

ABSTRACT
Neuromuscular disorders encompass a wide range of conditions often associated with a genetic component. In the present study, we report a patient with severe infantile-onset amyotrophy in whom two compound heterozygous variants in the gene CACNA1H encoding for Cav3.2 T-type calcium channels were identified. Functional analysis of Cav3.2 variants revealed several alterations of the gating properties of the channel that were in general consistent with a loss-of-channel function. Taken together, these findings suggest that severe congenital amyoplasia may be related to CACNA1H and would represent a new phenotype associated with mutations in this gene.
KEYWORDS: Congenital amyotrophy, CACNA1H, mutations, calcium channel, Cav3.2 channel, T-type channel
Introduction
Neuromuscular disorders (NMD) encompass a wide range of conditions including amyotrophic lateral sclerosis (ALS), congenital muscular dystrophies and myopathies, Duchenne muscular dystrophy, and spinal muscular atrophy. They are characterized by progressive muscle degeneration and weakness that primarily or secondarily impair skeletal muscles and their innervation. The onset of NMD ranges from in utero to old age, but primarily affect infants, children, and teenagers [1]. Although some NMD are acquired, the vast majority are genetic, and may display autosomal recessive, autosomal dominant, or X-linked inheritance [2]. It is also apparent that NMD phenotypes may vary widely even in individuals carrying the same disease-causing mutation, suggesting the existence of additional factors such as modifier genes that contribute to the onset, severity, and duration of the disease [3].
Recently, whole exome sequence analysis has identified two compound heterozygous recessive missense mutations in the gene CACNA1H in a patient with ALS [4,5]. CACNA1H encodes for Cav3.2 channel, a member of the voltage-gated calcium channel family [6]. Cav3.2 channels are widely expressed throughout the body including the central and peripheral nervous system, heart, kidney, smooth muscle, as well as in several neuroendocrine organs [7]. Through their ability to support low-threshold calcium influx (T-type current), they serve essential physiological processes including neuronal firing, hormone secretion, smooth muscle contraction, and myoblast fusion [8]. Their physiological implication is further exemplified by the existence of polymorphisms in CACNA1H associated with a number of human disorders including several forms of epilepsy [9], autism spectrum disorders [10,11], congenital pain [12], primary aldosteronism [13,14], and ALS [4,5].
In the present study, we report a patient with severe congenital amyotrophy in whom two compound heterozygous variants in CACNA1H were identified. Functional analysis of Cav3.2 variants revealed altered channel gating, strengthening the genetic association of CACNA1H with NMD.
Materials and methods
Whole exome sequencing
Whole exome sequencing was performed at a commercial laboratory (GeneDx). Using genomic DNA from the proband and both parents, exonic regions and flanking splice junctions were selected, sequenced, and analyzed as per established proprietary protocols using an Illumina sequencing system with 100 bp or greater paired end reads. Reads were aligned to human genome build GRCh37/UCSC hg19 and analyzed for sequence variants using a custom‐developed analysis tool (Xome Analyzer). Mean depth of coverage was 100x.
Plasmid cDNA constructs
The human wild-type Cav3.2 in pcDNA3.1 [15] was used as template to introduce separately the V681L and D1233H mutations by site-directed mutagenesis using the Q5® Site-Directed Mutagenesis Kit (NEB) and the following pairs of primers: p.V681L: 5‘-GGGCCTCAGTttgCCCTGCCC-3‘ (forward) and 5‘-GACAGATGGCCAGGGGCC-3‘ (reverse); p.D1233H: 5‘-CCTGCGCATCcacAGCCACCG-3‘ (forward) and 5‘-AAGAAGTCGCTGGGCAGG-3‘ (reverse). Final constructs were verified by sequencing.
Cell culture and heterologous expression
Human embryonic kidney tsA-201 cells were grown in DMEM medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (all media purchased from Invitrogen) and maintained under standard conditions at 37°C in a humidified atmosphere containing 5% CO2. Heterologous expression of Cav3.2 channels was performed by transfecting cells with plasmid cDNAs encoding for Cav3.2 channel variants using the calcium/phosphate method as previously described [16].
