

ABSTRACT
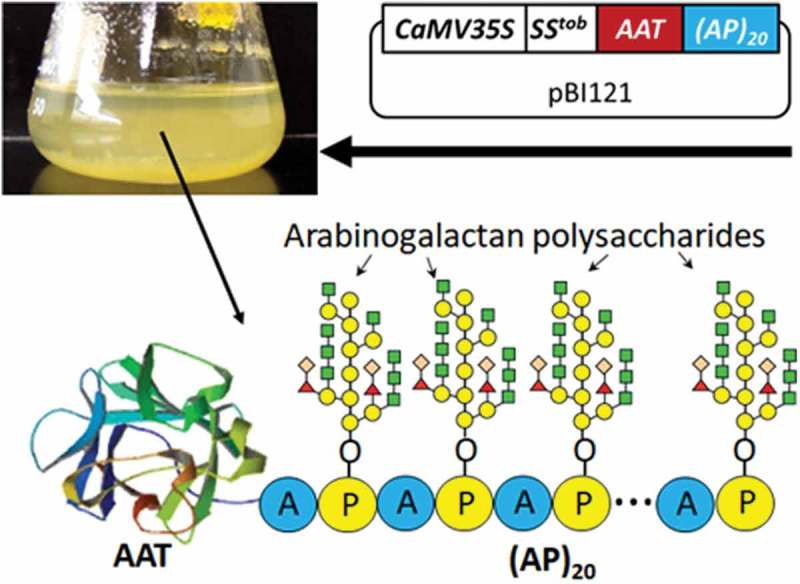
Expression of recombinant proteins fused to a novel glycomodule tag, termed hydroxyproline (Hyp)-O-glycosylated peptides (HypGP), was earlier found to boost secreted protein yields up to 500-fold in plant cell culture. Here, this technology was applied to the expression of human protease inhibitor α1-antitrypsin (AAT) in tobacco BY-2 cell culture. A designer HypGP tag composed of a ‘Ala-Pro’ motif of 20 units, or (AP)20, was engineered either at the N- or C-terminal end of AAT. The (AP)20 tag substantially increased the secreted yields of the recombinant AAT up to 34.7 mg/L. However, the (AP)20-tagged AAT products were frequently subjected to proteolytic processing. The intact AAT-(AP)20 along with some of the truncated AAT domains exhibited desired biological activity in inhibiting elastase. The results from this research demonstrated that the designer (AP)20 module engineered in BY-2 cells could function as a molecular carrier to substantially enhance the secreted yields of the recombinant AAT.
KEYWORDS: Plant cell culture, human α1-antitrypsin, recombinant protein, hydroxyproline-O-glycosylation, secretion
Graphical Abstract
Introduction
Human α1-antitrypsin (AAT) is a serine protease inhibitor (serpin) that functions in maintaining appropriate levels of neutrophil elastase in the respiratory system and other proteinases in the circulatory system [1]. Functional AAT binds irreversibly to neutrophil elastase in the lungs, protecting the lung tissue from being destroyed. A hereditary deficiency of AAT accounts for more than 1% of all chronic obstructive pulmonary cases and young persons with emphysema [2]. Those suffering from AAT deficiency can undergo replacement therapies with plasma-derived AAT to lessen the effects. In addition, AAT has also shown promise for the treatment of respiratory conditions related to cystic fibrosis, another genetic orphan disease characterized by elevated levels of elastase in sputum. However, problems arise with these treatments due to high demand for plasma donors, prohibitive cost to the patient at $100,000 per year [3], risk of unknown blood-borne pathogens/diseases, and the frequency of treatment at least once a week [3]. Thus, there is a pressing need for safer, lower cost and expandable sources of AAT.
Recombinant AAT has been produced in a number of expression systems, including E. coli [4], yeast [5], mammalian cell [6], and whole-plants such as Nicotiana benthamiana [7], tomato [8] and tobacco chloroplasts [9]. However, there have often been challenges associated with cost, safety, bioactivity and authenticity for each expression system. Plant cell suspension cultures are emerging as a promising alternative bioproduction system for recombinant pharmaceuticals as it integrates the merits of whole plant cultivation, microbial fermentation and mammalian cell culture [10,11]. Particularly, as eukaryotic organisms plant cells can produce complex proteins with correct post-translational modifications (e.g., glycosylation) without risk of contamination by human pathogens [12,13]. In fact, recombinant AAT has earlier been produced using plant cell culture, mainly in rice cells using an inducible α-amylase promoter–RAmy3D (induced by sugar starvation) [14–16], which generated a remarkably high secreted AAT yield of 247 mg/L [17]. However, the growth rates, characteristics, and stability of rice cell lines cannot compare with those of tobacco bright yellow-2 (BY-2) cell, and the viability of rice cell is substantially decreased when cultivated in a sucrose-starvation medium to induce gene expression [11]. Tobacco BY-2 cell line is extremely attractive as a bioproduction system due to its fast growth rate (doubling time as short as 11 hr) and ease of genetic transformation and cell cycle synchronization [18]. However, low protein productivity has been the bottleneck limiting the commercial application of the BY-2 cell culture system.
Engineering novel glycomodules composed of a hydroxyproline (Hyp)-O-glycosylated peptide (HypGP) was earlier found to dramatically increase the secreted yields of fused proteins as much as 500-fold in plant cell culture [19–21]. The HypGP is made of a proline-rich peptide backbone (e.g., tandem repeats of a dipeptide ‘Ser-Pro’ motif) subjected to intensive post-translational modifications in plant cells, including proline hydroxylation and subsequent Hyp-O-glycosylation with arabinogalactan polysaccharides [22,23]. Our previous studies indicated that the glycosylated HypGP module could function as a ‘molecular carrier’ to boost the secretion of fused proteins, including interferon α2b, human growth hormone and green fluorescent protein, presumably by facilitating efficient transport of the proteins into extracellular space and preventing proteolytic degradation of the proteins [20,21,24]. Most recently, the HypGP engineering technology was extended to green microalgae (Chlamydomonas reinhardtii), where engineering (SP)n tags (n = 10, 20) enhanced the secreted yields of a fused protein by up to 12-fold [25].
Inspired by early success, we continued to apply this technology to producing AAT, a relatively large-sized protein (52 kDa) compared with those in our previous studies (18 to 27 kDa). Furthermore, a different HypGP design comprised of a ‘Ala-Pro’ motif of 20 units, or (AP)20 was used in this study. Early studies indicated that both the ‘Ser-Pro’ and ‘Ala-Pro’ motifs were 100% hydroxylated and underwent 100% polysaccharide addition in plant cells [26,27]. However, compared with the (SP)n motifs, the (AP)20 lacks the amino acid Ser that might give rise to O-glycosylation [28,29], thus generating more homogenous Hyp-O-glycans. In this report, AAT was expressed in BY-2 cell with a (AP)20 tag, aiming to achieve high secreted protein yields. The Hyp-O-glycosylation, secreted protein yields, and biologic activity of the (AP)20-tagged AAT were characterized.
