

Abstract
CD73 converts AMP to immunosuppressive adenosine, and its inhibition was proposed as a new strategy for cancer treatment. We synthesized 5′-O-[(phosphonomethyl)phosphonic acid] derivatives of purine and pyrimidine nucleosides, which represent nucleoside diphosphate analogs, and compared their CD73 inhibitory potencies. In the adenine series, most ribose modifications and 1-deaza and 3-deaza were detrimental, but 7-deaza was tolerated. Uracil substitution with N3-methyl, but not larger groups, or 2-thio, was tolerated. 1,2-Diphosphono-ethyl modifications were not tolerated. N4-(Aryl)alkyloxy-cytosine derivatives, especially with bulky benzyloxy substituents, showed increased potency. Among the most potent inhibitors were the 5′-O-[(phosphonomethyl)phosphonic acid] derivatives of 5-fluorouridine (4l), N4-benzoyl-cytidine (7f), N4-[O-(4-benzyloxy)]-cytidine (9h), and N4-[O-(4-naphth-2-ylmethyloxy)]-cytidine (9e) (Ki values 5–10 nM at human CD73). Selected compounds tested at the two UDP-activated P2Y receptor subtypes showed high CD73-selectivity, especially those with large nucleobase substituents. These nucleotide analogs are among the most potent CD73 inhibitors reported and may be considered for development as parenteral drugs.
Keywords: Ecto-5′-nucleotidase, deazaadenine derivatives, inhibitors, phosphonic acids, synthesis, structure-activity relationships, uracil derivatives
Graphical Abstract
Introduction
Ecto-5′-nucleotidase (ecto-5′-NT, eN, CD73, EC 3.1.3.5) is a glycosylphosphatidylinositol (GPI)-linked cell surface enzyme that dephosphorylates extracellular nucleoside monophosphates.1–3 The enzyme can be cleaved from its GPI-linker and is also present in a soluble active form in serum. The vertebrate enzyme selectively hydrolyzes adenosine 5′-monophosphate (AMP) over adenosine 2′- or 3′-monophosphates leading to elevated extracellular concentrations of adenosine.1 The X-ray crystallographic structures of the enzyme in complex with either inhibitor or substrate have been reported.4,5 There is a marked conformational rearrangement of the structure as catalysis occurs. Two conformational classes have been determined, an open and a closed form.
Although the substrate, AMP, is not a potent agonist of adenosine receptors (ARs), the enzymatic reaction product, adenosine, activates four AR subtypes (A1AR, A2AAR, A2BAR and A3AR).6 One of the important activities of adenosine is the suppression of inflammation.6 Thus, CD73 upregulation and the increased production of adenosine are beneficial in chronic inflammatory diseases. A coexpression of CD73 and the A2AAR found in many tissues including the brain and immune cells, especially when inflammation is present, allows the concerted activation of this receptor.2,7,8 However, in the tumor microenvironment, elevated CD73 and adenosine counteract the body’s immune defense against the tumor.9–14 This is particularly important in cancer immunotherapy, for which coadministration of either an A2AAR antagonist or an inhibitor of CD73 offers synergistic anti-tumor activity. A CD73 inhibitor also potentiates the in vivo anticancer effect of inhibitors of nicotinamide phosphoribosyltransferase,15 which is required for the biosynthesis of intracellular NAD+. Antibodies against CD73 are currently undergoing clinical trials for cancer therapy.16,17 The development of CD73 assays18,19 has led to reports of diverse inhibitors of the enzyme.20–24 Among the first potent inhibitors to be identified was adenosine-5′-O-[(phosphonomethyl)phosphonic acid (I, α,β-methylene-ADP, AOPCP, Chart 1).25 Also, various anthraquinone21 and sulfonamide22 derivatives were found to be competitive inhibitors, while polyphenols26 and polyoxometalates (POMs)27 are non-competitive CD73 inhibitors. Recently, adenine nucleotide analogs of I that display low nM potency in inhibition of CD73 were identified.12 Modeling based on the X-ray structures of CD73 has aided in the design of novel inhibitors.28 The present work complements that study through the exploration of the structure-activity relationships (SAR) of both purine and pyrimidine nucleotides as inhibitors of CD73. An advantage of inhibitors derived from pyrimidines would be that if hydrolyzed to the parent nucleoside, they would not activate ARs, unlike adenine nucleotide I and its congeners.
Chart 1.

Selected inhibitors of ecto-5′-nucleotidase (eN, CD73).20
Results
Chemistry
There are several commonly used multi-step methods for the preparation of nucleoside-5′-O-[(phosphonomethyl)phosphonic acid] derivatives, i.e. either reacting the protected nucleoside with activated bisphosphonate or utilizing methylene diphosphonic acid and coupling reagents.29–31 However, despite the use of protecting groups these synthetic strategies suffer overall from very low yields. Previously, we were able to demonstrate that phosphonylation reactions of unprotected nucleosides using methylenebis(phosphonic dichloride) in trimethyl phosphate provided the nucleoside-5′-O-[(phosphonomethyl)phosphonic acids] as the main products under optimized conditions.20 Furthermore, reacting unprotected nucleosides with 1.5 eq. methylene diphosphonic acid and 3 eq. dicyclohexylcarbodiimide (DCC) in dimethylformamide (DMF) led to the formation of nucleoside-5′-O-[(phosphonomethyl)phosphonic acids] as the main products, as well. Therefore, we employed solely unprotected nucleosides as starting materials in our syntheses. When additional phosphonylation occurred at the 2′- and/or 3′-position, these side products were easily separated from the desired 5′-substituted product through a combination of ion exchange and reverse phase C18 chromatography. The SAR of the purine scaffold in AOPCP derivatives was extensively explored in a previous study, leading to CD73 inhibitors with potency in low nanomolar range.12 However, the SAR of the ribose moiety of AOPCP derivatives had not been explored. Therefore, we prepared 2′-deoxy (2a), 2′-amino-2′-deoxy (2b), and 3′-deoxy (2d) AOPCP derivatives by reacting the respective nucleoside with methylene diphosphonic acid in the presence of DCC in DMF (Scheme 1). In the case of 2b, the reaction proceeded very slowly, and the 3′-phosphonate side product (2c) was formed to an equal extent (0.5 % yield). Compound 2c was isolated, and its structure was unequivocally determined using 1H-1H-COSY NMR spectroscopy. To address ring variations of the adenine moiety, the 1-deaza (2e), 3-deaza (2f), and 7-deaza (2g) AOPCP derivatives were prepared by reaction of unprotected nucleosides with methylenebis(phosphonic dichloride) in trimethyl phosphate to increase yields (Scheme 1).
Scheme 1.

Synthesis of adenosine 2a–g and uridine derivatives 4a–y. Reagents and conditions: (a) DCC (3 eq.), methylene diphosphonic acid (1.5 eq.), DMF, room temp, 3–24 h; for compounds 4w and 4x: DCC (3 eq.), ethylene diphosphonic acid (1.5 eq.), DMF, room temp, 3 h (b) methylenebis(phosphonic dichloride) (3 eq.), trimethyl phosphate, 0 °C, 30 min, then triethylammonium hydrogencarbonate buffer pH 8.4–8.6, rt, 30 min.
Then, we turned our focus to pyrimidine-derived 5′-O-[(phosphonomethyl)phosphonic acids. e.g. UOPCP (4a), prepared by phosphonylation of uridine (3a). Furthermore, N3-substituted nucleosides 3b-e were prepared via nucleophilic substitution using 3a (Supporting information), and their subsequent phosphonylation afforded compounds 4b-e. Substitution of the uracil 5-position was explored with methyl 4f-h and halogen 4l-o derivatives. The reaction of 5-ethynyl-uridine with methylenebis(phosphonic dichloride) led, besides the desired 5-ethynyl-uridine derivative 4i, to the formation of 5-(1-chlorovinyl)uridine- (4j) and 5-(1-chlorovinyl)-3-methyluridine-5′-O-[(phosphonomethyl)phosphonic acid] (4k) through the addition of HCl to the alkyne bond. In order to explore the variations of the 2′-position at the ribose moiety 2′-deoxy (4p), 2′-amino-2′-deoxy (4q), 2′-azido-2′-deoxy (4r), and 2′-fluoro-2′-deoxy (4s) derivatives were prepared. The role of the stereochemistry at the 2′-position was addressed through the synthesis of 2′-ara-fluoro-2′-deoxyuridine- (4t) and 1-(β-D-arabinofuranosyl)-uridine-5′-O-[(phosphonomethyl)phosphonic acid (4u). Synthesis of 6-azauridine derivative (4v) allowed the introduction of an H-bond acceptor at the 6-position. In addition to the methylene group, the linker between the two phosphonate groups was extended to an ethylene moiety by reacting ethylene diphosphonic acid with the respective nucleosides 3a, 3f to afford uridine- (4w) and 5-methyluridine-5′-O-[(phosphonoethyl)phosphonic acid] (4x). 2-Thiouridine derivative 4y was prepared following a previously published procedure.41
Next, cytidine derivatives varying at the 2′-position (7a and 7b), as well as at the 5-position (7c-e), were prepared (Scheme 2). The N4-benzoyl COPCP derivative 7f displayed gradual decomposition when left at room temperature over several weeks in aqueous solution to UOPCP (4a) and COPCP (7a) (Supporting information). In order to introduce bulky aromatic substituents at the 4-position of the cytosine moiety without the inherent instability issues as seen with N4-benzoyl COPCP derivative 7f, we turned our focus towards alkoxyimino derivatives 9a-i. The required nucleosides were prepared by either reacting cytidine (6a), 2′-deoxycytidine (6b), 5-fluorocytidine (6d) or 5-methylcytidine (6e) with respective alkoxyamino derivatives in pyridine7 followed by phosphonylation reaction to afford compounds 9a-f, or by reacting cytidine (6a) with benzyloxyamine, followed by nucleophilic substitution at the 3-position (Supporting information) and subsequent 5′-O-phosphonylation of the nucleosides to afford compounds 9h and 9i (Scheme 2). Furthermore, 3-deazauridine-5′-α,β-methylene-diphosphate (10), (S)-methanocarba-5′-α,β-methylene-diphosphate (11) and the ethyl ester of 2-thio-UDP (12)32 were prepared to extend the exploration of SAR of the nucleotide analogs as CD73 inhibitors. All phosphonate-substituted nucleotides were purified to homogeneity by ion exchange chromatography followed by reverse phase C18 HPLC.
Scheme 2.
Synthesis of cytosine-derived 5′-O-[(phosphonomethyl)phosphonic acid] derivatives 7a–f and 9a-i. Reagents and conditions: (a) DCC (3 eq.), methylene diphosphonic acid (1.5 eq.), DMF, room temp, 3–24 h; (b) methylenebis(phosphonic dichloride) (3 eq.), trimethyl phosphate, 0 °C, 30 min, then triethylammonium hydrogencarbonate buffer pH 8.4–8.6, rt, 30 min: (c) R3-O-NH2xHCl, pyridine, 80 °C, 12 h. (d) alkyl iodide (ol, 1.5 eq.), K2CO3 (1.7 eq.) in DMF/acetone (1:1) at 50 °C for 3 d.
Pharmacological Evaluation
The potency of the compounds to inhibit CD73 was determined by a radiometric CD73 assay using [2,8-3H]AMP as a substrate and recombinant soluble rat CD73.29,33 After the enzymatic reaction, the substrate was separated from its product [2,8-3H]adenosine by precipitation with lanthanum chloride followed by filtration through glass fiber filters.34 For compounds that inhibited CD73 activity by more than 50% at an initial screening concentration of 1 µM, full concentration–response curves were determined in at least three separate experiments performed in duplicates using 10 different concentrations of inhibitor. Ki values were calculated from the obtained IC50 values using the Cheng-Prusoff equation.30 Results are summarized in Tables 1–3, and concentration–inhibition curves are shown in Figures 1 and 2. Rat CD73 is similar to the human isoform (87% sequence identity¸ BLAST algorithm35,36), and the active sites only differ in a single amino acid (Phe in the human enzyme is replaced by Tyr in rat).34 Previous studies had shown that the potency of competitive inhibitors targeting rat CD73 showed comparable or higher potency for human CD73.12 Nevertheless, the most potent compounds (4l, 7f, 9d, 9e, 9g and 9h) were analyzed using recombinant soluble human CD73. Furthermore, to study the inhibitors in a less artificial environment, they were investigated on membrane-anchored CD73 using membrane preparations derived from the triple-negative breast cancer (TNBC) cell line MDA-MB-231. These cells overexpress CD73, serve as an in vitro model for TNBC and had previously been used for the development of therapeutic antibodies against CD73.16,37–40
Table 1.
The inhibitory potency of adenine-based AOPCP analogs 2a-g at rat CD73. X, Y, Z = N, unless otherwise noted, compound 2c: 5′-OH.
 | ||||
|---|---|---|---|---|
| Compd. | Substitution | R1 | R2 | Ki ± SEM (nM) (% inhibition at indicated concentration), rat CD73 |
| I, (AOPCP) | OH | OH | 167 ± 53 | |
| 2a | OH | H | > 1000 (30%) | |
| 2b | OH | NH2 | > 1000 (2%) | |
| 2c | 5′-OH | OP2O5CH5 | NH2 | > 1000 (8%) |
| 2d | H | OH | 1970 ± 220 | |
| 2e | X = CH | OH | OH | > 1000 (36%) |
| 2f | Y = CH | OH | OH | > 1000 (36%) |
| 2g | Z = CH | OH | OH | 88.6 ± 4.0 |
Table 3.
The inhibitory potency of cytosine derivatives 7a-f and 9a-i at rat CD73.
 | ||||
|---|---|---|---|---|
| Compd. | Substitution | R1 | R2 | Ki ± SEM (nM) (% inhibition at indicated concentration) rat CD73 |
| COPCP 7a | OH | H | 898 ± 63 | |
| 7b | H | H | > 1000 (18%) | |
| 7c | X = I | OH | H | 502 ± 83 |
| 7d | X = F | OH | H | 349 ± 41 |
| 7e | X = CH3 | OH | H | 2030 ± 670 |
| 7f | OH | benzoyl | 13.9 ± 1.6 | |
| 9a | OH | CH3 | 257 ± 39 | |
| 9b | OH | benzyl | 112 ± 15 | |
| 9c | H | benzyl | 780 ± 15 | |
| 9d | OH | 4-trifluoromethylbenzyl | 30.3 ± 4.2 | |
| 9e | OH | naphth-2-ylmethyl | 18.8 ± 3.2 | |
| 9f | X = CH3 | OH | benzyl | 321 ± 9 |
| 9g | X = F | OH | benzyl | 85.1 ± 7.5 |
| 9h | Y = CH3 | OH | benzyl | 3.67 ± 0.26 |
| 9i | Y = C2H5 | OH | benzyl | 262 ± 46 |
Figure 1.
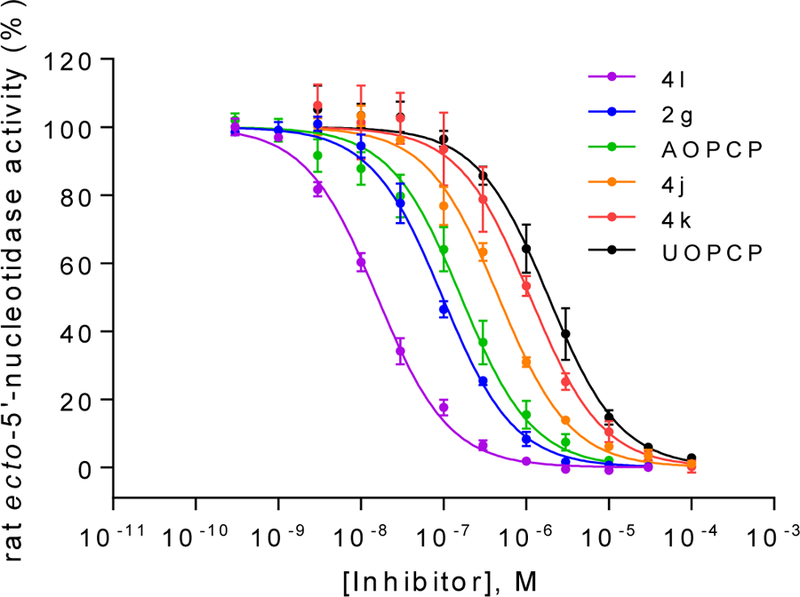
Concentration-inhibition curves of selected compounds at soluble rat CD73. Rat enzyme Km: 53 μM; AMP concentration: 5 μM; Data points are from three separate experiments performed in duplicates. For Ki values see Table 1 and 2.
Figure 2.
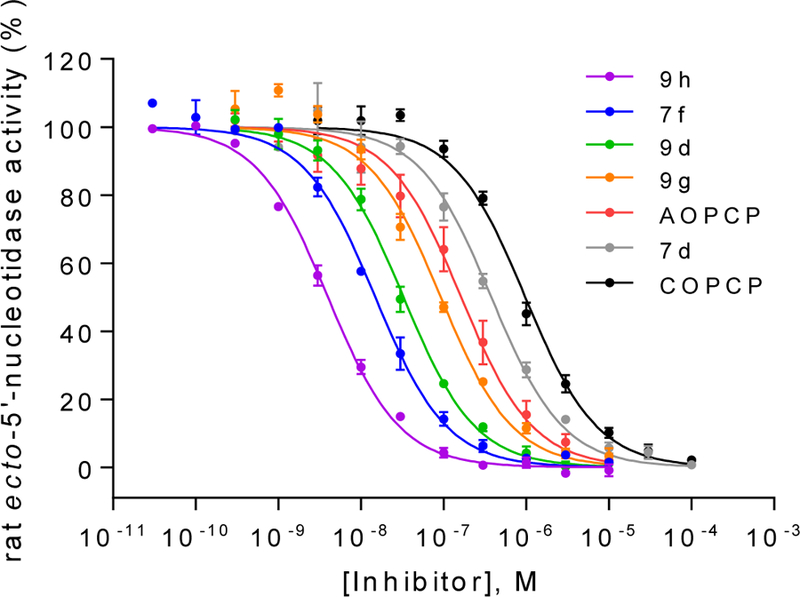
Concentration-inhibition curves of selected compounds at soluble rat CD73. Rat enzyme Km: 53 μM; AMP concentration: 5 μM. Data points are from three separate experiments performed in duplicates. For Ki values see Table 3.
Structure-Activity Relationships
AOPCP (I) displays moderate CD73 inhibitory activity (Ki, nM) (167, Table 1), whereas UOPCP (4a, 1830, Table 2) and COPCP (7a, 898, Table 3) are 11- to 5-fold less active. Variations at the ribose moiety 2a-2d, 4p-4u, and 7b are not tolerated, with the exception for 2′-ara-fluoro-2′-deoxyuridine 5′-α,β-methylene-diphosphate (4t, 1750 nM) that displays similar inhibitory activity compared to UOPCP (4a). 3-Deaza (2e) and 1-deaza (2f) derivatives of AOPCP are inactive, whereas the 7-deaza analog (2g, 88.6 nM) displays 2-fold higher CD73 inhibitory activity compared to AOPCP.
Table 2.
The inhibitory potency of uridine-derived nucleotides 4a-y as rat CD73 inhibitors. R2, X, Y = H, W = O, V = CH, Z = N, n = 1, unless otherwise noted.