Recording of T-type currents
Patch clamp recording of T-type currents in tsA-201 cells expressing Cav3.2 channels was performed 72 h after transfection in the whole-cell configuration at room temperature (22–24°C). The bath solution contained (in millimolar): 5 BaCl2, 5 KCl, 1 MgCl2, 128 NaCl, 10 TEA-Cl, 10 D-glucose, 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.2 with NaOH). Patch pipettes were filled with a solution containing (in millimolar): 110 CsCl, 3 Mg-ATP, 0.5 Na-GTP, 2.5 gCl2, 5 D-glucose, 10 EGTA, and 10 HEPES (pH 7.4 with CsOH), and had a resistance of 2–4 MΩ. Recordings were performed using an Axopatch 200B amplifier (Axon Instruments) and acquisition and analysis were performed using pClamp 10 and Clampfit 10software, respectively (Axon Instruments). The linear leak component of the current was corrected online, and current traces were digitized at 10 kHz and filtered at 2 kHz. The voltage dependence of activation of Cav3.2 channels was determined by measuring the peak T-type current amplitude in response to 150 ms depolarizing steps to various potentials applied every 10 s from a holding membrane potential of −100 mV. The current-voltage relationship (I/V) curve was fitted with the following modified Boltzmann Equation (1):
with I(V) being the peak current amplitude at the command potential V, Gmax the maximum conductance, Vrev the reversal potential, V0.5 the half-activation potential, and k the slope factor. The voltage dependence of the whole-cell Ba2+ conductance was calculated using the following modified Boltzmann Equation (2):
with G(V) being the Ba2+ conductance at the command potential V.
The voltage dependence of the steady-state inactivation of Cav3.2 channels was determined by measuring the peak T-type current amplitude in response to a 150 ms depolarizing step to −20 mV applied after a 5 s-long conditioning prepulse ranging from −120 mV to −30 mV. The current amplitude obtained during each test pulse was normalized to the maximal current amplitude and plotted as a function of the prepulse potential. The voltage dependence of the steady-state inactivation was fitted with the following two-state Boltzmann function (3):
| (3) |
with Imax corresponding to the maximal peak current amplitude and V0.5 to the half-inactivation voltage.
The recovery from inactivation was assessed using a double-pulse protocol from a holding potential of −100 mV. The cell membrane was depolarized for 2 s at 0 mV (inactivating prepulse) to ensure complete inactivation of the channel, and then to −20 mV for 150 ms (test pulse) after an increasing time period (interpulse) ranging between 0.1 ms and 2 s at −100 mV. The peak current from the test pulse was plotted as a ratio of the maximum prepulse current versus interpulse interval. The data were fitted with the following single-exponential function (4):
| (4) |
where τ the time constant of channel recovery from inactivation.
Statistical analysis
Values are presented as mean ± S.E.M. for n experiments. Statistical significance was determined using a one-way ANOVA test followed by Dunnett multiple comparison test. * p< 0.05, ** p< 0.005, *** p< 0.001.
Results
Clinical characteristics and genetic analysis
The patient is a 13-month-old girl who was born at term following a pregnancy complicated by severe polyhydramnios treated with amnioreduction at 31 weeks. Her birth weight was 2.9 kg (20%ile). Her Apgar score (Appearance, Pulse, Grimace, Activity, and Respiration) used to summarize the health of newborn children was critically low (2 at 1-min and 5-min) and was intubated for poor respiratory effort, absent gag and suck. She had arthrogryposis, areflexia, and no antigravity movements. Nerve conduction studies showed intact sensory but absent motor responses. Magnetic resonance imaging (MRI) of her brain was unremarkable. However, the MRI of her muscles noted severe and diffuse amyoplasia with largest muscles being right biceps femoris (8 x 5 mm at widest diameter) and medial gastrocnemius (8 x 3 mm) (Figure 1(a,b)). A biopsy of the vastus lateralis revealed no viable muscle and repeat biopsies of the gastrocnemius noted a strong predominance of type 1 fibers and a marked variability of fiber size. She remains fully ventilated via tracheostomy with no antigravity movement at 13 months old. Whole exome sequencing identified two compound heterozygous variants (c.2041G>T and c.3697G>C) in CACNA1H, causing the substitution of the valine (V) 681 by a leucine (L) (p.V681L) and of the aspartic acid (D) 1233 by a histidine (H) (p.D1233H) in the Cav3.2 T-type calcium channel, respectively. These mutations were inherited from the father and mother, respectively, who have no symptoms of muscle disease (Figure 1(c)). The p.V681L and p.D1233H mutations are located within the intracellular loops connecting domains I-II and domains II-III of Cav3.2 channel, respectively (Figure 1(d)).
Figure 1.