Materials and methods
Construction of expression vectors and transformation of BY-2 cell
The gene constructs encoding human AAT with a C-terminal (AP)20 tag, or AAT-(AP)20, was synthesized in pUC18 plasmid by GenScript (Piscataway, NJ). The native AAT gene sequence was used in this study, and its encoded amino acid sequence is shown in Supplementary Fig. 1. The AAT gene fragment was then amplified by PCR using the primer pair AAT-F1 and AAT-R (Supplementary Table 1), and subcloned into the plasmid pUC18-(AP)20-EGFP at the NcoI and BsrGI sites by the In-Fusion HD Cloning Kit (Takara Bio USA, Inc) to generate pUC18-(AP)20-AAT. Similarly, the AAT gene was PCR amplified using the primer pair AAT-F2 and AAT-R (Supplementary Table 1), and subcloned into the plasmid pUC18-EGFP at the XmaI and BsrGI sites to generate pUC18-AAT. All the three gene constructs were subcloned into the plant expression vector pBI121-SStob-EGFP [22,30] at the XmaI and BsrGI sites to generate pBI121-SStob-AAT, pBI121-SStob-AAT-(AP)20 and pBI121-SStob-(AP)20-AAT, respectively (Figure 1), and then stably transformed into BY-2 cell with the Agrobacterium-mediated approach [31]. Transgene expression was driven by the cauliflower mosaic virus 35S promoter (CaMV35S).
Figure 1.
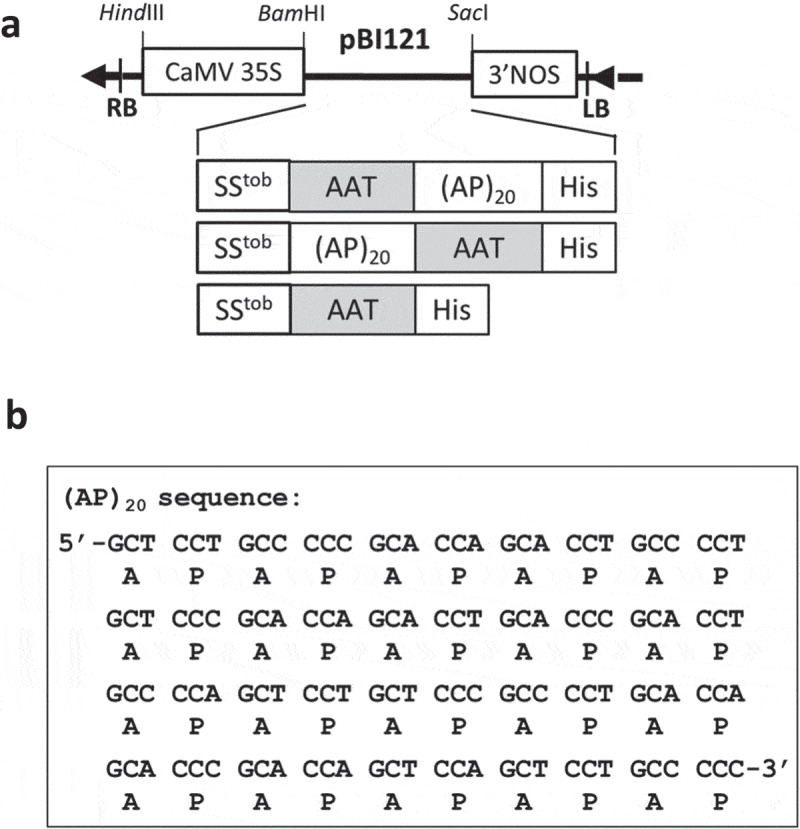
Schematic of the gene constructions in pBI121 expression vector (a) and the cDNA sequence of the designed synthetic (AP)20 gene (b). When the gene constructs encoding the (AP)20-tagged AAT are expressed in BY-2 cells, the clustered non-contiguous Pro (P) residues in the (AP)20 module are expected to hydroxylate to be Hyp, and subsequently O-glycosylated with arabinogalactan polysaccharides [22,30]. (AP)20: twenty tandem repeats of ‘Ala-Pro’ motif; SStob: tobacco extensin signal sequence; CaMV35S: 35S cauliflower mosaic virus promoter; 3ʹNOS: nopaline synthase terminator; His: 6 × His tag.
For verification of genome integration of the SStob-AAT, SStob-AAT-(AP)20 and SStob-(AP)20-AAT constructs in transgenic BY-2 cells, the genomic DNA was extracted from different transgenic cell lines and the target genes were amplified by PCR using the primer pair of SS-F and SS-R (Supplementary Table 1). The PCR amplicons were then separated by 1% (w/v) agarose gel electrophoresis.
Culture of BY-2 cells and determination of cell biomass
Transgenic BY-2 cells expressing three types of AAT proteins were grown in Schenk & Hildebrandt (SH) medium [32] supplemented with 0.4 mg/L 2,4-dichlorophenoxyacetic acid (2,4-D), 0.1 mg/L kinetin and 34 g/L sucrose. Cell suspension cultures were conducted in 250 ml flasks containing 75 mL medium, which were rotated at 90 rpm on a gyratory shaker at 25°C. Subcultures were carried out every week. For kinetic studies of the cell growth and protein secretion, suspension cultured cells were collected at an interval of 2 days for determination of cell biomass and secreted AAT products. Cultured cells were harvested by vacuum filtration and washed three times with distilled water before the determination of fresh weight (FW). The cells were then dried in an oven at 70°C for 48 hr to determine dry weight (DW). Cell biomass and secreted protein yields were analyzed for each sample, as described below.
Purification of protein from culture media
The media harvested after 10 days of BY-2 cell culture were pre-separated with 35% to 60% (w/v) ammonium sulfate precipitation. The fraction precipitated with 60% (w/v) ammonium sulfate was further purified with nickel affinity chromatography with the Ni-NTA Spin Columns (Qiagen, MA) following the manufacturer’s procedures.
Immunoblotting analysis
For Western blotting assay, the secreted AAT products accumulated in culture media were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as described earlier [24,33]. The protein bands were then electrotransferred onto a 0.2 μm nitrocellulose membrane (Thermo Scientific Pierce, Rockford, lL), and detected using the rabbit anti-AAT polyclonal antibody (Assaypro, St Charles, MO), and the goat anti-rabbit IgG (H + L) secondary antibody, peroxidase conjugated (Jackson Immuno Research labs, West Grove, PA). Protein blots were detected using the SuperSignal® West Pico Chemiluminescent Substrate (Thermo Scientific Pierce, Rockford, lL) in accordance with the manufacturers’ procedures. For dot blotting, 1.0–1.5 μl of medium sample was heated at 90°C for 10 min before being blotted onto a 0.2 μm nitrocellulose membrane, and the AAT products were detected using the same approach as that described for the Western blotting assay.
Monosaccharide composition assay
The monosaccharide compositions of purified AAT-(AP)20 fusion protein was determined at the Complex Carbohydrate Research Center, The University of Georgia (Athens, GA), as described earlier [34].
N-terminal peptide sequencing
Purified transgene products were separated on a 10% Tris-HCl gel and stained with Coomassie blue R-250. The target bands were cut from the gel for peptide sequencing by Edman degradation at the Protein Facility of the Iowa State University (Ames, IA).