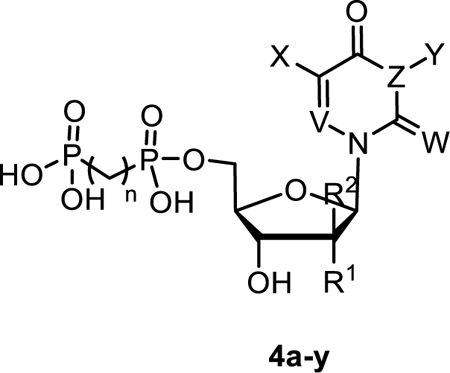 | ||||
|---|---|---|---|---|
| Compd. | Substitution | R1 | R2 | Ki ± SEM (nM) (% inhibition at indicated concentration), rat CD73 |
| UOPCP 4a | OH | H | 1830 ± 530 | |
| 4b | Y = CH3 | OH | H | 1860 ± 400 |
| 4c | Y = C2H5 | OH | H | > 1000 (4%) |
| 4d | Y = C3H7 | OH | H | > 1000 (7%) |
| 4e | Y = benzyl | OH | H | > 1000 (3%) |
| 4f | X = CH3 | OH | H | 338 ± 56 |
| 4g | X = CH3 | H | H | 639 ± 65 |
| 4h | X = CH3 | OCH3 | H | > 1000 (3%) |
| 4i | X = ethynyl | OH | H | 276 ± 37 |
| 4j | X = 1-chlorovinyl | OH | H | 424 ± 27 |
| 4k | X = 1-chlorovinyl, Y = CH3 | OH | H | 1050 ± 290 |
| 4l | X = F | OH | H | 14.8 ± 1.9 |
| 4m | X = Cl | OH | H | 86.7 ± 7.6 |
| 4n | X = Br | OH | H | 88.7 ± 12.5 |
| 4o | X = I | OH | H | 162 ± 4 |
| 4p | H | H | > 1000 (37%) | |
| 4q | NH2 | H | > 1000 (2%) | |
| 4r | N3 | H | > 1000 (9%) | |
| 4s | F | H | > 1000 (11%) | |
| 4t | H | F | 1750± 380 | |
| 4u | H | OH | > 1000 (8%) | |
| 4v | V = N | OH | H | > 1000 (21%) |
| 4w | n = 2 | OH | H | > 1000 (4%) |
| 4x | n= 2, X = CH3 | OH | H | > 1000 (9%) |
| 4y | W = S | OH | H | > 1000 (7%) |
Introduction of a substituent larger than a methyl group at the uridine 3-position is not tolerated: 3-methyluridine (4b, 1864 nM) is equipotent to UOPCP (4a), while 3-ethyl- (4c), 3-propyl- (4d) and the 3-benzyl-uridine-5′-α,β-methylene-diphosphate (4e) are inactive. These results indicate the limited size of the binding pocket. Substitution at the uridine 5-position showed the following rank order of potency (Ki, nM): 5-F (4l, 14.8) > 5-Cl (4m, 86.7) = 5-Br (4n, 88.7) > 5-I (4o, 162) ≥ 5-ethynyl (4i, 276) ≥ 5-methyl (4f, 338) ≥ 5-(1-chlorovinyl) (4j, 424). Combination of a 5-methyl group with variations at the ribose 2′-position (2′-deoxy (4g, 639 nM) and 2′-methoxy (4h, inactive)), as well as combining a 5-(1-chlorovinyl) group with a 3-methyl substituent (4k, 1050 nM) reduced inhibitory activity. Extending the distance between the phosphonate groups by introducing an ethylene linker (4w, 4x) led to a complete loss of CD73 inhibition. Furthermore, replacement of 2-oxo by 2-thio (4y) or the introduction of a nitrogen atom at the 6-position was not tolerated.
Since the SAR around the uridine scaffold appeared to be rather limited, tolerating only a small methyl group at the 3-position (4b) and rather small, highly electronegative substituents at the 5-position, such as fluoro (4l), we decided to further explore the cytidine scaffold. COPCP (7a) was twice as potent as UOPCP (4a). However, for the COPCP derivatives, introduction of a substituent at the 5-position had a less pronounced effect leading to an increase in potency in the following rank order (Ki, nM): 5-CH3 (7e, 2030) < 5-H (7a, COPCP, 898) ≤ 5-I (7c, 502) ≤ 5-F (7d, 349), but altogether rather moderate activity. Surprisingly, the introduction of a benzylamide at the 4-position (7f) provided a 5′-α,β-methylene-diphosphate derivative with a CD73 inhibitory potency of 13.9 nM. This high potency indicated the presence of a large hydrophobic subpocket that could be probed by further structural modification of the inhibitor. Unfortunately, quality control LC-MS experiments displayed decomposition of compound 7f to UOPCP, which was additionally confirmed via NMR experiments (Supporting information, Figure S1). In order to address the hydrophobic subpocket of CD73, we envisaged the introduction of an alkoxyimino group at the 4-position leading to an increase in potency in the following order (Ki, nM): H3CON= (9a, 257) < benzyloxy-N= (9b, 112) < 4-F3C-benzyloxy-N= (9d, 30.3) ≤ naphthalen-2-ylmethoxy-N= (9e, 18.8). Combination of a 4-benzyloxyimino group with a methyl group at the 5-position (9f, 321 nM) did not improve the CD73 inhibitory activity, and the introduction of a 5-F substituent had no effect (9g, 85.1 nM). Remarkably, the introduction of a methyl group at the 3-position of the benzyloxyimino derivative (9h, 3.67 nM) led to a 23-fold increase in inhibitory activity and provided the most potent compound of the series. However, the tolerated size of the substituent remained limited to a methyl group, as the ethyl derivative (9i, 262 nM) displayed a 100-fold drop in inhibitory potency. 3-Deazauridine-5′-α,β-methylene-diphosphate (10), (S)-methanocarba-uridine-5′-α,β-methylene-diphosphate (11) and the ethyl ester of 2-thio-UDP (12)32 were inactive at CD73.
Although it is well known that rat and human CD73 share identical orthosteric binding sites, and species differences for competitive inhibitors are therefore moderate, the most potent compounds (4l, 7f, 9d, 9e, 9g and 9h) were additionally evaluated for inhibition of human CD73. We employed recombinantly expressed soluble enzyme (Figure 3) as well as membrane preparations of CD73-expressing MDA-MB-231 cells (Figure 4). The comparison of potencies measured with the different sources of CD73 (see Table 4 and Figure 5) confirmed the observation that AOPCP analogs are usually slightly more potent at human CD73 as compared to the rat enzyme.4,20 Compounds 4l, 7f, 9d and 9e are 2 to 3-fold more potent while 9g is 5-fold more potent. However, the most potent compound at rat CD73 (9h, 3.67 nM) is somewhat less potent at the human enzyme (Ki, soluble CD73, 10.6 nM; membrane preparation, 7.96 nM). All tested inhibitors were similarly potent at soluble CD73 as compared to the membrane-bound enzyme.
Figure 3.
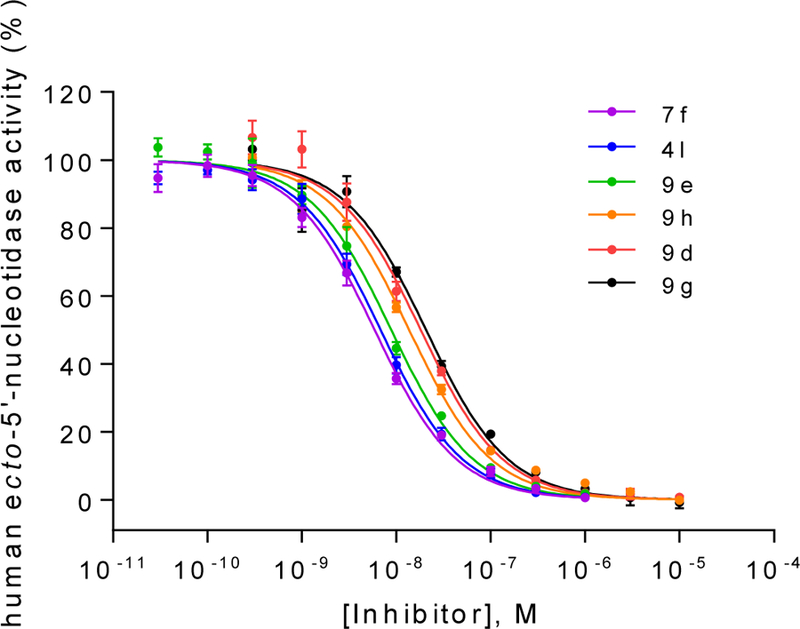
Concentration-inhibition curves of selected compounds at soluble human CD73. Human enzyme Km: 17 μM; AMP concentration: 5 μM. Data points are from three separate experiments performed in duplicates. For Ki values see Table 5.
Figure 4.
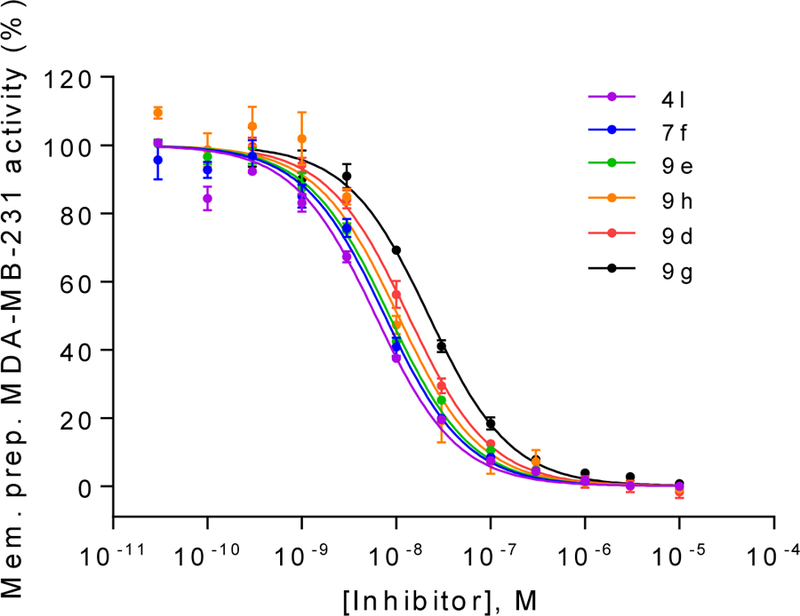
Concentration−inhibition curves of selected compounds at membrane preparations of the human triple-negative breast cancer cell line MDA-MB-231 which natively expresses CD73. Km: 14.8 μM; AMP concentration: 5 μM; Data points are from three separate experiments performed in duplicates. For Ki values see Table 5.
Table 4.
Inhibitory potency at rat ecto-5′-nucleotidase of 3-deazauridine-derived (10), (S)-methanocarba-based (11) nucleotides and ethyl ester 12.
 | |
|---|---|
| Compd. | Ki (nM) (% inhibition at indicated concentration) rat CD73 |
| 10 | > 1000 (19%) |
| 11 | > 1000 (12%) |
| 12 | > 1000 (10%) |
Figure 5.
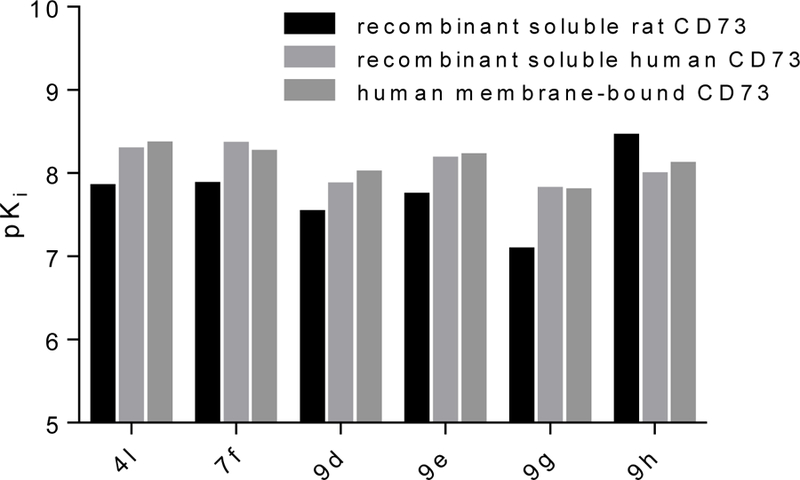
Comparison of pKi values of selected compounds 4l, 7f 9e, 9h, 9d and 9g determined at recombinant soluble rat and human and at membrane bound native CD73.
Selectivity
UDP-activated receptors
Various pyrimidine-derived nucleoside 5′-diphosphonates were previously shown to activate two subtypes of P2Y receptor, P2Y6 and P2Y14 receptors, both of which are UDP-activated.41 Therefore, the activity of representative compounds 4l, 7f, and 9h was tested at human P2Y6 and P2Y14 receptors, and the results are summarized in Table 5. A calcium assay of P2Y6 receptor activation in astrocytoma cells42 and a fluorescent binding competition assay in whole mammalian cells (CHO) expressing the P2Y14 receptor43 were used. 5-Fluorouridine-5′-α,β-methylene-diphosphate (4l) activated the P2Y6 receptor (EC50 203 nM) and also showed affinity for the P2Y14 receptor (IC50 362 nM). The 4-benzoylcytidine derivative was (7f) was less potent at P2Y6 and P2Y14 receptors with potencies in the micromolar range. However, the benzyloxyimino-3-methylcytidine-5′-α,β-methylene-diphosphate (9h) was completely inactive at P2Y6 and P2Y14 receptors even at high concentration (3.0 µM).
Table 5.
Potencies of selected compounds at human soluble and membrane-bound CD73.
| Compd. | Rat soluble CD73 Ki ± SEM (nM) | Human soluble CD73 Ki ± SEM (nM) | Human membrane-bound CD73 Ki ± SEM (nM) |
|---|---|---|---|
| 4l | 14.8 ± 1.9 | 5.33 ± 0.73 | 4.51 ± 0.13 |
| 7f | 13.9 ± 1.6 | 4.58 ± 0.55 | 5.68 ± 0.75 |
| 9d | 30.3 ± 4.2 | 14.0 ± 1.6 | 10.1 ± 1.4 |
| 9e | 18.8 ± 3.2 | 6.88 ± 1.05 | 6.29 ± 0.45 |
| 9g | 85.1 ± 7.5 | 15.9 ± 1.1 | 16.6 ± 0.7 |
| 9h | 3.67 ± 0.26 | 10.6 ± 0.4 | 7.96 ± 0.57 |
Cytosolic nucleotidases
The synthesized compounds are negatively charged under physiological conditions and membrane permeability is therefore expected to be low. However, cellular uptake via transporters cannot be completely excluded. To test whether the developed CD73 inhibitors can additionally block cytosolic nucleotidases, we utilized a cytosolic preparation of CD73 knockout melanoma cells44 and tested inhibition of AMP hydrolysis by selected compounds 4l, 7f, 9h, 9e. Results are collected in Table 7. While a mixture of the phosphatase inhibitors NaF45 and levamisole46 inhibited ATP hydrolysis under the applied conditions by 70%, none of the four investigated CD73 inhibitors showed any significant inhibition of AMP hydrolysis. Adding one of the CD73 inhibitors to the mixture of NaF and levamisole did not change the percentage of AMP hydrolysis. This clearly indicates that the investigated compounds selectively inhibit AMP hydrolysis by CD73, but not by cytosolic nucleotidases.
Table 7.
Inhibition of cytosolic AMP hydrolysis by selected CD73 inhibitors investigated in a CD73-knockout melanoma cell line.
| Compound (concentration) | Inhibition of AMP hydrolysis ± SEM (%)a |
|---|---|
| Levamisole (1 mM) and NaF (5 mM) | 70 ± 1 |
| 4l (100 µM) | 4 ± 13 |
| 7f (100 µM) | −3 ± 6 |
| 9e (100 µM) | −5 ± 5 |
| 9h (100 µM) | −2 ± 3 |
Determined in a malachite green assay using a cytosolic extract of MaMel.65-CD73ko cells (for details see Experimental Section). Data are from three independent experiments performed in duplicate.
In situ ecto-nucleotidase activity assay
Different analytic approaches have been employed for the measurement of ecto-nucleotidase activities, including colorimetric Pi-liberating assays, capillary electrophoresis, chromatography-based assays, and lead nitrate-based enzyme histochemistry.3 The latter technique is of particular importance in defining the distribution of ecto-nucleotidase activities within a tissue, taking advantage of the abilities of these enzymes to generate inorganic phosphorus (Pi) when incubated with appropriate nucleotide substrates in the presence of lead nitrate, Pb(NO3)2. This technique was originally employed by Wachstein and Meisel for the histochemical characterization of hepatic phosphatases.48 Using this approach, tissue localization of CD73 and other ecto-nucleotidases has been characterized as brown signal in images of the murine brain [3], thoracic aortas [4], as well as other human and rodent tissues.3
Figure 6 depicts representative staining images of human tonsillar CD73-mediated AMPase activity, showing selective and spatially distinct localization of enzymatic activity in the germinal centers and connective tissues. Compound 9h at 1, 10 and 100 nM concentrations inhibited catalytic activity of CD73 in a concentration-dependent manner, in situ more potently than AOPCP, but it is a highly reversible CD73 inhibitor. Therefore, it was necessary for 9h to remain in the assay medium during both the tissue pre-treatment with the inhibitor tested and also subsequent incubation with AMP substrate.
Figure 6.
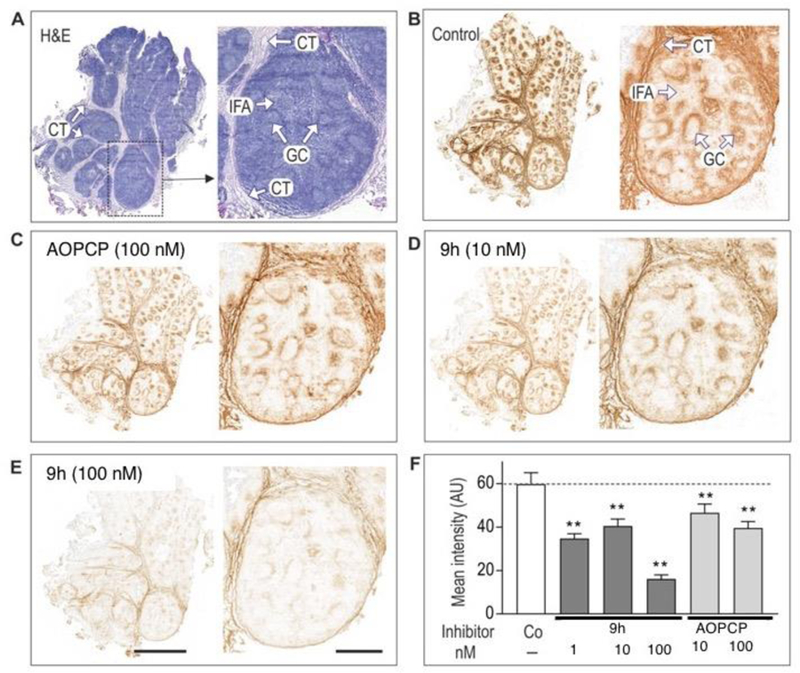
Histochemical analysis of the distribution of eN/CD73 in human tonsils. (A) Tissue samples were stained with hematoxylin and eosin (H&E). (B–E) Tonsillar eN/CD73 (AMPase) activity was assayed by incubating tissue cryosections with 100 µM AMP and 1.5 mM Pb(PO4)2 in the absence (Control) presence of the indicated concentrations of AOPCP and 9h, followed by microscopic detection of the nucleotide-derived Pi as a brown precipitate. All images were captured as tile scans of adjacent areas by using the Pannoramic 250 slide scanner. CT, connective tissue; GC, germinal center; IFA, inter-follicular area. Scale bars: 4 mm (left images) and 1 mm (right). (F) Quantification of the enzymatic activities using ImageJ are shown as mean pixel intensities (mean ± SE) of five germinal centers. **P<0.01 compared with control, determined by one-way ANOVA with Dunnett’s multiple comparison test.
