

ABSTRACT
Amongst the regulators of voltage-gated ion channels is the collapsin response mediator protein 2 (CRMP2). CRMP2 regulation of the activity and trafficking of NaV1.7 voltage-gated sodium channels as well as the N-type (CaV2.2) voltage-gated calcium channel (VGCC) has been reported. On the other hand, CRMP2 does not appear to regulate L- (CaV1.x), P/Q- (CaV2.1), and R- (CaV2.3) type high VGCCs. Whether CRMP2 regulates low VGCCs remains an open question. Here, we asked if CRMP2 could regulate the low voltage-gated (T-type/CaV3.x) channels in sensory neurons. Reducing CRMP2 protein levels with short interfering RNAs yielded no change in macroscopic currents carried by T-type channels. No change in biophysical properties of the T-type currents was noted. Future studies pursuing CRMP2 druggability in neuropathic pain will benefit from the findings that CRMP2 regulates only the N-type (CaV2.2) calcium channels.
KEYWORDS: CRMP2, T-type calcium channel, DRG sensory neuron, electrophysiology
Regulators of voltage-gated calcium channels (VGCCs) shape a diversity of biological functions within the nervous system 1,2. In chronic pain, proteins regulating the N-type (CaV2.2) VGCCs can modulate nociception [1]. One such protein is the collapsin response mediator protein 2 (CRMP2) [3–15]. Our continuing studies have established CRMP2 as a bona fide binding partner and regulator of the presynaptic trafficking of CaV2.2 [4,8,13,15–18]. In neuropathic pain, increased phosphorylation of CRMP2 by cyclin-dependent kinase 5 defines the presynaptic content for CaV2.2, but not for CaV2.3 (i.e. the R-type) VGCCs [3,13,19]. In a proteomic study, the L-type VGCC was shown to be part of the CRMP2 interactome [20] likely though a motif in the C-terminus in the CaV1.2 L-type channel [21]. While CRMP2-dependent regulation of CaV2.1 (i.e. the P/Q-type) VGCC has never been investigated, CRMP2 interaction domains found in CaV2.2 are poorly conserved in CaV2.1 or other channels [21]. In investigating CRMP2 phosphorylation with the small molecule (S)-lacosamide, we found that only calcium influx via N-type, but not other high-voltage gated channels, was blocked [5]. Collectively, these results hinted at a specific action of CRMP2 on N-type VGCCs.
Loss of CRMP2 expression abolishes pathological pain [11,19]. This reversal of pain was attributed to decreased excitability of dorsal root ganglion (DRG) neurons likely due to CRMP2-dependent effects on the trafficking and activity of the NaV1.7 voltage-gated sodium channel [22]. Further work from our group revealed that a peptide derived from the CRMP2/CaV2.2 interface (designated CBD3, for calcium binding domain 3) decreased the frequency of spontaneous excitatory postsynaptic potentials in spinal cord [18]. A single mutation that stabilized CBD3’s helical structure rendered the mutant peptide capable of inhibition of sensory neuron excitability [23]. This suppression of excitability was not related to inhibition of sodium channels but rather linked to impairment of T-type (CaV3.x) channel function [23]. T-type VGCCs are low voltage-activated calcium channels important for the initiation of an action potential [1,24,25]. In neuropathic pain, CaV3.2 channels were reported to be increased at presynaptic sites through a trafficking mechanism [26]. Thus, the observations that (1) a peptide derived from CRMP2 could inhibit T-type VGCCs [23] and (2) T-type VGCCs are increased in a model where CRMP2 is dysregulated, together support the hypothesis that CRMP2 could modulate T-type VGCCs.
