization and in vitro, as well as, in vivo evaluation of a platelet microparticle-inspired nanovesicle (PMIN) system for active platelet-directed thrombus site-selective delivery and enzyme-triggered release of thrombolytic drug for targeted fibrinolytic action while minimizing off-target systemic side-effects. ‘Targeted thrombolysis with minimal effect on systemic hemostatic capabilities’ is a clinically highly significant area of research that drives exploration of novel drugs, unique drug delivery systems and new thromboinflammatory mechanisms. Our studies fit in this metric by virtue of developing and utilizing bio-inspired drug delivery systems. Our studies reported here clearly establish the capability of the PMINs to actively anchor onto platelet-rich thrombi under flow, allow lipase-triggered vesicle degradation for encapsulated payload release and thereby allow targeted fibrinolytic action at the clot site. Also, per our aggregometry studies, the peptide ligands themselves do not activate and aggregate quiescent platelets, indicating that these PMINs should have minimal interaction with circulating resting platelets and hence minimal systemic pro-thrombotic risk. Our in vivo results further validate that these enzyme-responsive PMINs can render targeted thrombolysis effectively without affecting systemic hemostatic capabilities. Besides targeted thrombolysis, the PMIN system can also become an efficient delivery platform for other therapeutic payloads (e.g. anticoagulant and anti-inflammatory drugs) in the vascular compartment, for potential site-selective treatment of vaso-occlusive pathologies in acute myocardial infarction, ischemic stroke, pulmonary embolism, deep vein thrombosis, peripheral arterial occlusion and indwelling catheter occlusions.
Supplementary Material
Acknowledgments
The authors acknowledge Rekha Srinivasan at CWRU for help with SEM, and, Tolou Shokuhfar and Emre Firlar at University of Illinois Chicago for help with cryo-TEM. The authors also acknowledge Dr. Matthew Neal at University of Pittsburgh for his insightful comments during manuscript preparation. This work was supported by grant 5R01 HL129179 (PI: Sen Gupta) from the National Institutes of Health (NIH). The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.biomaterials.2017.03.012.
Conflict of interest
ASG is inventor on a patent related to the heteromultivalent ligand decoration of nanoparticle systems, US 9107963: Heteromultivalent Nanoparticle Compositions.
Supplementary video related to this article can be found at http://dx.doi.org/10.1016/j.biomaterials.2017.03.012.
References
- [1].Heart disease and stroke statistics—2016 update. A report from the American Heart Association, Circulation 133 (2016) e38–360. [DOI] [PubMed] [Google Scholar]
- [2].Lippi G, Franchini M, Targher G, Arterial thrombus formation in cardiovascular disease, Nat. Rev. Cardiol. 8 (2011) 502–512. [DOI] [PubMed] [Google Scholar]
- [3].Cesarman-Maus G, Hajjar KA, Molecular mechanisms of fibrinolysis, Br. J. Haematol. 129 (2005) 307–321. [DOI] [PubMed] [Google Scholar]
- [4].Verstraete M, Collen D, Pharmacology of thrombolytic drugs, J. Am. Coll. Cardiol. 8 (1986) 33B–40B. [DOI] [PubMed] [Google Scholar]