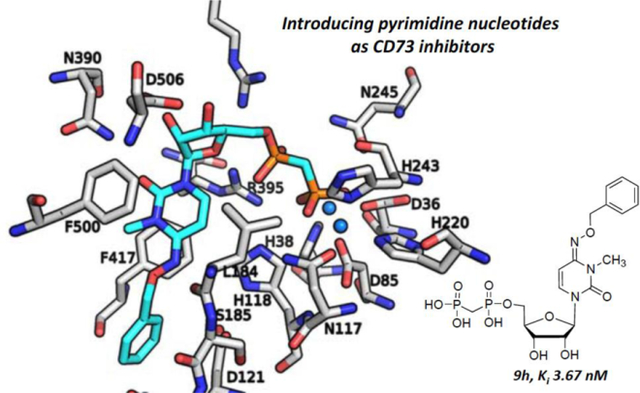
Molecular Modeling
Recently, a high resolution (2.0 Å, PDB ID: 4H2I) CD73-AOPCP complex structure in the closed form was reported, and this crystal structure provided insight into the interaction patterns of the purine derivative, AOPCP, with the amino acid residues and the two zinc ions in the binding site.49 In order to gain further insights into the molecular determinants involved in binding of the pyrimidine derivatives, we subsequently docked one of the potent and selective molecules, 9h, into the human CD73 binding site, and studied its interactions with the enzyme (Figure 7). Similar to AOPCP, the results from the docking studies with 9h showed that it occupies the same binding site (Supporting information, Figure S3), and the diphosphonate chain (PCP) is bound between the two zinc ions, the α-phosphonate forming H-bond interactions with Asn245, Arg354 and Arg395, and the β-phosphonate group with Asn117, His118, and Arg395 (Figure 7A–D). The binding orientation of AOPCP obtained from the crystal structure shows that the ribose hydroxyl groups form H-bond interactions with Arg354, Arg395, and Asp506. This interaction could be important for positioning the ligand in the correct orientation in the binding pocket of CD73. The importance of the ribose 2′- and 3′-hydroxyl groups was confirmed with derivatives 2a-d, where modification of the two hydroxyl groups in AOPCP was not tolerated. In particular, modification of the 2′-OH group leads to a large decrease in potency (2a) and indicates the importance of its interaction and presumably its role in ligand orientation in the substrate binding pocket. The docked pose of 9h shows the same interaction profile and binding orientation in the enzyme active site. Although the pyrimidine moiety of 9h (like the adenine ring of AOPCP) is stacked between Phe417 and Phe500 and stabilized through H-bonding interactions between the diphosphonate group (PCP) and the enzyme, it is expected to have weaker π-π interactions with the aromatic residues than the adenine ring of AOPCP. Due to the interactions of N3 (H-bond interaction with Asn390) and N1 (interaction with a water molecule) of the adenine ring with the enzyme, the N1- (2e) and N3-deaza analogs (2f) were not tolerated. In contrast, an N7-deaza-adenine modification (2g) is tolerated since N7 does not form any interaction with the amino acid residues or water molecules inside the binding pocket of CD73. The keto group at position 2 of the cytidine ring is oriented in the same direction as the N3 of the adenine ring and likely forms an interaction with the same residue, Asn390, as seen with AOPCP (Figure 7E). Adjacent to the exocyclic 6-amino group of the adenine ring, a large pocket is exposed to the surface with bound water molecules. This pocket was explored with different substitutions at the cytidine ring, and among them the benzyloxyimino residue at the 4-position of cytidine (9h) was found to confer high CD73-inhibitory activity (see Table 5). Upon exploring the pocket further, three loops (Asp121–Val124, Leu184–Asn190, Leu415–Thr420) were observed adjacent to the binding pocket of 9h which possibly provide hydrophobic as well as the polar or charged residues as potential interaction partners (Supplementary Figure S4). On the other hand, the small, electronegative substituent fluorine at the 5-position of the uracil moiety provided high potency in comparison to the other substituents including H, I and CH3. A possible explanation for this SAR pattern is that the electronegative fluorine atom is located at a distance of approximately 4 Å from His118 potentially forming a H-bond interaction directly with, or mediated through, the water molecules (located at a distance of ~3 Å) inside the binding pocket.
Figure 7.

Binding poses of AOPCP and 9h. (A) The binding pose of AOPCP (yellow) obtained from the X-ray structure (PDB ID: 4H2I used as a starting point) and (B) the docked pose of 9h (cyan) with important residues (gray) and zinc ions (marine blue) in the binding site of the human CD73. 2D interaction diagrams of (C) AOPCP and (D) 9h. (E) Binding pose comparison of AOPCP crystal structure (yellow) and docked 9h (cyan). The amino acid residues (gray), AOPCP (yellow) and 9h (cyan) are shown as stick models, and the zinc ions in the active site are represented as spheres (marine blue). Oxygen atoms are colored in red, nitrogen atoms in blue and phosphorus atoms in orange.
Discussion
We have prepared a range of nucleoside 5′-α,β-methylene-diphosphates by standard synthetic methods31,41,42,50–53 and evaluated them in vitro as CD73 inhibitors. The assay method using radiolabeled AMP substrate has been validated to have high reproducibility.34 This indirect approach to correcting an imbalance of excess adenosine that occurs in the microenvironment of a wide range of tumors holds promise as a co-therapy in the immunotherapy of cancer, and potent CD73 inhibitors (both small molecules and monoclonal antibodies) are already in preclinical and clinical development.12,17,32,54–58 Co-inhibition of CD73 and the A2AAR is also being considered for anti-cancer therapeutic development.59 In addition to cancer, CD73 inhibitors might have utility in preeclampsia, pulmonary edema, infectious disease, and other conditions.33,60,61
The current study presents important new SAR for AOPCP, UOCPC and COPCP analogues as inhibitors of CD73 (Figure 8). Furthermore, the recognition of a potent COPCP analogue 9h in the enzyme active site was modeled using a CD73 X-ray crystallographic structure, and this binding mode can be used to facilitate a structure-based approach in future SAR studies. While most of the ribose modifications resulted in weak inhibition or inactivity, substantial enhancement of CD73 inhibitory activity was observed with various nucleobase, both purine and pyrimidine, modifications. In the adenine series, most ribose modifications and 1-deaza and 3-deaza substitutions were detrimental, but 7-deaza was well tolerated. For example, on the ribose moiety 2′-methoxy and 2′-amino modifications or a [3.1.0]bicyclohexyl (S)-methanocarba modification resulted in loss of potency; only 2′-deoxy (intermediate) and 2′-deoxy-2′-fluoro (arabino, potent) analogues retained inhibitory activity. In the uracil series, the nucleobase N3 could be substituted with methyl but not larger alkyl groups. 2-Thiouracil, 3-deaza-uracil, 6-aza-uracil or 1,2-diphosphono-ethyl modifications were not tolerated. However, the uracil-5-position was amenable to various substitutions, with a rank order of potency of F > Cl, Br > I, Me.
Figure 8.
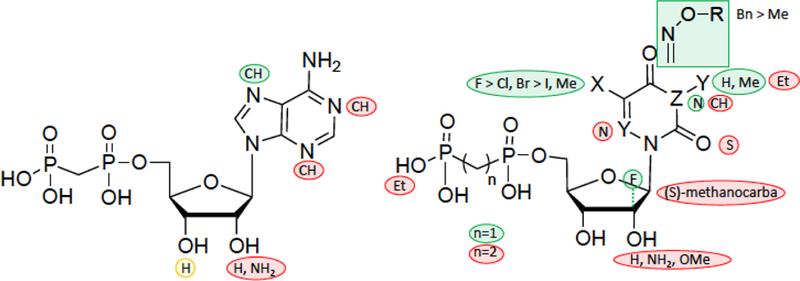
Summary of SAR for nucleotide derivatives as CD73 inhibitors in the purine (left) and pyrimidine series (right).
The most successful modification was found in N4-(aryl)alkyloxy-cytosine derivatives, especially with bulky benzyloxy substituents, which increased inhibitory potency at CD73. The α,β-methylene 5′-diphosphate derivatives of 5-fluorouridine (4l), 4-benzoylcytidine (7f), N4-[O-(naphthalen-2-ylmethoxy)]-cytidine (9e) and N4-[O-(4-benzyloxy)]-3-methyl-cytidine (9h) are the most potent CD73 inhibitors prepared in this work. At the human CD73 isoform, most of the potent inhibitors examined were at least several-fold more potent than at the rat isoform. Compound 9h displayed Ki values of (nM): 3.67 (soluble CD73, rat); 10.6 (soluble CD73, human); 7.96 (membrane-bound CD73, human). A major advantage of such pyrimidine-based inhibitors over adenine nucleotide analogues is that their hydrolysis products, the parent nucleosides, do not activate ARs, which would counteract the intended effects in cancer treatment. Also, 7-deazaadenosine derivatives are inactive or only very weakly active at adenosine receptors,47,71 and the 7-deazaadenine scaffold is therefore preferable to adenine for the development of CD73 inhibitors. In the CD73 crystal structure with bound AOPCP, the adenine N7 does not participate in any apparent stabilizing protein interactions, such as H-bonding. Thus, its replacement with CH in the more potent 7-deaza analogue 2g might conceivably displace a water molecule, but in the current X-ray structure no water is detected in this region.
The observed SAR was rationalized by docking 9h into the substrate binding site of human CD73. We were also concerned about off-target activity of the phosphonate analogues derived from uracil at the UDP-activated P2Y6 and P2Y14 receptors. The 5-F derivative 4l interacted with these two P2Y receptors at higher nanomolar concentrations, but the 4-benzoylcytidine derivative was (7f) less potent at P2Y6 and P2Y14 receptors. The potent inhibition by compound 9h of CD73 in situ in human tonsils was demonstrated, and it was more potent than the reference CD73 inhibitor AOPCP. Importantly, the benzyloxyimino-3-methyl-α,β-methylene-diphosphate (9h) was completely inactive at P2Y6 and P2Y14 receptors and in inhibition of cytosolic CD73, making it a particularly useful pharmacological tool compound for the in vitro and in vivo exploration of CD73 inhibition and an excellent starting point for future development of CD73 drugs that act parenterally.
Conclusions
A series of fifty purine- and pyrimidine-based nucleoside 5′-α,β-methylene-diphosphates was synthesized and obtained in high purity. The synthesized nucleosides were evaluated as inhibitors of CD73 in two species: rat and human (selected analogues). The AOPCP-derived nucleotides were modified at the ribose and the adenine moiety to complement the existing SAR in the adenine series, and to explore an alternative series of pyrimidine bases. These uridine- and cytosine-derived α,β-methylene diphosphonates represent an entirely new class of CD73 inhibitors. Analysis of their SAR allowed optimization leading to the development of inhibitors with Ki values in the low nanomolar range. N4-(Aryl)alkyloxy-cytosine derivatives, especially with bulky benzyloxy substituents, increased potency. The most potent inhibitors at rat CD73 were 5-fluorouridine (4l, 14.8 nM), the 4-benzoylcytidine (7f, 13.9 nM), N4-[O-(naphthalen-2-ylmethoxy)]-cytidine (9e, 18.8 nM) and N4-[O-(4-benzyloxy)]-3-methyl-cytidine (9h, 3.67 nM) 5′-α,β-methylene-diphosphates. Compound 9h displayed particularly high selectivity for CD73 compared to UDP-activated P2Y (P2Y6 and P2Y14) receptors and to cytosolic 5′-nucleotidase. Thus, we have expanded the SAR of both purine and pyrimidine nucleotide analogues as inhibitors of CD73 and achieved low nanomolar affinity. 5-Halo- and alkyl-uracil derivatives were also potent CD73 inhibitors. The presented compounds include the most potent CD73 inhibitors reported to date and are likely to become useful pharmacological tools to further elucidate the enzyme’s (patho)physiological role and its potential as a drug target in cancer immunotherapy and in other conditions.
Experimental Section
Chemical synthesis
General
Reagents and instrumentation.
All reagents were commercially obtained from various producers (Alfa Aesar, Carbosynth, and Sigma Aldrich) and used without further purification. The purity of all compounds including starting material was more than 95%, as determined using HPLC. Commercial solvents of specific reagent grades were used, without additional purification or drying. Analytical thin-layer chromatography was carried out on Sigma-Aldrich® TLC plates and compounds were visualized with UV light at 254 nm. Silica gel flash chromatography was performed using 230–400 mesh silica gel. Unless noted otherwise, reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). The 1H, 31P, and 13C NMR spectra were recorded using Bruker 400 MHz spectrometer, a DD2 400 MHz or DD2 600 MHz NMR spectrometer (Agilent). DMSO-d6, MeOD-d4, CDCl3 or D2O were used as solvents. Shifts are given in ppm relative to the remaining protons of the deuterated solvents used as internal standard (1H-, 13C-NMR). Purification of final compounds was performed by semi-preparative HPLC (Column: Luna 5 µm C18(2) 100 Å, LC Column 250 × 4.6 mm). Eluent: 10 mM triethylammonium acetate buffer - CH3CN from 80:20 to 20:80 in 40 min, with a flow rate of 5 mL/min. Purities of all tested compounds were ≥95%, as estimated by analytical HPLC: Method A: Eluent: 5 mM triethylammonium phosphate monobasic solution - CH3CN from 100:0 to 50:50 in 20 min, then triethylammonium phosphate monobasic solution - CH3CN to 100:0 in 5 min with a flow rate of 1 mL/min (Column: Zorbax SB-Aq 5 µm analytical column, 50 X 4.6 mm; Agilent Technologies, Inc). Method B: Eluent: 5 mM triethylammonium phosphate monobasic solution - CH3CN from 90:10 to 0:100 in 20 min, then triethylammonium phosphate monobasic solution - CH3CN from 0:100 to 90:10 in 5 min with a flow rate of 1 mL/min (Column: Zorbax SB-Aq 5 µm analytical column, 150 X 4.6 mm; Agilent Technologies, Inc). Method C: Eluent: 5 mM triethylammonium phosphate monobasic solution - CH3CN from 80:20 to 20:80 in 20 min, then triethylammonium phosphate monobasic solution - CH3CN from 20:80 to 80:20 in 10 min with a flow rate of 1 mL/min (Column: Zorbax SB-Aq 5 µm analytical column, 150 X 4.6 mm; Agilent Technologies, Inc). Peaks were detected by UV absorption (254 nm) using a diode array detector. All derivatives tested for biological activity showed >95% purity in the HPLC system. Low-resolution mass spectrometry was performed with a JEOL SX102 spectrometer with 6-kV Xe atoms following desorption from a glycerol matrix or on an Agilent LC/MS 1100 MSD, with a Waters (Milford, MA) Atlantis C18 column. High-resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine. For lyophilization, a freeze dryer (Labconco FreeZone 4.5) was used.
Preparation of triethylammonium hydrogen carbonate buffer
A 1 M solution of TEAC was prepared by adding dry ice slowly to 1 M triethylamine solution in deionized water for several hours until the pH of approximately 8.4–8.6 was indicated using a pH meter.
Purification of nucleotides
Ion exchange chromatography
The crude nucleoside-5′-O-[(phosphonomethyl)phosphonic acid] derivatives were purified by ion exchange chromatography on an HPLC instrument UltiMate 3000 (Dionex Corp.) with a HiScaleTM 26 20 BH, 26 mm x130 mm length column. The column was packed with Source 15Q® gel, swelled in a 20 % EtOH-solution. Before running purification, the column was washed and equilibrated with deionized water. The sample was prepared by dissolving crude product in 0.5–1 mL of aqueous triethylammonium hydrogen carbonate buffer. Separation was achieved by running a solvent gradient of triethylammonium hydrogen carbonate buffer: deionized water from 0 : 100 for 5 min, then from 0 : 100 to 100 : 0 in 25 min, followed by a gradient from 100 : 0 to 0 : 100 in 20 min, and holding 0 : 100 for 10 min with a flow rate of 5 mL/min. The UV absorption was detected at 254 nm, 210 nm and 280 nm. Fractions were collected, and appropriate fractions pooled, diluted in water, and lyophilized.
General procedure A for the synthesis of nucleotides
To a solution of DCC (3 eq.) and the unprotected nucleoside in DMF (2 mL) methylene diphosphonic acid (1.5 eq.) was added at rt and the mixture was allowed to stir at rt for 6–24 h. Samples were withdrawn at 3–12 h interval for LC-MS to check the disappearance of nucleosides and to monitor the formation of the desired nucleotide. On the disappearance of a nucleoside, 10 mL of cold TEAC-solution was added. The mixture was stirred at rt for 30 min followed by filtration and lyophilization of the aqueous solution. The mixture of nucleotide and dinucleotide was separated by ion-exchange chromatography on Source 15Q. Fractions containing the product were pooled and evaporated to dryness. The compound was then purified by RP-HPLC using a gradient of 10 mM triethylammonium acetate buffer - CH3CN from 80:20 to 20:80 in 40 min, suitable fractions were pooled and lyophilized to obtain the final product as glassy solid.
General procedure B for the synthesis of nucleotides
A solution of methylenebis(phosphonic dichloride) (3 eq.) in trimethyl phosphate (2 mL), cooled to 0°C was added to a suspension of the corresponding nucleoside in trimethyl phosphate at 0°C. The reaction mixture was stirred at 0°C and samples were withdrawn at 10 min interval for LC-MS to check the disappearance of nucleosides. After 30 min, on the disappearance of a nucleoside, 7 mL of cold 1 M aqueous triethylammonium hydrogen carbonate buffer solution (pH 8.4–8.6) was added. It was stirred at 0 °C for 15 min followed by stirring at rt for 30 min. Trimethyl phosphate was extracted using (2 × 100 mL) of tert-butyl methyl ether, and the aqueous layer was lyophilized. The mixture of mononucleotide and dinucleotide was separated by ion-exchange chromatography on Source 15Q. Fractions containing the mononucleotide product were pooled and evaporated to dryness. The compound was then purified by RP-HPLC using a gradient of 10 mM triethylammonium acetate buffer - CH3CN from 80:20 to 20:80 in 40 min, then 10 mM triethylammonium acetate buffer - CH3CN from 100:0 to 90:10 in 40 min, then 100:0 in 5 min, with a flow rate of 5 mL/min, suitable fractions were pooled and lyophilized to obtain final product as glassy solid.
2′-Deoxyadenosine-5′-O-[(phosphonomethyl)phosphonic acid] (2a).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 33.6 mg, 15 %). 1H NMR (400 MHz, D2O): δ 8.41 (s, 1H), 8.16 (s, 1H), 6.41 (t, J = 5.9 Hz, 1H), 4.17 (s, 1H), 4.08 – 3.90 (m, 3H), 3.10 (q, J = 7.3 Hz, 11H), 2.76 (dt, J = 13.3, 6.0 Hz, 1H), 2.55 – 2.43 (m, 1H), 2.04 (t, J = 18.6 Hz, 2H), 1.18 (t, J = 7.3 Hz, 16H). 31P NMR (160 MHz, D2O): δ 21.8, 11.8. MS (ESI, m/z) 408.0 [M-H]−; ESI-HRMS calcd. m/z for C11H16N5O8P2 408.0474, found 408.0479 [M-H]−. HPLC purity 95 % (Rt = 4.8 min, Method HPLC-A).
2′-Amino-2′-deoxyadenosine-5′-O-[(phosphonomethyl)phosphonic acid] (2b).
Method A. The product was obtained as colorless solid after lyophilization (0.5 eq Et3N-salt, 1.5 mg, 0.6 %). 1H NMR (400 MHz, D2O): δ 8.59 (s, 1H), 8.28 (s, 1H), 6.45 (d, J = 7.4 Hz, 1H), 4.88 – 4.84 (m, 1H), 4.64 (t, J = 5.6 Hz, 1H), 4.52 (s, 1H), 4.18 (q, J = 11.7, 10.7 Hz, 2H), 3.20 (q, J = 7.3 Hz, 3H), 2.19 (t, J = 19.0 Hz, 2H), 1.28 (t, J = 7.3 Hz, 5H). 13C NMR (100 MHz, D2O): δ 155.7, 153.0, 149.3, 140.1, 118.8, 86.0 (d, J = 6.0 Hz), 84.6, 70.1, 63.7, 56.3, 46.8 (2C), 27.6 (t, J = 124.3 Hz), 8.3 (2C). 31P NMR (160 MHz, D2O): δ 18.6, 14.6.MS (ESI, m/z) 423.1 [M-H]−; ESI-HRMS calcd. m/z for C11H17N6O8P2 423.0583, found 423.0590 [M-H]−. HPLC purity 98 % (Rt = 9.9 min, Method HPLC-B).