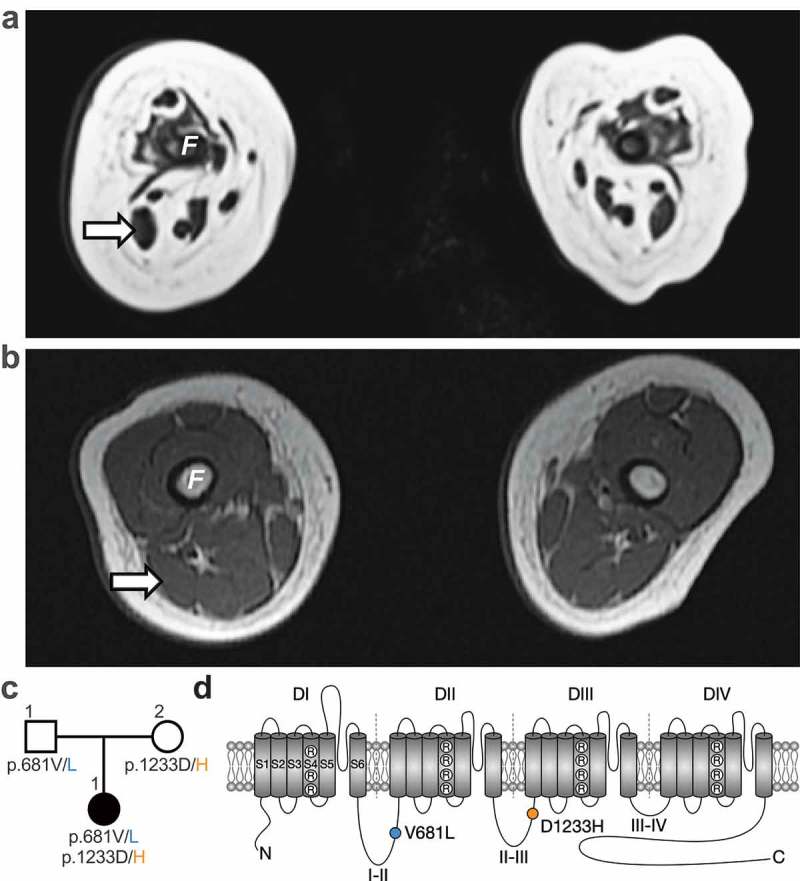
Whole exome sequencing identifies two CACNA1H missense mutations associated with severe congenital amyotrophy. (a) Whole-body magnetic resonance T1 weighted images (T1W1) 3T (axial view) of the thigh at 6 weeks old revealed severe muscle amyotrophy. No muscle was visualized in the anterior compartment. Her right biceps femoris (arrow) measured 8 × 5 mm at its widest. (b) For comparison, a T1WI (axial view) from a hypotonic boy with nemaline myopathy was performed at 4 months old. His biceps femoris measured 14 × 9 mm. F: femur. (c) Family pedigree chart. Filled and open symbols indicate affected and unaffected individuals, respectively. (d) Location of the p.V681L (blue circle) and p.D1233H mutations (orange circle) within the secondary membrane topology of Cav3.2 channel.
Expression of Cav3.2 channel variants
To assess the functional consequence of CACNA1H variants, the two mutations were introduced separately into the human Cav3.2 channel, and functional expression was assessed by patch clamp recording of T-type currents in tsA-201 cells expressing Cav3.2 channel variants. Cells expressing WT, V681L and D1233H Cav3.2 channel variants, or co-expressing the two-channel variants (V681L + D1233H) to resemble the heterozygous status of the patient for the two mutations, displayed characteristic low-threshold voltage-activated T-type currents (Figure 2(a,b)). The maximal T-type conductance in cells expressing Cav3.2 variants was similar (V681L: 756 ± 49 pS/pF, n = 45; D1233H: 640 ± 60 pS/pF, n = 48) and was not significantly different from cells expressing the WT channel (688 ± 59, n = 45) (Figure 2(c,d)) and Table (1)).
Figure 2.
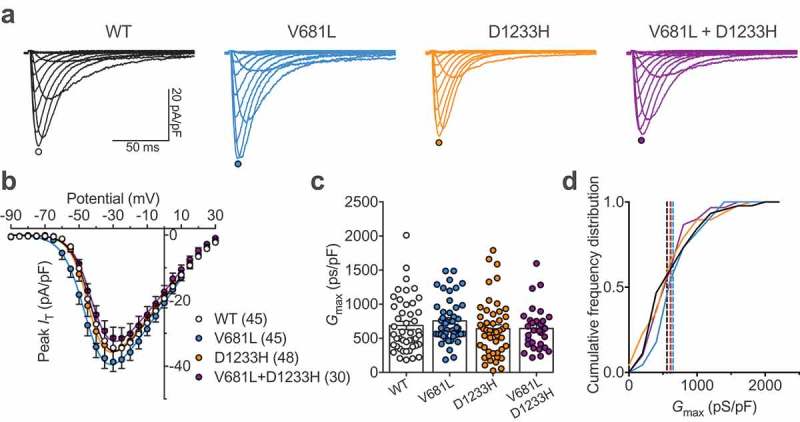
Functional expression of Cav3.2 variants. (a) Representative T-type current traces recorded from cells expressing WT (black traces), V681L (blue traces), D1233H (orange traces), and co-expressing V681L and D1233H channel variants (purple traces) in response to 150 ms depolarizing steps ranging between −90 mV and 30 mV from a holding potential of −100 mV. (b) Corresponding mean peak current-voltage (I/V) relationship. (c) Corresponding mean maximal macroscopic conductance (Gmax) values obtained from the fit of the I/V curves with the modified Boltzmann Equation (1). (d) Cumulative frequency plot of data shown in (C). Dotted lines indicate the median values for each distribution (WT: 558 pS/pF; V681L: 648 pS/pF; D1233H: 601 pS/pF; V681L + D1233H: 615 pS/pF).