- [5].Mackman N, Triggers, targets and treatments for thrombosis, Nature 451 (2008) 914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Miller DJ, Simpson JR, Silver B, Safety of thrombolysis in acute ischemic stroke: a review of complications, risk factors, and newer technologies, Neurohospitalist 1 (2011) 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gravanis I, Tsirka SE, tPA as a therapeutic target in stroke, Expert Opin. Ther. Targets 12 (2008), 10.1517/14728222.12.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wolf P, The nature and significance of platelet products in human plasma, Br. J. Haematol. 13 (1967) 269–288. [DOI] [PubMed] [Google Scholar]
- [9].Italiano JE, Mairuhu ATA, Flaumenhaft R, Clinical relevance of microparticles from platelets and megakaryocytes, Curr. Opin. Haematol. 17 (2010) 578–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Furie B, Furie BC, Role of platelet P-selectin and microparticle PSGL-1 in thrombus formation, Trends Mol. Med. 10 (2004) 171–178. [DOI] [PubMed] [Google Scholar]
- [11].Owens AP III, Mackman N, Microparticles in hemostasis and thrombosis, Circ. Res. 108 (2011) 1284–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Garcia BA, Smalley DM, Cho H,Shabanowitz J, Ley K, Hunt DF, The platelet microparticle proteome, J. Proteome Res. 4 (2005) 1516–1521. [DOI] [PubMed] [Google Scholar]
- [13].Ruggeri ZM, Platelets in atherothrombosis, Nat. Med. 8 (2002) 1227–1234. [DOI] [PubMed] [Google Scholar]
- [14].Plow EF, Pierschbacher MD, Ruoslahti E, Marguerie GA, Ginsberg MH, The effect of Arg-Gly-Asp-containing peptides on fibrinogen and von Willebrand factor binding to platelets, Proc. Natl. Acad. Sci. U. S. A. 82 (1985) 8057–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Appeldoorn CCM, Molenaar TJM, Bonnefoy A, van Leeuwen SH, Vandervoort PAH, Hoylaerts MF, van Berkel TJ, Biessen EA, Rational optimization of a short human P-selectin binding peptide leads to nanomolar affinity antagonists, J. Biol. Chem. 278 (2003) 10201–10207. [DOI] [PubMed] [Google Scholar]
- [16].Burke JE, Dennis EA, Phospholipase A2 structure/function, mechanism, and signaling, J. Lipid Res. 50 (2009) S237–S242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Takahashi S, Suzuki K, Watanabe Y, Watanabe K, Fujioka D, Nakamura T, Obata J, Kawabata K, Mishina H, Kugiyama K, Phospholipase A2 expression in coronary thrombus is increased in patients with recurrent cardiac events after acute myocardial infarction, Int. J. Cardiol. 168 (2013) 4214–4221. [DOI] [PubMed] [Google Scholar]
- [18].Zhu G, Mock JN, Aljuffali I, Cummings BS, Arnold RD, Secretory phospholipase A2 responsive liposomes, J. Pharm. Sci. 100 (2011) 3146–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Huang Z, Jaafari MR, Szoka FC, Disterolphospholipids: non-exchangeable lipids and their application to liposomal drug delivery, Angew. Chem. Int. Ed. 48 (2009) 4146–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].van Kruchten R, Cosemans JMEM, Heemskirk JWM, Measurement of whole blood thrombus formation using parallel-plate flow chambers — a practical guide, Platelets 23 (2012) 229–242. [DOI] [PubMed] [Google Scholar]
- [21].Arouri A, Trojnar J, Schmidt S, Hansen AH, Mollenhauer J, Mouritsen OG, Development of a cell-based bioassay for phospholipase A2-triggered liposomal drug release, PLoS One 10 (2015) e0125508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li W, Nieman M, Sen Gupta A, Ferric chloride induced murine thrombosis models, J. Vis. Exp. 115 (2016) e54479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cooley BC, In vivo fluorescence imaging of large vessel thrombosis in mice, Arterioscler. Thromb. Vasc. Biol. 31 (2011) 1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Turecek PL, Gritsch H, Richter G, Auer W, Pichler L, Schwarz HP, Assessment of bleeding for the evaluation of therapeutic preparations in small animal models of antibody-induced hemophilia and von Willebrand disease, Thromb. Haemost. 77 (1997) 591–599. [PubMed] [Google Scholar]