2′-Amino-2′-deoxyadenosine-3′-O-[(phosphonomethyl)phosphonic acid] (2c).
Method A. The product was obtained as colorless solid after lyophilization (0.66 eq Et3N-salt, 1.1 mg, 0.5 %). 1H NMR (400 MHz, D2O): δ 8.37 (s, 1H), 8.28 (s, 1H), 6.50 (d, J = 7.6 Hz, 1H), 5.22 (t, J = 5.6 Hz, 1H), 4.79 (s, 1H), 4.59 (s, 1H), 3.94 (d, J = 2.7 Hz, 2H), 3.21 (q, J = 7.3 Hz, 4H), 2.29 (t, J = 19.4 Hz, 2H), 1.28 (t, J = 7.3 Hz, 6H). 13C NMR (100 MHz, D2O): δ 155.9, 152.9, 148.7, 141.0, 119.5, 87.3, 86.4, 73.3, 61.3, 54.8, 46.8 (2C), 28.2 (t, J = 124.4 Hz), 8.3 (2C). 31P NMR (160 MHz, D2O): δ 18.8, 14.2. MS (ESI, m/z) 423.1 [M-H]−; ESI-HRMS calcd. m/z for C11H17N6O8P2 423.0583, found 423.0582 [M-H]−. HPLC purity 98 % (Rt = 6.9 min, Method HPLC-B).
3′-Deoxyadenosine-5′-O-[(phosphonomethyl)phosphonic acid] (2d).
Method A. The product was obtained as colorless solid after lyophilization (1.5 eq Et3N-salt, 2.5 mg, 6 %). 1H NMR (400 MHz, D2O): δ 8.49 (s, 1H), 8.25 (s, 1H), 6.10 (s, 1H), 4.77 – 4.65 (m, 1H), 4.25 (d, J = 10.2 Hz, 1H), 4.13 – 4.01 (m, 1H), 3.19 (q, J = 7.3 Hz, 10H), 2.42 (ddd, J = 14.5, 9.2, 5.7 Hz, 1H), 2.25 – 2.09 (m, 3H), 1.27 (t, J = 7.3 Hz, 15H). 13C NMR (100 MHz, D2O): δ 154.7, 151.4, 148.4, 140.3, 118.8, 90.6, 80.4 (d, J = 7.3 Hz), 75.4, 64.9, 46.8 (4C), 33.0, 27.5 (t, J = 124.5 Hz), 8.3 (4C). 31P NMR (160 MHz, D2O): δ 18.4, 14.9. MS (ESI, m/z) 408.0 [M-H]−; ESI-HRMS calcd. m/z for C11H16N5O8P2 408.0474, found 408.0471 [M-H]−. HPLC purity 99 % (Rt = 17.1 min, Method HPLC-B).
1-Deazaadenosine-5′-O-[(phosphonomethyl)phosphonic acid] (2e).
Method B. The product was obtained as colorless solid after lyophilization (1.5 eq Et3N-salt, 1.7 mg, 8 %). 1H NMR (400 MHz, D2O): δ 8.50 (s, 1H), 7.99 (d, J = 5.9 Hz, 1H), 6.67 (d, J = 5.9 Hz, 1H), 6.14 (d, J = 5.9 Hz, 1H), 4.53 (s, 1H), 4.37 (s, 1H), 4.15 (s, 2H), 3.18 (q, J = 7.3 Hz, 7H), 2.17 (t, J = 19.5 Hz, 2H), 1.26 (t, J = 7.3 Hz, 12H). 13C NMR (100 MHz, D2O): δ 147.9, 145.3, 144.2, 139.7, 104.3, 87.0, 84.0 (d, J = 6.2 Hz), 73.9, 70.4, 63.7, 46.7 (4C), 27.4, 8.3 (4C). 31P NMR (160 MHz, D2O): δ 18.6, 14.6. MS (ESI, m/z) 425.0 [M+H]+; ESI-HRMS calcd. m/z for C12H19N4O9P2 425.0622, found 425.0626 [M+H]+. HPLC purity 98 % (Rt = 8 min, Method HPLC-C).
3-Deazaadenosine-5′-O-[(phosphonomethyl)phosphonic acid] (2f).
Method B. 1H NMR (400 MHz, D2O): 8.58 (s, 1H), 7.75 (s, 1H), 7.32 (d, J = 6.2 Hz, 1H), 6.06 (d, J = 6.2 Hz, 1H), 4.69 (t, J = 5.9 Hz, 1H), 4.58 – 4.52 (m, 1H), 4.41 (s, 1H), 4.20 (s, 2H), 3.21 (q, J = 7.3 Hz, 3H), 2.17 (t, J = 19.3 Hz, 3H), 1.28 (t, J = 7.3 Hz, 6H). 31P NMR (160 MHz, D2O): δ 18.8, 14.4. MS (ESI, m/z) 423.1 [M-H]−; ESI-HRMS calcd. m/z for C12H17N4O9P2 423.0476, found 423.0473 [M-H]−. HPLC purity 97 % (Rt = 9.9 min, Method HPLC-C).
7-Deazaadenosine-5′-O-[(phosphonomethyl)phosphonic acid] (2g).
Method B. The product was obtained as colorless solid after lyophilization (1 eq Et3N-salt, 13.5 mg, 14 %). 1H NMR (600 MHz, D2O): δ 8.13 (bs, 1H), 7.61 (s, 1H), 6.70 (s, 1H), 6.14 (d, J = 5.4 Hz, 1H), 4.52 (t, J = 5.5 Hz, 1H), 4.47 (t, J = 4.4 Hz, 1H), 4.36–4.31 (m, 1H), 4.29 (d, J = 11.6 Hz, 1H), 4.17 (d, J = 11.5 Hz, 1H), 3.18 (q, J = 7.3 Hz, 6H), 2.24 (t, J = 20.5 Hz, 2H), 1.26 (t, J = 7.3 Hz, 9H). 13C NMR (100 MHz, D2O): δ 150.8, 146.9, 142.9, 124.6, 102.6, 102.1, 86.7, 83.7 (d, J = 5.4 Hz), 74.6, 70.3, 63.9, 46.7 (3C), 27.6 (t, J = 124.5 Hz), 8.3 (3C). 31P NMR (160 MHz, D2O): δ 18.5, 15.1. MS (ESI m/z) 423.1 [M-H]−; ESI-HRMS calcd. m/z for C12H17N4O9P2 423.0476, found 423.0476 [M-H]−. HPLC purity >99 % (Rt = 4.3 min, Method HPLC-C).
Uridine-5′-O-[(phosphonomethyl)phosphonic acid] (4a).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 6 mg, 5 %). 1H NMR (400 MHz, D2O): δ 7.91 (d, J = 8.1 Hz, 1H), 5.90 – 5.80 (m, 2H), 4.34 – 4.24 (m, 2H), 4.17 – 4.14 (m, 1H), 4.13 – 4.02 (m, 2H), 3.10 (q, J = 7.3 Hz, 12H), 2.04 (t, J = 19.6 Hz, 2H), 1.17 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 166.5 152.0, 141.9, 102.6, 88.7, 83.4 (d, J = 6.9 Hz), 73.9, 69.5, 63.3, 46.8 (6C), 27.8 (t, J = 122.4 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 20.6, 12.7. MS (ESI, m/z) 401.0 [M-H]−; ESI-HRMS calcd. m/z for C10H15N2O11P2 401.0157, found 401.0154 [M-H]−. HPLC purity 99 % (Rt = 5.6 min, Method HPLC-B).
3-Methyluridine-5′-O-[(phosphonomethyl)phosphonic acid] (4b).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 4.8 mg, 19 %). 1H NMR (400 MHz, D2O): δ 8.00 (d, J = 8.0 Hz, 1H), 6.02 (d, J = 8.0 Hz, 1H), 5.98 (d, J = 3.0 Hz, 1H), 4.38 (d, J = 3.6 Hz, 2H), 4.27 (s, 1H), 4.20 (s, 2H), 3.30 (s, 3H), 3.21 (q, J = 7.3 Hz, 12H), 2.12 (t, J = 19.3 Hz, 2H), 1.29 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 165.7, 152.3, 139.7, 101.8, 89.8, 83.1, 74.0, 69.3, 63.1, 46.8 (6C), 27.8, 8.3 (6C). 31P NMR (160 MHz, D2O): δ 20.2, 13.2. MS (ESI, m/z) 415.1 [M-H]−; ESI-HRMS calcd. m/z for C11H17N2O11P2 415.0308, found 415.0311 [M-H]−. HPLC purity 96 % (Rt = 3.7 min, Method HPLC-C).
3-Ethyluridine-5′-O-[(phosphonomethyl)phosphonic acid] (4c).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 26.1 mg, 11 %). 1H NMR (400 MHz, D2O): δ 8.01 (d, J = 7.1 Hz, 1H), 6.00 (d, J = 6.5 Hz, 1H), 5.97–5.94 (m, 1H), 4.47–4.34 (m, 2H), 4.30–4.16 (m, 3H), 3.93 (q, J = 6.9 Hz, 2H), 3.19 (q, J = 7.4 Hz), 2.19–1.94 (m, 2H), 1.27 (t, J = 7.3 Hz), 1.18 (t, J = 7.1 Hz). 13C NMR (100 MHz, D2O): δ 165.3, 151.8, 139.8, 102.0, 89.8, 83.1, 74.1, 69.0, 62.9, 46.7 (6C), 36.8, 11.8, 8.3 (6C). The signal for PCH2P could not be observed. 31P NMR (160 MHz, D2O): δ 21.1, 12.0. MS (ESI, m/z) 429.1 [M-H]−; ESI-HRMS calcd. m/z for C12H19N2O11P2 429.0470, found 429.0470 [M-H]−. HPLC purity 97 % (Rt = 9.8 min, Method HPLC-C).
3-Propyluridine-5′-O-[(phosphonomethyl)phosphonic acid] (4d).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 11 mg, 5 %). 1H NMR (400 MHz, D2O): δ 8.00 (d, J = 7.8 Hz, 1H), 6.03 (d, J = 7.7 Hz, 1H), 6.00 (d, J = 3.5 Hz, 1H), 4.43–4.39 (m, 2H), 4.32–4.27 (m, 1H), 4.26–4.13 (m, 2H), 3.87 (dd, J = 8.7, 6.6 Hz, 2H), 3.22 (q, J = 7.3 Hz, 12H), 2.30–2.11 (m, 2H), 1.64 (dq, J = 14.8, 7.5 Hz, 2H), 1.30 (t, J = 7.3 Hz, 18H), 0.92 (t, J = 7.5 Hz, 3H). 13C NMR (100 MHz, D2O): δ 165.5, 152.1, 139.8, 102.1, 89.5, 83.3, 73.9, 69.5, 63.3, 46.8 (6C), 43.2, 27.2 (1C, PCH2P), 20.3, 10.6, 8.3 (6C). The signal for PCH2P could not be observed in 1D experiment. 13C-NMR shift of PCH2P was determined using HSQC. 31P NMR (160 MHz, D2O): δ 18.4, 14.5. MS (ESI, m/z) 443.1 [M-H]−; ESI-HRMS calcd. m/z for C13H21N2O11P2 443.0626, found 443.0634 [M-H]−. HPLC purity 95 % (Rt = 10.3 min, Method HPLC-C).
3-Benzyluridine-5′-O-[(phosphonomethyl)phosphonic acid] (4e).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 3.7 mg, 2 %). 1H NMR (400 MHz, D2O): δ 8.07 (d, J = 7.9 Hz, 1H), 7.44–7.38 (m, 2H), 7.38–7.32 (m, 3H), 6.09 (d, J = 7.9 Hz, 1H), 5.99 (d, J = 3.7 Hz, 1H), 5.16 (d, J = 15.1 Hz), 5.11 (d, J = 15.2 Hz), 4.39 (d, J = 3.9 Hz, 2H), 4.30–4.26 (m, 1H), 4.20 (q, J = 11.4 Hz, 2H), 3.20 (q, J = 7.3 Hz, 12H), 2.20 (t, J = 18.3 Hz, 2H), 1.28 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 165.2, 152.1, 140.1, 136.1, 128.9 (2C), 127.8, 127.1 (2C), 102.1, 89.5, 83.3 (1C), 74.0, 69.5, 63.2, 46.7 (6C), 44.6, 27.5 (t, J = 124.1 Hz, 1C, PCH2P), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.4, 14.9. MS (ESI, m/z) 491.1 [M-H]−; ESI-HRMS calcd. m/z for for C17H21N2O11P2 491.0626, found 491.0622 [M-H]−. HPLC purity 98 % (Rt = 11.2 min, Method HPLC-C).
5-Methyluridine-5′-O-[(phosphonomethyl)phosphonic acid] (4f).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 54 mg, 24 %). 1H NMR (400 MHz, D2O): δ 7.77 (s, 1H), 5.99 (d, J = 5.0 Hz, 1H), 4.46 – 4.34 (m, 2H), 4.26 (s, 1H), 4.16 (q, J = 4.7, 4.1 Hz, 2H), 3.20 (q, J = 7.3 Hz, 12H), 2.20 (t, J = 19.7 Hz, 2H), 1.94 (s, 3H), 1.28 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 166.6, 152.1, 137.3, 111.9, 88.1, 83.6 (d, J = 8.0 Hz), 73.5, 69.9, 63.6 (d, J = 4.3 Hz), 59.0, 46.7 (6C), 27.6 (t, J = 124.2 Hz), 11.7, 8.3 (6C), 7.5. 31P NMR (160 MHz, D2O): δ 18.1 (d, J = 9.4 Hz), 14.7 (d, J = 9.4 Hz). MS (ESI, m/z) 415.0 [M-H]−; ESI-HRMS calcd. m/z for C11H17N2O11P2 415.0308, found 415.0302 [M-H]−. HPLC purity 95 % (Rt = 2.9 min, Method HPLC-B).
Thymidine-5′-O-[(phosphonomethyl)phosphonic acid] (4g).
Method B. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 28.35 mg, 12 %). 1H NMR (400 MHz, D2O): δ 7.74 (d, J = 1.3 Hz, 1H), 6.34 (dd, J = 7.5, 6.3 Hz, 1H), 4.62 (dt, J = 6.4, 3.4 Hz, 1H), 4.19 – 4.14 (m, 1H), 4.14 – 4.07 (m, 2H), 3.20 (q, J = 7.3 Hz, 12H), 2.45 – 2.30 (m, 2H), 2.18 (t, J = 19.8 Hz, 2H), 1.93 (s, 3H), 1.28 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 151.8, 137.5, 111.8, 109.9, 85.5 (d, J = 7.2 Hz), 85.0, 71.0, 63.9, 46.7 (6C), 38.4, 26.9 (t, J = 124.2 Hz), 11.7, 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.1, 14.7. MS (ESI, m/z) 399.0 [M-H]−; ESI-HRMS calcd. m/z for C11H17N2O10P2 399.0358, found 399.0357 [M-H]−. HPLC purity 97 % (Rt = 9.3 min, Method HPLC-B).
2′-O-Methyl-5-methyluridine-5′-O-[(phosphonomethyl)phosphonic acid] (4h).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 21 mg, 9 %). 1H NMR (400 MHz, D2O): δ 7.79 (d, J = 1.3 Hz, 1H), 6.03 (d, J = 5.1 Hz, 1H), 4.53 (t, J = 5.0 Hz, 1H), 4.26 – 4.22 (m, 1H), 4.22–4.14 (m, 2H), 4.13 (t, J = 5.2 Hz, 1H), 3.49 (s, 3H), 3.20 (q, J = 7.3 Hz, 12H), 2.20 (td, J = 19.5, 2.0 Hz, 2H), 1.94 (d, J = 1.2 Hz, 3H), 1.28 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 166.6, 151.8, 137.2, 111.9, 86.7, 83.6 (d, J = 8.1 Hz), 82.4, 68.3, 63.4 (d, J = 5.2 Hz), 58.1, 46.7 (6C), 27.5 (t, J = 124.0 Hz), 11.7, 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.0, 14.7. MS (ESI, m/z) 429.0 [M-H]−; ESI-HRMS calcd. m/z for C12H19N2O11P2 429.0464, found 429.0465 [M-H]−. HPLC purity 95 % (Rt = 9.5 min Method HPLC-B).
5-Ethynylyluridine-5′-O-[(phosphonomethyl)phosphonic acid] (4i).
Method B. The product was obtained as brown solid after lyophilization (2 eq Et3N-salt, 6.7 mg, 3 %). 1H NMR (600 MHz, D2O): δ 8.26 (s, 1H), 5.92 (d, J = 4.0 Hz, 1H), 4.37 (dt, J = 13.8, 4.9 Hz, 2H), 4.28–4.24 (m, 1H), 4.20 (d, J = 10.0 Hz, 1H), 4.14 (d, J = 11.6 Hz, 1H), 3.63 (s, 1H), 3.18 (q, J = 7.3 Hz, 12H), 2.25–2.12 (m, 2H), 1.26 (t, J = 7.3 Hz, 18H). 13C NMR (150 MHz, D2O): δ 164.7, 150.7, 145.4, 98.8, 89.2, 83.7, 83.4, 74.5, 74.1, 69.4, 62.9, 46.7 (6C), 27.3 (t, J = 126.5 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 17.1 (br). MS (ESI, m/z) 429.0 [M-H]−; ESI-HRMS calcd. m/z for C12H15N2O11P2 425.0157, found 425.0165 [M-H]− HPLC purity 99 % (Rt = 8.6 min Method HPLC-C).
5-(1-Chlorovinyl)uridine-5′-O-[(phosphonomethyl)phosphonic acid] (4j)
The compound was obtained as a side product during the synthesis of compound 4i via Method B. The product was obtained as brown solid after lyophilisation (3 eq Et3N-salt, 2.7 mg, 1 %). 1H NMR (600 MHz, D2O): δ 8.10 (d, J = 0.7 Hz, 1H), 6.01 (dd, J = 1.6, 0.7 Hz, 1H), 5.95 (d, J = 5.1 Hz, 1H), 5.71 (d, J = 1.7, 0.8 Hz, 1H), 4.42 (t, J = 5.2 Hz, 1H), 4.35 (t, J = 4.9 Hz, 1H), 4.27 (q, J = 3.8 Hz, 1H), 4.17–4.13 (m, 2H), 3.18 (q, J = 7.3 Hz, 18H), 2.18 (t, J = 19.9 Hz, 2H), 1.26 (t, J = 7.3 Hz, 27H). 13C NMR (150 MHz, D2O): δ 163.1, 151.0, 140.8, 130.3, 119.0, 113.1, 89.2, 83.7 (d, J = 8.1), 73.8, 69.8, 63.4 (d, J = 5.4), 46.7 (9C), 27.5 (t, J = 124.5), 8.3 (9C). 31P NMR (160 MHz, D2O): δ 18.1, 15.2. MS (ESI, m/z) 461.0 [M-H]−; ESI-HRMS calcd. m/z for C12H16ClN2O11P2 460.9923, found 460.9923 [M-H]−. HPLC purity 95 % (Rt = 9.8 min, Method HPLC-C).
5-(1-Chlorovinyl)-3-methyluridine-5′-O-[(phosphonomethyl)phosphonic acid] (4k)
The compound was obtained as a side product during the synthesis of compound 4i via Method B. The product was obtained as brown solid after lyophilisation (3 eq Et3N-salt, 5.6 mg, 2 %). 1H NMR (600 MHz, D2O): δ 8.10 (d, J = 0.8 Hz, 1H), 5.97 (dd, J = 4.6, 0.9 Hz, 1H), 5.96 (dd, J = 1.6, 0.7 Hz, 1H), 5.71 (dd, J = 1.6, 1.0 Hz, 1H), 4.41 (t, J = 5.0 Hz, 1H), 4.34 (t, J = 5.2 Hz, 1H), 4.27 (q, J = 3.9 Hz, 1H), 4.20–4.12 (m, 2H), 3.30 (s, 3H), 3.18 (q, J = 7.3 Hz, 18H), 2.18 (t, J = 19.6 Hz, 2H), 1.26 (t, J = 7.3 Hz, 27H). 13C NMR (150 MHz, D2O): δ 162.6, 151.4, 138.7, 131.0, 119.1, 112.5, 90.2, 83.4 (d, J = 8.0 Hz), 73.9, 69.6, 63.3 (d, J = 5.1Hz), 46.7 (9C), 28.1, 27.5 (t, J = 125.0 Hz), 8.3 (9C). 31P NMR (160 MHz, D2O): δ 18.0, 15.4. MS (ESI, m/z) 475.0 [M-H]−; ESI-HRMS calcd. m/z for C13H18ClN2O11P2 475.0080, found 475.0093 [M-H]−. HPLC purity 95 % (Rt = 10.4 min, Method HPLC-C).