Precipitation with (β-D-galactosyl)3-yariv reagent
(β-D-galactosyl)3-Yariv (Biosupplies Australia Pty Ltd, Australia) was dissolved in 2% (w/v) NaCl at the concentration of 1.0 mg/ml. (AP)20-tagged protein (50 and 100 µg) was dissolved in 300 µl of distilled H2O. An equal volume of protein sample and Yariv reagent was mixed and incubated at room temperature for 1 hr before the precipitates were pelleted in a microcentrifuge (Eppendorf MiniSpinTM) at full speed (13,200 rpm). The resulted pellets were washed with distilled H2O, dissolved in 0.1 M NaOH, and then the absorbance was measured at 420 nm. Tobacco BY-2 cell-secreted (SP)32-EGFP [33] was used as a positive control.
Quantification of recombinant AAT products
Secreted AAT and AAT equivalent of (AP)20-AAT or AAT-(AP)20 in BY-2 cell culture media was assayed using a sandwich Human AAT ELISA kit (Assaypro, St Charles, MO). For quantification of intracellular AAT products, the proteins were first extracted from harvested cells with the SDS extraction buffer (150 mM Tris-HCl, pH 6.8, 30% glycerol, 6% SDS, 5 mM EDTA) as described by Zhang et al. [33], and then assayed by the sandwich ELISA.
Biological activity assay of recombinant AAT products
Biological activity of BY-2 cell-secreted AAT products was assayed in vitro for inhibiting elastase, as described by Huang et al. [15] and McDonald et al. [17]. Briefly, 50 µl of human AAT standard (Assaypro, St Charles, MO) or recombinant AAT samples (crude media or purified protein) was added to 100 µL of assay buffer (0.15 M NaCl, 0.02 M Tris-HCl and 0.01% Tween 80, pH 8.1) in a 96-well microtiter plate. Fifty microliters of porcine pancreatic elastase (PPE, 2.8 µg/mL, Sigma, St. Louis, MO) was then added to individual wells, followed by incubation at 37°C for 15 min to initiate the inhibition of elastase. Finally, 50 µl of substrate consisting of 2 mM of N-succinyl-Ala-Ala-Ala-p-nitroanilide (Sigma, St. Louis, MO) was added to determine the active residual elastase that could cleave the substrate to generate chromogenic p-nitroanilide, which was detected at OD = 405 nm. Human AAT standard at concentrations ranging from 0 to 14 µg/mL was used to generate the calibration curve correlating the active AAT concentration to the residual PPE activity. The medium collected from wild type BY-2 cell culture was used as a negative control.
Statistical analysis
Sample assays were carried out with three replicates, and data were presented as the mean with standard deviation (SD). One-way analysis of variance (ANOVA) followed by a Tukey post hoc range test was used to determine differences among treatments with p < 0.05 being significant and P < 0.01 extremely significant.
Results and discussions
Plant expression vectors were constructed
We constructed the gene constructs encoding three AAT products: one untagged control and two with a (AP)20 tag at either N-terminus or C-terminus, all being targeted for extracellular secretion by a signal peptide of tobacco extensins SStob [30] (Figure 1). Based on the Hyp-O-glycosylation ‘code’ elucidated earlier and our previous results [21,22,27,35], we predicted that all the Pro residues in the (AP)20 tag, upon engineered into plant cells, would undergo hydroxylation and subsequent O-glycosylation with arabinogalactan polysaccharides. This would greatly increase the molecular size of the recombinant AAT from ~52 kDa to more than 100 kDa.
Engineered (AP)20 module enhanced extracellular secretion of recombinant AAT
With the Agrobacterium-mediated transformation, many stably transformed BY-2 cell colonies (calli) were obtained (Figure 2(a)). The integration of the corresponding gene construct into the genome of BY-2 calli was verified by PCR (Figure 2(b)), which showed a ~ 1.5 kb amplicon for SStob-AAT-(AP)20 and SStob-(AP)20-AAT and a ~ 1.3 kb amplicon for SStob-AAT. At least 30 cell lines for each gene construct were isolated and grown in liquid SH medium to screen for high-secretion lines by dot blotting assay (data not shown). Five lines for each gene construct were finally selected and subcultured for quantification of the protein yields by ELISA.
Figure 2.
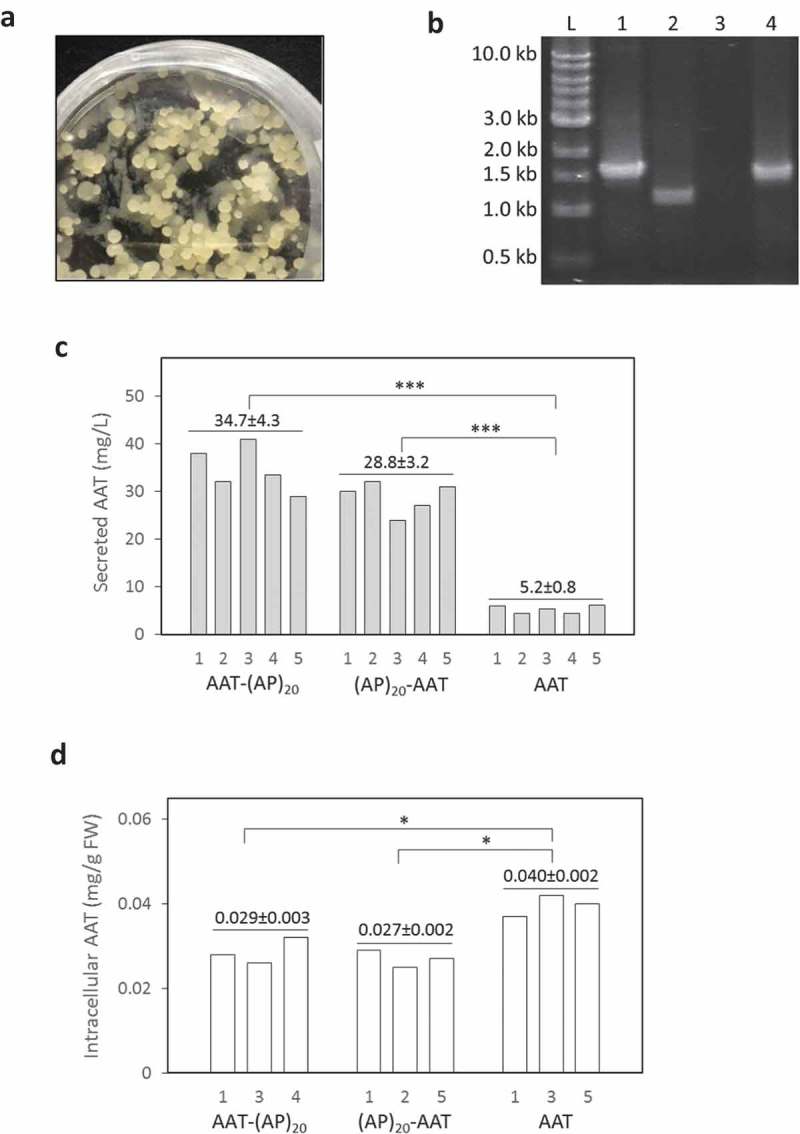
Expression of recombinant AAT products in tobacco BY-2 cell culture. (a) Transgenic BY-2 cell colonies appeared in a selection medium (MS medium with 100 mg/L kanamycin) 3 weeks after co-culture of wild-type BY-2 cell with Agrobacterium; (b) PCR detection of the target gene integration into the genome of transgenic BY-2 cell lines. L: DNA ladder, Lane 1, 2, 3, 4: PCR amplicons amplified from the genomic DNA extracted from the BY-2 cells expressing AAT-(AP)20, AAT control, wild type line, and (AP)20-AAT, respectively; (c) Secreted yields of AAT-(AP)20, (AP)20-AAT and AAT control in the transgenic BY-2 cell cultures. Five top-expression BY-2 colonies for each gene construct (# 1 to 5 on X-axis) selected by dot blotting were grown in SH medium for 10 days before the assay; (d) Intracellular AAT contents of the BY-2 cells. Three top-secretion BY-2 colonies for each construct identified in Panel (c) were assayed. Statistical significance is indicated by *(p < 0.05) and ***(p < 0.01).