- [25].Immordino ML, Dosio F, Cattel L, Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential, Int. J. Nanomed 1 (2006) 297–315. [PMC free article] [PubMed] [Google Scholar]
- [26].Gurman P, Miranda OR, Nathan A, Washington C, Rosen Y, Elman NM, Recombinant tissue plasminogen activators (rtPA): a review, Clin. Pharmcol. Ther. 97 (2015) 274–285. [DOI] [PubMed] [Google Scholar]
- [27].The GUSTO Investigators, An International randomized trial comparing four thrombolytic strategies for acute myocardial infarction, NEJM 329 (1993) 673–682. [DOI] [PubMed] [Google Scholar]
- [28].Butcher K, Shuaib A, Saver J, Donnan G, Davis SM, Norrving B, Wong KSL, Abd-Allah F, Bhatia R, Khan A, Thrombolysis in the developing world: is there a role for Streptokinase? Int. J. Stroke 8 (2013) 560–565. [DOI] [PubMed] [Google Scholar]
- [29].Wardlaw JM, Murray V, Berge E, Del Zoppo GJ, Thrombolysis for acute ischemic stroke, Cochrane Database Syst. Rev. 4 (2009) CD000213. [DOI] [PubMed] [Google Scholar]
- [30].Kim I-S, Choi H-G, Choi H-S, Kim B-K, Kim C-K, Prolonged systemic delivery of streptokinase using liposome, Arch. Pharm. Res. 21 (1998) 248–252. [DOI] [PubMed] [Google Scholar]
- [31].Leach JK, O’Rear EA, Patterson E, Miao Y, Johnson AE, Accelerated thrombolysis in a rabbit model of carotid artery thrombosis with liposome-encapsulated and microencapsulated streptokinase, Thromb. Haemost. 90 (2003) 64–70. [PubMed] [Google Scholar]
- [32].Bode C, Meinhardt G, Runge MS, Freitag M, Nordt T, Arens M, Newell JB, Kübler W, Haber E, Platelet-targeted fibrinolysis enhances clot lysis and inhibits platelet aggregation, Circulation 84 (1991) 805–813. [DOI] [PubMed] [Google Scholar]
- [33].Ding B-S, Hong N, Murciano J-C, Ganguly K, Gottstein C, Christofidou-Solomidou M, Albelda SM, Fisher AB, Cines DB, Muzykantov VR, Prophylactic thrombolysis by thrombin-activated latent prourokinase targeted to PECAM-1 in the pulmonary vasculature, Blood 111 (2008) 1999–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang X, Palasubramaniam J, Gkanatsas Y, Hohmann JD, Westein E, Kanojia R, Alt K, Huang D, Jia F, Ahrens I, Medcalf RL, Peter K, Hagemeyer CE, Towards effective and safe Thrombolysis and Thromboprophylaxis. Preclinical testing of a novel antibody-targeted recombinant plasminogen activator directed against activated platelets, Circ. Res. 114 (2014) 1083–1093. [DOI] [PubMed] [Google Scholar]
- [35].Fuentes RE, Zaitsev S, Ahn HS, Hayes V, Kowalska MA, Lambert MP, Wang Y, Siegel DL, Bougie DW, Aster RH, Myers DD, Stepanova V,Cines DB, Muzykantov VR, Poncz M, A chimeric platelet-targeted urokinase prodrug selectively blocks new thrombus formation, J. Clin. Investig. 126 (2016) 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lanza GM, Marsh JN, Hu G, Scott MJ, Schmieder AH, Caruthers SD, Pan D, Wickline SA, Rationale for a nanomedicine approach to thrombolytic therapy, Stroke 41 (2010) S42–S44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lippi G, Mattiuzzi C, Favaloro EJ, Novel and emerging therapies: thrombus-targeted fibrinolysis, Semin. Thromb. Hemost. 39 (2013) 48–58. [DOI] [PubMed] [Google Scholar]