Table 1.
Electrophysiological properties of human Cav3.2 channel variants expressed in tsA-201 cells. Activation and inactivation kinetic values are shown for a depolarization step to −40 mV from a holding potential of −100 mV.
| Activation |
Inactivation |
RFI |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cav3.2 variant | V0.5 (mV) | k | (n) | τ (ms) | (n) | Gmax (pS/pF) | (n) | V0.5 (mV) | k | (n) | τ (ms) | (n) | τ (ms) | (n) |
| WT | −39.9 ± 0.6 | 5.6 ± 0.2 | 45 | 5.8 ± 0.2 | 45 | 688 ± 59 | 45 | −69.8 ± 0.6 | 4.4 ± 0.1 | 32 | 21.9 ± 1.0 | 32 | 377 ± 35 | 11 |
| V681L | −43.4 ± 0.9** | 5.4 ± 0.2 | 45 | 5.1 ± 0.3 | 45 | 756 ± 49 | 45 | −75.3 ± 1.2*** | 4.4 ± 0.1 | 30 | 18.4 ± 0.9* | 30 | 373 ± 29 | 18 |
| D1233H | −41.1 ± 0.8 | 5.0 ± 0.2* | 48 | 5.8 + 0.3 | 48 | 640 ± 60 | 48 | −70.0 ± 0.7 | 4.1 ± 0.1 | 34 | 19.3 ± 1.1 | 34 | 343 ± 30 | 15 |
| V681L+D1233H | −40.8 ± 0.6 | 5.6 ± 0.1 | 30 | 5.7 ± 0.2 | 30 | 644 ± 58 | 30 | −74.0 ± 1.0** | 4.7 ± 0.2 | 31 | 20.6 ± 0.8 | 31 | 435 ± 24 | 27 |
These results indicate that the V681L and D1233H mutations do not alter the expression level of Cav3.2 channels.
Gating properties of Cav3.2 channel variants
To assess the functional properties of Cav3.2 variants, we analyzed the voltage-dependence of T-type current activation in cells expressing WT, V681L, D1233H, or co-expressing the V681L and D1233H channel variants (co-expression of the two separate cDNA constructs each carrying one mutation). The mean half-activation potential in cells expressing the V681L variant was shifted by 3.5 mV (p = 0.0039) toward hyperpolarized potentials (−43.4 ± 0.9 mV, n = 45) compared to cells expressing the WT channel (−39.9 ± 0.6 mV, n = 45) (Figure 3(a,b) and Table (1)). Although the mean half-activation potential of T-type currents measured in cells expressing the D1233H variant was not significantly altered, we observed a significant decrease (p = 0.0429) of the inactivation slope factor (5.0 ± 0.2, n = 48) compared to WT channels (5.6 ± 0.2, n = 45) (Figure 3(c) and Table (1)). To further investigate the gating properties of Cav3.2 variants, we assessed the voltage-dependence of the steady-state inactivation of the T-type current. We observed a hyperpolarized shift of the mean half-inactivation potential by 5.5 mV (p < 0.0001) in cells expressing the V681L variant alone (−75.3 ± 1.2 mV, n = 30) and by 4.2 mV (p = 0.0027) in cells co-expressing the V681L and D1233H variants (−74.0 ± 1.0 mV, n = 31) compared to cells expressing the WT channel (−69.8 ± 0.6 mV, n = 32) (Figure 3(d–f) and Table (1)). In contrast, the voltage-dependence of steady-state inactivation of the T-type current in cells expressing the D1233H remained unaltered (Figure 3(d–f) and Table (1)). In addition, recovery from inactivation (Figure 3(g,h) and Table (1)) as well as inactivation and inactivation kinetics remain similar between the channel variants, and only a mild acceleration of the inactivation kinetic of the V681L variant (18.4 ± 0.9, n = 30) was observed compared to the WT channel (21.9 ± 1.0, n = 45) (Figure 3(i) and Table (1)).