The engineered (AP)20 tag, either attached at the N-terminus or C-terminus of AAT, substantially enhanced the secreted yields of the recombinant protein (Figure 2(c)). The secreted AAT yields (AAT equivalent detected by anti-AAT ELISA) after 12 days of culture were determined to be 28.8 ± 3.2 mg/L for (AP)20-AAT and 34.7 ± 4.3 mg/L for AAT-(AP)20, which represented 5.5 to 6.7-fold greater than the expression of AAT control (5.2 ± 0.8 mg/L). This indicated that the ‘Ala-Pro’ motif-based HypGP tag design, as the (SP)n tag (n = 5, 10, 20, 32) used previously [20–22,24], could function as a molecular carrier in facilitating extracellular secretion of the fused AAT protein. Time course of AAT products secretion was further investigated. As shown in Figure 3(a), dramatic accumulation of the (AP)20-AAT or AAT-(AP)20 products started after 4 days of culture until day 12, which correlated with the rapid increase in cell biomass (Figure 3(b)). The growth curves of the three genotypes of BY-2 cells were similar, all showing a growth cycle of 12 to 14 days with an exponential growth phase occurring between day 4 and 10 (Figure 3(b)). The cell biomass harvested at the end of the cultures (7.2 to 7.5 gDW/L) were comparable among the three transgenic cell lines.
Figure 3.
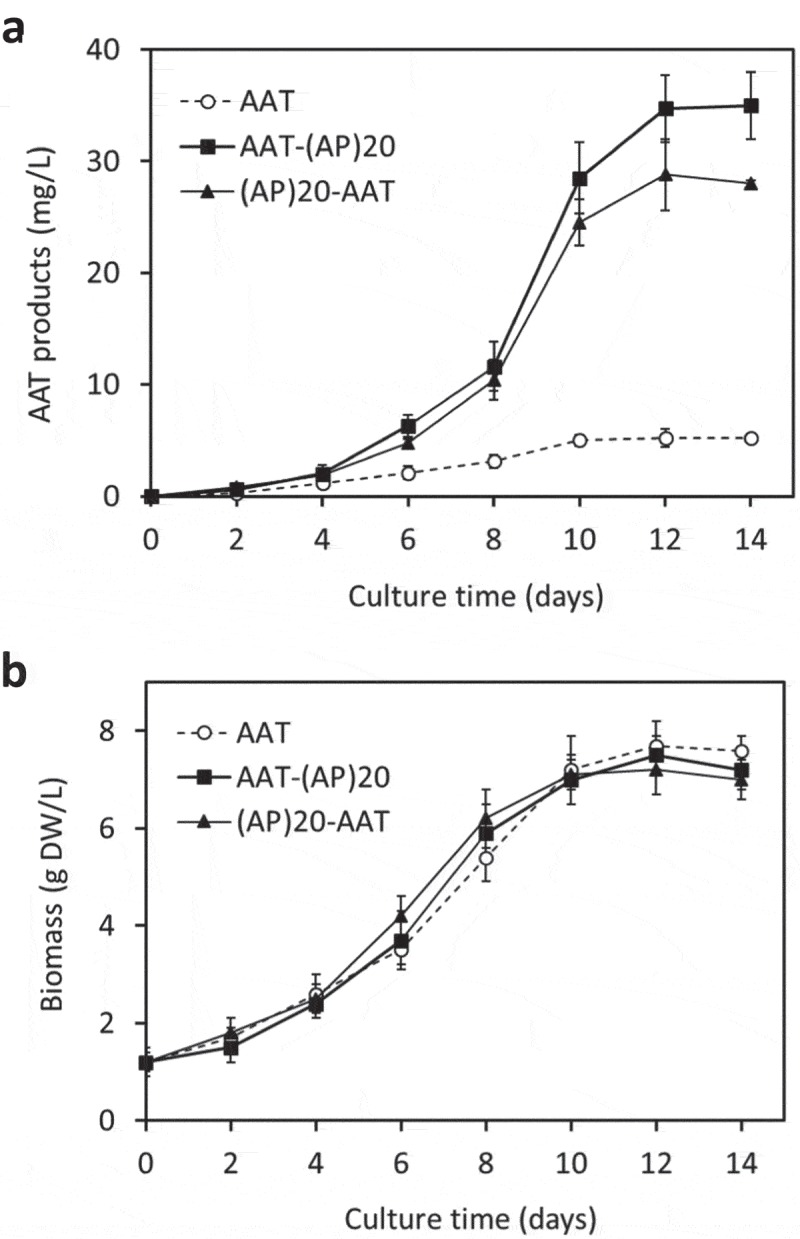
Time course of cell growth and AAT product secretion of the transgenic BY-2 cell cultures. (a) Secreted AAT products in the culture media; (b) Cell biomass accumulation. The error bars represent the standard deviation of three parallel cultures.
When the intracellular accumulation of AAT products was examined, more AAT control (0.040 mg/gFW) than the (AP)20-tagged AAT (0.027 to 0.029 mg/gFW) was observed (Figure 2(d)), which was consistent with the enhanced secretion of the (AP)20-tagged AAT as shown in Figure 2(c). In terms of spacious distribution of the synthesized AAT products in BY-2 cell cultures, the secreted AAT with a (AP)20 tag accounted for 87.1% to 89.5% of the total AAT products produced (intracellular and extracellular combined). In contrast, 47.0% of AAT control was extracellularly secreted. Compared with our previous studies in which less than 5% of recombinant proteins (including EGFP, hGH, and IFNα2 that is absent of glycosylation) was usually secreted from cultured BY-2 cell [20,21,24], AAT is naturally a secreted protein in plant cell culture, where up to 247 mg/L AAT was secreted in rice cell culture [17]. This is largely attributed to AAT being a glycoprotein that carries three N-linked glycosylation sites (46Asn, 83Asn, and 247Asn) [36]. All these sites were found to be fully occupied when AAT was expressed in N. benthamiana, which prompted extracellular secretion of the protein [7]. In this study about half of the synthesized AAT control (47%) was secreted into BY-2 cell culture media. However, the protein secretion was substantially increased to 89.5% when engineered with the (AP)20 tag, leading to a 6.7-fold increase in the final secreted protein yields. This demonstrated that the HypGP engineering, specifically engineering a (AP)20 module in this study, was applicable for improving the secreted yield of a relatively large glycoprotein (52 kDa) in plant cell culture.