5-Fluorouridine-5′-O-[(phosphonomethyl)phosphonic acid] (4l).
Method B. The product was obtained as a colorless solid after lyophilisation (2 eq Et3N-salt, 32.2 mg, 6 %). 1H NMR (600 MHz, D2O): δ = 8.19 (d, J = 6.5 Hz, 1H), 5.96 (dt, J = 2.7, 1.6 Hz, 1H), 4.41–4.37 (m, 2H), 4.31–4.26 (m, 1H), 4.23 (ddd, J = 11.7, 4.8, 2.7 Hz, 1H), 4.17 (ddd, J = 11.7, 5.6, 3.0 Hz, 1H), 3.22 (q, J = 7.3 Hz, 12H), 2.24 (t, J = 19.9 Hz, 2H, PCH2P), 1.29 (t, J = 7.3 Hz, 18H). 13C NMR (150 MHz, D2O): δ 159.5 (d, J = 26.0 Hz), 150.4, 140.9 (d, J = 233.4 Hz), 125.7 (d, J = 34.8 Hz), 88.8, 83.5 (d, J = 8.0 Hz), 73.9, 69.5, 63.2 (d, J = 5.3 Hz), 46.7 (6C), 27.4 (t, J = 125.0 Hz, PCH2P), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.0, 15.4. MS (ESI, m/z) 419.0 [M-H]−; ESI-HRMS calcd. m/z for C10H14FN2O11P2 419.0062, found 419.0057 [M-H]−. HPLC purity 97 % (Rt = 9.3 min, Method HPLC-C).
5-Chlorouridine-5′-O-[(phosphonomethyl)phosphonic acid] (4m).
Method B. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 23.1 mg, 10 %). 1H NMR (400 MHz, D2O): δ 8.19 (d, J = 0.7 Hz, 1H), 5.92 (d, J = 4.4 Hz, 1H), 4.40–4.34 (m, 2H), 4.28–4.24 (m, 1H), 4.19 (ddd, J = 11.6, 4.6, 2.7 Hz, 1H), 4.13 (ddd, J = 11.8, 5.7, 3.1 Hz, 1H), 3.18 (q, J = 7.3 Hz, 12H), 2.22 (t, J = 19.9 Hz, 2H), 1.26 (t, J = 7.4 Hz, 18H). 31P NMR (160 MHz, D2O): δ 18.2, 14.9. MS (ESI, m/z) 435.0 [M-H]−; ESI-HRMS calcd. m/z for C10H14ClN2O11P2 434.9767, found 434.9776 [M-H]−. HPLC purity 97 % (Rt = 8.9 min, Method HPLC-C).
5-Bromouridine-5′-O-[(phosphonomethyl)phosphonic acid] (4n).
Method B. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 4.1 mg, 2 %). 1H NMR (400 MHz, D2O): δ 8.29 (s, 1H), 5.96 (d, J = 4.5 Hz, 1H), 4.46–4.38 (m, 2H), 4.32–4.28 (m, 1H), 4.25–4.16 (m, 2H), 3.22 (q, J = 7.3 Hz, 12H), 2.25 (t, J = 18.7 Hz, 2H, PCH2P), 1.30 (t, J = 7.3 Hz, 18H). 13C NMR (150 MHz, D2O): δ 162.0, 151.2, 141.0, 97.0, 89.0, 83.6, 74.0, 69.6, 63.1, 46.8 (6C), 27.5 (PCH2P), 8.3 (6C). The signal for PCH2P could not be observed in 1D experiment. 13C-NMR shift of PCH2P was determined using HSQC. 31P NMR (160 MHz, D2O): δ 18.6, 14.8. MS (ESI, m/z) 479.0 [M-H]−; ESI-HRMS calcd. m/z for C10H1479BrN2O11P2 478.9262, found 478.9264 [M-H]−. HPLC purity 95 % (Rt = 9.3 min, Method HPLC-C).
5-Iodouridine-5′-O-[(phosphonomethyl)phosphonic acid] (4o).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 17.5 mg, 9 %). 1H NMR (400 MHz, D2O): δ 8.27 (s, 1H), 5.93 (d, J = 4.7 Hz, 1H), 4.39 (dt, J = 13.0, 5.1 Hz, 2H), 4.31 – 4.21 (m, 1H), 4.22 – 4.10 (m, 2H), 3.21 (q, J = 7.3 Hz, 12H), 2.27 (t, J = 19.7 Hz, 2H), 1.28 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 163.6, 151.9, 145.9, 88.9, 83.6 (d, J = 8.4 Hz), 73.9, 69.7, 68.7, 63.2, 46.7 (6C), 28.0 (t, J = 124.0 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.4, 14.7. MS (ESI, m/z) 526.9 [M-H]−; ESI-HRMS calcd. m/z for C10H14N2O11IP2 526.9118, found 526.9123 [M-H]−. HPLC purity 98 % (Rt = 9.6 min Method HPLC-B).
2′-Deoxyuridine-5′-O-[(phosphonomethyl)phosphonic acid] (4p).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 16.2 mg, 13 %). 1H NMR (400 MHz, D2O): δ 7.98 (d, J = 8.1 Hz, 1H), 6.31 (t, J = 6.8 Hz, 1H), 5.94 (d, J = 8.1 Hz, 1H), 4.65 – 4.51 (m, 1H), 4.21 – 4.14 (m, 1H), 4.15 – 4.07 (m, 2H), 3.20 (q, J = 7.3 Hz, 12H), 2.39 (dd, J = 6.8, 5.0 Hz, 2H), 2.17 (t, J = 19.9 Hz, 2H), 1.28 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 166.4, 151.7, 142.2, 102.5, 85.8 (d, J = 7.5 Hz), 85.4, 70.9, 63.8 (d, J = 4.7 Hz), 46.7 (6C), 38.8, 27.5 (t, J = 124.0 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.3, 14.7. MS (ESI, m/z) 385.0 [M-H]−; ESI-HRMS calcd. m/z for C10H15N2O10P2 385.0202, found 385.0201 [M-H]−. HPLC purity 99 % (Rt = 16 min Method HPLC-B).
2′-Amino-2′-deoxyuridine-5′-O-[(phosphonomethyl)phosphonic acid] (4q).
Method A. The product was obtained as colorless solid after lyophilization (1 eq Et3N-salt, 4 mg, 8 %), containing 8 % of methylenediphosphonic acid. 1H NMR (400 MHz, D2O): δ 8.02 (d, J = 8.1 Hz, 1H), 6.28 (d, J = 7.6 Hz, 1H), 6.00 (d, J = 8.1 Hz, 1H), 4.70 (d, J = 4.6 Hz, 1H), 4.44 (s, 1H), 4.23 – 4.08 (m, 3H), 3.21 (q, J = 7.3 Hz, 6H), 2.18 (td, J = 19.8, 4.0 Hz, 2H), 1.28 (t, J = 7.3 Hz, 9H). 31P NMR (160 MHz, D2O): δ 18.5, 15.9 (8 % methylenediphosphonic acid), 14.4. MS (ESI, m/z) 400.0 [M-H]−; ESI-HRMS calcd. m/z for C10H16N3O10P2 400.0311, found 400.0304 [M-H]−. HPLC purity 96 % (Rt = 1.8 min, Method HPLC-A).
2′-Azido-2′-deoxyuridine-5′-O-[(phosphonomethyl)phosphonic acid] (4r).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 11 mg, 8 %). 1H NMR (400 MHz, D2O): δ 8.02 (d, J = 8.1 Hz, 1H), 6.02 (d, J = 5.2 Hz, 1H), 5.97 (d, J = 8.1 Hz, 1H), 4.62 (t, J = 5.2 Hz, 1H), 4.40 (t, J = 5.4 Hz, 1H), 4.27 – 4.09 (m, 3H), 3.20 (q, J = 7.3 Hz, 12H), 2.19 (t, J = 19.7 Hz, 2H), 1.28 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 166.3, 151.7, 141.6, 102.7, 87.1, 83.7 (d, J = 8.0 Hz), 70.1, 65.4, 63.0 (d, J = 3.8 Hz), 46.7 (6C), 27.5 (t, J = 124.6 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.4, 14.6. MS (ESI, m/z) 426.0 [M-H]−; ESI-HRMS calcd. m/z for C10H14N5O10P2 426.0216, found 426.0222 [M-H]−. HPLC purity 99 % (Rt = 17.5 min, Method HPLC-B).
2′-Fluoro-2′-deoxyuridine-5′-O-[(phosphonomethyl)phosphonic acid] (4s).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 25.3 mg, 10 %). 1H NMR (400 MHz, D2O): δ 7.97 (d, J = 8.1 Hz, 1H), 6.08 (dd, J = 17.8, 1.8 Hz, 1H), 5.93 (d, J = 8.1 Hz, 1H), 5.52 (ddd, J = 52.5, 4.6, 1.8 Hz), 4.52 (ddd, J = 21.6, 7.8, 4.6 Hz, 1H), 4.38 – 4.25 (m, 2H), 4.19 (ddd, J = 11.7, 5.8, 2.8 Hz, 1H), 3.20 (q, J = 7.3 Hz, 12H), 2.20 (t, J = 19.7 Hz, 2H), 1.28 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 166.4, 151.4, 142.1, 102.3, 93.5 (d, J = 185.9 Hz), 88.4 (d, J = 35.1 Hz), 81.6 (d, J = 7.9 Hz), 67.7 (d, J = 15.9 Hz), 62.1 (d, J = 4.9 Hz), 46.7 (6C), 27.5 (t, J = 124.6 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.4, 14.7. MS (ESI, m/z) 403.0 [M-H]−; ESI-HRMS calcd. m/z for C10H14N2O10FP2 403.0108, found 403.0105 [M-H]−. HPLC purity 98 % (Rt = 9.5 min, Method HPLC-B).
2′-ara-Fluoro-2′-deoxyuridine-5′-O-[(phosphonomethyl)phosphonic acid] (4t).
Method A. The product was obtained as colorless solid after lyophilization (1.5 eq Et3N-salt, 11 mg, 8 %). 1H NMR (400 MHz, D2O): δ 7.93 (dd, J = 8.1, 1.7 Hz, 1H), 6.32 (dd, J = 15.5, 4.3 Hz, 1H), 5.92 (d, J = 8.1 Hz, 1H), 5.23 (td, J = 51.7, 3.6 Hz, 1H), 4.57 (dt, J = 19.8, 3.6 Hz, 1H), 4.25 – 4.08 (m, 3H), 3.21 (q, J = 7.3 Hz, 9H), 2.19 (t, J = 19.7 Hz, 2H), 1.28 (t, J = 7.3 Hz, 14H). 13C NMR (100 MHz, D2O): δ 166.3, 151.4, 142.9, 101.9, 94.6 (d, J = 191.8 Hz), 83.6 (d, J = 16.9 Hz), 82.1, 73.2 (d, J = 26.0 Hz), 62.7, 46.8 (4C), 27.5, 8.3 (4C). 31P NMR (160 MHz, D2O): δ 20.6, 12.8. MS (ESI, m/z) 403.0 [M-H]−; ESI-HRMS calcd. m/z for C10H14N2O10FP2 403.0108, found 403.0112 [M-H]−. HPLC purity 98 % (Rt = 9.6 min, Method HPLC-B).
1(β-D-Arabinofuranosyl)-uridine-5′-O-[(phosphonomethyl)phosphonic acid] (4u).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 21.3 mg, 9 %). 1H NMR (400 MHz, D2O): δ = 7.96 (d, J = 8.1 Hz, 1H), 6.23 (d, J = 5.5 Hz, 1H), 5.94 (d, J = 8.1 Hz, 1H), 4.46 (t, J = 5.5 Hz, 1H), 4.29 (t, J = 6.0 Hz, 1H), 4.21 (tt, J = 11.6, 6.6 Hz), 4.10 (dt, J = 7.4, 3.9 Hz, 1H), 3.22 (q, J = 7.3 Hz, 12H), 2.21 (t, J = 19.7 Hz, 2H), 1.30 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 166.2, 151.4, 143.0, 101.3, 84.7, 81.2, 75.2, 73.8, 62.4, 46.7 (6C), 27.4 (t, J = 124.8 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.4, 14.9. MS (ESI, m/z) 401.0 [M-H]−; ESI-HRMS calcd. m/z for C10H15N2O11P2 401.0157, found 401.0144 [M-H]−. HPLC purity 99 % (Rt = 8.6 min, Method HPLC-C).
6-Azauridine-5′-O-[(phosphonomethyl)phosphonic acid] (4v).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 17.7 mg, 7 %). 1H NMR (400 MHz, D2O): δ 7.66 (s, 1H), 6.15 (d, J = 3.7 Hz, 1H), 4.65 (dd, J = 5.1, 3.7 Hz, 1H), 4.48 (t, J = 5.2 Hz, 1H), 4.30–4.21 (m, 1H), 4.16–3.98 (m 2H), 3.22 (q, J = 7.3 Hz, 12H), 2.13 (t, J = 17.8 Hz, 2H, PCH2P), 1.30 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 158.6, 150.0, 137.2, 89.7, 83.1, 72.6, 70.5, 64.0, 46.7 (6C), 27.1 (1C, PCH2P), 8.3 (6C). The signal of PCH2P could not be observed in 1D experiment. 13C-NMR shift of PCH2P was determined using HSQC. 31P NMR (160 MHz, D2O): δ 18.3, 15.0. MS (ESI, m/z) 403.0 [M-H]−; ESI-HRMS calcd. m/z for C9H14N3O11P2 402.0109, found 402.0098 [M-H]−. HPLC purity 96 % (Rt = 9.1 min, Method HPLC-C).
Uridine-5′-O-[(phosphonoethyl)phosphonic acid] (4w).
Method A. The product was obtained as colorless solid after lyophilization (3 eq Et3N-salt and 1 eq H3CCO2H, 60 mg, 8 %). 1H NMR (400 MHz, D2O): δ 7.93 (d, J = 8.1 Hz, 1H), 6.14 – 5.71 (m, 2H), 4.32 (p, J = 5.1 Hz, 2H), 4.28 – 4.22 (m, 1H), 4.17 – 4.01 (m, 2H), 3.18 (q, J = 7.3 Hz, 18H), 1.90 (s, 3H, H3CCO2H), 1.85 – 1.64 (m, 4H), 1.26 (t, J = 7.3 Hz, 27H). 13C NMR (100 MHz, D2O): δ 181.0, 166.2, 151.8, 141.6, 102.6, 88.6, 83.4 (d, J = 7.7 Hz), 73.9, 69.7, 63.1 (d, J = 5.5 Hz), 46.7 (9C), 23.2, 22.1 (dd, J = 133.5, 5.5 Hz), 20.4 (dd, J = 135.4, 4.2 Hz), 8.3 (9C). 31P NMR (160 MHz, D2O): δ 27.2 (d, J = 73.5 Hz), 24.0 (d, J = 73.5 Hz). MS (ESI, m/z) 415.0 [M-H]−; ESI-HRMS calcd. m/z for C11H17N2O11P2 415.0308, found 415.0311 [M-H]−. HPLC purity 99 % (Rt = 8.7 min, Method HPLC-B).
5-Methyluridine-5′-O-[(phosphonoethyl)phosphonic acid] (4x).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 68 mg, 10 %). 1H NMR (400 MHz, D2O): δ 7.74 (s, 1H), 5.99 (d, J = 5.3 Hz, 1H), 4.44 – 4.32 (m, 2H), 4.30 – 4.23 (m, 1H), 4.16–4.03 (m, 2H), 3.20 (q, J = 7.3 Hz, 12H), 1.94 (s, 3H), 1.90 – 1.65 (m, 4H), 1.28 (t, J = 7.3 Hz, 17H). 13C NMR (100 MHz, D2O): δ 166.6, 152.0, 137.1, 112.0, 88.1, 83.6 (d, J = 7.9 Hz), 73.6, 70.0, 63.4 (d, J = 5.5 Hz), 46.7 (6C), 22.0 (dd, J = 133.4, 5.0 Hz), 20.4 (dd, J = 134.8, 4.8 Hz), 11.8, 8.3 (6C). 31P NMR (160 MHz, D2O): δ 27.3 (d, J = 73.5 Hz), 24.1 (d, J = 73.5 Hz). MS (ESI, m/z) 429.0 [M-H]−; ESI-HRMS calcd. m/z for C12H19N2O11P2 429.0464, found 429.0472 [M-H]−. HPLC purity 99 % (Rt = 9.1 min, Method HPLC-B).
2-Thiouridine-5′-O-[(phosphonomethyl)phosphonic acid] (4y).7
The product was obtained as colorless solid after lyophilization (1.5 eq Et3N-salt). 1H NMR (400 MHz, D2O): δ 8.21 (d, J = 8.2 Hz, 1H), 6.63 (d, J = 2.8 Hz, 1H), 6.24 (d, J = 8.1 Hz, 1H), 4.51–4.40 (m, 1H), 4.35 (t, J = 5.7 Hz, 1H), 4.33–4.26 (m, 2H), 4.25–4.16 (m, 1H), 3.20 (q, J = 7.4 Hz, 10H), 2.17 (t, J = 19.6 Hz, 2H), 1.28 (t, J = 7.3 Hz, 15H). 31P NMR (160 MHz, D2O): δ 19.2, 14.0.
Cytidine-5′-O-[(phosphonomethyl)phosphonic acid] (7a).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 10.3 mg, 8 %). 1H NMR (400 MHz, D2O): δ 8.02 (d, J = 7.6 Hz, 1H), 6.13 (d, J = 7.5 Hz, 1H), 5.98 (d, J = 4.1 Hz, 1H), 4.35 (dq, J = 9.2, 5.0 Hz, 2H), 4.30 – 4.10 (m, 3H), 3.20 (q, J = 7.3 Hz, 12H), 2.19 (t, J = 19.7 Hz, 2H), 1.28 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 166.0, 157.4, 141.8, 96.5, 89.4, 82.9 (d, J = 7.3 Hz), 74.3, 69.3, 63.1, 46.8 (6C), 27.5 (t, J = 124.3 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.4, 14.6. MS (ESI, m/z) 403.0 [M-H]−; ESI-HRMS calcd. m/z for C10H16N3O10P2 400.0311, found 400.0309 [M-H]−. HPLC purity 96 % (Rt = 15.6 min, Method HPLC-B).
2′-Deoxycytidine-5′-O-[(phosphonomethyl)phosphonic acid] (7b).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 34 mg, 14 %). 1H NMR (400 MHz, D2O): δ 8.14 (d, J = 7.7 Hz, 1H), 6.28 (t, J = 6.5 Hz, 1H), 6.21 (d, J = 7.7 Hz, 1H), 4.60 (dt, J = 7.1, 3.8 Hz, 1H), 4.31 – 4.18 (m, 1H), 4.16 – 4.06 (m, 2H), 3.20 (q, J = 7.3 Hz, 9H), 2.51 – 2.28 (m, 2H), 2.17 (t, J = 19.8 Hz, 2H), 1.28 (t, J = 7.3 Hz, 14H). 13C NMR (100 MHz, D2O): δ 162.2, 152.4, 143.5, 95.7, 86.4, 86.1 (d, J = 7.3 Hz), 70.7, 63.6 (d, J = 4.3 Hz), 46.7 (4C), 39.5, 27.5 (t, J = 124.7 Hz), 8.3 (4C). 31P NMR (160 MHz, D2O): δ 18.3, 14.7. MS (ESI, m/z) 384.0 [M-H]−; ESI-HRMS calcd. m/z for C10H16N3O9P2 384.0362, found 384.0365 [M-H]−. HPLC purity 99 % (Rt = 8.8 min, Method HPLC-B).