Figure 3.
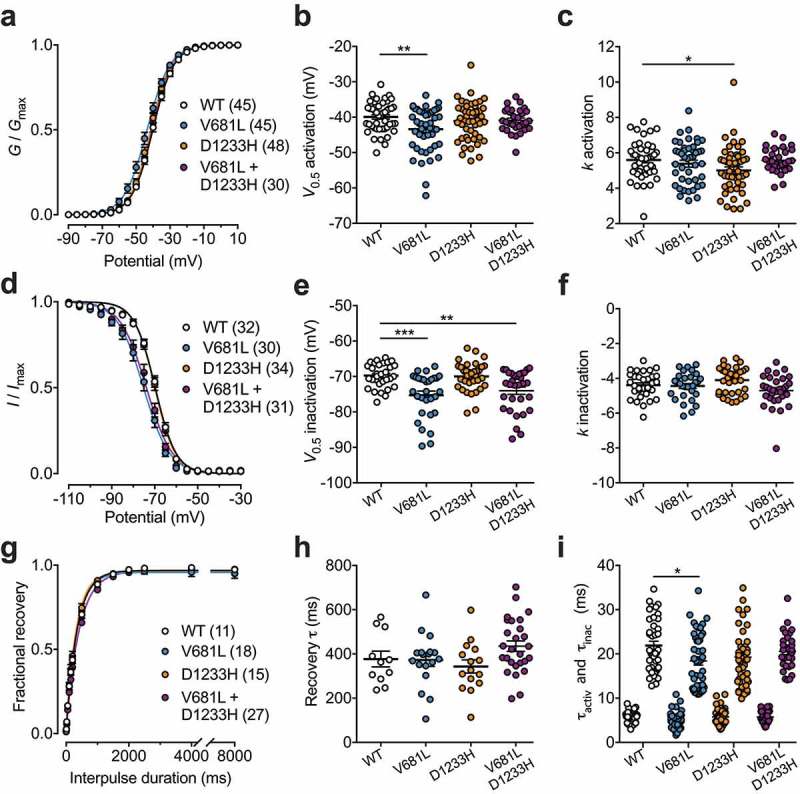
Electrophysiological properties of Cav3.2 variants. (a) Mean normalized voltage-dependence of T-type current activation for cells expressing WT (white circles), V681L (blue circles), D1233H (orange circles), and co-expressing V681L and D1233H channel variants (purple circles). (b) Corresponding mean half-activation potential values obtained from the fit of the activation curves with the modified Boltzmann Equation (2). (c) Corresponding mean activation slope factor. (d) Mean normalized voltage-dependence of steady-state inactivation. (e) Corresponding mean half-inactivation potential values obtained from the fit of the inactivation curves with the two-state Boltzmann function (3). (f) Corresponding mean inactivation slope factor. (g) Mean normalized recovery from inactivation kinetics. (h) Corresponding mean time constant τ values of recovery from inactivation obtained by fitting recovery curves with the single-exponential function (4). (i) Mean time constant τ values of activation and inactivation of T-type currents obtained by fitting the rising and decay phase of the current with a single exponential function, respectively.
Collectively, these data indicate that the V681L and D1233H mutations produce mild but significant alterations of the gating of Cav3.2 by altering the voltage dependence of activation and inactivation of the channel.
Window current of Cav3.2 channel variants
Considering the effect of the V681L and D1233H mutations on the voltage dependence of activation and inactivation of Cav3.2 channels, we analyzed the window current of Cav3.2 variants as the area under the overlap between the activation and inactivation curves (Figure 4(a)). We observed a decrease of the window current generated by Cav3.2 variants. For instance, the mean area of the window current was decreased by 22% (p = 0.0055) and 19% (p = 0.0188) in cells expressing the V681L and D1233H variants, respectively (Figure 4(b,c)). Consistent with this observation, the window current in cells co-expressing the V681L and D1233H channel variants were decreased by 22% (p = 0.0079) compared to cells expressing the WT channel (Figure 4(b,c)). In addition, we observed a hyperpolarized shift of the peak-voltage of the window current by 4.6 mV (p < 0.0001) in cells expressing the V681L variant (Figure 4(d)).
Figure 4.