As far as we know this is the first report on AAT protein expression in tobacco BY-2 cell. However, compared with the AAT expression in rice cell with an inducible RAmy3D promoter, the secreted protein yield obtained in this study was still low. However, BY-2 cell has major advantages over rice cell in terms of growth rate, characteristics, and stability. Particularly, the growth rate of BY-2 cell with a doubling time as short as 11 hr [18] is much higher than rice cell whose growth doubling time was reported to be 6 to 7 days [14,16]. In addition, exchanging medium to create sucrose-starvation environments in rice cell culture to induce the gene expression (under the RAmy3D promoter) imposes a major technical difficulty on a large scale and advanced bioreactor culture strategies that could potentially enhance protein productivity, e.g., perfusion cultures, cannot be readily implemented [11]. The major bottleneck preventing commercial applications of the BY-2 cell culture system lies in low protein productivity, particularly low secreted protein yields. This, to a large extent, could be overcome by the HypGP engineering technology, with which secreted EGFP yields up to 168 mg/L was previously achieved [33]. Boosting protein secretion in plant cell culture by the HypGP engineering is of practical significance to the biotechnology industry, as protein products can be easily isolated from simple plant cell culture media at a substantially reduced cost compared with the protein expression in whole plants.
Secreted AAT products were subjected to proteolytic cleavage
The secreted AAT and (AP)20-tagged AAT products were analyzed by Western blotting with a polyclonal anti-AAT antibody (Figure 4(a)). The AAT with a C-terminal (AP)20 tag (AAT-(AP)20), migrated as two distinct bands at molecular size of ~110 kDa (upper band) and 40–46 kDa (lower band), respectively. According to calculation based on the polypeptide sequence, the engineered Hyp-O-glycosylated (AP)20 module would increase the AAT size from 52 kDa to ~106 kDa, which resulted from the (AP)20 peptide backbone (3.7 kDa) and the attached 20 Hyp-glycan (~2.5 kDa per glycan as disclosed earlier [26,27]). This indicated the upper band detected in Figure 4(a) corresponded to the AAT-(AP)20 fusion glycoprotein. Then, the lower bands (40–46 kDa) must be the truncated AAT domains absent of the (AP)20 tag. Right above the lower band occurred a faint band (~52 kDa) that presumably corresponded to the full-length AAT domain as it migrated at the same position as the AAT standard. With the Ni-NTA affinity chromatography, only the intact AAT-(AP)20 fusion protein (with a C-terminal 6× His tag) could be purified from culture media (Figure 5(a)). No any lower-band products were recovered by the Ni-NTA columns due to the 6× His tag being cleaved. N-terminal peptide sequencing of the purified protein showed the sequence of ‘EDPQGDAAQKTD…’ (Figure 4(b)), same as that of the native AAT (Supplementary Figure 1), which confirmed that this product (upper band) was the intact AAT-(AP)20 fusion protein. It accounted for ~38.5% (w/w) of the total secreted AAT products as estimated by the Western blot densitometry using the Quant-1 software (Bio-Rad, CA).
Figure 4.
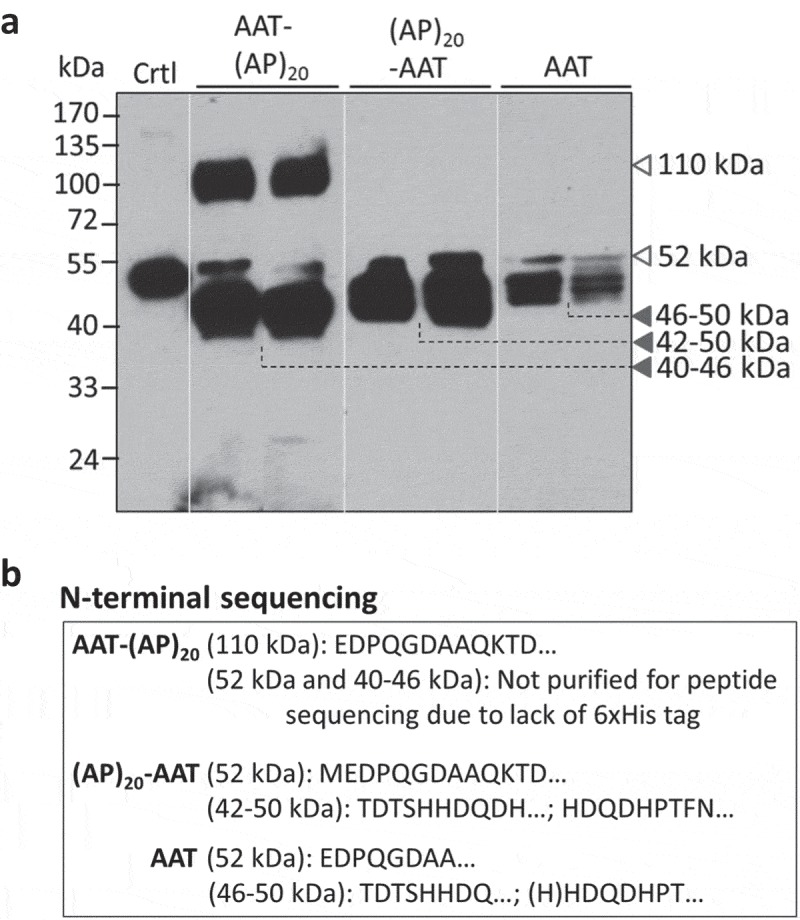
Detection of (AP)20-AAT, AAT-(AP)20 and AAT products accumulated in BY-2 cell culture medium. (a) Western blotting detection of the transgenic products. Two transgenic BY-2 cell lines for each gene construct were grown in liquid medium for 10 days before the assay with an anti-AAT polyclonal antibody. Crtl: AAT standard (100 ng). Sixteen microliter of culture media was loaded into each well. (b) N-terminal peptide sequencing of transgenic products. Those with a C-terminal 6 × His tag were able to be purified with the Ni-NTA Spin Columns for sequencing.
Figure 5.
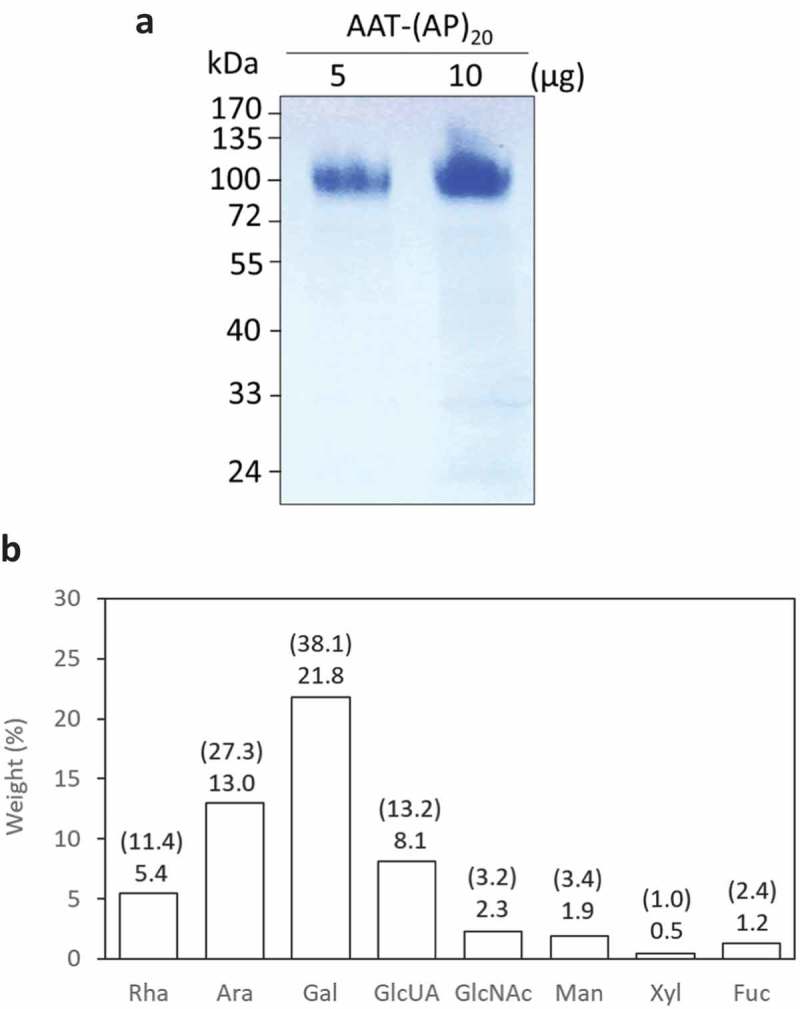
Biochemical characterization of recombinant AAT-(AP)20 fusion proteins. (a) SDS-PAGE separation of AAT-(AP)20 purified from the culture media. (b) Monosaccharide compositions of the AAT-(AP)20 fusion proteins. The weight percentage (wt %) of each sugar residue accounts for the total fusion protein is presented and the value indicated on the top of each bar. The number in brackets indicates the molar ratio of each sugar residue in the glycans attached to the AAT-(AP)20 polypeptide. Rha, Ara, Gal, and GlcUA are typical sugar residues of Hyp-glycans; GlcNAc, Man, Xyl and Fuc are typical sugar residues of N-glycans.