To directly test whether CRMP2 could regulate T-type calcium currents in DRG sensory neurons, we used a previously validated short interfering RNA (siRNA) against CRMP2 [19,27] to knockdown CRMP2 protein levels. Rat DRG neurons were co-transfected with CRMP2 siRNA or control siRNA and (~50 ng) of green fluorescent protein (GFP) expressing plasmid to identify the transfected cells prior to electrophysiological recordings. We used electrophysiology protocols described before [24] to record T-type currents. From a holding potential of −90 mV, we used 200-ms depolarization steps to change the membrane potential from −70 to +60 mV (10 mV increments) to evoke prototypical T-type calcium currents (Figure 1(a)). After transfection with CRMP2 (n = 15) or control (n = 17) siRNA, we recorded low voltage-activated calcium currents (Figure 1(a)) from DRG neurons with an average diameter between 20 and 30 µm. We measured current–voltage (I–V) relationships (Figure 1(b)) and observed that loss of CRMP2 expression had no effect on T-type calcium current amplitudes at all test potentials tested (Figure 1(b)). At peak current density (−10 mV), no difference was found between CRMP2 siRNA or control siRNA transfected DRG neurons (Figure 1(c) and Table 1). Knockdown of CRMP2 did not alter the channel gating properties as we measured a similar half-maximal activation (V0.5) of T-type calcium channels in both transfected conditions (Figure 1(d)). The kinetics of macroscopic current inactivation (Figure 1(e)) were unchanged at all membrane potentials tested (−40 mV, Figure 1(f)). The time-dependent activation (10–90% rise time) of T-type currents was not affected by loss of CRMP2 expression (Figure 1(g–h)). We next tested whether CRMP2 could control the voltage-dependent kinetics of channel inactivation (Figure 1(i)) and found this property to also not be affected by CRMP2 siRNA transfection. Deactivating tail currents calculated using the single exponential function: y = A1 × e(−x/τ1) + y0, where A1 is the amplitude, τ1 is the decay constant, and y0 is the offset. The resulting τ values (Figure 1(j)), showed no differences irrespective of CRMP2 expression. Finally, because upon long membrane hyperpolarizations in DRG neurons T-type calcium channels can recover from inactivation, we tested if this biophysical parameter could be contingent on CRMP2. This property has important consequences on the firing properties of sensory neurons expressing T-type calcium channels. Thus, we tested the recovery from inactivation using a double-pulse protocol with a variable interpulse duration at −90 mV (Figure 1(k)) after a 500-ms-long inactivating pulse (Vh = −90 mV; Vt = −30 mV). T-type currents recovered fully, independently of the transfection condition (Figure 1(k)). Taken together, our results show that CRMP2 has no role to play on regulation of T-type calcium currents and gating properties of these T-type calcium channels. We previously reported that CRMP2 has no effect on the R-type VGCC [13]. The present results showing the lack of CRMP2 regulation of T-type VGCC suggest now to support the assertion that CRMP2 is a specific regulatory protein for CaV2.2 but for no other type of VGCCs. This specificity is an important step in furthering our understanding of the contribution of CRMP2 in neuropathic pain.
Figure 1.
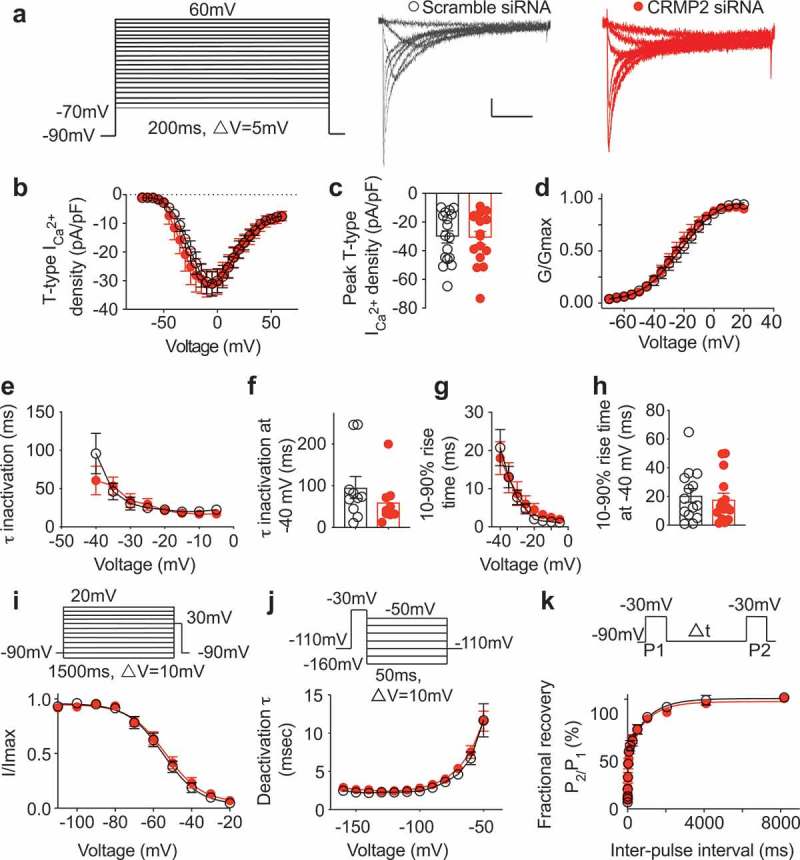