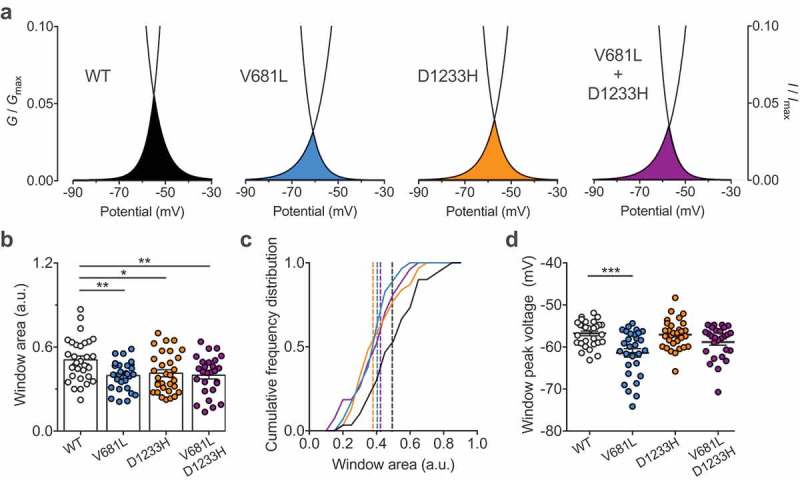
Window current of Cav3.2 variants. (a) Representative activation (G/Gmax) – inactivation curves (I/Imax) showing the window current for cells expressing WT (black), V681L (blue), D1233H (orange), and co-expressing V681L and D1233H channel variants (purple). (b) Corresponding mean window current measured as the area of the overlap between the activation and inactivation curves. (c) Corresponding frequency plot for each distribution. The dotted lines indicate the median values (WT: 0.4936; V681L: 0.4047; D1233H: 0.3792; V681L + D1233H: 0.4233). (D) Corresponding mean peak-voltage of the window currents. (d) Corresponding mean peak-voltage of the window currents.
Altogether, these data indicate that alteration of the voltage-dependence of activation and inactivation caused by the V681L and D1233H mutations ultimately translates into a decrease of the window current supported by Cav3.2 variants.
Discussion
Here we describe two compounds heterozygous CACNA1H mutations identified in a case-unaffected-parents trio with infantile-onset NMD. The two mutations, V681L and D1233H, are located within the intracellular loops connecting domains I and II (I-II loop) and II and III (II-III loop) of the Cav3.2 T-type calcium channel. Previous structure-function studies have documented the role of the I-II loop of Cav3.2 in the control of the gating and surface expression of the channel [17,18]. Additionally, several human mutations associated with idiopathic generalized epilepsy and located within the I-II loop and II-III-loop of Cav3.2 alter channel gating [19–22]. Consistent with these observations, our functional analysis of Cav3.2 missense variants revealed several alterations of the gating properties of the channel. For instance, the V681L mutation produced a hyperpolarized shift of the voltage dependence of activation, as well as a concomitant hyperpolarized shift of the voltage dependence of inactivation. From a mechanistic viewpoint, this apparent mix gain- and loss-of-channel function can be explained by a model in which the V681L mutation may destabilize the closed state of the channel. This notion has recently been proposed for Cav3.3 channels [23] and is consistent with the observation that T-type channels inactivate from both open and closed states [24,25]. Of particular importance is the observation that as a result of the effect on the voltage-dependence of activation and inactivation, the V681L mutation caused a marked decrease of the window current supported by Cav3.2 channel. Likewise, decrease of the window current was also observed with the D1233H as a result of the increased slope of the activation curve without alteration of the mid-voltage.
The question then arises as to how channel defects caused by the V681L and D1233H mutations may contribute to the disease phenotype. Alteration of the window current appears particularly relevant as an underlying mechanism of the amyotrophy observed in the proband for several reasons. First, Cav3.2 channels are functionally expressed in embryonic skeletal muscle fibers where they present a developmental expression pattern consistent with a role in early-stage muscle differentiation. For instance, the T-type current density in muscle fibers increases transiently during prenatal myogenesis to reach a maximum expression at embryonic day E16 before to decrease dramatically until birth [26]. Second, extracellular calcium is required for the fusion of myoblasts [27] and a rise in intracellular calcium level precedes fusion [28,29]. Third, it was proposed that rise in intracellular calcium levels occurs via the window current supported by Cav3.2 channels in response to the hyperpolarization of the plasma membrane that proceeds the fusion of the myoblasts [30]. Therefore, our observation that the p.V681L and p.D1233H mutations caused a reduction of the window current could have an important consequence at early stages of muscle differentiation and may represent an underlying mechanism of infantile-onset amyotrophy (Figure 5(a)). Additionally, several studies have reported the expression of T-type channels in motor neurons [31,32]. Although the role of T-type channels in motor neurons has not been investigated yet, it is likely that they contribute to some forms of neuronal excitability. Therefore, alteration of T-type channel activity is likely to affect muscle afferent signaling which may participate to some extend to motor symptoms.