When the AAT with a N-terminal (AP)20 tag was expressed in BY-2 cells, hardly any intact (AP)20-AAT fusion protein at ~110 kDa was detected by the anti-AAT Western blot (Figure 4(a)). The major products found were the truncated AAT domains (42–50 kDa) lacking the (AP)20 tag, whose molecular sizes were slightly larger than the AAT domains cleaved from the AAT-(AP)20 fusion. As in the AAT-(AP)20 expression, a faint ~52-kDa band was also observed. N-terminal peptide sequencing detected the sequence of ‘MEDPQGDAAQKTD…’ for the ~52 kDa product and the mixture of ‘TDTSHHDQDH…’ and ‘HDQDHPTFN…’ for the 42–50 kDa products (Figure 4(b)), indicating the (AP)20 tag was cleaved at multi-sites from the N-terminus of the fusion protein. Obviously, the ~52 kDa product corresponded to a full-length AAT domain. The first and extra amino acid ‘M’ detected in this domain was introduced while the AAT-(AP)20 gene construct was cloned.
For the expression of AAT control without a (AP)20 tag, the products recovered in media existed as multiple truncated polypeptides with molecular sizes ranging from 46 to 50 kDa (Figure 4(a)), similar to those reported earlier in rice cell cultures [14,15]. Again, a faint ~52-kDa band was observed, which could be purified by the Ni-NTA columns and was identified as the full-length AAT based on the peptide sequencing assay (Figure 4(b)). Some of the 46–50 kDa products could also be purified and their N-terminal peptide sequences read almost the same as those of the (AP)20-AAT products (Figure 4(b)).
N- and C-terminal truncations of recombinant AAT have been previously reported in plant-produced AAT. In transient expression of AAT in N. Benthamiana, both full-length AAT and truncated variants at both termini were found. However, only the truncated version of AAT was detected in intercellular fluids [7]. In inducible expression in rice cells, considerable amounts of recombinant AAT were found to be truncated. In fact, the truncated version of AAT dominated the population of the AAT products secreted into culture media [14–17]. In this study, all the three types of recombinant AAT products secreted from BY-2 cells were subjected to proteolytic degradation (Figure 4). Although the truncation was found to happen at both terminal ends of the expressed AAT, however, the N-terminus was more prone to proteolytic cleavage than the C-terminus, because no intact (AP)20-AAT fusion was detected in culture media while ~38.5% secreted AAT-(AP)20 fusion protein remaining intact. In addition, the observed smaller sizes of the truncated AAT domains cleaved from the AAT-(AP)20 fusion (C-terminal cleavage) than from the (AP)20-AAT fusion (N-terminal cleavage) conformed to the frequent cleavage sites of AAT reported earlier: up to 15 amino acids at the N-terminus and up to 41 amino acids at the C- terminus [7].
Engineered (AP)20 was yp-o-glycosylated with arabinogalactan polysaccharides
This was the first time that ‘Ala-Pro’ motif, rather than ‘Ser-Pro’ motif, was used as a HypGP design to facilitate the secretion of fused protein from cultured plant cells. Our previous studies indicated that extensive Hyp-O-glycosylation of the designer HypGP modules was essential in facilitating secretion of the fused protein [21,22,24]. The Hyp-O-glycosylation of the engineered (AP)20 was then investigated. In this study, only the AAT-(AP)20 fusion protein could be purified from culture media at sufficient amount for monosaccharide composition assay (Figure 5(a)). As shown in Figure 5(b), a total of eight sugar residues were detected, which included rhamnose (Rha), arabinose (Ara), galactose (Gal) and glucuronic acid (GlcUA) residues that constitute the Hyp-glycans, and N-acetylglucosamine (GlcNAc), mannose (Man), xylose (Xyl) and fucose (Fuc) residues that are the typical sugar residues of N-glycans. The detected monosaccharide profile demonstrated the AAT-(AP)20 fusion protein was both Hyp-O-glycosylated and N-glycosylated. The total sugars accounted for 54.2% (w/v) of the fusion protein, of which ~47.1% was contributed by the Hyp-glycans and the rest ~7.1% contributed by the N-glycans, as estimated based on the Hyp-glycan structure elucidated earlier [21,26].
The purified AAT-(AP)20 was also tested for its ability to precipitate (β-D-galactosyl)3-Yariv reagent, which specifically binds AGPs [37]. Like the (SP)32-EGFP containing a synthetic (SP)32 module, the AAT-(AP)20 could react with the Yariv reagent, but not for the AAT control though it bears three N-glycans (Table 1). This confirmed that the glycans attached to the (AP)20 module were Type II arabinogalactans typical of classical plant AGPs [26,27]. Compared with the (SP)32-EGFP fusion, the AAT-(AP)20 generated less amounts of Yariv precipitates, which resulted from the smaller percentage of AGP module present in the AAT-(AP)20 ((AP)20 accounts for ~51%) than in the (SP)32-EGFP ((SP)32 accounts for ~77%).
Table 1.
Yariv reagent co-precipitation of the AAT-(AP)20 fusion protein compared with the (SP)32-EGFP. The data presented represent the mean of three parallel samples ± standard deviation. (SP)32-EGFP isolated from BY-2 cell culture medium [24] and AAT standard without the (AP)20 module served as positive and negative control, respectively. Twenty-five µg of AAT is roughly equal to the amount of AAT in the AAT-(AP)20 fusion glycoprotein.
| Absorbance (420 nm) |
|||
|---|---|---|---|
| Sample weight (µg) | AAT-(AP)20 | (SP)32-EGFP | AAT (25 µg) |
| 50 | 0.32 ± 0.01 | 0.92 ± 0.04 | 0.01 |
| 100 | 0.72 ± 0.03 | 1.68 ± 0.06 | Not assayed |
BY-2 cell secreted AAT products exhibited biological activity
Biological activity of the secreted AAT products was determined by their ability to inhibit the activity of PPE, a natural target protease. As shown in Figure 6, the active AAT accounted for 38%, 55% and 52% of the total secreted AAT-(AP)20, (AP)20-AAT and AAT products, respectively. This was similar to the expression of AAT in rice cell cultures, where substantial amounts of secreted recombinant AAT were truncated and lost activity [7,14,15]. For example, [14], reported that only 10% to 20% of the recombinant AAT produced by rice cell culture was functionally active.