CRMP2 does not affect T-type Ca2+ currents in dorsal root ganglion (DRG) sensory neurons. (a) Representative family of traces of T-type Ca2+ currents from DRG sensory transfected with either control or CRMP2 siRNA. Voltage protocol used to evoke the currents is shown. Summary of the normalized (pA/pF) T-type calcium current density versus voltage relationship (b) and peak T-type Ca2+ current density at −10 mV (mean ± SEM) (c) from DRG sensory neurons transfected as indicated. (d) Boltzmann fits for normalized conductance G/Gmax voltage relations for voltage-dependent activation of T-type currents. (e) Inactivation τ (single-exponential fit of decaying portion of the current waveforms using a single-exponential equation: y = A1 × e(−x/τ1) + y0, where A1 is the amplitude, τ1 is the decay constant, and y0 is the offset), isolated at −40 mV (f) and (g) time-dependent activation (10–90% rise time) from I–V curves and at −40 mV (h) in DRG cells shown in (b). Boltzmann fits for normalized conductance G/Gmax voltage relations for voltage-dependent inactivation (i) of sensory neurons treated as indicated. (j) Deactivating tail currents in DRG neurons transfected with control or CRMP2 siRNA were fit with a single-exponential function. The resulting τ values are plotted. (k) Recovery from inactivation in indicated groups. Data are averaged and fitted by double exponential association (P > 0.05, n > 12 per condition). All graphs show mean ± SEM with individual data points showed when possible.
Table 1.
Gating properties of T-type calcium channels in DRG neurons.a
| Control | siRNA | |
|---|---|---|
| Activation | ||
| V1/2 (P = 0.10) | −22.7 ± 1.6(17) | −26.3 ± 1.4(16) |
| k (P = 0.42) | 12.8 ± 1.7(17) | 11.0 ± 1.4(16) |
| Inactivation | ||
| V1/2 (P = 0.41) | −54.9 ± 1.1(18) | −53.6 ± 1.1(18) |
| k (P = 0.94) | −9.4 ± 1.0(18) | −9.5 ± 1.0(18) |
| Recovery | ||
| τ1 (ms) (P = 0.87) | 1036.0 ± 217.2(15) | 981.0 ± 255.1(18) |
| τ2 (ms) (P = 0.82) | 25.2 ± 4.8(15) | 27.2 ± 6.9(18) |
aValues are means ± SEM calculated from fits of the data from the indicated number of individual cells (in parentheses) to the Boltzmann equation; V1/2 midpoint potential (mV) for voltage-dependent activation or inactivation; k, slope factor. τ1 and τ2, time constants; the data could only be fit with a double exponential equation as reported by the Todorovic group [24] and P > 0.05; Student’s t test.
We conclude that CRMP2 regulation of NaV1.7 [9,10,12,13,22,28] is responsible for controlling DRG neurons excitability and that CRMP2 regulation of CaV2.2 is responsible for increased neurotransmitter release at the primary afferent [11,13,17,18]. For future therapeutic targeting, the specificity of action of CRMP2 toward CaV2.2 will reduce the potential for unwanted effects of novel CRMP2-targeted compounds.
Methods
Preparation of rat DRG cultures (acutely dissociated neurons)
DRG neurons were cultured using methods as described previously [29]. Collected dorsal root ganglia were digested in an enzymatic combination containing bicarbonate-free, serum-free, sterile DMEM (Cat# 11965, Thermo Fisher Scientific) solution, neutral protease (3.125 mg/mL, Cat#LS02104; Worthington, Lakewood, NJ), and collagenase type I (5 mg/mL, Cat# LS004194, Worthington, Lakewood, NJ). After incubation for approximately 1 h with gentle agitation under 37°C, DRG neurons were centrifuged post-dissociation and isolated from DRG media (DMEM containing 1% penicillin/streptomycin sulfate from 10,000 μg/mL stock, 30 ng/mL nerve growth factor, and 10% fetal bovine serum [Hyclone]). Dissociated DRG neurons were subsequently plated onto 12- mm laminin and poly-d-lysine-coated coverslips. Cultures were utilized before 2-days’ time.