Figure 5.
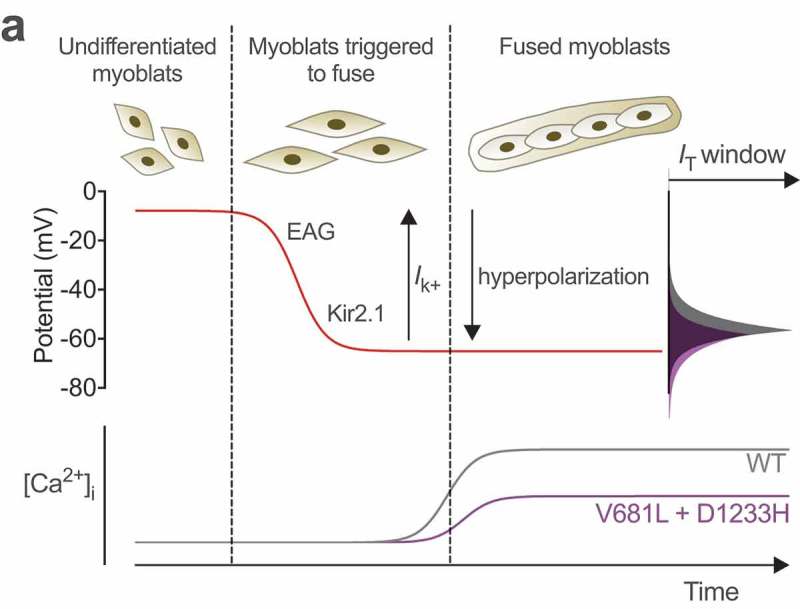
Hypothetical model for altered myogenesis by Cav3.2 variants. While the membrane potential of undifferentiated myoblast is about −8 mV, sequential expression of the slow-inactivating voltage-gated ether-a-go-go (EAG) and inward-rectifying Kir2.1 potassium channels brings the membrane potential to approximately −65 mV (red line), allowing window calcium influx through Cav3.2 channels (grey line) required for the fusion of myoblasts. Reduced window current caused the by V681L and D1233H mutations results in a decreased window calcium influx (purple line) which potentially compromises early stage myogenesis. Adapted from [30].
In summary, we identified new CACNA1H variants in an individual with infantile-onset NMD. Clinical, histopathological, and electrophysiological findings indicating a loss-of-Cav3.2 channel function suggest that severe congenital amyoplasia may be related to CACNA1H, which add to the growing list of CACNA1H channelopathies and would represent a new phenotype associated with mutations in this gene.
Funding Statement
A.T. is supported by a postdoctoral fellowship from the Institute of Organic Chemistry and Biochemistry (IOCB). N.W. is supported by the IOCB.
Acknowledgments
We thank family members for participating in this study.
Disclosure statement
The authors report no conflict of interest.
References
- [1].Zatz M, Passos-Bueno MR, Vainzof M.. Neuromuscular disorders: genes, genetic counseling and therapeutic trials. Genet Mol Biol. 2016;39(3):339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Laing NG. Genetics of neuromuscular disorders. Crit Rev Clin Lab Sci. 2012;49(2):33–48. [DOI] [PubMed] [Google Scholar]
- [3].Lamar KM, McNally EM. Genetic modifiers for neuromuscular diseases. J Neuromuscul Dis. 2014;1(1):3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Steinberg KM, Yu B, Koboldt DC, et al. Exome sequencing of case-unaffected-parents trios reveals recessive and de novo genetic variants in sporadic ALS. Sci Rep. 2015;5:9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rzhepetskyy Y, Lazniewska J, Blesneac I, et al. CACNA1H missense mutations associated with amyotrophic lateral sclerosis alter Cav3.2 T-type calcium channel activity and reticular thalamic neuron firing. Channels (Austin). 2016;466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Weiss N, Zamponi GW. T-type calcium channels: from molecule to therapeutic opportunities. Int J Biochem Cell Biol. 2019;108:34–39. [DOI] [PubMed] [Google Scholar]
- [7].Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83(1):117–161. [DOI] [PubMed] [Google Scholar]
- [8].Zamponi GW, Striessnig J, Koschak A, et al. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. 2015;67(4):821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gambardella A, Labate A. The role of calcium channel mutations in human epilepsy. Prog Brain Res. 2014;213:87–96. [DOI] [PubMed] [Google Scholar]
- [10].Splawski I, Yoo DS, Stotz SC, et al. CACNA1H mutations in autism spectrum disorders. J Biol Chem. 2006;281(31):22085–22091. [DOI] [PubMed] [Google Scholar]
- [11].Lu AT, Dai X, Martinez-Agosto JA, et al. Support for calcium channel gene defects in autism spectrum disorders. Mol Autism. 2012;3(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Souza IA, Gandini MA, Wan MM, et al. Two heterozygous Cav3.2 channel mutations in a pediatric chronic pain patient: recording condition-dependent biophysical effects. Pflugers Arch. 2016;468(4):635–642. [DOI] [PubMed] [Google Scholar]
- [13].Scholl UI, Stölting G, Nelson-Williams C, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife. 2015;4:e06315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Daniil G, Fernandes-Rosa FL, Chemin J, et al. CACNA1H mutations are associated with different forms of primary aldosteronism. EBioMedicine. 2016;13:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dubel SJ, Altier C, Chaumont S, et al. Plasma membrane expression of T-type calcium channel alpha(1) subunits is modulated by high voltage-activated auxiliary subunits. J Biol Chem. 2004;279(28):29263–29269. [DOI] [PubMed] [Google Scholar]