- [38].Greineder CF, Howard MD, Carnemolla R, Cines DB, Muzykantov VR, Advanced drug delivery systems for antithrombotic agents, Blood 122 (2013) 1565–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Absar S, Gupta N, Nahar K, Ahsan F, Engineering of plasminogen activators for targeting to thrombus and heightening thrombolytic efficacy, J. Thromb. Haemost. 13 (2015) 1545–1556. [DOI] [PubMed] [Google Scholar]
- [40].Mura S, Nicolas J, Couvreur P, Stimuli-responsive nanocarriers for drug delivery, Nat. Mater. 12 (2013) 991–1003. [DOI] [PubMed] [Google Scholar]
- [41].Marsh JN, Senpan A, Hu G, Scott MJ, Gaffney PJ, Wickline SA, Lanza GM, Fibrin-targeted perfluorocarbon nanoparticles for targeted thrombolysis, Nanomed. (Lond) 2 (2007) 533–543. [DOI] [PubMed] [Google Scholar]
- [42].Gunawan ST, Kempe K, Bonnard T, Cui J, Alt K, Law LS, Wang X,Westein E, Such GK, Peter K, Hagemeyer CE, Caruso F, Multifunctional thrombin-activatable polymer capsules for specific targeting to activated platelets, Adv. Mater 27 (2015) 5153–5157. [DOI] [PubMed] [Google Scholar]
- [43].Murciano J-C, Medinilla S, Eslin D, Atochina E, Cines DB, Muzykantov VR, Prophylactic fibrinolysis through selective dissolution of nascent clots by tPA-carrying erythrocytes, Nat. Biotech. 21 (2003) 891–896. [DOI] [PubMed] [Google Scholar]
- [44].Cheng R, Huang W, Huang L, Yang B, Mao L, Jin K, ZhuGe Q, Zhao Y, Acceleration of tissue plasminogen activator-mediated thrombolysis by magnetically powered nanomotors, ACS Nano 8 (2014) 7746–7754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hagisawa K, Nishioka T, Suzuki R, Maruyama K, Takase B, Ishihara M, Kurita A, Yoshimoto N, Nishida Y, Iida K, Luo H, Siegel RJ, Thrombus-targeted perfluorocarbon-containing liposomal bubbles for enhancement of ultrasonic thrombolysis: in vitro and in vivo study, J. Thromb. Haemost. 11 (2013) 1565–1573. [DOI] [PubMed] [Google Scholar]
- [46].Hua X, Zhou L, Liu P, He Y, Tan K, Chen Q, Gao Y, Gao Y, In vivo thrombolysis with targeted microbubbles loading tissue plasminogen activator in a rabbit femoral artery thrombus model, J. Thromb. Thrombolysis 38 (2014) 57–64. [DOI] [PubMed] [Google Scholar]
- [47].Wang X, Gkanatsas Y, Palasubramaniam J, Hohmann JD, Chen YC, Lim B, Hagemeyer CE, Peter K, Thrombus-targeted theranostic microbubbles: a new technology towards concurrent rapid ultrasound diagnosis and bleeding free fibrinolytic treatment of thrombosis, Theranostics 6 (2016) 726–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sen Gupta A, Huang G, Lestini BJ, Sagnella S, Kottke-Marchant K, Marchant RE, RGD-modified liposomes targeted to activated platelets as a potential vascular drug delivery system, Thromb. Haemost. 93 (2005) 106–114. [DOI] [PubMed] [Google Scholar]
- [49].Modery CL, Ravikumar M, Wong TL, Dzuricky MJ, Durongkaveroj N, Sen Gupta A, Heteromultivalent liposomal nanoconstructs for enhanced targeting and shear-stable binding to active platelets for site-selective vascular drug delivery, Biomaterials 32 (2011) 9504–9514. [DOI] [PubMed] [Google Scholar]
- [50].Anselmo AC, Modery-Pawlowski CL, Menegatti S, Kumar S, Vogus DR, Tian LL, Chen M, Squires TM, Sen Gupta A, Mitragotri S, Platelet-like nanoparticles: mimicking shape, flexibility, and surface biology of platelets to target vascular injuries, ACS Nano 8 (2014) 11243–11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Vaidya B, Agarwal GP, Vyas SP, Platelets directed liposomes for the delivery of streptokinase: development and characterization, Eur. J. Pharm. Sci. 44 (2011) 589–594. [DOI] [PubMed] [Google Scholar]