Transfection of rat primary DRG neurons
Collected cells were resuspended in nucleofector transfection reagent containing 6 μL (50 nM) scrambled or CRMP2 siRNA plus 6 µL GFP (0.5 μg/μL). Then, cells were subjected to electroporation protocol O-003 in an Amaxa Biosystems (Lonza) and plated onto 12- mm poly-d-lysine- and laminin-coated glass coverslips.
Whole-cell electrophysiological recordings of calcium currents in acutely dissociated DRG neurons
The protocol for isolating T-type calcium currents was previously described by Choe et al. [24]. The extracellular recording solution used to isolate T currents consisted of the following (in millimolar): 2CaCl2, 152 TEA-Cl, 10 HEPES, pH adjusted to 7.4 with TEA-OH. The intracellular recording solution consisted of (in millimolar) 135 tetramethylammonium hydroxide, 10 EGTA, 40 HEPES, and 2 MgCl2, pH adjusted to 7.2 with hydrofluoric acid. As previously described, the majority of acutely dissociated small DRG cells express T currents [24]. Thus, in order to record T-type calcium currents, we focused on small DRG neurons with an average soma diameter of 20–30 μm.
To avoid contamination by residual High Voltage Activated (HVA) currents which are present at more positive membrane potentials, we measured the T-type Ca2+ current from the peak, which was subtracted from the current at the end of the depolarizing test potential. Activation of ICa-T was measured by using a holding voltage of −90 mV with voltage steps 200 ms in duration applied at 500-ms intervals in 10 mV increments from −70 to +60 mV. Inactivation of ICa-T was determined by applying an 1500-ms conditioning prepulse (−110 to +20 mV in 10 mV increments) after which the voltage was stepped to −30 mV for 20 ms; a 40-ms interval with a holding voltage of −90 mV separated each acquisition. In the deactivation tau protocol, the neuron was first hold at −110 mV, then the voltage jumped to −30 mV for 10 ms followed by a 50-ms conditioning prepulse (−160 to −40 mV in 10 mV increments). A 2-s interval with a holding voltage of −90 mV separated each acquisition. ICa-T recovery from inactivation was obtained by using our standard double-pulse protocol with variable interpulse duration at −90 mV after a 500-ms-long inactivating pulse (Vh = −90 mV; Vt = −30 mV).
The Boltzmann relation was used to determine the voltage dependence for activation of ICa wherein the conductance–voltage curve was fit by the equation G/Gmax = 1/[1+ exp (V0.5 − Vm)/k], where G is the conductance G = I/(Vm − ECa), Gmax is the maximal conductance obtained from the Boltzmann fit under control conditions, V0.5 is the voltage for half-maximal activation, Vm is the membrane potential, and k is a slope factor. ECa is the reversal potential for ICa. The values of ICa around the reversal potential were fit with a linear regression line to establish the voltage at which the current was zero. The Boltzmann parameters were determined for each individual neuron and then used to calculate the mean ± SEM.
Funding Statement
This work was supported by the Guangdong Medical Research Foundation: [Grant Number A2017047], National Institute of Neurological Disorders and Stroke: [Grant Number R01NS098772], and National Institute on Drug Abuse: [Grant Number R01DA042852].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Bourinet E, Altier C, Hildebrand ME, et al. Calcium-permeable ion channels in pain signaling. Physiol Rev. 2014;94:81–140. [DOI] [PubMed] [Google Scholar]
- [2].Park J, Luo ZD.. Calcium channel functions in pain processing. Channels. 2010;4:510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Moutal A, White KA, Chefdeville A, et al. Dysregulation of CRMP2 post-translational modifications drive its pathological functions. Mol Neurobiol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Francois-Moutal L, Wang Y, Moutal A, et al. A membrane-delimited N-myristoylated CRMP2 peptide aptamer inhibits CaV2.2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain. 2015;156:1247–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Moutal A, Chew LA, Yang X, et al. (S)-lacosamide inhibition of CRMP2 phosphorylation reduces postoperative and neuropathic pain behaviors through distinct classes of sensory neurons identified by constellation pharmacology. Pain. 2016;157:1448–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Moutal A, Eyde N, Telemi E, et al. Efficacy of (S)-Lacosamide in preclinical models of cephalic pain. Pain Rep. 2016;1:pii: e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Moutal A, Francois-Moutal L, Perez-Miller S, et al. Lacosamide binding to collapsin response mediator protein 2 (CRMP2) regulates CaV2.2 activity by subverting its phosphorylation by Cdk5. Mol Neurobiol. 2016;53:1959–1976. [DOI] [PubMed] [Google Scholar]