- [16].Proft J, Rzhepetskyy Y, Lazniewska J, et al. The Cacna1h mutation in the GAERS model of absence epilepsy enhances T-type Ca2+ currents by altering calnexin-dependent trafficking of Cav3.2 channels. Sci Rep. 2017;7(1):11513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vitko I, Bidaud I, Arias JM, et al. The I-II loop controls plasma membrane expression and gating of Ca(v)3.2 T-type Ca2+ channels: a paradigm for childhood absence epilepsy mutations. J Neurosci. 2007;27(2):322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Arias-Olguín II, Vitko I, Fortuna M, et al. Characterization of the gating brake in the I-II loop of Ca(v)3.2 T-type Ca(2+) channels. J Biol Chem. 2008;283(13):8136–8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Khosravani H, Altier C, Simms B, et al. Gating effects of mutations in the Cav3.2 T-type calcium channel associated with childhood absence epilepsy. J Biol Chem. 2004;279(11):9681–9684. [DOI] [PubMed] [Google Scholar]
- [20].Khosravani H, Bladen C, Parker DB, et al. Effects of Cav3.2 channel mutations linked to idiopathic generalized epilepsy. Ann Neurol. 2005;57(5):745–749. [DOI] [PubMed] [Google Scholar]
- [21].Peloquin JB, Khosravani H, Barr W, et al. Functional analysis of Ca3.2 T-type calcium channel mutations linked to childhood absence epilepsy. Epilepsia. 2006;47(3):655–658. [DOI] [PubMed] [Google Scholar]
- [22].Eckle VS, Shcheglovitov A, Vitko I, et al. Mechanisms by which a CACNA1H mutation in epilepsy patients increases seizure susceptibility. J Physiol. 2014;592(4):795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jurkovicova-Tarabova B, Cmarko L, Rehak R, et al. Identification of a molecular gating determinant within the carboxy terminal region of Cav3.3 T-type channels. Mol Brain. 2019;34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Frazier CJ, Serrano JR, George EG, et al. Gating kinetics of the alpha1I T-type calcium channel. J Gen Physiol. 2001;12(1):457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Serrano JR, Perez-Reyes E, Jones SW. State-dependent inactivation of the alpha1G T-type calcium channel. J Gen Physiol. 1999;114(2):185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Berthier C, Monteil A, Lory P, et al. Alpha(1H) mRNA in single skeletal muscle fibres accounts for T-type calcium current transient expression during fetal development in mice. J Physiol. 2002;539(3):681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wakelam MJ. The fusion of myoblasts. Biochem J. 1985;228(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Entwistle A, Zalin RJ, Bevan S, et al. The control of chick myoblast fusion by ion channels operated by prostaglandins and acetylcholine. J Cell Biol. 1988;106(5):1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rapuano M, Ross AF, Prives J. Opposing effects of calcium entry and phorbol esters on fusion of chick muscle cells. Dev Biol. 1989;134(2):271–278. [DOI] [PubMed] [Google Scholar]
- [30].Bijlenga P, Liu JH, Espinos E, et al. T-type alpha 1H Ca2+ channels are involved in Ca2+ signaling during terminal differentiation (fusion) of human myoblasts. Proc Natl Acad Sci U S A. 2000;97(13):7627–7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Canto-Bustos M, Loeza-Alcocer E, González-Ramírez R, et al. Functional expression of T-type Ca2+ channels in spinal motoneurons of the adult turtle. PLoS One. 2014;9(9):e108187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang Z, David G. Stimulation-induced Ca(2+) influx at nodes of Ranvier in mouse peripheral motor axons. J Physiol. 2016;594(1):39–57. [DOI] [PMC free article] [PubMed] [Google Scholar]