5-Iodocytidine-5′-O-[(phosphonomethyl)phosphonic acid] (7c).
Method B. The product was obtained as colorless solid after lyophilization (1.75 eq Et3N-salt, 9.4 mg, 5 %). 1H NMR (400 MHz, D2O): δ 8.26 (s, 1H), 5.93 (d, 1H, J = 3.2 Hz), 4.41–4.33 (m, 2H), 4.31–4.27 (m, 1H), 4.21 (m, 2H), 3.22 (q, J = 7.3 Hz, 10.5H), 2.29 (t, J = 17.9 Hz, 2H, PCH2P), 1.30 (t, J = 7.3 Hz, 15.75H).13C NMR (100 MHz, D2O): δ 164.6, 156.6, 147.6, 89.8, 83.1, 74.4, 69.2, 62.9, 46.7 (5.25C), 27.7 (PCH2P), 8.3 (5.25C). The signal for PCH2P could not be observed in 1D experiment. 13C-NMR shift of PCH2P was determined using HSQC. 31P NMR (160 MHz, D2O): δ 18.5, 15.1. MS (ESI, m/z) 525.9 [M-H]−; ESI-HRMS calcd. m/z for C10H16IN3O10P2 525.9283, found 525.9275 [M-H]−. HPLC purity 99 % (Rt = 9.0 min, Method HPLC-C.
5-Fluorocytidine-5′-O-[(phosphonomethyl)phosphonic acid] (7d).
Method B. The product was obtained as colorless solid after lyophilisation (1.5 eq Et3N-salt, 15.5 mg, 7 %). 1H NMR (600 MHz, D2O): δ 8.15 (d, J = 6.3 Hz, 1H), 5.90 (dd, J = 3.9, 1.4 Hz, 1H), 4.34 (t, J = 5.3 Hz, 1H), 4.30 (dd, J = 5.1, 3.8 Hz, 1H), 4.27–4.19 (m, 2H), 4.14 (d, J = 11.8 Hz, 1H), 3.18 (q, J = 7.3 Hz, 9H), 2.19 (t, J = 19.2 Hz, 2H), 1.26 (t, J = 7.3 Hz, 15H). 13C NMR (150 MHz, D2O): δ 158.1 (d, J = 15.1 Hz), 155.4, 137.6 (d, J = 248.3 Hz), 126.0 (d, J = 33.0 Hz), 89.7, 82.9 (d, J = 6.7 Hz), 74.4, 69.1, 62.9 (d, J = 3.6 Hz), 46.7 (4.5C), 27.5 (t, J = 119.5 Hz), 8.3 (4.5C). 31P NMR (160 MHz, D2O): δ 18.9, 15.1. MS (ESI, m/z) 418.0 [M-H]−; ESI-HRMS calcd. m/z for C10H15FN3O10P2 418.0222, found 418.0235 [M-H]−. HPLC purity 98 % (Rt = 8.8 min, Method HPLC-C).
5-Methylcytidine-5′-O-[(phosphonomethyl)phosphonic acid] (7e).
Method B. The product was obtained as colorless solid after lyophilisation (1.5 eq Et3N-salt, 47.7 mg, 22 %). 1H NMR (600 MHz, D2O): δ 7.92 (s, 1H), 5.99 (d, 1H, J = 3.9 Hz), 4.42–4.35 (m, 2H), 4.32–4.25 (m, 1H), 4.25–4.13 (m, 2H, J = 11.6 Hz), 3.21 (q, J = 7.3 Hz, 9H), 2.21 (t, J = 19.0 Hz, 2H, PCH2P), 2.07 (s, 3H), 1.29 (t, J = 7.3 Hz, 13.5H). 13C NMR (150 MHz, D2O): δ 163.2, 153.8, 139.7, 89.2, 83.3, 74.2, 69.4, 63.3, 46.7 (4.5C), 27.5 (t, J = 122.7 Hz, 1C, PCH2P), 12.3, 8.3 (4.5C). 31P NMR (160 MHz, D2O): δ 18.0, 14.8. MS (ESI, m/z) 414.0 [M-H]−; ESI-HRMS calcd. m/z for C11H19N3O10P2 414.0473, found 414.0484 [M-H]−. HPLC purity 99 % (Rt = 7.0 min, Method HPLC-C).
4-Benzoylcytidine-5′-O-[(phosphonomethyl)phosphonic acid] (7f).
Method B. The product was obtained as colorless solid after lyophilisation (5 eq Et3N-salt, 39.2 mg, 13 %). 1H NMR (600 MHz, D2O): δ 8.47 (d, J = 7.0 Hz, 1H), 7.89 (d, J = 7.7 Hz, 2H), 7.67 (t, J = 7.4 Hz, 1H), 7.55 (t, J = 7.7 Hz, 2H), 7.52–7.44 (m, 1H), 5.96–5.94 (m, 1H), 4.39–4.33 (m, 2H), 4.33–4.28 (m, 2H), 4.20 (d, J = 11.6 Hz, 1H), 3.18 (q, J = 7.3 Hz, 30H), 2.29–2.10 (m, 2H), 1.26 (t, J = 7.3 Hz, 45H). 13C NMR (150 MHz, D2O): δ 169.5, 163.2, 156.7, 145.8, 133.6, 132.6, 129.0 (2C), 128.1 (2C), 98.8, 90.9 (d, J = 3.4 Hz), 82.8, 74.8, 68.7, 62.7, 46.7 (15C), 27.4 (t, J = 120.2 Hz), 8.3 (15C). 31P NMR (160 MHz, D2O): δ 18.5, 15.1. MS (ESI, m/z) 504.0 [M-H]−; ESI-HRMS calcd. m/z for C17H20N3O11P2 504.0579, found 504.0588 [M-H]−. HPLC purity 90 % (Rt = 10.5 min, Method HPLC-C). However, the compound displays decomposition in aqueous solution (Supporting information) and was tested for its CD73 inhibition at a purity of 75 %.
N4-[O-(Benzyloxy)]-2′-deoxycytidine-5′-O-[(phosphonomethyl)phosphonic acid] (9c).
Method B. The product was obtained as colorless solid after lyophilization (1.5 eq Et3N-salt, 4.3 mg, 4.4 %). 1H NMR (400 MHz, D2O): δ 7.50 – 7.37 (m, 5H), 7.20 (d, J = 8.3 Hz, 1H), 6.28 (t, J = 7.1 Hz, 1H), 5.71 (d, J = 8.3 Hz, 1H), 5.03 (s, 2H), 4.57 (dt, J = 5.9, 2.8 Hz, 1H), 4.16 – 3.97 (m, 3H), 3.19 (q, J = 7.3 Hz, 9H), 2.33 (dt, J = 14.1, 7.0 Hz, 1H), 2.23 (ddd, J = 14.1, 6.4, 3.3 Hz, 1H), 2.14 (t, J = 19.8 Hz, 2H), 1.27 (t, J = 7.3 Hz, 14H). 13C NMR (100 MHz, D2O): δ 147.2, 137.3, 132.3, 130.2, 128.8 (2C), 128.5, 128.4 (2C), 98.2, 85.1 (d, J = 7.8 Hz), 84.3, 75.6, 71.2, 63.9, 46.7 (4C), 37.7, 13.6, 8.3 (4C). 31P NMR (160 MHz, D2O): δ 21.3, 12.0. MS (ESI, m/z) 492.1 [M+H]ˉ; ESI-HRMS calcd. m/z for C17H24N3O10P2 492.0937, found 492.0928 [M+H]ˉ. HPLC purity 99 % (Rt = 10.9 min, Method HPLC-B).
N4-[O-(4-Trifluoromethylbenzyloxy)]-cytidine-5′-O-[(phosphonomethyl)phosphonic acid] (9d).
Method B. The product was obtained as colorless solid after lyophilisation (2 eq Et3N-salt, 32.1 mg, 17 %). 1H NMR (600 MHz, D2O): δ 7.69 (d, J = 8.0 Hz, 2H), 7.54 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 8.1 Hz, 1H), 5.91 (d, J = 4.8 Hz, 1H), 5.71 (d, J = 8.1 Hz, 1H), 5.10 (s, 2H), 4.35–4.31 (m, 2H), 4.22–4.19 (m, 1H), 4.12–4.07 (m, 2H), 3.18 (q, J = 7.3 Hz, 12H), 2.16 (t, J = 18.8 Hz, 2H), 1.26 (t, J = 7.3 Hz, 18H). 13C NMR (150 MHz, D2O): δ 151.0, 147.0, 141.8, 132.2, 129.5 (q, J = 31.9 Hz), 128.3 (2C), 125.5 (q, J = 3.8 Hz, 2C), 124.3 (q, J = 271.5 Hz), 98.3, 87.3, 83.4 (d, J = 6.6 Hz), 74.5, 72.7, 70.2, 63.8, 46.7 (6C), 27.4 (t, J = 124.4 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.2, 15.1. MS (ESI, m/z) 574.1 [M+H]ˉ; ESI-HRMS calcd. m/z for C18H21F3N3O11P2 574.0609, found 574.0616 [M-H]−. HPLC purity >99 % (Rt = 12.5 min, Method HPLC-C).
N4-[O-(Naphthalen-2-ylmethoxy)]-cytidine-5′-O-[(phosphonomethyl)phosphonic acid] (9e).
Method B. The product was obtained as colorless solid after lyophilisation (2 eq Et3N-salt, 4.5 mg, 2 %).
1H NMR (600 MHz, D2O): δ 7.97–7.94 (m, 3H), 7.93–7.91 (m, 1H), 7.60–7.57 (m, 3H), 7.20 (d, J = 8.2 Hz, 1H), 5.90 (d, J = 5.5 Hz, 1H), 5.72 (d, J = 8.2 Hz, 1H), 5.20 (s, 2H), 4.34–4.30 (m, 2H), 4.21–4.19 (m, 1H), 4.10–4.07 (m, 2H), 3.17 (q, J = 7.3 Hz, 12H), 2.14 (t, J = 19.2 Hz, 2H), 1.26 (t, J = 7.3 Hz, 18H). 13C NMR (150 MHz, D2O): δ 151.1, 147.0, 135.1, 133.0, 132.9, 132.1, 128.4, 128.0, 127.8, 127.2, 126.7, 126.6, 126.2, 98.4, 87.3, 83.4 (d, J = 7.2 Hz), 75.5, 72.6, 70.2, 63.8, 46.7 (6C), 27.5 (t, J = 127.0 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.3, 14.6. MS (ESI, m/z) 556.1 [M+H]ˉ; ESI-HRMS calcd. m/z for C21H24N3O11P2 556.0892, found 556.0901 [M-H]−. HPLC purity 99 % (Rt = 12.2 min, Method HPLC-C).
N4-[O-(4-Benzyloxy)]-5-methyl-cytidine-5′-O-[(phosphonomethyl)phosphonic acid] (9f).
Method B. The product was obtained as colorless solid after lyophilisation (2 eq Et3N-salt, 17.5 mg, 9 %). 1H NMR (400 MHz, D2O): δ 7.48–7.36 (m, 5H), 7.03–7.00 (m, 1H), 5.92–5.88 (m, 1H), 5.07 (s, 2H), 4.37–4.32 (m, 2H), 4.21–4.17 (m, 1H), 4.11–4.06 (m, 2H), 3.18 (q, J = 7.3 Hz, 12H), 2.16 (t, J = 19.6 Hz, 2H), 1.80 (s, 3H), 1.26 (t, J = 7.3 Hz, 18H). 31P NMR (160 MHz, D2O): δ 18.0, 14.7. MS (ESI, m/z) 520.1 [M+H]ˉ; ESI-HRMS calcd. m/z for C18H24N3O11P2 520.0892, found 520.0911 [M-H]−. HPLC purity >99 % (Rt = 10.9 min, Method HPLC-C).
N4-[O-(4-Benzyloxy)]-5-fluoro-cytidine-5′-O-[(phosphonomethyl)phosphonic acid] (9g).
Method B. The product was obtained as colorless solid after lyophilisation (2 eq Et3N- and 1 H2CO3- salt, 40.0 mg, 11 %). 1H NMR (600 MHz, D2O): δ 7.45–7.36 (m, 6H), 5.89 (d, J = 5.3 Hz, 1H), 5.09 (s, 2H), 4.35–4.31 (m, 1H), 4.29 (t, J = 5.5 Hz, 1H), 4.24–4.17 (m, 1H), 4.14–4.07 (m, 2H), 3.57 (q, J = 7.2 Hz, 6H), 3.17 (q, J = 7.3 Hz, 6H), 2.24–2.05 (m, 2H), 1.33 (t, J = 7.2 Hz, 9H), 1.26 (t, J = 7.3 Hz, 9H). 13C NMR (150 MHz, D2O): δ 149.7, 140.6 (d, J = 21.7 Hz), 137.9 (d, J = 235.3 Hz), 137.0, 128.8 (2C), 128.5, 128.3 (2C), 116.4 (d, J = 35.1 Hz), 87.6, 83.4, 75.9, 72.8, 70.1, 63.7, 58.5 (3C), 46.7 (3C), 27.2 (t, J = 124.4 Hz), 8.3 (3C), 7.19 (3C). 31P NMR (160 MHz, D2O): δ 16.5. MS (ESI, m/z) 524.1 [M+H]ˉ; ESI-HRMS calcd. m/z for C17H21FN3O11P2 524.0641, found 524.0645 [M-H]−. HPLC purity 99 % (Rt = 11.4 min, Method HPLC-C).
N4-[O-(4-Benzyloxy)]-3-methyl-cytidine-5′-O-[(phosphonomethyl)phosphonic acid] (9h).
Method B. The product was obtained as colorless solid after lyophilisation (2 eq Et3N-salt, 8.5 mg, 4 %). 1H NMR (600 MHz, D2O): δ 7.48–7.44 (m, 2H), 7.44–7.41 (m, 2H), 7.40–7.37 (m, 1H), 7.32 (d, J = 8.4 Hz, 1H), 6.44 (d, J = 8.3 Hz, 1H), 5.94 (d, J = 5.2 Hz, 1H), 5.01 (s, 2H), 4.34–4.30 (m, 2H), 4.20 (q, J = 3.1 Hz, 1H), 4.12–4.08 (m, 2H), 3.21–3.14 (m, 15H), 2.16 (t, J = 19.1 Hz, 2H), 1.26 (t, J = 7.4 Hz, 18H). 13C NMR (150 MHz, D2O): δ 153.9, 151.5, 136.9, 132.7, 128.9 (2C), 128.7 (2C), 128.5, 94.1, 88.4, 83.2 (d, J = 7.3 Hz), 75.6, 73.0, 70.0, 63.7 (d, J = 4.4 Hz,), 46.7 (6C), 29.3, 27.4 (t, J = 124.2 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.4, 15.0. MS (ESI, m/z) 520.1 [M+H]ˉ; ESI-HRMS calcd. m/z for C18H24N3O11P2 520.0892, found 520.0889 [M-H]−. HPLC purity >99 % (Rt = 11.6 min, Method HPLC-C).The synthesis was further optimized: A solution of methylenebis(phosphonic dichloride) (719 mg, 2.88 mmol, 1.5 eq.) in trimethyl phosphate (10.5 mL), cooled to 0°C was added to a suspension of 8h (697 mg, 1.92 mmol, 1.0 eq.) in trimethyl phosphate at 0°C. The reaction mixture was stirred at 0°C and samples were withdrawn at 10 min intervals for LC-MS. After 45 min, on the disappearance of 8h formate [M+CHO2-]- ([M-H+]+ disappeared after ~30 min) 7 mL of cold 1 M aqueous triethylammonium hydrogen carbonate buffer solution (pH 8.4−8.6) was added. It was stirred at 0 °C for 15 min followed by stirring at rt for 30 min. Due to the high lipophilicity of the product, no extraction was performed and instead the aqueous layer was lyophilized directly. Separation of the mixture of nucleotide and dinucleotide was done following method B to obtain final product as glassy solid (2 eq Et3N-salt, 423 mg, 0.584 mmol, 30 %).
N4-[O-(4-Benzyloxy)]-3-ethyl-cytidine-5′-O-[(phosphonomethyl)phosphonic acid] (9i).
Method B. The product was obtained as colorless solid after lyophilisation (2 eq Et3N-salt, 9.0 mg, 5 %). 1H NMR (600 MHz, D2O): δ 7.46 (dt, J = 7.2, 1.3 Hz, 2H), 7.42 (tq, J = 6.4, 1.1 Hz, 2H) 7.40–7.36 (m, 1H), 7.27 (dd, J = 8.4, 1.1 Hz, 1H), 6.41 (dd, J = 8.5, 1.2 Hz, 1H), 5.92 (dd, J = 5.4, 1.2 Hz, 1H), 5.00 (s, 2H), 4.34–4.29 (m, 2H), 4.19 (q, J = 3.2 Hz, 1H), 4.12–4.08 (m, 2H), 3.18 (q, J = 7.3 Hz, 12H), 2.16 (t, J = 18.9 Hz, 2H), 1.26 (t, J = 7.3 Hz, 18H). 13C NMR (150 MHz, D2O): δ 153.0, 151.1, 137.0, 132.7, 129.1 (2C), 128.7 (2C), 128.5, 94.3, 88.3, 83.2 (d, J = 7.4 Hz), 75.6, 72.9, 70.0, 63.7 (d, J = 4.6 Hz), 46.7 (6C), 27.4 (t, J = 124.4 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.3, 15.0. MS (ESI, m/z) 534.1 [M+H]ˉ; ESI-HRMS calcd. m/z for C19H26N3O11P2 534.1048, found 534.1074 [M-H]−. HPLC purity >99 % (Rt = 11.9 min, Method HPLC-B).
3-Deazauridine-5′-O-[(phosphonomethyl)phosphonic acid] (10).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 19 mg, 15 %). 1H NMR (400 MHz, D2O): δ 7.92 (d, J = 7.8 Hz, 1H), 6.31 (d, J = 7.6 Hz, 1H), 6.19 (d, J = 4.3 Hz, 1H), 5.87 (d, J = 2.6 Hz, 1H), 4.37 (dt, J = 18.0, 5.1 Hz, 2H), 4.33 – 4.27 (m, 1H), 4.27 – 4.09 (m, 2H), 3.21 (q, J = 7.3 Hz, 12H), 2.20 (t, J = 19.7 Hz, 2H), 1.28 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 169.7, 165.7, 134.7, 104.2, 88.4, 82.9 (d, J = 7.8 Hz), 74.7, 69.4, 63.2, 46.7 (6C), 27.5 (t, J = 124.3 Hz), 8.3 (6C). 31P NMR (160 MHz, D2O): δ 18.3, 14.7. MS (ESI, m/z) 400.0 [M-H]−; ESI-HRMS calcd. m/z for C11H16NO11IP2 400.0199, found 400.0203 [M-H]−. HPLC purity 96 % (Rt = 9.4 min Method HPLC-C).
(S)-Methanocarbauridine-5′-O-[(phosphonomethyl)phosphonic acid] (11).