- [8].Xie JY, Chew LA, Yang X, et al. Sustained relief of ongoing experimental neuropathic pain by a CRMP2 peptide aptamer with low abuse potential. Pain. 2016;157:2124–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Moutal A, Wang Y, Yang X, et al. Dissecting the role of the CRMP2-neurofibromin complex on pain behaviors. Pain. 2017;158:2203–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Moutal A, Yang X, Li W, et al. CRISPR/Cas9 editing of Nf1 gene identifies CRMP2 as a therapeutic target in neurofibromatosis type 1-related pain that is reversed by (S)-Lacosamide. Pain. 2017;158:2301–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Moutal A, Cai S, Luo S, et al. CRMP2 is necessary for neurofibromatosis type 1 related pain. Channels. 2018;12:47–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Moutal A, Sun L, Yang X, et al. CRMP2-neurofibromin interface drives NF1-related Pain. Neuroscience. 2018;381:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yu J, Moutal A, Dorame A, et al. Phosphorylated CRMP2 regulates spinal nociceptive neurotransmission. Mol Neurobiol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Khanna R, Yu J, Yang X, et al. Targeting the CaValpha-CaVbeta interaction yields an antagonist of the N-type CaV2.2 channel with broad antinociceptive efficacy. Pain. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Moutal A, Li W, Wang Y, et al. Homology-guided mutational analysis reveals the functional requirements for antinociceptive specificity of collapsin response mediator protein 2-derived peptides. Br J Pharmacol. 2018;175:2244–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brittain JM, Piekarz AD, Wang Y, et al. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem. 2009;284:31375–31390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chi XX, Schmutzler BS, Brittain JM, et al. Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J Cell Sci. 2009;122:4351–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Brittain JM, Duarte DB, Wilson SM, et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca(2)(+) channel complex. Nat Med. 2011;17:822–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Moutal A, Luo S, Largent-Milnes TM, et al. Cdk5-mediated CRMP2 phosphorylation is necessary and sufficient for peripheral neuropathic pain. Neurobiol Pain. 201. 9;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Martins-de-Souza D, Cassoli JS, Nascimento JM, et al. The protein interactome of collapsin response mediator protein-2 (CRMP2/DPYSL2) reveals novel partner proteins in brain tissue. Proteomics Clin Appl. 2015;9:817–831. [DOI] [PubMed] [Google Scholar]
- [21].Wilson SM, Schmutzler BS, Brittain JM, et al. Inhibition of transmitter release and attenuation of AIDS therapy-induced and tibial nerve injury-related painful peripheral neuropathy by novel synthetic Ca2+ channel peptides. J Biol Chem. 2012;287:35065–35077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dustrude ET, Moutal A, Yang X, et al. Hierarchical CRMP2 posttranslational modifications control NaV1.7 function. Proc Natl Acad Sci USA. 2016;113:E8443–E8452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Piekarz AD, Due MR, Khanna M, et al. CRMP-2 peptide mediated decrease of high and low voltage-activated calcium channels, attenuation of nociceptor excitability, and anti-nociception in a model of AIDS therapy-induced painful peripheral neuropathy. Mol Pain. 2012;8:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Choe W, Messinger RB, Leach E, et al. TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. MolPharmacol. 2011;80:900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Todorovic SM, Jevtovic-Todorovic V. Regulation of T-type calcium channels in the peripheral pain pathway. Channels. 2007;1:238–245. [DOI] [PubMed] [Google Scholar]
- [26].Garcia-Caballero A, Gadotti VM, Stemkowski P, et al. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron. 2014;83:1144–1158. [DOI] [PubMed] [Google Scholar]
- [27].Brustovetsky T, Pellman JJ, Yang XF, et al. Collapsin response mediator protein 2 (CRMP2) interacts with N-methyl-D-aspartate (NMDA) receptor and Na+/Ca2+ exchanger and regulates their functional activity. J Biol Chem. 2014;289:7470–7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dustrude ET, Perez-Miller S, Francois-Moutal L, et al. A single structurally conserved SUMOylation site in CRMP2 controls NaV1.7 function. Channels. 2017;11:316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cai S, Bellampalli SS, Yu J, et al. (-)-Hardwickiic acid and Hautriwaic acid induce antinociception via blockade of tetrodotoxin-sensitive voltage-dependent sodium channels. ACS Chem Neurosci. 2019;10:1716–1728. [DOI] [PubMed] [Google Scholar]