- [52].Ruoslahti E, RGD and other recognition sequences for integrins, Annu. Rev. Cell Dev. Biol. 12 (1996) 697–715. [DOI] [PubMed] [Google Scholar]
- [53].Humphries JD, Byron A, Humphries MJ, Integrin ligands at a glance, J. Cell Sci. 119 (2006) 3901–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kapp TG, Rechenmacher F, Neubauer S, Maltsev OV, Cavalcanti-Adam EA, Zarka R, Reuning U, Notni J, Wester H-J, Mas-Moruno C, Spatz J, Geiger B, Kessler H, A comprehensive evaluation of the activity and selectivity profile of ligands for RGD-binding integrins, Sci. Rep. 7 (2017) 39805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lee I, Marchant RE, Force measurements on the molecular interaction between ligand (RGD) and human platelet aIIbb3 receptor system, Surf. Sci. 491 (2001) 433–443. [Google Scholar]
- [56].Lee I, Marchant RE, Molecular interaction studies of hemostasis: fibrinogen ligand—human platelet receptor interactions, Ultramicroscopy 97 (2003) 341–352. [DOI] [PubMed] [Google Scholar]
- [57].Huang G, Zhou Z, Srinivasan R, Penn MS, Kottke-Marchant K, Marchant RE, Sen Gupta A, Affinity manipulation of surface-conjugated RGD peptide to modulate binding of liposomes to activated platelets, Biomaterials 29 (2008) 1676–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Baldwin AD, Kiick KL, Reversible maleimide—thiol adducts yield glutathione-sensitive poly(ethylene glycol)—heparin hydrogels, Polym. Chem. 4 (2013) 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ross PL, Wolfe JL, Physical and chemical stability of antibody-drug conjugates: current status, J. Pharm. Sci. 105 (2016) 391–397. [DOI] [PubMed] [Google Scholar]
- [60].Nguyen PD, O’Rear EA, Johnson AE, Patterson E, Whitsett TL, Bhakta R, Accelerated thrombolysis and reperfusion in a canine model of myocardial infarction by liposomal encapsulation of streptokinase, Circ. Res. 66 (1990) 875–878. [DOI] [PubMed] [Google Scholar]
- [61].Perkins WR, Vaughan DE, Plavin SR, Daley WL, Rauch J, Lee L, Janoff AS, Streptokinase entrapment in interdigitation-fusion liposomes improves thrombolysis in an experimental rabbit model, Thromb. Haemost. 77 (1997) 1174–1178. [PubMed] [Google Scholar]
- [62].Jin S-E, Kim I-S, Kim C-K, Comparative effects of PEG-containing liposomal formulations on in vivo pharmacokinetics of streptokinase, Arch. Pharm. Res. 38 (2015) 1822–1829. [DOI] [PubMed] [Google Scholar]
- [63].Vaidya B, Nayak MK, Dash D, Agrawal GP, Vyas SP, Development and characterization of highly selective target-sensitive liposomes for the delivery of streptokinase: in vitro/in vivo studies, Drug Deliv. 23 (2016) 791–797. [DOI] [PubMed] [Google Scholar]
- [64].Holt BD, Sen Gupta A, Streptokinase loading in liposomes for vascular targeted nanomedicine applications: encapsulation efficiency and effects of processing, J. Biomater. Appl. 26 (2012) 509–527. [DOI] [PubMed] [Google Scholar]
- [65].Hu C-MJ, Fang RH, Wang K-C, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L, Nanoparticle biointerfacing by platelet membrane cloaking, Nature 526 (2015) 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hu Q, Sun W, Qian C, Wang C, Bomba HN, Gu Z, Anti-cancer platelet-mimicking nanovehicles, Adv. Mater 27 (2015) 7043–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Li J, Ai Y, Wang L, Bu P, Sharkey CC, Wu Q, Wun B, Roy S, Shen X, King MR, Targeted drug delivery to circulating tumor cells via platelet membrane-functionalized particles, Biomaterials 76 (2016) 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.