Method A. The product was obtained as colorless solid after lyophilization (2 eq Et3N-salt, 1.4 mg, 5 %). 1H NMR (400 MHz, D2O): δ 7.76 (d, J = 7.8 Hz, 1H), 5.83 (d, J = 7.8 Hz, 1H), 4.67 (d, J = 6.3 Hz, 1H), 4.12 (d, J = 6.5 Hz, 1H), 4.10 – 3.98 (m, 2H), 3.21 (q, J = 7.3 Hz, 12H), 2.35 (s, 1H), 2.22 – 2.02 (m, 2H), 1.86 (dd, J = 9.5, 4.8 Hz, 1H), 1.68 (t, J = 5.5 Hz, 1H), 1.28 (t, J = 7.3 Hz, 18H). 13C NMR (100 MHz, D2O): δ 166.9, 152.7, 148.2, 101.8, 75.0, 71.9, 65.4, 51.6, 48.3, 46.8 (6C), 24.8, 15.7, 8.3 (6C). 31P NMR (160 MHz, D2O): δ 19.3, 14.1. MS (ESI, m/z) 411.0 [M-H]−; ESI-HRMS calcd. m/z for C12H17N2O10P2 411.0358, found 411.0363 [M-H]−. HPLC purity 96 % (Rt = 2.9 min, Method HPLC-A).
Soluble CD73 enzyme preparations
Soluble rat CD73 was expressed in Spodoptera frugiperda 9 (Sf9) insect cells and purified as previously described.29 The cDNA for the soluble human CD73 (Genbank accession no. NM_002526) was obtained from Prof. Dr. Norbert Sträter (University of Leipzig, Germany).49 In order to generate a soluble enzyme the signaling sequence for anchoring the protein to the membrane via a GPI-anchor had been omitted (N-terminal residues: 1–27, C-terminal residues: 550–574 including GPI-anchor attachment site).49 In addition, a 6xHis-Tag was fused to the C-terminus and the construct was cloned into the vector pACGP67B, which provides an N-terminal signal peptide for the protein secretion. Sf9 insect cells were grown in Insect-XPRESS™ media (#: BE12–730Q, Lonza, Switzerland) with 10 mg/l gentamicin and split at a ratio of 1:3 every fourth day. For transfection, cells were seeded into cell culture flasks (25 cm2) at 60–70% confluence. 100 µl of cell medium and 1 µl of vector DNA (1000 ng/µl) were mixed with 2.5 µl of baculovirus genomic ProEasy™ vector DNA (AB vector, CA, USA) and combined with premixed 100 µl of cell medium and 8 µl of Cellfectin™ II Reagent (Thermo Fisher Scientific, MA, USA). The transfection mixture was left for 30 min at rt and then dropwise added to the cells into the cell culture flasks. The cells were incubated for 30 min at rt, and for further 4 days at 27°C. Cells from the transfection procedure were detached from the bottom of the flasks and centrifuged for 5 min at 2000g. 1.5 mL of the supernatant (viral stock) was added to 75 cm2 cell culture flasks containing Sf9 cells (60–70% confluence), and the cells were incubated for four days at 27°C. Then 1.5 mL of the supernatant were taken and added to uninfected Sf9 cells in a 75 cm2 flask. This was repeated five more times, using more cells and larger flasks after the third round of infections (175 cm2 to which 3.0 ml of supernatant were added).
The final stock solution was used for infection of the cells. For protein expression, 3 ml of the virus solution were used to infect 150 ml of cell media containing 2×106 cells/ml in a 500 ml Erlenmeyer flask, and they were incubated for 4 days at 27°C with shaking (150 rpm). Then, cell suspensions were transferred to 50 ml Falcon tubes and centrifuged at 15 min at 5000g at 4°C. The supernatants were subjected to ultrafiltration using Amicon® Ultra-15, 10 kDa cut-off (Merck Millipore, MA, USA) at 5000g for 15–30 min at 4°C. The concentrated protein was purified with HisPur™ Ni2+-NTA spin columns (#: 88226, Thermo Fisher Scientific, MA, USA). The elution of the columns was performed as recommended in the instruction manual with adjusting the incubation time for protein binding to 1 h at 4°C with an end-over-end mixer and an additional incubation step of 5 min with the elution buffer before eluting. Eluates were pooled and dialyzed (Membra-Cel™, 14 kDa cut-off, 250 mm x 44 mm x 0.02 mm; Carl Roth, Germany) at 4°C in 25 mM Tris buffer, pH 7.4, with a volume adjusted to 40 times the volume of the elution fraction. The buffer was exchanged after 8 h. The enzyme was aliquoted and stored at −80°C until use.
Cell culture
Triple-negative breast cancer cells (MDA-MB-231), which natively express CD73, were grown in Dulbecco’s Modified Eagle Medium (DMEM, #: 41966, Thermo Fisher Scientific, MA, USA) and melanoma cancer cells with CRISPR-Cas9 knockout of CD7370 (MaMel.65-CD73ko) were cultivated in Roswell Park Memorial Institute (RPMI) medium 1640, (Thermo Fisher Scientific, Langerwehe, Germany) plus 2 mM of L-glutamine (PAN Biotech, Germany). Both media were supplemented with 100 U/mL penicillin/100 µg/mL streptomycin (PAN Biotech, Germany) and 10% of fetal bovine serum (PAN Biotech, Germany) and cultivated at 37°C with 5% CO2. MDA-MB-231 and MaMel.65-CD73ko cells were split 1:20, and 1:5, respectively, every 72 h (at 80–90% cell confluence). To detach the adherent cells, growth media was removed, cells were washed with phosphate-buffered saline (PBS, 25 cm2 flask: 2.5 mL, for larger flasks correspondingly larger amounts) and incubated with trypsin/EDTA (0.05%/0.6 mM, PAN Biotech, Germany; 1 mL for 25 cm2 flask) for 5 min in the incubator at 37°C. Detached cells were diluted with growth media (2 mL for 25 cm2 flask) and transferred to new culture flasks containing growth media (5 mL for 25 cm2 flask).
Membrane preparation of CD73 from MDA-MB-231 and preparation of cytosolic extract from MaMel.65-CD73ko cells
For both preparations, cells were expanded in 175 cm2 culture flasks to 80–90% cell confluence. After detachment by trypsin/EDTA (0.05%/0.6 mM), 106 cells per dish were transferred to cell culture dishes (150 cm2) and incubated for 4 days at 37°C with 5% CO2. The culture medium was removed, cells were washed with 10 mL of PBS and frozen at -20°C. For membrane preparation, cells were treated with 1 mL of ice-cold buffer (50 mM Tris, 2 mM EDTA, pH 7.4), scraped off, collected in a conical tube and centrifuged for 10 min at 1000g (4°C). The pellet was resuspended in membrane buffer (0.5 mL/dish; 25 mM Tris, 1 mM EDTA, 320 mM sucrose, 1:1000 protease inhibitor cocktail (Sigma-Aldrich, MO, USA), pH 7.4, and homogenized three times for 30 s each (20,500 rpm, Ultraturrax, IKA-Labortechnik, Germany). After centrifugation for 10 min, at 1000g (4°C), the supernatants were collected and centrifuged for 30 min at 48,000g (4°C). The resulting supernatants containing the cytosolic proteins were discarded, and the pellet was resuspended in washing puffer (0.5 mL/dish; 50 mM Tris, pH 7.4) and centrifuged again (same conditions). This step was repeated three times. Finally, the pellet was resuspended in washing buffer (0.1 mL/dish), aliquoted and stored at –80°C until use. For producing the cytosolic extract from MaMel.65-CD73ko cells, a published procedure was adapted.62 The protein of the supernatant that was obtained after ultracentrifugation was precipitated with 40% aq. ammonium sulfate and centrifuged for 10 min at 12.000g (4°C). The resulting pellet was solubilized in 4 mL of HEPES buffer (40 mM HEPES, 1 mM EDTA, 5 mM MgCl2, 1 mM dithiothreitol 0.2 mM phenylmethylsulfonyl fluoride, 15% glycerol, pH 7.0), transferred to Amicon® Ultra-0.5 tubes (10 kDa cut-off, Merck Millipore, MA, USA) and centrifuged for 20 min at 16.000g (Mikro 200R, Hettich, Germany) for the removal of ammonium sulfate, phosphate and nucleotide contaminants. The protein samples were washed three times with 4 mL of the HEPES buffer by using the Amicon® filtration tubes under the described centrifugation conditions, aliquoted and stored at −80°C.
CD73 enzyme inhibition assays
The assay was performed essentially as previously described.34 Stock solutions (10 mM) of the compounds were prepared in demineralized water, and further dilutions were performed in assay reaction buffer (25 mM Tris, 140 mM sodium chloride, 25 mM sodium dihydrogen phosphate, pH 7.4). 10 µl of the inhibitor solution were added to 70 µl of assay reaction buffer. After the addition of 10 µl of CD73-containing solution or suspension (rat CD73: 1.63 ng; human CD73: 0.365 ng; membrane preparation of MDA-MB-231 cells expressing CD73: 7.4 ng of protein per vial), the reaction was initiated by the addition of 10 µl of [2,8-3H]AMP (specific activity 7.4×108 Bq/mmol (20 mCi/mmol)), American Radio-labeled Chemicals, MO, USA, distributed by Hartman Analytic, Germany) resulting in a final substrate concentration of 5 µM. The enzymatic reaction was performed for 25 min at 37°C in a shaking water bath. Then, 500 µl of cold precipitation buffer (100 mM lanthanum chloride, 100 mM sodium acetate, pH 4.0) were added to stop the reaction and to facilitate precipitation of free phosphate and unconverted [2,8-3H]AMP. After the precipitation was completed (after at least 30 min on ice), the mixture was separated by filtration through GF/B glass fiber filters using a cell harvester (M-48, Brandel, Gaithersburg, MD, USA). After washing each reaction vial three times with 400 µl of cold (4°C) demineralized water, 5 mL of the scintillation cocktail (ULTIMA Gold XR, PerkinElmer, MA, USA) was added and radioactivity was measured by scintillation counting (TRICARB 2900 TR, Packard/PerkinElmer; counting efficacy: 49–52%). Two controls were included and measured as duplicates. One reaction was performed without the inhibitor resulting in 100% enzyme activity (positive control) and one was incubated without the inhibitor and the enzyme and served as background control. The resulting data were subtracted from the background and were normalized to the positive control. The results were plotted, and concentration-inhibition curves were fitted with GraphPad Prism 5 (GraphPad Software, La Jolla, USA). The mean IC50 ± SEM from three independent experiments was used to calculate the Ki value with the Cheng-Prusoff equation30 (Km, rat CD73: 53.0 ± 4.1 µM; Km, human CD73: 17.0 ± 2.1 µM; Km, (MDA-MB-231): 14.8 ± 2.1 µM; Supporting information, Figure S2).
To show that the triethylammonium salt form of the inhibitors did not affect is inhibitory activity, triethylammonium chloride was tested at 500 µM concentration and no reduction of enzymatic activity was detected (data not shown).
Analysis of AMP hydrolysis by cytosolic extract derived from MaMel.65-CD73ko cells
Reactions were performed in 50 mM Tris, pH 7.0, buffer containing 100 mM KCl, 5 mM MgCl2 and 15.25 µg of the cytosolic extract derived from MaMel.65-CD73ko cells.70 After preincubation of 5 min at 37°C, the reactions were initiated by the addition of 5 mM AMP as a substrate. After 60 min, the reactions were terminated by heat inactivation (5 min at 95°C) and samples were placed on ice. Reaction tubes were centrifuged for 5 min at 23,000g (4°C), and the supernatant was transferred into the wells of a 96-well half-area microplate (clear, Greiner Bio-One, Austria). To enable phosphate detection, 20 µl of detection reagent I (final concentration 120 µM malachite green oxalate, 0.06% polyvinyl alcohol) and 30 µl of the detection reagent II (final concentration: 6 mM ammonium heptamolybdate, 0.45 M H2SO4) was added. The plate was incubated at room temperature and 500 rpm for 20 min. The absorption of the formed colorimetric complex was analyzed at 600 nm (PHERAstar FS, BMG Labtech, Ortenberg, Germany). Compounds 4l, 7f, 9g, 9h were analyzed at 100 µM, and a mixture of the phosphatase inhibitors levamisole (1 mM) and sodium fluoride (5 mM) was used as a positive control. As negative controls, reactions in the absence of any inhibitor, or without AMP were performed. In addition, to the mixture of levamisole and NaF, one of the CD73 inhibitors (4l, 7f, 9g, or 9h, respectively, 100 µM) was added, however, the CD73 inhibitors did not result in increased inhibition of AMP hydrolysis as compared to levamisole and NaF alone, even after an extended incubation time of up to 120 min.
Human P2Y6 receptor assay
Calcium mobilization induced by the nucleotide derivatives was measured in a human astrocytoma cell line (1321N1) expressing the human P2Y6 receptor, as previously described.42
Human P2Y14 receptor assay
Inhibition of binding of a high affinity fluorescent antagonist was measured using flow cytometry in a CHO cell line expressing the human P2Y14 receptor, as previously described.37
In situ ecto-5′-nucleotidase activity assay
For localization of AMPase activities in human tonsils, a modification of the lead nitrate method was employed.63,64 In brief, palatine tonsils were obtained from adult patients with chronic tonsillitis undergoing routine tonsillectomy at Turku University Hospital (permission # TO6/033/18 from the Hospital Research Council). The tonsils were washed with physiological salt solution, embedded in the cryo-mold with Tissue-Tek® O.C.T. compound (Sakura Finetek Europe B.V. The Netherlands), cut using a cryostat and stored at −80 °C. Tonsil cryosections were pre-incubated for 30 min in Trizma-maleate sucrose buffer (TMSB; 40 mM Trizma® maleate; 0.25 M sucrose, pH 7.4) supplemented with the alkaline phosphatase inhibitor levamisole (2 mM) and different concentrations of CD73 inhibitor AOPCP or 9h. The enzymatic reaction was performed then for 45 min at 37°C in a final volume of 20 mL TMSB-buffered substrate solution containing 1.5 mM Pb(NO3)2, 1 mM CaCl2, 100 μM AMP, and tested CD73 inhibitor at the same concentration. The lead orthophosphate precipitated in the course of nucleotidase activity was visualized as a brown deposit by incubating sections in 0.5% (NH4)2S for 30 sec followed by three washes in Trizma-maleate buffer for 5 min each. Slides were mounted with Aquatex medium (Merck, Germany). Multiple images of adjacent tissue areas were captured using Pannoramic 250 slide scanner (3DHistech Ltd., Budapest, Hungary), and further stitched to a larger overview using the accompanying Pannoramic Viewer 1.15.4 software. The images of control and treated tissue were captured at identical exposure times and other settings and further acquired in parallel using Adobe Photoshop CS6 software. Nucleotidase activities were quantified as mean pixel intensities after grayscale conversion using ImageJ 1.52h software.
CD73 is highly expressed in the germinal centers, connective tissues and, to lesser extent, in the inter-follicular area. Such heterogeneity precludes proper quantification of “global” AMPase staining in the whole tissue. Therefore, we selected the same matched areas of germinal centers in the control and treated tonsils and further compared their mean pixel intensities. Also, the inhibitor concentrations are given as nM for matched volumes, as we have noticed that the actual amount of inhibitor per tissue slide is an important factor.
Molecular docking studies
The recent co-crystal structure of the human CD73 (PDB ID: 4H2I)49 with the antagonist AOPCP was obtained from the RCSB (Research Collaboratory for Structural Bioinformatics) Protein Data Bank (PDB).65 The downloaded crystal structures were prepared using the protein preparation tool, and the hydrogen atoms were assigned according to Protonate-3D implemented in Molecular Operation Environment (MOE 2018.01).66 The crystal structure of the human CD73 was applied for flexible ligand docking using AutoDock 4.2.67–69 During the docking simulations, the ligands were fully flexible while the residues of the enzyme were treated as rigid. The selected potent compound 9f was docked into the active site of the enzyme to predict the binding mode of the compounds. The atomic partial charges were added and three-dimensional energy scoring grids for a box of 60 × 60 × 60 points with a spacing of 0.375 Å were computed using AutoDockTools.67,68 The grids were centered based on the co-crystallized ligand, AOPCP. Fifty independent docking calculations using the varCPSO-ls algorithm from PSO@Autodock implemented in AutoDock4.2 were performed and terminated after 500,000 evaluation steps.69 Parameters of varCPSO-ls algorithm, the cognitive and social coefficients c1 and c2, were set at 6.05 with 60 individual particles as swarm size. All the other parameters of the algorithm were set at their default values. Possible binding modes of the compounds were explored by visual inspection of the resulting docking poses.
Supplementary Material
Table 6.
Potency of α,β-methylene analogues of UDP and CDP as agonists at the human P2Y6R and inhibitors of fluorescent ligand binding at the human P2Y14R.
| Compd. | hP2Y6 EC50 ± SEM (nM) (% activation at indicated concentration) | hP2Y14 IC50 ± SEM (nM) (% inhibition at indicated concentration) | CD73 Ki ± SEM (nM) rat eN |
|---|---|---|---|
| 4l | 203 ± 30 | 362 ± 90 | 14.8 ± 1.9 |
| 7f | 1390 ± 220 | 6660 ± 4740 | 13.9 ± 1.6 |
| 9h | > 3000 (3.6 %) | >3000 (inactive) | 3.67 ± 0.26 |
Acknowledgments
We thank Dr. John Lloyd and Dr. Noel Whittaker (NIDDK) for mass spectral determinations, Prof. Dr. Norbert Sträter for the gift of human CD73 cDNA, Prof. Dr. Henrik Ditzel for MDA-MB-231 and Prof. Dr. Michael Hölzel for MaMel.65-CD73ko cells. Dr. Ali El-Tayeb and Dr. Sanjay Bhattarai are acknowledged for introduction into the synthesis of AOPCP analogs. We acknowledge support from the NIDDK Intramural Research Program (ZIADK031116). Financial support of Anna Junker by the Deutsche Forschungsgemeinschaft (German Research Foundation, JU 2966/1–1) and the Cells-in-Motion (CiM) cluster of excellence (Münster, Germany) is gratefully acknowledged.
List of abbreviations:
- AR
adenosine receptor
- ACN
acetonitrile
- ADP
adenosine 5′diphosphate
- AMP
adenosine 5′monophosphate
- ATP
adenosine 5′triphosphate
- AOPCP
α,β-methylene-ADP, adenosine-5′-O-[(phosphonomethyl)phosphonic acid], [{5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl}methoxyhydroxyphosphoryl]methylphosphonic acid
- CD73
cluster of differentiation 73
- CE
capillary electrophoresis
- DMEM
Dulbecco’s Modified Eagle Medium
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- EC
enzyme commission
- EDTA
ethylenediaminetetraacetic acid
- ESI
electrospray ionization
- FBS
fetal bovine serum
- GPI
glycophosphatidylinositol
- HPLC
high performance liquid chromatography
- LC-MS
liquid chromatography-mass spectrometry
- MeOD-d4
deuterated methanol
- MOE
Molecular Operating Environment
- eN
ecto-5′-nucleotidase
- eNPPs
ecto-nucleoside pyrophosphatases/phosphodiesterases
- eNTPDases
ecto-nucleoside triphosphate diphosphohydrolases
- PBS
phosphate-buffered saline
- pdb
protein data bank
- POMs
polyoxometalates
- PSB
Pharmaceutical Sciences Bonn
- SAR
structure-activity relationship
- SF9
Spodoptera frugiperda 9
- TEA
trimethylamine
- TEAC
triethylammonium hydrogencarbonate
- THF
tetrahydrofuran, TLC, thin layer chromatography, TNBC, triple-negative breast cancer
- Tris
tris(hydroxymethyl)aminomethane
Footnotes
Supporting Information Available
1H, 13C, and 31P NMR spectra of all compounds, plots of pharmacological data, molecular modeling figures and the atomic coordinates of the model of 9h bound to CD73 are included in Supporting Information. This material is available free of charge in the Internet at http://pubs.acs.org.