Figure 6.
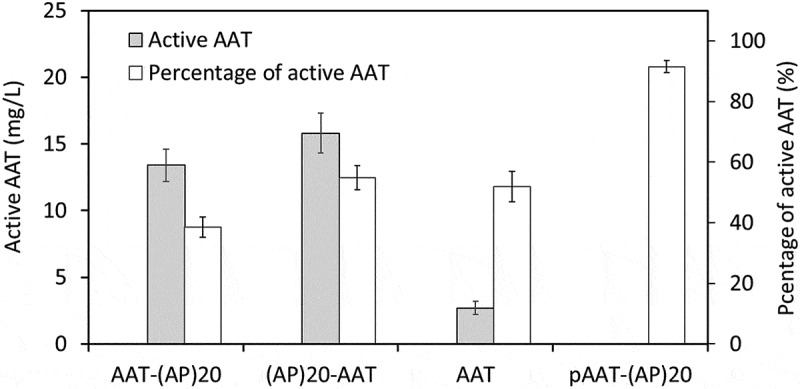
Biological activities of the recombinant AAT products secreted into BY-2 cell culture media. The medium samples harvested after 10-day of BY-2 cell culture as well as the purified AAT-(AP)20 fusion protein (pAAT-(AP)20) were assayed. The percentage of the active AAT accounting for the total AAT products in the samples was then calculated as: active AAT content/total AAT concentration×100%. The error bars represent the standard deviation of three top-expression cell lines for each gene construct (for the medium samples) or three parallel samples (for the pAAT-(AP)20 protein).
Of the two types of (AP)20-tagged AAT expressed in BY-2 cell, only the intact AAT-(AP)20 fusion protein could be recovered from culture media, and the purified fusion protein exhibited protease inhibitory activity up to 92.5% of the AAT standard. This indicated the engineered (AP)20 tag, though heavily O-glycosylated with arabinogalactan polysaccharides, did not adversely affect the biological activity of the fused AAT. This was consistent with our previous studies where the engineered (SP)n (n = 5, 10, 20) glycomodule showed limited effects on the bioactivity of two fused therapeutic proteins: interferon α2 and human growth hormone [20,21].
AAT is known to inhibit serine proteases. It contains an exposed and mobile reactive center loop (RCL) at C-terminus with a methionine (358M) residue acting as bait for target proteinases [38]. Since the active site of the AAT is located near the C-terminal end, proteolytic cleavage at C-terminal of AAT, particularly occurring within RCL, destroys the RCL structure and thereby renders AAT inactive [7]. For the recombinant AAT secreted from rice cell cultures, the AAT having molecular sizes around 48–50 kDa was found to remain biologically active, but those with molecular size around 42–44 kDa were inactive [15]. In this study, most of the BY-2 cell secreted AAT products were truncated at either terminal ends. Not only the (AP)20 tag was cleaved from the fusion proteins, but also the cleaved AAT domains were truncated. It seemed that the AAT domains cleaved from the AAT-(AP)20 fusion lost more amino acids, most possibly at the C-terminal end, than those from the (AP)20-AAT fusion or the AAT control, because the former migrated to a lower position (40–46 kDa) than the latter (42–50 kDa) on the SDS-PAGE (Figure 4(a)). This also implied that the C-terminal (AP)20 tag might facilitate the proteolytic degradation at sites preceding RCL. Due to loss of the active site (RCL) at the C-terminal end, most of the AAT domains cleaved from the AAT-(AP)20 became inactive. This was reflected by the finding that less active AAT populations in the AAT-(AP)20 cell culture media (38%) than in the (AP)20-AAT or AAT media (52–55%) (Figure 6). In fact, the active AAT population in the AAT-(AP)20 media was largely contributed by the intact AAT-(AP)20 fusion protein that retained a high biological activity (92.5%). This also indicated that many truncated AAT domains from the (AP)20-AAT or AAT still kept the RCL site at their C-terminal ends and thus remained biologically active.
Conclusions
The present study is the first report on the expression of human AAT in tobacco BY-2 cells. Engineering a designer HypGP tag composed of the (AP)20 motif at either N- or C-terminal end of AAT substantially increased the secreted protein yields from 5.2 mg/L up to 34.7 mg/L (a 6.7-fold increase). The secreted recombinant AAT products, including the intact AAT-(AP)20 fusion protein and some of the truncated AAT domains, exhibited desired protease inhibitory activity. Our results demonstrated that the HypGP engineering technology could be applied to the expression of a relatively large protein-AAT in BY-2 cell to substantially increase the secreted yields.
Highlights
Synthetic (AP)20 module is extensively Hyp-O-glycosylated in BY-2 cell
(AP)20 module facilitates extracellular secretion of fused α1-antitrypsin
BY-2 secreted α1-antitrypsin are subjected to proteolytic processing
(AP)20–tagged α1-antitrypsin exhibits biological activity in inhibiting elastase
Funding Statement
This work was supported by the National Science Fundation Directorate for Engineering [1605564]; National Institutes of Health [P20GM103429].
Acknowledgements
This work was supported by the National Science Foundation under Grant No. 1605564, the Arkansas IDeA Network of Biomedical Research Excellence-Research Development Grant under Grant No. P20GM103429, and the Arkansas Biosciences Institute, the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Materials
Supplemental data for this article can be accessed https://www.tandfonline.com/doi/full/10.1080/21655979.2019.1604037.
References
- [1].Blank CA, Brantly M.. Clinical features and molecular characteristics of alpha 1-antitrypsin deficiency. Ann Allergy. 1994;72(2): 105–120. [PubMed] [Google Scholar]
- [2].Brode SK, Ling SC, Chapman KR. Alpha-1 antitrypsin deficiency: a commonly overlooked cause of lung disease. CMAJ. 2012;184(12):1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Alkins SA, O‘Malley P. Should health-care systems pay for replacement therapy in patients with alpha(1)-antitrypsin deficiency? A critical review and cost-effectiveness analysis. Chest. 2000;117(3):875–880. [DOI] [PubMed] [Google Scholar]
- [4].Krishnan B, Hedstrom L, Hebert DN, et al. Expression and purification of active recombinant human alpha-1 antitrypsin (aat) from Escherichia coli. Methods Mol Biol. 2017;1639:195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Khatami M, Hosseini SN, Hasannia S. Co-expression of alpha-1 antitrypsin with cytoplasmic domain of v-SNARE in Pichia pastoris: preserving biological activity of alpha-1 antitrypsin. Biotechnol Appl Biochem. 2018;65(2):181-187. [DOI] [PubMed] [Google Scholar]
- [6].Paterson T, Innes J, Moore S. Approaches to maximizing stable expression of alpha 1-antitrypsin in transformed CHO cells. Appl Microbiol Biotechnol. 1994;40(5):691–698. [DOI] [PubMed] [Google Scholar]
- [7].Castilho A, Windwarder M, Gattinger P, et al. Proteolytic and N-glycan processing of human alpha1-antitrypsin expressed in Nicotiana benthamiana. Plant Physiol. 2014;166(4):1839–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Agarwal S, Singh R, Sanyal I, et al. Expression of modified gene encoding functional human alpha-1-antitrypsin protein in transgenic tomato plants. Transgenic Res. 2008;17(5):881–896. [DOI] [PubMed] [Google Scholar]
- [9].Nadai M, Bally J, Vitel M, et al. High-level expression of active human alpha1-antitrypsin in transgenic tobacco chloroplasts. Transgenic Res. 2009;18(2):173–183. [DOI] [PubMed] [Google Scholar]
- [10].Hellwig S, Drossard J, Twyman RM, et al. Plant cell cultures for the production of recombinant proteins. Nat Biotechnol. 2004;22(11):1415–1422. [DOI] [PubMed] [Google Scholar]
- [11].Huang TK, McDonald KA. Bioreactor engineering for recombinant protein production in plant cell suspension cultures. Biochem Eng J. 2009;45:168–184. [Google Scholar]
- [12].Santos RB, Abranches R, Fischer R, et al. Putting the spotlight back on plant suspension cultures. Front Plant Sci. 2016;7:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xu J, Dolan MC, Medrano G, et al. Green factory: plants as bioproduction platforms for recombinant proteins. Biotechnol Adv. 2012;30(5):1171–1184. [DOI] [PubMed] [Google Scholar]
- [14].Terashima M, Murai Y, Kawamura M, et al. Production of functional human alpha 1-antitrypsin by plant cell culture. Appl Microbiol Biotechnol. 1999;52(4):516–523. [DOI] [PubMed] [Google Scholar]
- [15].Huang J, Sutliff TD, Wu L, et al. Expression and purification of functional human alpha-1-antitrypsin from cultured plant cells. Biotechnol Prog. 2001;17(1):126-133. [DOI] [PubMed] [Google Scholar]
- [16].Trexler MM, McDonald KA, Jackman AP. Bioreactor production of human alpha(1)-antitrypsin using metabolically regulated plant cell cultures. Biotechnol Prog. 2002;18(3):501–508. [DOI] [PubMed] [Google Scholar]
- [17].McDonald KA, Hong LM, Trombly DM, et al. Production of human alpha-1-antitrypsin from transgenic rice cell culture in a membrane bioreactor. Biotechnol Prog. 2005;21(3):728-734. [DOI] [PubMed] [Google Scholar]
- [18].Xu J, Ge X, Dolan MC. Towards high-yield production of pharmaceutical proteins with plant cell suspension cultures. Biotechnol Adv. 2011;29(3):278–299. [DOI] [PubMed] [Google Scholar]
- [19].Kieliszewski MJ, Xu J, Meyer G (2015) Nucleic acid for plant expression of a fusion protein comprising hydroxyproline O-glycosylation glycomodule. US Patent No. 9006410
- [20].Xu J, Okada S, Tan L, et al. Human growth hormone expressed in tobacco cells as an arabinogalactan-protein fusion glycoprotein has a prolonged serum life. Transgenic Res. 2010;19(5):849–867. [DOI] [PubMed] [Google Scholar]
- [21].Xu J, Tan L, Goodrum KJ, et al. High-yields and extended serum half-life of human interferon alpha2b expressed in tobacco cells as arabinogalactan-protein fusions. Biotechnol Bioeng. 2007;97(5):997–1008. [DOI] [PubMed] [Google Scholar]
- [22].Xu J, Tan L, Lamport DTA, et al. The O-Hyp glycosylation code in tobacco and Arabidopsis and a proposed role of Hyp-glycans in secretion. Phytochem. 2008;69(8):1631–1640. [DOI] [PubMed] [Google Scholar]
- [23].Kieliszewski MJ, Shpak E. Synthetic genes for the elucidation of glycosylation codes for arabinogalactan-proteins and other hydroxyproline-rich glycoproteins. Cell Mol Life Sci. 2001;58(10):1386–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang N, Gonzalez M, Savary B, et al. High-yield secretion of recombinant proteins expressed in tobacco cell culture with a designer glycopeptide tag: process development. Biotechnol J. 2016b;11(4):497–506. [DOI] [PubMed] [Google Scholar]
- [25].Ramos-Martinez EM, Fimognari L, Sakuragi Y. High-yield secretion of recombinant proteins from the microalga Chlamydomonas reinhardtii. Plant Biotechnol J. 2017;15(9):1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tan L, Varnai P, Lamport DTA, et al. Plant O-hydroxyproline arabinogalactans are composed of repeating trigalactosyl subunits with short bifurcated side chains. J Biol Chem. 2010;285(32):24575–24583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tan L, Leykam JF, Kieliszewski MJ. Glycosylation motifs that direct arabinogalactan addition to arabinogalactan-proteins. Plant Physiol. 2003;132(3):1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kieliszewski MJ. The latest hype on Hyp-O-glycosylation codes. Phytochem. 2001;57(3):319–323. [DOI] [PubMed] [Google Scholar]
- [29].Showalter AM. Structure and function of plant cell wall proteins. Plant Cell. 1993;5(1):9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shpak E, Leykam JF, Kieliszewski MJ. Synthetic genes for glycoprotein design and the elucidation of hydroxyproline-O-glycosylation codes. Proc Natl Acad Sci USA. 1999;96(26):14736–14741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].An G. High efficiency transformation of cultured tobacco cells. Plant Physiol. 1985;79(2):568–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schenk RU, Hildebrandt AC. Medium and techniques for induction and growth of monocotyledonous and dicotyledonous plant cell cultures. Can J Bot. 1972;50:199–204. [Google Scholar]
- [33].Zhang N, Dolan M, Wu D, et al. Dramatic secretion of recombinant protein expressed in tobacco cells with a designer glycopeptide tag is highly impacted by medium composition. Plant Cell Rep. 2016a;35(12):2513–2522. [DOI] [PubMed] [Google Scholar]
- [34].Xu J, Kieliszewski M. Enhanced accumulation of secreted human growth hormone by transgenic tobacco cells correlates with the introduction of an N-glycosylation site. J Biotechnol. 2011;154(1):54–59. [DOI] [PubMed] [Google Scholar]
- [35].Shpak E, Barbar E, Leykam JF, et al. Contiguous hydroxyproline residues direct hydroxyproline arabinosylation in Nicotiana tabacum. J Biol Chem. 2001;276(14):11272–11278. [DOI] [PubMed] [Google Scholar]
- [36].Kolarich D, Weber A, Turecek PL, et al. Comprehensive glyco-proteomic analysis of human alpha1-antitrypsin and its charge isoforms. Proteomics. 2006;6(11):3369–3380. [DOI] [PubMed] [Google Scholar]
- [37].Kitazawa K, Tryfona T, Yoshimi Y, et al. beta-galactosyl Yariv reagent binds to the beta-1,3-galactan of arabinogalactan proteins. Plant Physiol. 2013;161(3):1117–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Travis J, Salvesen GS. Human plasma proteinase inhibitors. Annu Rev Biochem. 1983;52:655–709. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.