References
- 1.Zimmermann H 5′-Nucleotidase: Molecular structure and functional aspects. Biochem. J 1992, 285 (Pt 2), 345–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sträter N Ecto-5′-nucleotidase: Structure function relationships. Purinergic Signalling 2006, 2 (2), 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yegutkin GG Enzymes involved in metabolism of extracellular nucleotides and nucleosides: functional implications and measurement of activities. Crit. Rev. Biochem. Mol. Biol 2014, 49, 473–497. [DOI] [PubMed] [Google Scholar]
- 4.Knapp K; Zebisch M; Pippel J; El-Tayeb A; Müller Christa E.; Sträter N. Crystal structure of the human ecto-5′-nucleotidase (CD73): Insights into the regulation of purinergic signaling. Structure 2012, 20 (12), 2161–2173. [DOI] [PubMed] [Google Scholar]
- 5.Heuts DPHM; Weissenborn MJ; Olkhov RV; Shaw AM; Gummadova J; Levy C; Scrutton NS Crystal structure of a soluble form of human CD73 with ecto-5′-nucleotidase activity. ChemBioChem 2012, 13, 2384–2391. [DOI] [PubMed] [Google Scholar]
- 6.Cekic C; Linden J Purinergic regulation of the immune system. Nature Rev. Immunol 2016, 16, 177–192. [DOI] [PubMed] [Google Scholar]
- 7.Augusto E; Matos M; Sévigny J; El-Tayeb A; Bynoe MS; Müller CE; Cunha RA; Chen JF Ecto-5′-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J. Neurosci 2013, 33, 11390–11399. doi: 10.1523/JNEUROSCI.5817-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flögel U; Burghoff S; van Lent PL.; Temme S; Galbarz L; Ding Z; El-Tayeb A; Huels S; Bönner F; Borg N; Jacoby C; Müller CE; van den Berg WB; Schrader J Selective activation of adenosine A2A receptors on immune cells by a CD73-dependent prodrug suppresses joint inflammation in experimental rheumatoid arthritis. Sci. Transl. Med 2012, 4, 10.1126/scitranslmed.3003717. [DOI] [PubMed] [Google Scholar]
- 9.Corbelini PF; Figueir F; Machado das Neves G; Andrade S; Kawano DF; Oliveira Battastini AM; Eifler-Lima VL Insights into ecto-5′-nucleotidase as a new target for cancer therapy: A medicinal chemistry study. Curr. Med. Chem 2015, 22, 1776–1792. [DOI] [PubMed] [Google Scholar]
- 10.Xu S; Shao QQ; Sun JT; Yang N; Xie Q; Wang DH; Huang QB; Huang B; Wang XY; Li XG; Qu X Synergy between the ectoenzymes CD39 and CD73 contributes to adenosinergic immunosuppression in human malignant gliomas. Neuro. Oncol 2013, 15, 1160–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stagg J; Divisekera U; McLaughlin N; Sharkey J; Pommey S; Denoyer D; Dwyer KM; Smyth MJ Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci 2010, 107, 1547–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yegutkin GG; Marttila-Ichihara F; Karikoski M; Niemelä J; Laurila JP; Elima K; Jalkanen S; Salmi M Altered purinergic signaling in CD73-deficient mice inhibits tumor progression. Eur. J. Immunol 2011, 41, 1231–1241. [DOI] [PubMed] [Google Scholar]
- 13.Beavis PA; Stagg J; Darcy PK; Smyth MJ CD73: a potent suppressor of antitumor immune responses. Trends Immunol 2012, 33, 231–237. [DOI] [PubMed] [Google Scholar]
- 14.Antonioli L; Yegutkin GG; Pacher P; Blandizzi C; Haskó G Anti-CD73 in cancer immunotherapy: awakening new opportunities. Trends Cancer 2016, 2, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sociali G, Raffaghello L, Magnone M, Zamporlini F, Emionite L, Sturla L, Bianchi G, Vigliarolo T, Nahimana A, Nencioni A, et al. Antitumor effect of combined NAMPT and CD73 inhibition in an ovarian cancer model. Oncotarget 2016, 7, 2968–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hay CM; Sult E; Huang Q; Mulgrew K; Fuhrmann SR; McGlinchey KA; Hammond SA; Rothstein R; Rios-Doria J; Poon E; Holoweckyj N; Durham NM; Leow CC; Diedrich G; Damschroder M; Herbst R; Hollingsworth RE; Sachsenmeier KF Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 2016, 5, e1208875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MEDI9447 Alone and in Combination with MEDI4736 in Adult Subjects with Select Advanced Solid Tumors https://clinicaltrials.gov/ct2/show/NCT02503774, accessed July 23, 2018.
- 18.Freundlieb M; Zimmermann H; Müller CE A new, sensitive ecto-5′-nucleotidase assay for compound screening. Anal. Biochem 2014, 446, 53–58. [DOI] [PubMed] [Google Scholar]
- 19.Iqbal J; Jirovsky D; Lee S-Y; Zimmermann H; Müller CE Capillary electrophoresis-based nanoscale assays for monitoring ecto-5′-nucleotidase activity and inhibition in preparations of recombinant enzyme and melanoma cell membranes. Anal. Biochem 2008, 373, 129–140. [DOI] [PubMed] [Google Scholar]
- 20.Bhattarai S; Freundlieb M; Pippel J; Meyer A; Abdelrahman A; Fiene A; Lee SY; Zimmermann H; Yegutkin GG; Sträter N; El-Tayeb A; Müller CE α,β-Methylene-ADP (AOPCP) derivatives and analogues: Development of potent and selective ecto-5’-nucleotidase (CD73) inhibitors. J. Med. Chem 2015, 58, 6248–6263. [DOI] [PubMed] [Google Scholar]
- 21.Baqi Y; Lee S-Y; Iqbal J; Ripphausen P; Lehr A; Scheiff A; Zimmermann H; Bajorath J; Müller C Development of potent and selective inhibitors of ecto-5′-nucleotidase based on an anthraquinone scaffold. J. Med. Chem 2010, 53, 2076–2086. [DOI] [PubMed] [Google Scholar]
- 22.Ripphausen P; Freundlieb M; Brunschweiger A; Zimmermann H; Müller CE; Bajorath J Virtual screening identifies novel sulfonamide inhibitors of ecto-5′-nucleotidase. J. Med. Chem 2012, 55, 6576–6581. [DOI] [PubMed] [Google Scholar]
- 23.Lee S-Y; Fiene A; Li W; Hanck T; Brylev KA; Fedorov VE; Lecka J; Haider A; Pietzsch H-J; Zimmermann H; Sévigny J; Kortz U; Stephan H; Müller CE Polyoxometalates - potent and selective ecto-nucleotidase inhibitors. Biochem. Pharmacol 2015, 93, 171–181. [DOI] [PubMed] [Google Scholar]
- 24.al-Rashida M; Iqbal J Therapeutic potentials of ecto-nucleoside triphosphate diphosphohydrolase, ecto-nucleotide pyrophosphatase/phosphodiesterase, ecto-5’-nucleotidase, and alkaline phosphatase inhibitors. Med. Res. Rev 2014, 34, 703–743. [DOI] [PubMed] [Google Scholar]
- 25.Bruns RF Adenosine receptor activation by adenine nucleotides requires conversion of the nucleotides to adenosine. Naunyn Schmiedebergs Arch. Pharmacol 1980, 315, 5–13. [DOI] [PubMed] [Google Scholar]
- 26.Braganhol E; Tamajusuku AS; Bernardi A; Wink MR; Battastini AM Ecto-5′- nucleotidase/CD73 inhibition by quercetin in the human U138MG glioma cell line. Biochim. Biophys. Acta 2007, 1770, 1352–1359. [DOI] [PubMed] [Google Scholar]
- 27.Lee S-Y; Fiene A; Li W; Hanck T; Brylev KA; Fedorov VE; Lecka J; Haider A; Pietzsch H-J; Zimmermann H; Sévigny J; Kortz U; Stephan H; Müller CE Polyoxometalates - potent and selective ecto-nucleotidase inhibitors. Biochem. Pharmacol 2015, 93, 171–181. [DOI] [PubMed] [Google Scholar]
- 28.Furtmann N; Bajorath J Structural and modeling studies on ecto-5’-nucleotidase aiding in inhibitor design. Mini Rev. Med. Chem 2015, 15, 34–40 [DOI] [PubMed] [Google Scholar]
- 29.Servos J; Reiländer H; Zimmermann H Catalytically active soluble ecto-5′-nucleotidase purified after heterologous expression as a tool for drug screening. Drug Dev. Res 1998, 45, 269–276. [Google Scholar]
- 30.a) Marquardt DW An algorithm for least squares estimation of non-linear parameters. J. Soc. Ind. Appl. Math 1963, 11, 431–441. [Google Scholar]; b) Cheng Y-C; Prusoff WH Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- 31.Williamson DS; Borgognoni J; Clay A; Daniels Z; Dokurno P; Drysdale MJ; Foloppe N; Francis GL; Graham CJ; Howes R; Macias AT; Murray JB; Parsons R; Shaw T; Surgenor AE; Terry L; Wang Y; Wood M; Massey AJ Novel adenosine-derived inhibitors of 70 kDa heat shock protein, discovered through structure-based design. J. Med. Chem 2009, 52, 1510–1513. [DOI] [PubMed] [Google Scholar]
- 32.Yegutkin GG; Auvinen K; Karikoski M; Rantakari P; Gerke H; Elima K; Maksimow M; Quintero IB; Vihko P; Salmi M; Jalkanen S Consequences of the lack of CD73 and prostatic acid phosphatase in the lymphoid organs. Mediators Inflamm [online] Volume 2014, Article ID 485743, 10 pages. doi: 10.1155/2014/485743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colgan SP; Eltzschig HK; Eckle T; Thompson LF Physiological roles for ecto-5’-nucleotidase (CD73). Purinergic Signalling, 2006, 2, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freundlieb M; Zimmermann H; Müller CE A new, sensitive ecto-5’-nucleotidase assay for compound screening. Anal. Biochem 2014, 446, 53–58. [DOI] [PubMed] [Google Scholar]
- 35.Altschul SF; Madden TL; Schäffer AA; Zhang J; Zhang Z; Miller W; Lipman DJ Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altschul SF; Wootton JC; Gertz EM; Agarwala R; Morgulis A; Schäffer AA; Yu Y-K Protein database searches using compositionally adjusted substitution matrices. FEBS J 2005, 272, 5101–5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loi S; Pommey S; Haibe-Kains B; Beavis PA; Darcy PK; Smyth MJ; Stagg J CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci U S A 2013, 110, 11091–11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terp MG; Olesen KA; Arnspang EC; Lund RR; Lagerholm BC; Ditzel HJ; Leth-Larsen R Anti-human CD73 monoclonal antibody inhibits metastasis formation in human breast cancer by inducing clustering and internalization of CD73 expressed on the surface of cancer cells. J Immunol 2013, 191, 4165–4173. [DOI] [PubMed] [Google Scholar]
- 39.Young A; Ngiow SF; Barkauskas DS; Sult E; Hay C; Blake SJ; Huang Q; Liu J; Takeda K; Teng MWL; Sachsenmeier K; Smyth MJ Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell 2016, 30, 391–403. [DOI] [PubMed] [Google Scholar]
- 40.Allard D; Chrobak P; Allard B; Messaoudi N; Stagg J Targeting the CD73-adenosine axis in immuno-oncology. Immunol Lett 2019, 205, 31–39, doi: 10.1016/j.imlet.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 41.Das A; Ko H; Burianek LE; Barrett MO; Harden TK; Jacobson KA Human P2Y14 receptor agonists: Truncation of the hexose moiety of uridine-5′-diphosphoglucose and its replacement with alkyl and aryl groups. J. Med. Chem 2010, 53, 471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toti KS; Jain S; Ciancetta A; Balasubramanian R; Charkaborty S; Surujdin R; Shi ZD; Jacobson KA Pyrimidine nucleotides containing a (S)-methanocarba ring as P2Y6 receptor agonists. Med. Chem. Comm 2017, 8, 1897–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Junker A; Balasubramanian R; Ciancetta A; Uliassi E; Kiselev E; Martiriggiano C; Trujillo K; Mtchedlidze G; Birdwell L; Brown KA; Harden TK; Jacobson KA Structure-based design of 3-(4-aryl-1H-1,2,3-triazol-1-yl)-biphenyl derivatives as P2Y14 receptor antagonists. J. Med. Chem 2016, 59, 6149–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reinhardt J; Landsberg J; Schmid-Burgk JL; Ramis BB; Bald T; Glodde N; Lopez-Ramos D; Young A; Ngiow SF; Nettersheim D; Schorle H; Quast T; Kolanus W; Schadendorf D; Long GV; Madore J; Scolyer RA; Ribas A; Smyth MJ; Tumeh PC; Tüting T; Hölzel M MAPK signaling and inflammation link melanoma phenotype switching to induction of CD73 during immunotherapy. Cancer Res 2017, 77, 4697–4709. [DOI] [PubMed] [Google Scholar]
- 45.NagDas SK; Bhattacharyya AK The kinetics of inhibition of human seminal plasma acid phosphatase by sodium fluoride. Biochem Int 1984, 9, 659–668. [PubMed] [Google Scholar]
- 46.Khodaparast-Sharifi SH; Snow LD Levamisole inhibition of alkaline phosphatase and 5’-nucleotidase of bovine milk fat globule membranes. Int J Biochem 1989, 21, 401–405. [DOI] [PubMed] [Google Scholar]
- 47.Perlíková P, Hocek M. Pyrrolo[2,3‐d]pyrimidine (7‐deazapurine) as a privileged scaffold in design of antitumor and antiviral nucleosides. Med Res Rev 2017, 37, 1429–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wachstein M; Meisel E Histochemistry of hepatic phosphatases of a physiologic pH; with special reference to the demonstration of bile canaliculi. Am. J. Clin. Pathol 1957, 27, 13–23. [DOI] [PubMed] [Google Scholar]
- 49.Knapp K; Zebisch M; Pippel J; El-Tayeb A; Muller CE; Strater N Crystal structure of the human ecto-5’-nucleotidase (CD73): insights into the regulation of purinergic signaling. Structure 2012, 20 (12), 2161–2173. [DOI] [PubMed] [Google Scholar]
- 50.Lesiak K; Watanabe KA; George J; Pankiewicz KW 2-(4-Nitrophenyl)ethyl methylenebis(phosphonate): A versatile reagent for the synthesis of nucleoside 5’-methylenebis(phosphonate)s. J. Org. Chem 1998, 63, 1906–1909. [Google Scholar]
- 51.a) Myers TC; Nakamura K; Danielzadeh AB Synthesis of Diimidotriphosphoric Acid and Related Esters. J. Org. Chem 1965, 30, 1517–1520. [DOI] [PubMed] [Google Scholar]; b) Saady M; Lebeau L; Mioskowski C Synthesis of diimidotriphosphoric acid and related esters. Tetrahedron Lett 1995, 36, 4237–4240. [Google Scholar]
- 52.Davisson VJ; Davis DR; Dixit VM; Poulter CD Synthesis of nucleotide 5’-diphosphates from 5’-O-tosyl nucleosides. J. Org. Chem 1987, 52, 1794–1801. [Google Scholar]
- 53.Jayasekara PS; Jacobson KA Rapid synthesis of alkoxyamine hydrochloride derivatives from alkyl bromide and N,N′-di-tert-butoxycarbonylhydroxylamine ((Boc)2NOH). Synth. Commun 2014, 44, 2344–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schindler U; Becker A; Lawson K; Jin L; Jeffrey J; Kalisiak J; Yin F; Zhang K; Chen A; Swinarski D; Walters MJ; Young S; Powers JP; Tan J AB680, a potent and selective CD73 small molecule inhibitor, reverses the AMP/adenosine-mediated impairment of immune effector cell activation by immune checkpoint inhibitors. Eur. J. Cancer, 2018, 92, S14 10.1016/j.ejca.2018.01.036 [DOI] [Google Scholar]
- 55.Gong Y-P; Wan R-Z; Liu Z-P Evaluation of WO2017098421: GSK’s benzothiazine compounds as CD73 inhibitor filings. Expert Opin. Therap. Patents, 2018, 28(2), 167–171. 10.1080/13543776.2018.1407756 [DOI] [PubMed] [Google Scholar]
- 56.Cacatian S; Claremon DA; Jia L; Morales-Ramos M; Singh SB; Venkatraman S; Xu Z; Zheng Y Purine Derivatives as CD73 Inhibitors for the Treatment of Cancer US 20170044203A1, Vitae Pharmaceuticals, Inc., Fort Washington, PA (US). [Google Scholar]
- 57.Debien LPP; Jaen JC; Kalisiak J; Lawson KV; Leleti MR ; Lindsey EA; Miles DH; Newcomb E; Powers JP; Rosen BR; Sharif EUI Modulators of 5’-Nucleotidase, Ecto and the Use Thereof US2017/0267710 (Sept. 1, 2017), WO 2017/120508 Al (July 13, 2017), Arcus Biosciences, Inc. [Google Scholar]
- 58.Long A; Dominguez D; Qin L; Chen S; Fan J; Zhang M, Fang D; Zhang Y; Kuzel TM; Zhang B Type 2 Innate lymphoid cells impede IL-33-mediated tumor suppression. J. Immunol 2018, 201, 3456–3464. 10.4049/jimmunol.1800173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Young A; Ngiow SF; Barkauskas DS; Sult E; Hay C; Blake SJ; Huang Q; Liu J; Takeda K; Teng MWL; Sachsenmeier K; Smyth MJ Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell, 2016, 30(3), 391–403. [DOI] [PubMed] [Google Scholar]
- 60.Iriyama T; Sun K; Parchim NF; Li J; Zhao C; Song A; Hart LA; Blackwell SC; Sibai BM; Chan L-NL; Chan T-S; Hicks MJ; Blackburn MR; Kellems RE; Xia Y Elevated placental adenosine signaling contributes to the pathogenesis of preeclampsia. Circulation 2015, 131, 730–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mahamed DA; Toussaint LE; Bynoe MS CD73-generated adenosine is critical for immune regulation during toxoplasma gondii infection. Infection and Immunity, 2015, 83, 721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hunsucker SA; Spychala J; Mitchell BS Human cytosolic 5’-nucleotidase I. Characterization and role in nucleoside analog resistance. J Biol Chem 2001, 276, 10498–10504. [DOI] [PubMed] [Google Scholar]
- 63.Langer D; Hammer K;, Koszalka P; Schrader J; Robson S; Zimmermann H Distribution of ectonucleotidases in the rodent brain revisited. Cell. Tissue. Res 2008, 334, 199–217. [DOI] [PubMed] [Google Scholar]
- 64.Mercier N; Kiviniemi TO; Saraste A; Miiluniemi M; Silvola J; Jalkanen S; Yegutkin GG Impaired ATP-induced coronary blood flow and diminished aortic NTPDase activity precede lesion formation in apolipoprotein E-deficient mice. Am. J. Pathol 2012, 180, 419–428. [DOI] [PubMed] [Google Scholar]
- 65.Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res 2000, 28 (1), 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Molecular Operating Environment (MOE), 2018.08; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2018.
- 67.Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem 2009, 30 (16), 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sanner MF Python: a programming language for software integration and development. J. Mol. Graph. Model 1999, 17 (1), 57–61. [PubMed] [Google Scholar]
- 69.Namasivayam V; Gunther R pso@autodock: a fast flexible molecular docking program based on Swarm intelligence. Chem. Biol. Drug Des 2007, 70 (6), 475–484. [DOI] [PubMed] [Google Scholar]
- 70.Reinhardt J; Landsberg J; Schmid-Burgk JL; Ramis BB; Bald T; Glodde N; Lopez-Ramos D; Young A; Ngiow SF; Nettersheim D; Schorle H; Quast T; Kolanus W; Schadendorf D; Long GV; Madore J; Scolyer RA; Ribas A; Smyth MJ; Tumeh PC; Tüting T; Hölzel M MAPK Signaling and inflammation link melanoma phenotype switching to induction of CD73 during immunotherapy. Cancer Res 2017, 77, 4697–4709. [DOI] [PubMed] [Google Scholar]
- 71.a) Müller CE; Scior T Adenosine receptors and their modulators. Pharm. Acta Helv 1993, 68, 77–111. [DOI] [PubMed] [Google Scholar]; b) Müller CE; Stein B Adenosine receptor antagonists: structures and potential therapeutic applications. Curr. Pharm. Des 1996, 2, 501–530. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.